
Abstract
The SAMPL challenges focus on testing and driving progress of computational methods to help guide pharmaceutical drug discovery. However, assessment of methods for predicting binding affinities is often hampered by computational challenges such as conformational sampling, protonation state uncertainties, variation in test sets selected, and even lack of high quality experimental data. SAMPL blind challenges have thus frequently included a component focusing on host–guest binding, which removes some of these challenges while still focusing on molecular recognition. Here, we report on the results of the SAMPL7 blind prediction challenge for host–guest affinity prediction. In this study, we focused on three different host–guest categories—a familiar deep cavity cavitand series which has been featured in several prior challenges (where we examine binding of a series of guests to two hosts), a new series of cyclodextrin derivatives which are monofunctionalized around the rim to add amino acid-like functionality (where we examine binding of two guests to a series of hosts), and binding of a series of guests to a new acyclic TrimerTrip host which is related to previous cucurbituril hosts. Many predictions used methods based on molecular simulations, and overall success was mixed, though several methods stood out. As in SAMPL6, we find that one strategy for achieving reasonable accuracy here was to make empirical corrections to binding predictions based on previous data for host categories which have been studied well before, though this can be of limited value when new systems are included. Additionally, we found that alchemical free energy methods using the AMOEBA polarizable force field had considerable success for the two host categories in which they participated. The new TrimerTrip system was also found to introduce some sampling problems, because multiple conformations may be relevant to binding and interconvert only slowly. Overall, results in this challenge tentatively suggest that further investigation of polarizable force fields for these challenges may be warranted.













Similar content being viewed by others
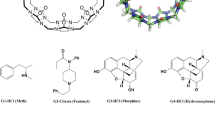
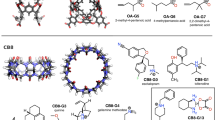
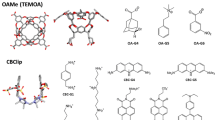
Abbreviations
- SAMPL:
-
Statistical Assessment of the Modeling of Proteins and Ligands
- AM1-BCC:
-
Austin model 1 bond charge correction
- RESP:
-
Restrained electrostatic potential
- REST:
-
Replica exchange with solute tempering
- FSDAM:
-
Fast switching double annihilation method
- B2PLYPD3:
-
Beck 2-parameter Lee–Yang–Parr D3 exchange-correlation functional [1]
- B3PW91:
-
Becke 3-parameter Perdew–Wang 91 exchange-correlation functional [2]
- GAFF:
-
Generalized AMBER force field
- CGenFF:
-
CHARMM generalized force field
- AMOEBA:
-
Atomic multipole optimized energetics for biomolecular simulations
- DDM:
-
Double decoupling method
- DFT:
-
Density functional theory
- QM/MM:
-
Mixed quantum mechanics and molecular mechanics
- MMPBSA:
-
Molecular mechanics Poisson–Boltzmann/solvent accessible surface area
- MMGBSA:
-
Molecular mechanics generalized born/solvent accessible surface area
- TIP3P:
-
Transferable interaction potential three-point
- TIP4PEw:
-
Transferable interaction potential four-point Ewald
- OPC3:
-
Optimal 3-point charge
- SEM:
-
Standard error of the mean
- RMSE:
-
Root mean squared error
- MAE:
-
Mean absolute error
- ME:
-
Mean signed error
- \(\tau \) :
-
Kendall’s rank correlation coefficient (Tau)
- \({R}^{2}\) :
-
Coefficient of determination (R-squared)
- QM:
-
Quantum Mechanics
- MM:
-
Molecular Mechanics
References
Goerigk L, Grimme S (2011) Efficient and accurate double-hybrid-meta-GGA density functionals–evaluation with the extended GMTKN30 database for general main group thermochemistry, kinetics, and noncovalent interactions. J Chem Theory Comput. https://doi.org/10.1021/ct100466k
Grimme S, Ehrlich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32(7):1456–1465. https://doi.org/10.1002/jcc.21759
Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL, Gilson MK (2017) Overview of the SAMPL5 Host–Guest Challenge: are we doing better? J Comput Aided Mol Des 31(1):1–19. https://doi.org/10.1007/s10822-016-9974-4
Rizzi A, Murkli S, McNeill JN, Yao W, Sullivan M, Gilson MK, Chiu MW, Isaacs L, Gibb BC, Mobley DL, Chodera JD (2018) Overview of the SAMPL6 host–guest binding affinity prediction challenge. J Comput Aided Mol Des 32(10):937–963. https://doi.org/10.1007/s10822-018-0170-6
Rizzi A, Jensen T, Slochower DR, Aldeghi M, Gapsys V, Ntekoumes D, Bosisio S, Papadourakis M, Henriksen NM, de Groot BL, Cournia Z, Dickson A, Michel J, Gilson MK, Shirts MR, Mobley DL, Chodera JD (2020) The SAMPL6 SAMPLing Challenge: assessing the reliability and efficiency of binding free energy calculations. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-020-00290-5
Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, Romero DL, Masse C, Knight JL, Steinbrecher T, Beuming T, Damm W, Harder E, Sherman W, Brewer M, Wester R et al (2015) Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc 137(7):2695–2703. https://doi.org/10.1021/ja512751q
Cournia Z, Allen B, Sherman W (2017) Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J Chem Inf Model 57(12):2911–2937. https://doi.org/10.1021/acs.jcim.7b00564
Rocklin GJ, Mobley DL, Dill KA, Hünenberger PH (2013) Calculating the binding free energies of charged species based on explicit-solvent simulations employing lattice-sum methods: an accurate correction scheme for electrostatic finite-size effects. J Chem Phys 139(18):184103. https://doi.org/10.1063/1.4826261
Mobley DL, Gilson MK (2017) Predicting binding free energies: frontiers and benchmarks. Annu Rev Biophys 46(1):531–558. https://doi.org/10.1146/annurev-biophys-070816-033654
Işık M, Bergazin TD, Fox T, Rizzi A, Chodera JD, Mobley DL (2020) Assessing the accuracy of octanol–water partition coefficient predictions in the SAMPL6 Part II Log P Challenge. J Comput Aided Mol Des 34(4):335–370. https://doi.org/10.1007/s10822-020-00295-0
Laury ML, Wang Z, Gordon AS, Ponder JW (2018) Absolute binding free energies for the SAMPL6 cucurbit[8]uril host-guest challenge via the AMOEBA polarizable force field. J Comput Aided Mol Des 32(10):1087–1095. https://doi.org/10.1007/s10822-018-0147-5
Gapsys V, de Groot BL (2017) Pmx webserver: a user friendly interface for alchemistry. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00498
Schindler C, Baumann H, Blum A, Böse D, Buchstaller HP, Burgdorf L, Cappel D, Chekler E, Czodrowski P, Dorsch D, Eguida M, Follows B, Fuchß T, Grädler U, Gunera J, Johnson T, Jorand Lebrun C, Karra S, Klein M, Kötzner L et al (2020) Large-scale assessment of binding free energy calculations in active drug discovery projects. ChemRxiv. https://doi.org/10.26434/chemrxiv.11364884.v1
Gapsys V, Pérez-Benito L, Aldeghi M, Seeliger D, van Vlijmen H, Tresadern G, de Groot BL (2020) Large scale relative protein ligand binding affinities using non-equilibrium alchemy. Chem Sci 11(4):1140–1152. https://doi.org/10.1039/C9SC03754C
Muddana HS, Varnado CD, Bielawski CW, Urbach AR, Isaacs L, Geballe MT, Gilson MK (2012) Blind prediction of host-guest binding affinities: a new SAMPL3 challenge. J Comput Aided Mol Des 26(5):475–487. https://doi.org/10.1007/s10822-012-9554-1
Skillman AG (2012) SAMPL3: blinded prediction of host–guest binding affinities, hydration free energies, and trypsin inhibitors. J Comput Aided Mol Des 26(5):473–474. https://doi.org/10.1007/s10822-012-9580-z
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) The SAMPL4 host–guest blind prediction challenge: an overview. J Comput Aided Mol Des 28(4):305–317. https://doi.org/10.1007/s10822-014-9735-1
Peat TS, Dolezal O, Newman J, Mobley DL, Deadman JJ (2014) Interrogating HIV integrase for compounds that bind—a SAMPL challenge. J Comput Aided Mol Des 28(4):347–362. https://doi.org/10.1007/s10822-014-9721-7
Gathiaka S, Liu S, Chiu M, Yang H, Stuckey JA, Kang YN, Delproposto J, Kubish G, Dunbar JB, Carlson HA, Burley SK, Walters WP, Amaro RE, Feher VA, Gilson MK (2016) D3R Grand Challenge 2015: evaluation of protein–ligand pose and affinity predictions. J Comput Aided Mol Des 30(9):651–668. https://doi.org/10.1007/s10822-016-9946-8
Gaieb Z, Liu S, Gathiaka S, Chiu M, Yang H, Shao C, Feher VA, Walters WP, Kuhn B, Rudolph MG, Burley SK, Gilson MK, Amaro RE (2018) D3R Grand Challenge 2: blind prediction of protein-ligand poses, affinity rankings, and relative binding free energies. J Comput Aided Mol Des 32(1):1–20. https://doi.org/10.1007/s10822-017-0088-4
Gaieb Z, Parks CD, Chiu M, Yang H, Shao C, Walters WP, Lambert MH, Nevins N, Bembenek SD, Ameriks MK, Mirzadegan T, Burley SK, Amaro RE, Gilson MK (2019) D3R Grand Challenge 3: blind prediction of protein-ligand poses and affinity rankings. J Comput Aided Mol Des 33(1):1–18. https://doi.org/10.1007/s10822-018-0180-4
Parks CD, Gaieb Z, Chiu M, Yang H, Shao C, Walters WP, Jansen JM, McGaughey G, Lewis RA, Bembenek SD, Ameriks MK, Mirzadegan T, Burley SK, Amaro RE, Gilson MK (2020) D3R Grand Challenge 4: blind prediction of protein-ligand poses, affinity rankings, and relative binding free energies. J Comput Aided Mol Des 34(2):99–119. https://doi.org/10.1007/s10822-020-00289-y
Sherborne B, Shanmugasundaram V, Cheng AC, Christ CD, DesJarlais RL, Duca JS, Lewis RA, Loughney DA, Manas ES, McGaughey GB, Peishoff CE, van Vlijmen H (2016) Collaborating to improve the use of free-energy and other quantitative methods in drug discovery. J Comput Aided Mol Des 30(12):1139–1141. https://doi.org/10.1007/s10822-016-9996-y
Reif MM, Hünenberger PH (2011) Computation of methodology-independent single-ion solvation properties from molecular simulations. III. Correction terms for the solvation free energies, enthalpies, entropies, heat capacities, volumes, compressibilities, and expansivities of solvated ions. J Chem Phys 134(14):144103. https://doi.org/10.1063/1.3567020
Öhlknecht C, Lier B, Petrov D, Fuchs J, Oostenbrink C (2020) Correcting electrostatic artifacts due to net-charge changes in the calculation of ligand binding free energies. J Comput Chem 41(10):986–999. https://doi.org/10.1002/jcc.26143
Hünenberger PH, McCammon JA (1999) Ewald artifacts in computer simulations of ionic solvation and ion-ion interaction: a continuum electrostatics study. J Chem Phys 110(4):1856–1872. https://doi.org/10.1063/1.477873
Lin YL, Aleksandrov A, Simonson T, Roux B (2014) An overview of electrostatic free energy computations for solutions and proteins. J Chem Theory Comput 10(7):2690–2709. https://doi.org/10.1021/ct500195p
Simonson T, Roux B (2016) Concepts and protocols for electrostatic free energies. Mol Simul 42(13):1090–1101. https://doi.org/10.1080/08927022.2015.1121544
Ji C, Mei Y (2014) Some practical approaches to treating electrostatic polarization of proteins. Acc Chem Res 47(9):2795–2803. https://doi.org/10.1021/ar500094n
Zhang C, Lu C, Wang Q, Ponder JW, Ren P (2015) Polarizable multipole-based force field for dimethyl and trimethyl phosphate. J Chem Theory Comput 11(11):5326–5339. https://doi.org/10.1021/acs.jctc.5b00562
Kognole AA, Aytenfisu AH, MacKerell AD (2020) Balanced polarizable Drude force field parameters for molecular anions: phosphates, sulfates, sulfamates, and oxides. J Mol Model 26(6):152. https://doi.org/10.1007/s00894-020-04399-0
Cerutti DS, Swope WC, Rice JE, Case DA (2014) Ff14ipq: a self-consistent force field for condensed-phase simulations of proteins. J Chem Theory Comput 10(10):4515–4534. https://doi.org/10.1021/ct500643c
Zhou A, Schauperl M, Nerenberg PS (2020) Benchmarking electronic structure methods for accurate fixed-charge electrostatic models. J Chem Inf Model 60(1):249–258. https://doi.org/10.1021/acs.jcim.9b00962
Schauperl M, Nerenberg PS, Jang H, Wang LP, Bayly CI, Mobley DL, Gilson MK (2020) Non-bonded force field model with advanced restrained electrostatic potential charges (RESP2). Commun Chem 3(1):1–11. https://doi.org/10.1038/s42004-020-0291-4
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general Amber force field. J Comput Chem 25(9):1157–1174. https://doi.org/10.1002/jcc.20035
Wang J, Wang W, Kollman PA, Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25(2):247–260. https://doi.org/10.1016/j.jmgm.2005.12.005
Mobley DL, Bannan CC, Rizzi A, Bayly CI, Chodera JD, Lim VT, Lim NM, Beauchamp KA, Slochower DR, Shirts MR, Gilson MK, Eastman PK (2018) Escaping atom types in force fields using direct chemical perception. J Chem Theory Comput. https://doi.org/10.1021/acs.jctc.8b00640
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD (2009) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. https://doi.org/10.1002/jcc.21367
Vanommeslaeghe K, MacKerell AD (2012) Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J Chem Inf Model 52(12):3144–3154. https://doi.org/10.1021/ci300363c
Vanommeslaeghe K, Raman EP, MacKerell AD (2012) Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 52(12):3155–3168. https://doi.org/10.1021/ci3003649
Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 105(28):6474–6487. https://doi.org/10.1021/jp003919d
Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA (2016) OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput 12(1):281–296. https://doi.org/10.1021/acs.jctc.5b00864
Mobley DL, Amezcua M, Ponder J, Khalak Y, Yigitkan Eken E, Almeida N, Isaacs L, Gibb B, Kellett K, Serrilon D (2020) The SAMPL7 host-guest challenge virtual workshop. Zenodo. https://doi.org/10.5281/zenodo.3674155
Saric D, Kohns M, Vrabec J (2020) Dielectric constant and density of aqueous alkali halide solutions by molecular dynamics: a force field assessment. J Chem Phys 152(16):164502. https://doi.org/10.1063/1.5144991
Vega C (2015) Water: one molecule, two surfaces. One mistake. Mol Phys 113(9–10):1145–1163. https://doi.org/10.1080/00268976.2015.1005191
González MA (2011) Force fields and molecular dynamics simulations. JDN 12:169–200. https://doi.org/10.1051/sfn/201112009
Guillot B (2002) A reappraisal of what we have learnt during three decades of computer simulations on water. J Mol Liq 101(1):219–260. https://doi.org/10.1016/S0167-7322(02)00094-6
Henriksen NM, Gilson MK (2017) Evaluating force field performance in thermodynamic calculations of cyclodextrin host-guest binding: water models, partial charges, and host force field parameters. J Chem Theory Comput 13(9):4253–4269. https://doi.org/10.1021/acs.jctc.7b00359
Yin J, Henriksen NM, Muddana HS, Gilson MK (2018) Bind3P: optimization of a water model based on host-guest binding data. J Chem Theory Comput 14(7):3621–3632. https://doi.org/10.1021/acs.jctc.8b00318
Warshel A (1978) Energetics of enzyme catalysis. Proc Natl Acad Sci USA 75(11):5250–5254. https://doi.org/10.1073/pnas.75.11.5250
Howard AE, Singh UC, Billeter M, Kollman PA (1988) Many-body potential for molecular interactions. J Am Chem Soc 110(21):6984–6991. https://doi.org/10.1021/ja00229a009
Humphreys DD, Friesner RA, Berne BJ (1995) Simulated annealing of a protein in a continuum solvent by multiple-time-step molecular dynamics. J Phys Chem 99(26):10674–10685. https://doi.org/10.1021/j100026a035
Grossfield A, Ren P, Ponder JW (2003) Ion solvation thermodynamics from simulation with a polarizable force field. J Am Chem Soc 125(50):15671–15682. https://doi.org/10.1021/ja037005r
Gibb CLD, Gibb BC (2011) Anion binding to hydrophobic concavity is central to the salting-in effects of Hofmeister chaotropes. J Am Chem Soc 133(19):7344–7347. https://doi.org/10.1021/ja202308n
Thormann E (2012) On understanding of the Hofmeister effect: how addition of salt alters the stability of temperature responsive polymers in aqueous solutions. RSC Adv. https://doi.org/10.1039/c2ra20164j
Gao K, Yin J, Henriksen NM, Fenley AT, Gilson MK (2015) Binding enthalpy calculations for a neutral host-guest pair yield widely divergent salt effects across water models. J Chem Theory Comput 11(10):4555–4564. https://doi.org/10.1021/acs.jctc.5b00676
Carnegie RS, Gibb CLD, Gibb BC (2014) Anion complexation and the Hofmeister effect. Angew Chem 126(43):11682–11684. https://doi.org/10.1002/ange.201405796
Gibb CLD, Gibb BC (2004) Well-defined, organic nanoenvironments in water: the hydrophobic effect drives a capsular assembly. J Am Chem Soc 126(37):11408–11409. https://doi.org/10.1021/ja0475611
Saltzman A, Tang D, Gibb BC, Ashbaugh HS (2020) Emergence of non-monotonic deep cavity cavitand assembly with increasing portal methylation. Mol Syst Des Eng 5(3):656–665. https://doi.org/10.1039/C9ME00076C
Brown A (2009) Analysis of cooperativity by isothermal titration calorimetry. Int J Mol Sci 10(8):3457–3477. https://doi.org/10.3390/ijms10083457
Ma YL, Ke H, Valkonen A, Rissanen K, Jiang W (2018) Achieving strong positive cooperativity through activating weak non-covalent interactions. Angew Chem Int Ed 57(3):709–713. https://doi.org/10.1002/anie.201711077
Ndendjio SZ, Liu W, Yvanez N, Meng Z, Zavalij PY, Isaacs L (2019) Triptycene walled glycoluril trimer: synthesis and recognition properties. N J Chem 44(2):338–345. https://doi.org/10.1039/C9NJ05336K
Suating P, Nguyen TT, Ernst EN, Wang Y, Jordan HJ, Gibb DCL, Ashbaugh SH, Gibb CB (2020) Proximal charge effects on guest binding to a non-polar pocket. Chem Sci 11(14):3656–3663. https://doi.org/10.1039/C9SC06268H
Kellett K, Slochower D, Schauperl M, Duggan BM, Gilson M (2020) Experimental characterization of the association of nine novel cyclodextrin derivatives with two guest compounds. ChemRxiv. https://doi.org/10.26434/chemrxiv.12663065.v1
Lee J, Tofoleanu F, Pickard FC, König G, Huang J, Damjanović A, Baek M, Seok C, Brooks BR (2017) Absolute binding free energy calculations of CBClip host-guest systems in the SAMPL5 Blind Challenge. J Comput Aided Mol Des 31(1):71–85. https://doi.org/10.1007/s10822-016-9968-2
Ma D, Zavalij PY, Isaacs L (2010) Acyclic cucurbit[n]uril congeners are high affinity hosts. J Org Chem 75(14):4786–4795. https://doi.org/10.1021/jo100760g
Biedermann F, Rauwald U, Cziferszky M, Williams KA, Gann LD, Guo BY, Urbach AR, Bielawski CW, Scherman OA (2010) Benzobis(imidazolium)-cucurbit[8]uril complexes for binding and sensing aromatic compounds in aqueous solution. Chem Eur J 16(46):13716–13722. https://doi.org/10.1002/chem.201002274
Gallicchio E, Levy RM (2012) Prediction of SAMPL3 host-guest affinities with the binding energy distribution analysis method (BEDAM). J Comput Aided Mol Des 26(5):505–516. https://doi.org/10.1007/s10822-012-9552-3
Naïm M, Bhat S, Rankin KN, Dennis S, Chowdhury SF, Siddiqi I, Drabik P, Sulea T, Bayly CI, Jakalian A, Purisima EO (2007) Solvated interaction energy (SIE) for scoring protein–ligand binding affinities. 1. Exploring the parameter space. J Chem Inf Model 47(1):122–133. https://doi.org/10.1021/ci600406v
Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL, Gilson MK (2017) Overview of the SAMPL5 Host-Guest Challenge: are we doing better? J Comput Aided Mol Des 31(1):1–19. https://doi.org/10.1007/s10822-016-9974-4
Liu W, Lu X, Xue W, Samanta SK, Zavalij PY, Meng Z, Isaacs L (2018) Hybrid molecular container based on glycoluril and triptycene: synthesis, binding properties, and triggered release. Chem Eur J 24(53):14101–14110. https://doi.org/10.1002/chem.201802981
Ndendjio SAZ, Isaacs L (2019) Molecular recognition properties of acyclic cucurbiturils toward amino acids, peptides, and a protein. Supramol Chem 31(7):432–441. https://doi.org/10.1080/10610278.2019.1619737
Biedermann F, Uzunova VD, Scherman OA, Nau WM, De Simone A (2012) Release of high-energy water as an essential driving force for the high-affinity binding of cucurbit[n]urils. J Am Chem Soc 134(37):15318–15323. https://doi.org/10.1021/ja303309e
Monroe JI, Shirts MR (2014) Converging free energies of binding in cucurbit[7]uril and octa-acid host-guest systems from SAMPL4 using expanded ensemble simulations. J Comput Aided Mol Des 28(4):401–415. https://doi.org/10.1007/s10822-014-9716-4
Liu W, Lu X, Meng Z, Isaacs L (2018) A glycoluril dimer-triptycene hybrid receptor: synthesis and molecular recognition properties. Org Biomol Chem 16(35):6499–6506. https://doi.org/10.1039/C8OB01575A
Barnett JW, Sullivan MR, Long JA, Tang D, Nguyen T, Ben-Amotz D, Gibb BC, Ashbaugh HS (2020) Spontaneous drying of non-polar deep-cavity cavitand pockets in aqueous solution. Nat Chem. https://doi.org/10.1038/s41557-020-0458-8
Gibb CLD, Gibb BC (2009) Guests of differing polarities provide insight into structural requirements for templates of water-soluble nano-capsules. Tetrahedron 65(35):7240–7248. https://doi.org/10.1016/j.tet.2009.01.106
Gibb CLD, Gibb BC (2014) Binding of cyclic carboxylates to octa-acid deep-cavity cavitand. J Comput Aided Mol Des 28(4):319–325. https://doi.org/10.1007/s10822-013-9690-2
Ewell J, Gibb BC, Rick SW (2008) Water inside a hydrophobic cavitand molecule. J Phys Chem B 112(33):10272–10279. https://doi.org/10.1021/jp804429n
Kellett K, Kantonen SA, Duggan BM, Gilson MK (2018) Toward expanded diversity of host-guest interactions via synthesis and characterization of cyclodextrin derivatives. J Solut Chem 47(10):1597–1608. https://doi.org/10.1007/s10953-018-0769-1
Slochower DR, Henriksen NM, Wang LP, Chodera JD, Mobley DL, Gilson MK (2019) Binding thermodynamics of host–guest systems with SMIRNOFF99Frosst 1.0.5 from the Open Force Field Initiative. J Chem Theory Comput. 15(11):6225–6242. https://doi.org/10.1021/acs.jctc.9b00748
Carrazana J, Jover A, Meijide F, Soto VH, Vázquez Tato J (2005) Complexation of adamantyl compounds by \(\beta \)-cyclodextrin and monoamino derivatives. J Phys Chem B 109(19):9719–9726. https://doi.org/10.1021/jp0505781
Rizzi A, Grinaway P, Parton D, Shirts M, Wang K, Eastman P, Friedrichs M, Pande V, Branson K, Mobley D, Chodera J (2020) YANK: a GPU-accelerated platform for alchemical free energy calculations
Wang K, Chodera JD, Yang Y, Shirts MR (2013) Identifying ligand binding sites and poses using GPU-accelerated Hamiltonian replica exchange molecular dynamics. J Comput Aided Mol Des 27(12):989–1007. https://doi.org/10.1007/s10822-013-9689-8
Friedrichs MS, Eastman P, Vaidyanathan V, Houston M, Legrand S, Beberg AL, Ensign DL, Bruns CM, Pande VS (2009) Accelerating molecular dynamic simulation on graphics processing units. J Comput Chem 30(6):864–872. https://doi.org/10.1002/jcc.21209
Eastman P, Pande V (2010) OpenMM: a hardware-independent framework for molecular simulations. Comput Sci Eng 12(4):34–39. https://doi.org/10.1109/MCSE.2010.27
Eastman P, Pande VS (2010) Constant constraint matrix approximation: a robust, parallelizable constraint method for molecular simulations. J Chem Theory Comput 6(2):434–437. https://doi.org/10.1021/ct900463w
Eastman P, Pande VS (2010) Efficient nonbonded interactions for molecular dynamics on a graphics processing unit. J Comput Chem 31(6):1268–1272. https://doi.org/10.1002/jcc.21413
Eastman P, Friedrichs MS, Chodera JD, Radmer RJ, Bruns CM, Ku JP, Beauchamp KA, Lane TJ, Wang LP, Shukla D, Tye T, Houston M, Stich T, Klein C, Shirts MR, Pande VS (2013) OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J Chem Theory Comput 9(1):461–469. https://doi.org/10.1021/ct300857j
Shirts MR, Chodera JD (2008) Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys 129(12):124105. https://doi.org/10.1063/1.2978177
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
Khalak Y, Tresadern G, de Groot BL, Gapsys V (2020) Non-equilibrium approach for binding free energies in cyclodextrins in SAMPL7: force fields and software. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-020-00359-1
Procacci P, Guarnieri G (2020) SAMPL7 blind predictions using nonequilibrium alchemical approaches. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-020-00365-3
Shi Y, Laury ML, Wang Z, Ponder JW (2020) AMOEBA binding free energies for the SAMPL7 TrimerTrip Host-Guest Challenge. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-020-00358-2
Eken Y, Almeida NMS, Wang C, Wilson AK (2020) SAMPL7: host-guest binding prediction by molecular dynamics and quantum mechanics. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-020-00357-3
Serillon D, Barril X (in press) Testing automatic methods to predict free binding energy of host–guest complexes in SAMPL7 Challenge. J Comput Aided Mol Des
Rizzi A, Murkli S, McNeill JN, Yao W, Sullivan M, Gilson MK, Chiu MW, Isaacs L, Gibb BC, Mobley DL, Chodera JD (2018) Overview of the SAMPL6 host-guest binding affinity prediction challenge. J Comput Aided Mol Des 32(10):937–963. https://doi.org/10.1007/s10822-018-0170-6
Procacci P (2019) Precision and computational efficiency of nonequilibrium alchemical methods for computing free energies of solvation. II. Unidirectional estimates. II. Unidirectional Estimates. J Chem Phys. 151(14):144115. https://doi.org/10.1063/1.5120616
Izadi S, Onufriev AV (2016) Accuracy limit of rigid 3-point water models. J Chem Phys 10(1063/1):4960175. https://doi.org/10.1063/1.4960175
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396. https://doi.org/10.1021/jp810292n
Acknowledgements
MA and DLM gratefully acknowledge support from NIH Grant R01GM124270 supporting the SAMPL Blind Challenges. We appreciate the laboratories of Michael K. Gilson (UCSD), Lyle Isaacs (Maryland) and Bruce Gibb (Tulane) for providing experimental data for the challenge. We are also grateful to OpenEye Scientific for providing a free academic software license for use in this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclaimers
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest
DLM is a Member of the Scientific Advisory Board of OpenEye Scientific Software, and DLM is an Open Science Fellow with Silicon Therapeutics.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Amezcua, M., El Khoury, L. & Mobley, D.L. SAMPL7 Host–Guest Challenge Overview: assessing the reliability of polarizable and non-polarizable methods for binding free energy calculations. J Comput Aided Mol Des 35, 1–35 (2021). https://doi.org/10.1007/s10822-020-00363-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-020-00363-5