
Abstract
As part of the SAMPL6 host–guest blind challenge, the AMOEBA force field was applied to calculate the absolute binding free energy for a cucurbit[8]uril host complexed with 14 diverse guests, ranging from small, rigid structures to drug molecules. The AMOEBA results from the initial submission prompted an investigation into aspects of the methodology and parameterization employed. Lessons learned from the blind challenge include: a double annihilation scheme (electrostatics and van der Waals) is needed to obtain proper sampling of guest conformations, annihilation of key torsion parameters of the guest are recommended for flexible guests, and a more thorough analysis of torsion parameters is warranted. When put in to practice with the AMOEBA model, the lessons learned improved the MUE from 2.63 to 1.20 kcal/mol and the RMSE from 3.62 to 1.68 kcal/mol, respectively. Overall, the AMOEBA protocol for determining absolute binding free energies benefitted from participation in the SAMPL6 host–guest blind challenge and the results suggest the implementation of the methodology in future host–guest calculations.





Similar content being viewed by others
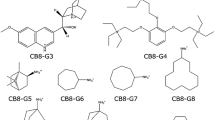
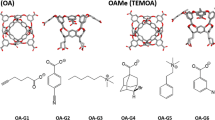
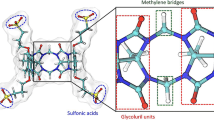
References
Williams-Noonan BJ, Yuriev E, Chalmers DK (2018) Free energy methods in drug design: prospects of “Alchemical Perturbation” in medicinal chemistry. J Med Chem 61:638
Mobley DL, Klimovich PV (2012) Perspective: alchemical free energy calculations for drug discovery. J Chem Phys 137:230901
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) The SAMPL4 host–guest blind prediction challenge: an overview. J Comput Aided Mol Des 28:305
Muddana HS, Varnado CD, Bielawski CW, Urbach AR, Isaacs L, Geballe MT, Gilson MK (2012) Blind prediction of host–guest binding AffinitiesL a new SAMPL3 challenge. J Comput Aided Mol Des 26:475
Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL (2017) Overview of the SAMPL5 host–guest challenge: are we doing better? J Comput Aided Mol Des 31:1
Barrow SJ, Kasera S, Rowland MJ, del Barrio J, Scherman OA (2015) Cucurbituril-bassed molecular recognition. Chem Rev 115:12320
Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic. Molecules J Am Chem Soc 117:5179
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671
Robertson MJ, Tirado-Rives J, Jorgensen WL (2015) Improved peptide and protein torsional energetics with the OPLSAA force field. J Chem Theory Comput 11:3499
Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Mobley DL, Schnieders MJ, Haque I, Lambrecht DS, DiStasio J, Head-Gordon RA, Clark M, Johnson GNI, Head-Gordon ME T (2010) Current status of the AMOEBA polarizable force. Field J Phys Chem B 114:2549
Laury ML, Wang L-P, Pande VS, Head-Gordon T, Ponder JW (2015) Revised parameters for the AMOEBA polarizable atomic multipole water model. J Phys Chem B 119:9423
Ren P, Ponder JW (2003) Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B 107:5933
Ren P, Wu C, Ponder JW (2011) Polarizable atomic multipole-based molecular mechanics for organic molecules. J Chem Theory Comput 7:3143
Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, Ren P (2013) Polarizable atomic multipole-based AMOEBA force field for proteins. J Chem Theory Comput 9:4046
Xiang JY, Ponder JW (2014) An angular overlap model for Cu(II) ion in the AMOEBA polarizable force. Field J Chem Theory Comput 10:298
Zhang C, Lu C, Jing Z, Wu C, Piquemal J-P, Ponder JW, Ren P (2018) AMOEBA polarizable atomic multpole force field for nucleic acids. J Chem Theory Comput 14:2084
Bell DR, Qi R, Jing Z, Xiang JY, Meijas C, Schnieders MJ, Ponder JW, Ren P (2016) Calculating binding free energies of host–guest systems using the AMOEBA polarizable force. Field Phys Chem Chem Phys 18:30261
Liu C, Ponder JW, Marshall GR (2014) Helix stability of oligoglycine, oligoalanine, and oligo-beta-alanine dodecamers reflected by hydrogen-bond. Persistence Proteins 82:3043
Murkli S, McNeil J, Isaacs L (2018) CB[8]-guest binding affinities: a blinded dataset for the SAMPL6 challenge. Supramolecular Chem https://doi.org/10.1101/371724
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery J, Peralta JA, Ogliaro JE, Bearpark F, Heyd M, Brothers JJ, Kudin E, Staroverov KN, Keith VN, Kobayashi T, Normand R, Raghavachari J, Rendell K, Burant A, Iyengar JC, Tomasi SS, Cossi J, Millam M, Klene JM, Adamo M, Cammi C, Ochterski R, Martin JW, Morokuma RL, Farkas K, Foresman O, Fox JB (2016) Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford CT
Parrish RM, Burns LA, Smith DGA, Simmonett AC, DePrince AE III, Hohenstein EG, Bozkaya U, Sokolov AY, Di Remigio R, RIchard RM, Gonthier JR, James AM, McAlexander HR, Kumar A, Saitow M, Wang X, Pritchard BP, Verma P, Schaefer HF III, Patkowski K, King RA, Valeev EF, Evangelista FA, Turney JM, Crawford TD, Sherrill CD (2017) Psi4 1.1: an open-source electronic structure program emphasizing automation, advanced libraries, and interoperability. J Chem Theory Comput 13:3185
Stone AJ (1981) Distributed multipole analysis, or how to describe a molecular charge. Distribution Chem Phys Lett 83:233
Stone AJ, Alderton M (2002) Distributed multipole analysis: methods and applications. Mol Phys 100:221
van Duijnen PT, Swart MJ (1998) Molecular and atomic polarizabilities: thole’s model revisited. J Phys Chem A 102:2399
Thole BT (1981) Molecular polarizabilities calculated with a modified dipole. Interaction Chem Phys 59:341
Rackers JA, Wang Z, Lu C, Laury ML, Lagardere L, Schnieders MJ, Piquemal J-P, Ren P, Ponder JW (2018) Tinker 8: software tools for molecular design. J Chem Theory Comput 14:xxx
Halgren TA (1995) Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17:490
Halgren TA (1995) Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J Comput Chem 17:520
Halgren TA (1995) Merck molecular force field. III. Molecular geometries and vibrational frequencies for MMFF94. J Comput Chem 17:553
Halgren TA (1995) Merck molecular force field. V. Extension of MMFF94 using experimental data additional computational data. J Comput Chem 17:616
Halgren TA, Nachbar RB (1995) Merck molecular force field. IV. Conformational energies and geometries for MMFF94. J Comput Chem 17:587
Zwanzig RW (1954) High-Temperature equation of state by a perturbation method. I. Nonpolar gases. J Chem Phys 22:1420
Bennett CH (1976) Efficient esimation of free energy differences from Monte Carlo Data. J Comput Phys 22:245
Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity-rescaling. J Chem Phys 126:014101
Bussi G, Parrinello M (2008) Stochastic thermostats: comparison of local and global schemes. Comput Phys Commun 179:26
Bussi G, Zykova-Timan T, Parrinello M (2009) Isothermal–isobaric molecular dynamics using stochastic velocity rescaling. J Chem Phys 130:074101
Frenkel D, Smit B (2001) Understanding molecular simulation: from algorithms to applications. Academic Press, New York
Faller R, de Pablo JJ (2002) Constant pressure hybrid molecular dynamics-Monte Carlo simulations. J Chem Phys 116:55
Tuckerman ME, Berne BJ (1991) Molecular dynamics in systems with multiple time scales: systems with stiff and soft degrees of freedom and with short and long range forces. J Chem Phys 95:8362
Tuckerman ME, Berne BJ (1992) Reversible multiple time scale molecular dynamics J Chem Phys 97:1990
Tuckerman ME, Berne BJ, Rossi A (1990) Molecullar dynamics algorithm for multiple time scales: systems with disparate masses. J Chem Phys 94:1465
Harger M, Li D, Wang Z, Dalby K, Lagardere L, Piquemal J-P, Ponder JW, Ren P (2017) Tinker-OpenMM: absolute and relative alchemical free energies using AMOEBA on GPUs. J Comput Chem 38:2047
Jiao D, Golubkov PA, Darden TA, Ren P (2008) Calculation of protein–ligand binding free energy by using a polarizable potential. Proc Nat Acad Sci USA 105:6290
Hamelberg D, McCammon JA (2004) Standard free energy of releasing a localized water molecule from the binding pockets of proteins: double-decoupling method. J Am Chem Soc 126:7683
Acknowledgements
The authors would like to thank the organizers of the D3R/SAMPL6 challenge. JWP wishes to thank the National Institutes of Health NIGMS for financial support via awards R01 GM106137 and R01 GM114237.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Laury, M.L., Wang, Z., Gordon, A.S. et al. Absolute binding free energies for the SAMPL6 cucurbit[8]uril host–guest challenge via the AMOEBA polarizable force field. J Comput Aided Mol Des 32, 1087–1095 (2018). https://doi.org/10.1007/s10822-018-0147-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-018-0147-5