
Abstract
The ability to computationally predict protein-small molecule binding affinities with high accuracy would accelerate drug discovery and reduce its cost by eliminating rounds of trial-and-error synthesis and experimental evaluation of candidate ligands. As academic and industrial groups work toward this capability, there is an ongoing need for datasets that can be used to rigorously test new computational methods. Although protein–ligand data are clearly important for this purpose, their size and complexity make it difficult to obtain well-converged results and to troubleshoot computational methods. Host–guest systems offer a valuable alternative class of test cases, as they exemplify noncovalent molecular recognition but are far smaller and simpler. As a consequence, host–guest systems have been part of the prior two rounds of SAMPL prediction exercises, and they also figure in the present SAMPL5 round. In addition to being blinded, and thus avoiding biases that may arise in retrospective studies, the SAMPL challenges have the merit of focusing multiple researchers on a common set of molecular systems, so that methods may be compared and ideas exchanged. The present paper provides an overview of the host–guest component of SAMPL5, which centers on three different hosts, two octa-acids and a glycoluril-based molecular clip, and two different sets of guest molecules, in aqueous solution. A range of methods were applied, including electronic structure calculations with implicit solvent models; methods that combine empirical force fields with implicit solvent models; and explicit solvent free energy simulations. The most reliable methods tend to fall in the latter class, consistent with results in prior SAMPL rounds, but the level of accuracy is still below that sought for reliable computer-aided drug design. Advances in force field accuracy, modeling of protonation equilibria, electronic structure methods, and solvent models, hold promise for future improvements.






Similar content being viewed by others
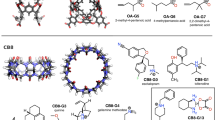
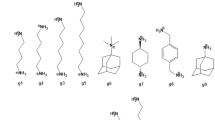
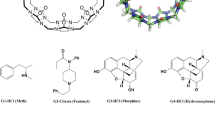
References
Borhani DW, Shaw DE (2012) The future of molecular dynamics simulations in drug discovery. J Comput Aided Mol Des 26:15–26. doi:10.1007/s10822-011-9517-y
Martin E, Ertl P, Hunt P, Duca J, Lewis R (2012) Gazing into the crystal ball; The future of computer-aided drug design. J Comput Aided Mol Des 26:77–79. doi:10.1007/s10822-011-9487-0
Chen L, Morrow JK, Tran HT, Phatak SS, Du-Cuny L, Zhang S (2012) From laptop to benchtop to bedside: structure-based drug design on protein targets. Curr Pharm Des 18:1217–1239. doi:10.2174/138920012799362837
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3:935–949. doi:10.1038/nrd1549
Klebe G (2006) Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today 11:580–594. doi:10.1016/j.drudis.2006.05.012
Lavecchia A, Di Giovanni C (2013) Virtual screening strategies in drug discovery: a critical review. Curr Med Chem 20:2839–2860. doi:10.2174/09298673113209990001
Mortier J, Rakers C, Bermudez M, Murgueitio MS, Riniker S (2015) The impact of molecular dynamics on drug design: applications for the characterization of ligand-macromolecule complexes. Drug Discov Today 20:686–702. doi:10.1016/j.drudis.2015.01.003
Gilson MK, Zhou H-X (2007) Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct 36:21–42. doi:10.1146/annurev.biophys.36.040306.132550
Adcock SA, Mccammon JA (2006) Molecular dynamics : survey of methods for simulating the activity of proteins. Proteins 106:1589–1615. doi:10.1021/cr040426m
Gumbart JC, Roux B, Chipot C (2013) Standard binding free energies from computer simulations: what is the best strategy? J Chem Theory Comput 9:794–802. doi:10.1021/ct3008099
Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, Romero DL (2015) Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc 137:2695–2703. doi:10.1021/ja512751q
Buch I, Giorgino T, De Fabritiis G (2011) Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc Natl Acad Sci USA 108:10184–10189. doi:10.1073/pnas.1103547108
Chodera JD, Mobley DL, Shirts MR, Dixon RW, Branson K, Pande VS (2011) Alchemical free energy methods for drug discovery: progress and challenges. Curr Opin Struct Biol 21:150–160. doi:10.1016/j.sbi.2011.01.011
Lu Y, Yang CY, Wang S (2006) Binding free energy contributions of interfacial waters in HIV-1 protease/inhibitor complexes. J Am Chem Soc 128:11830–11839. doi:10.1021/ja058042g
Cram DJ, Cram JM (1974) Host-guest chemistry. Science 183(80):803–809
Freeman WA, Mock WL, Shih N-Y (1981) Cucurbituril. J Am Chem Soc 103:7367–7368. doi:10.1021/ja00414a070
Lee JW, Samal S, Selvapalam N, Kim HJ, Kim K (2003) Cucurbituril homologues and derivatives: new opportunities in supramolecular chemistry. Acc Chem Res 36:621–630. doi:10.1021/ar020254k
Jeon YJ, Kim S-Y, Ko YH, Sakamoto S, Yamaguchi K, Kim K (2005) Novel molecular drug carrier: encapsulation of oxaliplatin in cucurbit[7]uril and its effects on stability and reactivity of the drug. Org Biomol Chem 3:2122–2125. doi:10.1039/b504487a
Lagona J, Mukhopadhyay P, Chakrabarti S, Isaacs L (2005) The cucurbit[n]uril family. Angew Chem Int Ed Engl 44:4844–4870. doi:10.1002/anie.200460675
Rekharsky MV, Inoue Y (1998) Complexation thermodynamics of cyclodextrins. Chem Rev 98:1875–1918. doi:10.1021/cr970015o
Zimmerman SC, Vanzyl CM (1987) Rigid molecular tweezers: synthesis, characterization, and complexation chemistry of a diacridine. J Am Chem Soc 109:7894–7896. doi:10.1021/ja00259a055
Zimmerman SC (1993) Rigid molecular tweezers as hosts for the complexation of neutral guests. Top Curr Chem 165:71–102. doi:10.1007/BFb0111281
Klärner FG, Kahlert B (2003) Molecular tweezers and clips as synthetic receptors. Molecular recognition and dynamics in receptor-substrate complexes. Acc Chem Res 36:919–932. doi:10.1021/ar0200448
Sinha S, Lopes DHJ, Du Z, Pang ES, Shanmugam A, Lomakin A, Talbiersky P, Tennstaedt A, McDaniel K, Bakshi R, Kuo PY (2011) Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc 133:16958–16969. doi:10.1021/ja206279b
Linton B, Hamilton AD (1999) Host-guest chemistry: combinatorial receptors. Curr Opin Chem Biol 3:307–312
Baron R, McCammon JA (2013) Molecular recognition and ligand association. Annu Rev Phys Chem 64:151–175. doi:10.1146/annurev-physchem-040412-110047
Yin J, Fenley AT, Henriksen NM, Gilson MK (2015) Toward improved force-field accuracy through sensitivity analysis of host-guest binding thermodynamics. J Phys Chem B. doi:10.1021/acs.jpcb.5b04262
Wickstrom L, Deng N, He P, Mentes A, Nguyen C, Gilson MK, Kurtzman T, Gallicchio E, Levy RM (2016) Parameterization of an effective potential for protein-ligand binding from host-guest affinity data. J Mol Recognit 29:10–21. doi:10.1002/jmr.2489
Geballe MT, Skillman AG, Nicholls A, Guthrie JP, Taylor PJ (2010) The SAMPL2 blind prediction challenge: introduction and overview. J Comput Aided Mol Des 24:259–279. doi:10.1007/s10822-010-9350-8
Muddana HS, Varnado CD, Bielawski CW, Urbach AR, Isaacs L, Geballe MT, Gilson MK (2012) Blind prediction of host-guest binding affinities: a new SAMPL3 challenge. J Comput Aided Mol Des 26:475–487. doi:10.1007/s10822-012-9554-1
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) The SAMPL4 host-guest blind prediction challenge: an overview. J Comput Aided Mol Des 28:305–317. doi:10.1007/s10822-014-9735-1
Mobley DL, Wymer KL, Lim NM, Guthrie JP (2014) Blind prediction of solvation free energies from the SAMPL4 challenge. J Comput Aided Mol Des 28:135–150. doi:10.1007/s10822-014-9718-2
Gibb CLD, Gibb BC (2014) Binding of cyclic carboxylates to octa-acid deep-cavity cavitand. J Comput Aided Mol Des 28:319–325. doi:10.1007/s10822-013-9690-2
Gan H, Gibb BC (2013) Guest-mediated switching of the assembly state of a water-soluble deep-cavity cavitand. Chem Commun 49:1395–1397. doi:10.1039/c2cc38227j
Jordan JH, Gibb BC (2014) Molecular containers assembled through the hydrophobic effect. Chem Soc Rev 44:547–585. doi:10.1039/c4cs00191e
Zhang B, Isaacs L (2014) Acyclic cucurbit[n]uril-type molecular containers: influence of aromatic walls on their function as solubilizing excipients for insoluble drugs. J Med Chem 57:9554–9563. doi:10.1021/jm501276u
Sullivan MR, Sokkalingam P, Nguyen T, Donahue JP, Gibb BC (2016) Binding of carboxylate and trimethylammonium salts to octa-acid and TEMOA deep-cavity cavitands. J Comput Aided Mol Des. doi:10.1007/s10822-016-9925-0
She N, Moncelet D, Gilberg L, Lu X, Sindelar V, Briken V, Isaacs L (2016) Glycoluril-derived molecular clips are potent and selective receptors for cationic dyes in water. Chem A Eur J 22:1–11. doi:10.1002/chem.201601796
Molecular Operating Environment (MOE) 2013.08 (2016) Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7
Bansal N, Zheng Z, Cerutti DS, Merz KM (2016) On the fly estimation of host-guest binding free energies using the movable type method: participation in the SAMPL5 blind challenge. J Comput Aided Mol Des (in press)
Yin J, Henriksen NM, Slochower DR, Gilson MK (2016) The SAMPL5 host-guest challenge: computing binding free energies and enthalpies from explicit solvent simulations using attach-pull-release (APR) approach. J Comput Aided Mol Des. doi:10.1007/s10822-016-9970-8
Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham DS III, Darden TA, Duke TA, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossvary I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, SalomonFerrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA (2014) AMBER 14. University of California, San Francisco
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. doi:10.1016/j.softx.2015.06.001
Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY (2016) Maestro-Desmond Interoperability Tools. Schrödinger, New York, NY, p 2016
Plimpton S (1995) Fast parallel algorithms for short-range molecular dynamics. J Comput Phys 117:1–19. doi:10.1006/jcph.1995.1039
Shirts MR, Klein C, Swails JM, Yin J, Gilson MK, Mobley DL, Case DA, Zhong ED (2016) Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J Comput Aided Mol Des. doi:10.1007/s10822-016-9977-1
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. doi:10.1063/1.445869
Grimme S (2012) Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem A Eur J 18:9955–9964. doi:10.1002/chem.201200497
Kirkwood JG (1935) Statistical mechanics of fluid mixtures. J Chem Phys 3:300–313. doi:10.1063/1.1749657
Bennett CH (1976) Efficient estimation of free energy differences from Monte Carlo data. J Comput Phys 22:245–268. doi:10.1016/0021-9991(76)90078-4
Laio A, Parrinello M (2002) Escaping free-energy minima. Proc Natl Acad Sci USA 99:12562–12566. doi:10.1073/pnas.202427399
Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA (1998) Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J Am Chem Soc 120:9401–9409. doi:10.1021/ja981844+
Zheng Z, Ucisik MN, Merz KM (2013) The movable type method applied to protein-ligand binding. J Chem Theory Comput 9:5526–5538. doi:10.1021/ct4005992
Pal RK, Haider K, Kaur D, Flynn W, Xia J, Levy RM, Taran T, Wickstrom L, Kurtzman T, Gallicchio E (2016) A combined treatment of hydration and dynamical effects for the modeling of host-guest binding thermodynamics: the SAMPL5 blinded challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9956-6
Caldararu O, Olsson MA, Riplinger C, Neese F, Ryde U (2016) Binding free energies in the SAMPL5 octa-acid host-guest challenge calculated with DFT-D3 and CCSD(T). J Comput Aided Mol Des. doi:10.1007/s10822-016-9957-5
Bhakat S, Söderhjelm P (2016) Resolving the problem of trapped water in binding cavities: prediction of host-guest binding free energies in the SAMPL5 challenge by funnel metadynamics. J Comput Aided Mol Des. doi:10.1007/s10822-016-9948-6
Tofoleanu F, Lee J, Pickard IV FC, König G, Huang J, Baek M, Seok C, Brooks BR (2016) Absolute binding free energies for octa-acids and guests in SAMPL5. J Comput Aided Mol Des. doi:10.1007/s10822-016-9965-5
Bosisio S, Mey ASJS, Michel J (2016) Blinded predictions of host-guest standard free energies of binding in the SAMPL5 challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9933-0
Lee J, Tofoleanu F, Pickard IV FC, König G, Huang J, Damjanović A, Baek M, Seok C, Brooks B (2016) Absolute binding free energy calculations of CBClip host–guest systems in the SAMPL5 blind challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9968-2
Henriksen NM, Fenley AT, Gilson MK (2015) Computational calorimetry: high-Precision calculation of host-guest binding thermodynamics. J Chem Theory Comput 11:4377–4394. doi:10.1021/acs.jctc.5b00405
Izadi S, Anandakrishnan R, Onufriev AV (2014) Building water models: a different approach. JPhysChemLett 5:3863–3871. doi:10.1021/jz501780a
Gallicchio E, Lapelosa M, Levy RM (2010) The binding energy distribution analysis method (BEDAM) for the estimation of protein-ligand binding affinities. J Chem Theory Comput 6:2961–2977. doi:10.1021/ct1002913
Riplinger C, Sandhoefer B, Hansen A, Neese F (2013) Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J Chem Phys 139(13):134101
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. doi:10.1063/1.3382344
Woods CJW, Mey A, Calabro G, Michel J (2016) Sire molecular simulations framework. http://siremol.org. Accessed 28 July
Eastman P, Friedrichs MS, Chodera JD, Radmer RJ, Bruns CM, Ku JP, Beauchamp KA, Lane TJ, Wang LP, Shukla D, Tye T (2013) OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J Chem Theory Comput 9:461–469. doi:10.1021/ct300857j
Straatsma TP, McCammon JA (1991) Multiconfiguration thermodynamic integration. J Chem Phys 95:1175. doi:10.1063/1.461148
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general Amber Force Field. J Comput Chem 25:1157–1174. doi:10.1002/jcc.20035
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671–690. doi:10.1002/jcc.21367
Bayly CCI, Cieplak P, Cornell WD, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 97:10269–10280. doi:10.1021/j100142a004
Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 105:6474–6487. doi:10.1021/jp003919d
Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R (2005) Integrated modeling program, applied chemical theory (IMPACT). J Comput Chem 26:1752–1780. doi:10.1002/jcc.20292
Zheng Z, Wang T, Li P, Merz KM (2014) KECSA-movable type implicit solvation model (KMTISM). J Chem Theory Comput 11:667–682. doi:10.1021/ct5007828
Gallicchio E, Paris K, Levy RM (2009) The AGBNP2 implicit solvation model. J Chem Theory Comput 5:2544–2564. doi:10.1021/ct900234u
Klamt A (1995) Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena. J Phys Chem 99:2224–2235. doi:10.1021/j100007a062
Velez-Vega C, Gilson MK (2013) Overcoming dissipation in the calculation of standard binding free energies by ligand extraction. J Comput Chem 34:2360–2371. doi:10.1002/jcc.23398
Hamelberg D, McCammon JA (2004) Standard free energy of releasing a localized water molecule from the binding pockets of proteins: double-decoupling method. J Am Chem Soc 126:7683–7689. doi:10.1021/ja0377908
Limongelli V, Bonomi M, Parrinello M (2013) Funnel metadynamics as accurate binding free-energy method. Proc Natl Acad Sci 110:6358–6363. doi:10.1073/pnas.1303186110
Gallicchio E, Levy RM (2012) Prediction of SAMPL3 host-guest affinities with the binding energy distribution analysis method (BEDAM). J Comput Aided Mol Des 26:505–516. doi:10.1007/s10822-012-9552-3
Mikulskis P, Cioloboc D, Andrejić M, Khare S, Brorsson J, Genheden S, Mata RA, Söderhjelm P, Ryde U (2014) Free-energy perturbation and quantum mechanical study of SAMPL4 octa-acid host-guest binding energies. J Comput Aided Mol Des 28:375–400. doi:10.1007/s10822-014-9739-x
Hsiao YW, Söderhjelm P (2014) Prediction of SAMPL4 host-guest binding affinities using funnel metadynamics. J Comput Aided Mol Des 28:443–454. doi:10.1007/s10822-014-9724-4
Fenley AT, Henriksen NM, Muddana HS, Gilson MK (2014) Bridging calorimetry and simulation through precise calculations of cucurbituril-guest binding enthalpies. J Chem Theory Comput 10:4069–4078. doi:10.1021/ct5004109
Acknowledgments
We thank Dr. Pär Söderhjelm for helpful discussion on the error analysis and Dr. Ulf Ryde for helpful comments on the manuscript. M.K.G. thanks the National Institutes of Health (NIH) for Grants GM061300 and U01GM111528 and the Air Force Office of Scientific Research (AFOSR) for Basic Research Initiative (BRI) Grant (FA9550-12-1-6440414). D.L.M appreciates financial support from the National institutes of Health (1R01GM108889-01) and the National Science Foundation (CHE 1352608). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, NSF or AFOSR. M.K.G. has an equity interest in, and is a cofounder and scientific advisor of VeraChem LLC.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yin, J., Henriksen, N.M., Slochower, D.R. et al. Overview of the SAMPL5 host–guest challenge: Are we doing better?. J Comput Aided Mol Des 31, 1–19 (2017). https://doi.org/10.1007/s10822-016-9974-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-016-9974-4