
Abstract
Genetic manipulation of Haematococcus pluvialis is difficult because of the lack of a stable and convenient transformation system. The pH124-EGFP-Ble vector containing ble as a selective gene and EGFP as a reporter gene was constructed and employed for effective transformation. H. pluvialis protoplasts were obtained by treating with cellulase and macerozeme. Then polyethylene glycol-mediated transformation was established by incubating the protoplast with the vector. To improve the transformation efficiency of H. pluvialis protoplasts, the transformation system was optimized in consideration of different influencing factors, including zeomycin concentration, growth stage, amount of transformed vector, linearization of the vector, and duration of low-intensity illumination. The integration and expression of ble and EGFP was confirmed in the transformants. Moreover, the optimal combination for protoplast transformation of H. pluvialis was determined to be 5 µg of the linearized vector used to transform cells in the log growth phase, and then the transformed protoplasts allowed to recover under low-intensity illumination for 6 h. This study represents and describes the successful development of an H. pluvialis transformation protocol using protoplasts, which will enable convenient genetic manipulation of this important algal species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Human health can be improved and various diseases potentially may be prevented by astaxanthin, such as cancer (Emiko et al. 2008), diabetes, diabetic nephropathy, eye fatigue and chronic inflammatory, cardiovascular, gastrointestinal, liver, neurodegenerative, and ophthalmic diseases (Chew et al. 1999; Wang et al. 2000; Gross and Lockwood 2005; Izumi-Nagai et al. 2008; Chang et al. 2010; Curek et al. 2010). Haematococcus pluvialis has been proven to be the most effective producer of astaxanthin among microalgae, bacteria, and fungi (Hagen et al. 2002). Haematococcus pluvialis is highly adaptable to environmental changes, such as temperature, light, and salinity, when compared to other algae. Under favorable conditions, H. pluvialis is in the motile flagellar stage and has a gelatinous extracellular matrix. When exposed to various stress conditions, such as nitrogen and phosphate limitations, as well as salt stress and high light intensities, the cells change into the aplanospore stage in which astaxanthin accumulates significantly. In the aplanospore stage, the cell wall is thickened which greatly hinders the extraction of astaxanthin and the transformation of H. pluvialis (Damiani et al. 2006).
Although stable transformation of foreign genes has been very successful in the model microalga Chlamydomonas reinhardtii (Rochaix 1995; Pratheesh et al., 2013; Doron et al. 2016), convenient and effective transformation in H. pluvialis is still difficult to achieve. The Agrobacterium-mediated method was adopted to transform the hptII gene driven by the CaMV35S promoter into H. pluvialis (Kathiresan and Sarada 2009; Kathiresan et al. 2009). Unfortunately, the false-positive rate of the Agrobacterium-mediated transformation method is too high and the method has poor repeatability (Sharon-Gojman et al. 2015). An endogenous pds gene resistant to norflurazon and the aadA gene driven by the tub promoter have been successfully transferred into the H. pluvialis genome by biolistic bombardment (Sharon-Gojman et al. 2015; Yuan et al. 2019). However, biolistic bombardment has several deficiencies, such as complex operations, high cost, low transformation efficiency, and poor repeatability. Hence, it is necessary to establish a more convenient and stable transformation method for the reconstruction of H. pluvialis.
Protoplast fusion involves obtaining descendants which contain all genes of the two-parent strains. In the presence of Ca2+, polyethylene glycol (PEG) promotes the fusion of protoplasts and improves the fusion rate (Kao and Michayluk 1974). Since the first tobacco somatic hybrid plant was obtained in 1972, protoplast fusion technology has been widely used for the fusion of higher plant cells (Vasil, and Indre, 1987). Based on the fusion of plant protoplasts, a versatile cell system for transient gene expression analysis has been introduced in Arabidopsis mesophyll protoplasts (Yoo et al. 2007). Recently, crisper/cas9 technology combined with protoplast transformation technology has been used to efficiently edit the Fusarium oxysporum genome. The PEG-mediated protoplast fusion method is simple in operation and has high efficiency and no species specificity (Wang et al. 2018). However, the protoplast transformation method has seldom been used for the transformation of microalgae.
In the present study, a protoplast transformation mediated by PEG in H. pluvialis was established. In addition, we optimized the transformation method by considering different influencing factors including zeomycin concentration and growth stage, thereby increasing the transformation efficiency and positive rate. This efficient and stable transformation protocol could contribute to modifying the commercial microalgae H. pluvialis.
Materials and methods
H. pluvialis strain and culture conditions
Haematococcus pluvialis strain 192.80 from our laboratory was originally purchased from the Experimental Phycology and Culture Collection of Algae, Göttingen University, Göttingen, Germany. The algal cells were kept in ESP Ag medium at 25 °C with continuous illumination of 30 µmol photons m−2 s−1 in a growth chamber, as recommended by EPSAG (Grünewald et al. 1997). The low-intensity illumination during recovery steps was continuous at 5–10 µmol photons m−2 s−1.
Construction of transformation vector pH124-EGFP-Ble
The expression vector pH124, which was used as the expression vector backbone, contained the ble gene driven by the RBCS2 promoter, which leads to algal zeomycin resistance. Based on the pH124 vector, we designed the sequence by adding an expression box that included EGFP promoted by the PSAD promoter upstream of the ble expression box. HA and His flags were observed upstream and downstream of EGFP, respectively. Expression cassettes were inserted into the pH124 vector to obtain pH124-EGFP-Ble.
Protoplast isolation and basic transformation procedure
Plant Protoplast Preparation and Transformation Kit Plus (Real-Times (Beijing) Biotech Co., Ltd., China) was used for protoplast preparation according to instructions with a few modifications. Briefly, algal cells were collected during the logarithmic growth period by centrifugation at 1600 × g for 5 min. After removing the supernatant, the remaining algal cells were resuspended in 10 mL of enzyme solution containing 5 mL of solution I, 0.15 g cellulase, 0.04 g macerozyme R-10, reductant (5 µL), and 200 µL BSA (50 mg mL−1). Macerozyme R-10, derived from Rhizopus sp, is a multi-enzymatic system consisting of high pectinase and hemicellulase activities and low cellulase activities. Macerozyme R-10 is useful in isolating single plant cells by degrading the plant cell wall (Suzuki et al, 1967). Digestion was protected from light. After approximately 5 h, 10 µL of the suspension was observed under an optical microscope. Once the walls of most algal cells walls were removed, 10 mL of solution II was added to the protoplast solution and mixed gently to inactivate enzyme activity. Protoplasts were collected by centrifugation at 100 × g for 2 min and resuspended in 1 mL of solution II. After incubation on ice for 30 min, the upper liquid layer was carefully removed. The remaining protoplast pellets were resuspended in 1 mL solution III.
For transformation, 10 µL of pH124-EGFP-Ble plasmid (1 μg μL−1) was added to 100 µL of protoplast solution and gently mixed in a 1.5-mL centrifuge tube. An equal volume of solution IV (110 µL) was gently added to the mixture and maintained at room temperature for 20 min. To end the transformation process, double reaction volume of solution II (440 µL) was added to the centrifuge tube and the protoplasts were pelleted by centrifugation at 100 × g for 2 min. Subsequently, the transformed protoplasts were resuspended in 1 mL of solution V and incubated overnight at room temperature in the dark. To obtain transformants, protoplasts were collected by centrifugation at 100 × g for 2 min and cultured in 1 mL of ESP medium at 5–10 µmol m−2 s−1 for 6 h, followed by overnight incubation at low-intensity illumination of 30 µmol photons m−2 s−1 at room temperature. Protoplasts were spread on ESP medium (containing 2% agar) with zeomycin resistance (5 µg mL−1) and a monoclonal of algal cells was observed for approximately 7–20 days.
Optimization of H. pluvialis protoplast transformation
To improve protoplast transformation efficiency, several factors were evaluated, including zeomycin concentration, growth stage, number of plasmids, plasmid linearization, and duration of low-intensity illumination.
To determine the best zeomycin concentration in the ESP medium for screening transformants, zeomycin concentrations of 0, 5, 10, 15, and 20 µg mL−1 were prepared. To select the best algal cell growth state for protoplast transformation, different growth periods, including early logarithmic phase, log growth phase, and late logarithmic phase, were chosen. pH124-EGFP-Ble at 1, 5, 10, and 20 µg was added to 9 × 106 protoplasts, respectively, based on a previous study (Yuan et al. 2019). The plasmid was extracted using a Plasmid Mini Kit II (Omega Bio-Tek, USA), and the DNA concentration was at least 1 µg µL−1.
Transformation efficiency can be improved by linearizing the expression vector in C. reinhardtii (Kindle et al. 1989). Hence, transformation with pH124-EGFP-Ble linearized by Not I was compared with that of pH124-EGFP-Ble.
After transformation, the protoplasts were incubated overnight in the dark. Before being transferred to normal-intensity illumination 30 µmol photons m−2 s−1, the protoplasts were subjected to low-intensity illumination 5–10 µmol photons m−2 s−1 to recover the cells. To determine the duration of the low intensity illumination, the recovery times were set at 0, 6, 12, and 24 h.
DNA/RNA extraction and first-strand cDNA synthesis
Total genomic DNA of H. pluvialis was isolated using the cetyltrimethylammonium bromide method (Murray and Thompson, 1980). The cells were collected by centrifugation at the speed of 5000 × g for 5 min and cracked with CTAB cracking liquid (2%) at 65 °C for approximately 1 h. Subsequently, an equal volume of chloroform-isopentyl alcohol (24:1, v/v) was added. The supernatant was then transferred into a new 1.5-mL centrifuge tube after centrifugation at 12,000 × g for 10 min. An equal volume of isopropyl alcohol was added, followed by placing the samples at − 20 °C for 30 min. Genomic DNA was then washed with 70% ethanol and resuspended in distilled water.
The total RNA of the transformants was extracted according to the instructions of the Plant Total RNA Extraction Kit (Tiangen Biotech (Beijing) Co., Ltd, China) and frozen in liquid nitrogen. Total RNA was stored at − 80 °C for further experiments. First-strand cDNA was synthesized using the PrimeScript TM II 1st Strand cDNA Synthesis Kit (TaKaRa Bio Inc., Japan), according to the manufacturer’s instructions. The resulting cDNA samples were stored at − 80 °C.
Regular PCR, reverse-transcript PCR, and quantitative real-time PCR
To confirm the transformation of the target gene, regular PCR was performed using the genomic DNA of putative transformants as a template. The ble primers were Hpble-F: ATATCAAGCTTATCGATACCGT, Hpble-R: CCAGGGTTTTCCCAGTCACGACGTT; the EGFP primers were Hpegfp-F: CGATGTTCCAGATTACGCTATGGTCAGCAAGGGC, Hpegfp-F: TGGTGATGGTGATGATGCTTGTACAGCTCGTCC; and the internal reference primers were Hpact-F: ACCTCAGCGTTCAGCCTTGT, Hpact-R: TGGTCCACGACACCATCAAC. PCR was conducted using 10-µL samples containing ~ 10 ng of genomic DNA, 0.5 µM each primer, 200 µM of dNTPs, 1 × reaction buffer, and 0.5 U Taq DNA polymerase (Aidlab Biotechnologies, Beijing, China). The PCR protocol was as follows: initial denaturation at 95 °C for 5 min, followed by 32 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 60 s, and a final 5-min extension step at 72 °C. In PCR amplification, 2 × Taq Plus PCR MasterMix (Real-Times (Beijing) Biotech Co., Ltd., China) was utilized.
To investigate the expression of the transferred genes, reverse-transcription PCR was performed using the cDNA of positive transformants as a template. In the reverse-transcription PCR, the ble primers were RTble-F: GTCGAGTTCTGGACCGACCG, RTble-R: GCGCTGATGAACAGGGTCAC; the EGFP primers were RTegfp-F: GAAGGGCATCGACTTCAAGGA, RTegfp-R: CTGGGTGCTCAGGTAGTGGT; and the internal reference primers were Hpact-F: ACCTCAGCGTTCAGCCTTGT, Hpact-R: TGGTCCACGACACCATCAAC. RT-PCR was conducted using 20-µL samples containing ~ 10 ng of cDNA, 0.5 µM each primer, 200 µM of dNTPs, 1 × reaction buffer, and 0.5 U Taq DNA polymerase (Aidlab Biotechnologies). The PCR protocol was as follows: initial denaturation at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, extension at 72 °C for 10 s, and a final 5-min extension step at 72 °C. In PCR amplification, 2 × Taq Plus PCR Master Mix (Real-Times (Beijing) Biotech Co., Ltd.) was applied.
To determine the expression level of ble and EGFP, the primer pairs RTble-F/R and RTegfp-F/R were used. The primer sequences used were RTble-F: GTCGAGTTCTGGACCGACCG, RTble-R: GCGCTGATGAACAGGGTCAC and RTegfp-F: GAAGGGCATCGACTTCAAGGA, RTegfp-R: CTGGGTGCTCAGGTAGTGGT. The internal reference primers were Hpact-F: ACCTCAGCGTTCAGCCTTGT and Hpact-R: TGGTCCACGACACCATCAAC. In a total volume of 25 µL, containing 2 µL of cDNA, 12.5 µL SYBR mix, 0.5 µL gene-specific primers, and 10 µL ddH2O, the qRT-PCR was performed with a Roche Light Cycler 480 system. qRT-PCR was conducted using the SYBR Premix Ex Taq II Kit (TaKaRa Bio Inc). The PCR protocol was as follows: initial denaturation at 95 °C for 20 s, then 40 cycles of denaturation at 95 °C for 5 s, and annealing and extension at 60 °C for 15 s each. Relative quantification analysis was performed using 2−∆∆Ct method.
For all PCRs a negative control using ddH2O as a template was used to check for the presence of PCR contaminants, and a positive control was performed using plasmid DNA as a template to confirm the success of PCR. At least three biological replicates were performed for each testing sample.
Statistical analysis
The number of transformants and positive transformants was counted according to the following formula:
In formula (1), T represents transformation efficiency, m is the number of transformants, and M is the number of total protoplasts. In formula (2), P represents a positive rate, n is the number of positive transformants, and m is the number of transformants.
For the Person’s T-test to compare two means, P-value less than 0.05 is considered the statistical significance.
Results
Construction of transformation vector pH124-EGFP-Ble and establishment of protoplast transformation method in H. pluvialis
The expression vector pH124-EGFP-Ble, containing ble and EGFP expression boxes, was obtained by inserting the EGFP expression box into the pH124 vector. The total length of the expression vector pH124-EGFP-Ble was 6691 bp. The algal transformants conferred the zeomycin resistance and the characteristics of the fluorescent protein. There was one specific restriction endonuclease site, Not I, upstream of the EGFP expression box for linearization of the expression vector (Fig. 1).

The map of constructed vector pH124-EGFP-Ble
Following the transformation of pH124-EGFP-Ble into H. pluvialis protoplasts, which were treated with cellulase and macerozeme, transformed cells were poured onto the ESP culture containing zeomycin (5 µg mL−1). Monoclonal colonies were obtained (Fig. 2A) and transferred to new ESP medium (5 µg mL−1zeomycin) with sterilized toothpicks (Fig. 2B). The PCR results showed that the ble fragment at 1289 bp could be amplified from the genomic DNA, similar to the positive control, with primers Hpble-F/R. EGFP was also amplified from the transformants using the primers Hpegfp-F/R. Moreover, none of the fragments was observed in the genomic DNA of the wild type (Fig. 3A, B). The above results indicate that both ble and EGFP were successfully inserted into the genomic DNA of H. pluvialis. Furthermore, the transcription of the transferred genes was detected by RT-PCR. The results showed that ble with primers RTble-F/R generated 98 bp fragments and EGFP with primers RTegfp-F/R generated 238 bp fragments in all positive transformants, similar to the positive control, which verified the expression of ble and EGFP in transgenic H. pluvialis (Fig. 3D, E). Here, β-actin from H. pluvialis was used as an internal reference (Fig. 3C, F).

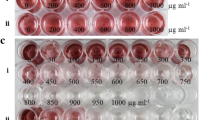
The transformants screened by zeomycin. A The transformants on the ESP plates with 5 μg mL−1 zeomycin. B The transformants transferred to a new ESP plate with 5 μg mL−1 zeomycin

Molecular confirmation of transformants by PCR and RT-PCR. A M represents the DNA ladder, WT is the wild type 192.80, and P is the positive control of pH124-EGFP-Ble plasmid DNA. The PCR products of ble using transformant DNA as templates. B The PCR products of EGFP using transformant DNA as templates. C The PCR products of Hpactin using transformant DNA as templates. D The RT-PCR products of ble using the cDNA from positive transformant as templates. E The RT-PCR products of EGFP using the cDNA from positive transformant as templates. F The RT-PCR products of Hpactin using the cDNA from positive transformant as templates
Optimization of the protoplast transformation method
To define the proper concentration of zeomycin for screening transformants, we designed the screening concentrations of zeomycin at 0, 5, 10, 15, and 20 µg mL−1. The results showed that green algal cells grew on the culture with no zeomycin, whereas there were only a few green colonies on the medium with 5 µg mL−1zeomycin (Fig. 4A, B). No colonies were observed on the medium with 10, 15, or 20 µg mL−1zeomycin (Fig. 4C–E). Considering the difficulty of transformation and low expression of the foreign gene in H. pluvialis, we chose 5 µg mL−1zeomycin for screening the transformants.

The concentration of zeomycin for transformants selection. A The algal cells on ESP culture with no zeomycin. B 5 μg mL−1 zeomycin. C 10 μg mL−1 zeomycin. D 15 μg mL−1 zeomycin. E 20 μg mL−1 zeomycin
The cell growth periods of H. pluvialis were designed as the early logarithmic phase (day 4), middle logarithmic phase (day 5), and late logarithmic phase (day 6), the cell concentrations were 2.47 × 105, 4.03 × 105, and 6.63 × 105 cells mL−1, respectively (Fig. 5). The numbers of transformants at early, kid, and late logarithmic phase were 161.7 ± 4.5, 138.3 ± 5.3, and 94.3 ± 6.8, and the positive transformants were 41.3 ± 5.8, 44.7 ± 2.1, and 18.7 ± 1.7, respectively. The results indicated that transformation efficiency decreased from early logarithmic phase to late logarithmic phase. The positive rate of the mid logarithmic phase was the highest compared to that of the early and late logarithmic phases (Fig. 6). We concluded that cells in the mid logarithmic growth phase might be the best phase for protoplast transformation.

The growth curve of H. pluvialis. Data are expressed as the average and standard deviation in biological replicates (n = 3)

Effects of different culture state of algal cells on the transformation of H. pluvialis. A Transformation efficiency of H. pluvialis. B Positive rate of H. pluvialis. *Indicates a significant difference between the first group and other groups, respectively. Data are expressed as the average and standard deviation in biological replicates (n = 3)
To provide the optimum proportion of the expression vector to protoplast for transformation, the number of vectors was set at 1, 5, 10, and 15 µg per 9 × 106 protoplasts. The number of transformants of 1, 5, 10, and 15 µg per 9 × 106 protoplasts was 135.7 ± 3.3, 163.7 ± 3.4, 107.3 ± 3.7, and 108.3 ± 3.9, and the positive transformants were 30.0 ± 0.9, 31 ± 0.9, 36.3 ± 1.2, and 33.7 ± 4.2, respectively. The results indicated that the 5 µg vector for 9 × 106 protoplasts had the highest transformation efficiency, which significantly declined as the vector amount increased to 10 µg and 15 µg. As for the positive rate, 10 µg of vector for 9 × 106 protoplasts were found to be the best proportion (Fig. 7). We concluded that most transformants were obtained at a proportion of 5 µg vector for 9 × 106 protoplasts, whereas the most positive transformants were accessible when the proportion was 10 µg vector for 9 × 106 protoplasts.

Effects of different amounts of vectors per 9 × 106 protoplasts on transformation of H. pluvialis. A The transformation efficiency of H. pluvialis. B The positive rate of H. pluvialis. *Indicates a significant difference between the first group and other groups, respectively. Data are expressed as the average and standard deviation in biological replicates (n = 3)
Restriction endonuclease Not I was used to linearize the transformation vector pH124-EGFP-Ble. The number of transformants obtained by pH124-EGFP-Ble and that linearized by Not I was 112.7 ± 6.1 and 155.3 ± 5.8, and the positive transformants were 37.3 ± 4.9 and 20.8 ± 2.1, respectively. The transformation efficiency of the circular vector pH124-EGFP-Ble was higher than that of linearized pH124-EGFP-Ble by Not I. However, the positive rate of circular pH124-EGFP-Ble was much lower than that of linearized pH124-EGFP-Ble. We hypothesize that the linearized vector would be more suitable for protoplast transformation of H. pluvialis (Fig. 8).

Effects of linearization by Not I on transformation of H. pluvialis. A The transformation efficiency of H. pluvialis. B The positive rate of H. pluvialis. *Indicates significant difference between pH124-EGFP-Ble and linearized pH124-EGFP-Ble. Data are expressed as the average and standard deviation in biological replicates (n = 3)
The number of transformants treated with low intensity light for 0, 6, 12, and 24 h was 27 ± 5.7, 134.3 ± 8.2, and 72.3 ± 7.6, and the positive transformants were 2.0 ± 0.8, 43.3 ± 2.1, 28.2.7 ± 1.2, and 24.9 ± 2.6, respectively. The results indicated that the transformation efficiency and positive rate of no low intensity of illumination (i.e., the normal light intensity of 30 µmol photon m−2 s−1) were the lowest and that of low intensity light of 5–10 µmol photon m−2 s−1 for 6 h was the highest. The transformation efficiency and positive rate gradually declined as the duration of low intensity light was extended (Fig. 9). We concluded that a long duration or no low intensity of illumination was harmful to the PEG-mediated protoplasts, and 6 h was optimal for the recovery of protoplasts.

Effects of low intensity of illumination for different duration time on the transformation of H. pluvialis. A The transformation efficiency of H. pluvialis. B The positive rate of H. pluvialis. * and **indicate a significant difference between first with other groups, respectively. Data are expressed as the average and standard deviation in biological replicates (n = 3)
Analysis of expression stability of ble and EGFP in positive transformants
Transformants were kept on resistant ESP medium (5 µg mL−1 zeomycin). The expression of exogenous genes in the positive transformants was successfully detected, with no expression in the wild type H. pluvialis 192.80 as a negative control. ble and EGFP were consistently detected to be stably expressed after being kept on the screening medium for an extended duration, although the transcriptional levels varied (Fig. 10). Thus, an improved protoplast transformation of H. pluvialis was established, which is convenient, stable, and simple in operation, and ensured that there were sufficient available transformants to support our research.

The relative expression level of ble and EGFP in the transformants. The blank bar graphs represent the expression level of ble, and the bar graphs filled with slash represent the expression level of EGFP. Data are expressed as the average and the standard deviation in biological replicates (n = 3)
Discussion
When exposed to external stressors, H. pluvialis enters the aplanospore stage during which astaxanthin content significantly increases and the algal cell wall is thickened at the same time. The structure of the cell wall in the aplanospore stage is mainly composed of the secondary wall, which contains mainly mannose and much less cellulose (Damiani et al. 2006). The cell wall of H. pluvialis may play an important role in recognizing and responding to external stimuli. Therefore, mechanical damage to the cell wall of H. pluvialis might be caused by bombardment during transformation with the biolistic method, which might result in the procedural death of the algal cells and, finally, the failure of transformation. In this study, cellulase was used to treat H. pluvialis cells in the logarithmic growth period to eliminate the cell wall which is a barrier to the cell. The cell membrane has good permeability mediated by PEG, so that the expression vector easily transforms into protoplasts.
In the previous transformation of H. pluvialis with biolistic bombardments, endogenous gene promoters, including rbcL, CaMV35S, and tub, were functional. Belonging to the photosystem complex, the RBCS2 promoter might interact with light, and the gene RBCS2 requires the induction of light for expression. The RBCS2 promoter has been widely used to promote gene expression in the nuclei of C. reinhardtii. It has been proven that insertion of the first intron of the RBCS2 into ble coding sequence greatly increases the expression level of the RBCS2 promoter (Kindle and Sodeinde, 1994; liu et al., 2005; Lumbreras et al., 2010). Therefore, the RBCS2 promoter was used in the present study.
We evaluated the effect of low intensity light for different time periods before being transferred to the normal light intensity. These results indicated that the transformation of protoplasts required a low light intensity to adapt gradually to changes in the light environment. The promoters RBCS2 and PsaD were from C. reinhardtii, which might have led to a low expression level of the exogenous resistance gene ble and EGFP when transformed into the protoplasts of H. pluvialis (Fig. 10). The endogenous promoter tub was utilized to promote the resistance gene aadA, and the expression level tended to be higher in a former study reported by Yuan et al. (2019). However, the biolistic method exhibited a low repeatability.
We observed that there was a relatively low positive rate in the transformants, which may be related to the low concentration of zeomycin. To improve the efficiency of the transformation, it would be optimal to select an appropriate concentration of 5–10 µg mL−1. Although the ble gene is one of the most successful screening markers for C. reinhardtii transformation, it has previously not been successfully expressed for screening transformants in H. pluvialis (Yuan et al. 2019). In this study, we used protoplast transformation to transform the ble and EGFP expression boxes into the genome of H. pluvialis, which were successfully expressed under the influence of the RBCS2 promoter and PsaD promoter, and confer zeomycin resistance. More than 100 transformants were obtained in each transformation using our method, and foreign genes were stably expressed. This provided a simple and efficient transformation system for subsequent research.
Introducing an expression vector into protoplasts and integrating foreign genes into the genomic DNA of H. pluvialis were the key technologies in the transformation experiment. In our study, one cleavage site of the restriction endonuclease Not I, which has been frequently used, was introduced into the vector. We concluded that the circular expression vector might access the protoplasts with less barrier, but the linear expression vector appeared to be integrated into the nuclear genome better. These results are consistent with those of C. reinhardtii transformation (Kindle et al., 1989). There are problems, such as multiple copy integration and gene silencing, in the nuclear genome transformation of C. reinhardtii using the biolistic bombardment method (Wu et al., 2000; Cerutti 2003). However, low-copy integration is usually detected in the transformation of C. reinhardtii using the glass-bead method, showing stable expression of foreign genes (Wang et al. 2018). In our study, neither gene silencing nor abnormal growth was observed in the transformants, indicating that the protoplast transformation method helped to obtain stable transformants. Furthermore, removing the cell wall that would prevent the formation of spores also significantly prevented the transformation of foreign genes. The plasma membrane of protoplasts possesses the ability to accept foreign plasmids. Considering its lower cost and convenient operation, the method provided here is simple and efficient with more applicability.
In conclusion, PEG-mediated protoplast transformation of H. pluvialis might be a viable solution to the difficulties in transformation, which has been troubling scientists for a long time. Based on the development of this transformation system, many important studies involving genetic engineering to regulate the synthesis of carotenoids, including astaxanthin, gene functions, such as cell wall formation and transcription regulation mechanism, as well as gene editing for the silencing genes, and new characteristic strains are expected to follow which will further the study of H. pluvialis and its most important metabolite astaxanthin. The protoplast transformation method will surely be a significant prospect for the study of microalgae.
Availability of data and materials
The authors promise that all data generated or analyzed in the present study are included in this article and in additional information.
References
Izumi-Nagai K, Nagai N, Ohgami K, Satofuka S, Ozawa Y, Tsubota K, Ohno S, Oike Y, Ishida S (2008) Inhibition of choroidal neovascularization with an anti-inflammatory carotenoid astaxanthin. Invest Opthalmol Vis Sci 49:1679–1685
Liu QQ, Yu HX, Zhang WJ, Wang HM, Gu MH (2005) Specific expression of the foreign gene regulated by the rice rbcS promoter in transgenic rice. J Plant Physiol Mol Biol 31:247–253
Lumbreras V, Stevens DR, Purton S (2010) Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron. Plant J 14:441–447
Cerutti HD (2003) RNA interference: traveling in the cell and gaining functions. Trends Genet 19:39–46
Chang CH, Chen CY, Chiou JY, Peng RY, Peng CH (2010) Astaxanthine secured apoptotic death of PC12 cells induced by beta-amyloid peptide 25–35: its molecular action targets. J Med Food 13:548–556
Chew BP, Wong MW, Park JS, Wong TS (1999) Dietary beta-carotene and astaxanthin but not canthaxanthin stimulate splenocyte function in mice. Anticancer Res 19:5223–5227
Curek GD, Cort A, Yucel G, Demir N, Ozturk S, Elpek GO, Savas B, Aslan M (2010) Effect of astaxanthin on hepatocellular injury following ischemia/reperfusion. Toxicology 267:147–153
Damiani MC, Leonardi PI, Pieroni OI, Cáceres EJ (2006) Ultrastructure of the cyst wall of Haematococcus pluvialis (Chlorophyceae): wall development and behaviour during cyst germination. Phycologia 45:616–623
Doron L, Segal N, Shapira M (2016) Transgene expression in microalgae-from tools to applications. Front Plant Sci 7:1–24
Emiko M, Osamu H, Yuji N, Katsura M, Satomi A, Satoko A, Tomohisa T, Satoshi K, Takashi M, Toshikazu Y (2008) Astaxanthin protects mesangial cells from hyperglycemia-induced oxidative signaling. J C Biochem 103:1925–1937
Gross GJ, Lockwood SF (2005) Acute and chronic administration of disodium disuccinate astaxanthin (Cardax TM) produces marked cardioprotection in dog hearts. Mol Cell Biochem 272:221–227
Grünewald K, Hagen C, Braune W (1997) Secondary carotenoid accumulation in flagellates of the green alga Haematococcus lacustris. Eur J Phycol 32:387–392
Hagen C, Siegmund S, Braune W (2002) Ultrastructural and chemical changes in the cell wall of Haematococcus pluvialis (Volvocales, Chlorophyta) during aplanospore formation. Eur J Phycol 37:217–226
Kao KN, Michayluk MR (1974) A method for high-frequency intergeneric fusion of plant protoplasts. Planta 115:355–367
Kathiresan S, Chandrashekar A, Ravishankar GA, Sarada R (2009) Agrobacterium-mediated transformation in the green alga Haematococcus pluvialis (Chlorophyceae. Volvocales). J Appl Phycol 45:642–649
Kathiresan S, Sarada R (2009) Towards genetic improvement of commercially important microalga Haematococcus pluvialis for biotech applications. J Appl Phycol 21:553–558
Kindle KL, Sodeinde OA (1994) Nuclear and chloroplast transformation in Chlamydomonas reinhardtii: strategies for genetic manipulation and gene expression. J Appl Phycol 6:231–238
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Pratheesh PT, Vineetha M, Kurup GM (2013) An efficient protocol for the agrobacterium-mediated genetic transformation of microalga Chlamydomonas reinhardtii. Mol Biotechnol 56:507–515
Rochaix J (1995) Chlamydomonas reinhardtii as the photosynthetic yeast. Annu Rev Genet 29:209–230
Sharon-Gojman R, Maimon E, Leu S, Zarka A, Boussiba S (2015) Advanced methods for genetic engineering of Haematococcus pluvialis (Chlorophyceae, Volvocales). Algal Res 10:8–15
Suzuki H, Abe T, Urade M, Nisizawa K, Kuroda A (1967) Nature of the macerating enzymes from Rhizopus sp. J Ferment Technol 45:73–85
Vasil J, Indre K (1987) Developing cell and tissue culture systems for the improvement of cereal and grass crops. J Plant Physiol 128:193–218
Wang Q, Cobine PA, Coleman JJ (2018) Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fungal Genet Biol 117:21–29
Wang X, Willén R, Wadström T (2000) Astaxanthin-rich algal meal and vitamin C inhibit Helicobacter pylori infection in BALB/cA mice. Antimicrob Agents Chemother 44:2452–2457
Wu SD, Jeong B, Zhang C, Cerutti H (2000) Transgene and transposon silencing in Chlamydomonas reinhardtii by a DEAH-box RNA helicase. Science 290:1159–1162
Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2:1565–1572
Yuan G, Xu X, Zhang W, Zhang W, Cui Y, Qin S, Liu T (2019) Biolistic transformation of Haematococcus pluvialis with constructs based on the flanking sequences of its endogenous alpha tubulin gene. Front Microbiol 10:1–13
Acknowledgements
We thank Dr. Danqiong Huang for proposing advises to prepare the manuscript and word editing.
Funding
This work was supported by China National Key R&D Project for Synthetic Biology (2018YFA0902500), Shenzhen Basic Research Projects (JCYJ20180507182405562), the National Natural Science Foundation of China (41876188), the National Natural Science Foundation of China (32000268), Guangxi Innovation Drive Development Special Funding (Gui Ke AA18242047), and Grant Plan for Demonstration City Project for Marine Economic Development in Shenzhen to Dr. Zhangli HU.
Author information
Authors and Affiliations
Contributions
Writing—original draft preparation, CG; data curation, MA; formal analysis, RM; methodology, JZ and XL; investigation, YJ and DZ; methodology, CL; supervision, CW and ZH; project administration and funding acquisition, Z.H. All authors agree with the authorship and have approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, C., Anwar, M., Mei, R. et al. Establishment and optimization of PEG-mediated protoplast transformation in the microalga Haematococcus pluvialis. J Appl Phycol 34, 1595–1605 (2022). https://doi.org/10.1007/s10811-022-02718-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-022-02718-x