
Abstract
Chronic pain is not only one of the most common health problems, it is often challenging to treat adequately. Chronic pain has a high prevalence globally, affecting approximately 20% of the adult population. Chronic inflammatory pain and neuropathic (nerve) pain conditions are areas of large unmet medical need because analgesic/adjuvant agents recommended for alleviation of these types of chronic pain often lack efficacy and/or they produce dose-limiting side effects. Recent work has implicated the NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome in the pathobiology of chronic pain, especially neuropathic and inflammatory pain conditions. NLRP3 is activated by damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). This in turn leads to recruitment and activation of caspase-1 an enzyme that cleaves the inactive IL-1β and IL-18 precursors to their respective mature pro-inflammatory cytokines (IL-1β and IL-18) for release into the cellular milieu. Caspase-1 also cleaves the pyroptosis-inducing factor, gasdermin D, that leads to oligomerization of its N-terminal fragment to form pores in the host cell membrane. This then results in cellular swelling, lysis and release of cytoplasmic contents in an inflammatory form of cell death, termed pyroptosis. The ultimate outcome may lead to the development of neuropathic pain and/or chronic inflammatory pain. In this review, we address a role for NLRP3 inflammasome activation in the pathogenesis of various chronic pain conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
According to the International Association for the Study of Pain (IASP), pain is defined as: “An unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage” (Raja et al. 2020). Pain can be defined according to its duration (acute or chronic), type (nociceptive, inflammatory, neuropathic) and intensity (mild, moderate, severe). In this review, we have focused on neuropathic pain and chronic inflammatory pain.
Prevalence
Chronic pain is a condition where pain lasts for more than 3 months and it occurs at least once a week (Debono et al. 2013). It imposes a significant burden on 30% of the world's individuals and economies (Cohen et al. 2021). The prevalence of chronic pain ranges from 11 to 40%, and the cure rate is low (Dahlhamer et al. 2018; Elliott et al. 2002). In a pan-European epidemiological study, the 1-month prevalence of moderate and severe chronic non-cancer pain was 19% (Reid et al. 2011), which was similar to that in Australia (15.0% in males, 16.9% in females (Economics 2019), Denmark (16%) (Harker et al. 2012) and Sweden (18%) (Harker et al. 2012). A study in the United States reported a point prevalence of approximately 20.4% (Dahlhamer et al. 2018). In a systematic review of the burden of chronic pain in the UK, the prevalence of chronic pain was 43.5% and that of moderate-to-severe disabling pain was in the range 10% to 14% (Fayaz et al. 2016). Compared with the young, the elderly have a much higher prevalence of chronic pain at 66% (Reid et al. 2011). Chronic pain is underpinned not only by pathological changes due to disease and neuroplastic changes in the somatosensory nervous system, but also by psychological factors and other influences such as region, social culture, lifestyle and behavior (Edwards et al. 2016; Diatchenko et al. 2013; Mills et al. 2019). Patients suffering from chronic pain have a higher incidence of co-morbid conditions such as depression, anxiety and insomnia (Debono et al. 2013), affecting their ability to work and impairing their quality of life. The annual medical expenses due to chronic pain are high. In Australia, approximately 15.4% of the population suffer from chronic pain, and the annual cost per person is AUD 22,588–42,979 (Economics 2019). From a macro-economic perspective, the direct costs of medical treatments and the associated socioeconomic costs are huge at USD560-635 billion per annum in the USA alone (Institute of Medicine Committee on Advancing Pain Research and Education 2011).
Unmet medical need
Medications recommended for the pharmacological management of chronic pain depend upon the type of pain that needs to be treated. For example, drugs used to relieve chronic inflammatory pain include nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen and COX2 inhibitors such as celecoxib (Eccleston et al. 2017). However, treatment with NSAIDs may induce acute hemorrhagic gastritis, peptic ulcer and an increased cardiovascular and renal risk (Shah and Mehta 2012; Enthoven et al. 2016). Additionally, long-term use of COX2 inhibitors is associated with increased risk of cardiovascular side-effects, such as heart attack and stroke (Labianca et al. 2012). First-line drug treatments for the relief of neuropathic pain include tricyclic antidepressants (e.g. amitriptyline), duloxetine and some anticonvulsants (e.g. pregabalin and gabapentin) (Finnerup et al. 2015), but these drugs often lack efficacy and/or have dose-limiting side effects such as orthostatic hypotension, tachycardia, sedation and dizziness (Labianca et al. 2012; Shah and Mehta 2012). Although opioids also relieve pain, they are only recommended as third-line treatments for the relief of neuropathic pain (Vowles et al. 2015). Opioids can be addictive and they evoke a plethora of side effects when used repeatedly (Vowles et al. 2015; Finnerup et al. 2015). Since chronic pain is underpinned by complex mechanisms, pain treatment should ideally be based not only on the severity of pain, but also on its underlying pathogenesis. To address the large unmet medical need for new highly effective and well-tolerated analgesics, many researchers over the past three decades have investigated the pathogenesis of various chronic pain states aimed at identifying novel drug targets for use in novel non-opioid analgesic drug discovery programs. One such target is the NLRP3 inflammasome and so in the following sections, we review a role for the NLRP3 inflammasome in the pathogenesis of neuropathic and chronic inflammatory pain.
NLRP3 inflammasome structure
The NLRP3 inflammasome is a macromolecular protein complex with a molecular weight in the range 500–700 kDa (Broz and Monack 2011). It is comprised of a sensor (NLRP3), an adaptor (ASC; also known as PYCARD) and an effector (caspase 1) (Bauernfeind and Hornung 2013; Tang et al. 2018; Swanson et al. 2019) (Fig. 1). NLRP3 is a tripartite protein that contains an amino-terminal pyrin domain (PYD), a central NACHT domain and a carboxy-terminal leucine-rich repeat domain (LRR domain) (Swanson et al. 2019; Sutterwala et al. 2014; Tang et al. 2018) (Fig. 1). ASC is an important intracellular junction protein comprising 195 amino acid residues and a molecular weight of 21.5 kDa. It comprises the Pyrin domain (PYD) and the caspase recruitment domain (CARD) (Wen et al. 2011). ASC can connect NLRP3 upstream to Caspase-1 downstream, and interact with PYD to recruit pro-caspase-1 through the CARD domain (Wen et al. 2011). This in turn facilitates NLRP3 to activate the cysteine protease, caspase-1 (Wu et al. 2010). ASC is mainly located in the nucleus of human monocytes/macrophages, but it can be rapidly distributed into the cytoplasm as needed, promoting the activation of the NLRP3 inflammasome (Gris et al. 2010). Caspase-1, also called IL-1 converting enzyme (ICE), is the effector of the NLRP3 inflammasome (Franchi et al. 2009).

Structure of the NLRP3 inflammasome
Activation of the NLRP3 inflammasome
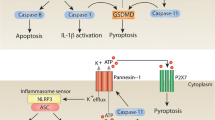
The NLRP3 inflammasome which is part of the innate immune system, is primed and activated by a broad array of sterile and microbial stimuli (Swanson et al. 2019). Priming (signal 1) upregulates expression of the inflammasome components, NLRP3, caspase 1 and pro-IL-1β (Fig. 2) (Swanson et al. 2019). This activity is induced via recognition of various pathogen-associated molecular patterns (PAMPs) (Franchi et al. 2009) via pattern recognition receptors (PRR) that include toll-like receptors (TLRs) or nucleotide-binding oligomerization domain-containing protein 2 (NOD2) or cytokines such as tumor necrosis factor (TNF) and Il-1β that leads to nuclear factor-κB (NF-κB) activation and gene transcription (Swanson et al. 2019). The 2nd function of priming is stabilization of NLRP3 in an auto-suppressed inactive, but signal-competent state, by induction of post-translational modifications such as phosphorylation, ubiquitylation and sumoylation (Swanson et al. 2019).
Signal 2 (activation) may be provided by numerous PAMPs such as bacteria, viruses and fungi, or damage-associated molecular patterns (DAMPs) including crystals (e.g. urea, cholesterol, silica), particulates (β-amyloid, environmental irritants) and adenosine triphosphate (ATP) (Fig. 2) (Swanson et al. 2019). Once formed, the NLRP3 inflammasome recruits and activates caspase-1 that cleaves pro-IL-1β and pro-IL-18 to produce the corresponding mature cytokines, namely IL-1β and IL18 (Franchi et al. 2009). These cytokines contribute to the removal of the pathogens and activation of the adaptive immune response (Franchi et al. 2009) via induction of cellular stress (Swanson et al. 2019). Precisely how the NLRP3 senses cellular stress and which pathways are induced to culminate in NLRP3 activation and inflammasome formation remain to be fully elucidated (Swanson et al. 2019). Because IL-1β and IL-18 are upstream components of the immune response, they can stimulate the production of a variety of inflammatory mediators, but their excessive production can lead to inflammatory disease (Swanson et al. 2019). Caspase-1 also cleaves the pyroptosis-inducing factor gasdermin D (Wu et al. 2010). The N-terminal fragment of gasdermin D oligomerizes and forms pores in the host cell membrane, leading to cellular swelling, lysis, and release of cytoplasmic contents in an inflammatory form of cell death, called pyroptosis (Wu et al. 2010).
The NLRP3 inflammasome is particularly prominent in inflammatory diseases and it is a Fig. 2 potential target for discovery of novel analgesics as inflammatory mechanisms contribute to the pathophysiology of multiple chronic pain conditions (Tang et al. 2018).

Priming and activation of the NLRP3 inflammasome (TLR: Toll-like receptors; TNF: Tumor necrosis factor; IL-1β: interleukin-1β; NOD2: Nucleotide Binding Oligomerization Domain Containing 2; TWIK2, two-pore domain weak inwardly rectifying K+ channel 2; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; IFN-α: interferon-α; TLR4: toll-like receptors 4; ATP: adenosine triphosphate; IRF3: interferon regulatory factor 3; CLIC: chloride intracellular channel protein; ER: endoplasmic reticulum; P2X7: P2X purinoceptor 7; PtdIns4P: phosphatidylinositol-4-phosphate; ROS: reactive oxygen species; GlcNAc: N-acetylglucosamine; HK: hexokinase)
There are multiple mechanisms that may activate the NLRP3 inflammasome. These include potassium (K+) or chloride (Cl−) efflux, calcium (Ca2+) flux, lysosomal disruption, mitochondrial dysfunction, metabolic changes and trans-Golgi disassembly (Swanson et al. 2019).
K+ efflux
Almost all pathways that activate the NLRP3 inflammasome are associated with K+ efflux and the NLRP3 inflammasome can be activated in a simple hypokalemic environment (Muñoz-Planillo et al. 2013), which suggests that K+ outflow may be a common mechanism (Mathur et al. 2018). NLRP3 inflammasome agonists, such as extracellular ATP, stimulate translocation to the cell surface and activation of purinergic P2X7 receptors which are non-selective channels for Na+, K+ and Ca2+ ions (Franceschini et al. 2015). Interestingly, ATP-induced activation of the P2X7 receptor promoted K+ efflux via the K+ channel two-pore domain weak inwardly rectifying K+ channel 2 (TWIK2) (Di et al. 2018). Additionally, LPS-induced NLRP3 inflammasome activation is dependent upon TWIK2 (Di et al. 2018). Other stimuli that activate the NLRP3 inflammasome by inducing K+ efflux include particulates such as alum, silica, sodium urate crystals and calcium pyrophosphate crystals (Muñoz-Planillo et al. 2013). More specifically, K+ efflux drives NLRP3 oligomerization (Green et al. 2018).
Cl− efflux
A role for Cl− efflux in NLRP3 activation is based upon work showing that ATP-induced IL-1β secretion increased or decreased in response to extracellular Cl− concentrations that were low or high respectively, and that chloride intracellular channel proteins (CLICs) are needed for NLRP3 activation by multiple stimuli (Domingo-Fernández et al. 2017; Tang et al. 2017). Translocation of CLICs from the cytosol to the plasma membrane where they form anion channels, is dependent upon the release of mitochondrial reactive oxygen species (mtROS) whereas Cl− efflux occurs downstream of K+ efflux (Tang et al. 2017). The role of Cl− efflux is to promote ASC polymerization during NLRP3 inflammasome formation (Green et al. 2018).
Ca2+ flux
A role for Ca2+ mobilization in NLRP3 activation is supported by work suggesting that it may occur downstream of both NLRP3 and caspase 1 activation (Katsnelson et al. 2015). However, others showed that K+ efflux induced Ca2+ flux was transduced by the opening of plasma membrane channels or by the release of endoplasmic reticulum (ER)-linked intracellular Ca2+ stores (Di et al. 2018). For example, ATP mobilized Ca2+ influx weakly via the P2X7 receptor that was coordinated with K+ efflux (Di et al. 2018). This in turn promoted the release of ER-linked Ca2+ stores that was followed by the opening of membrane Ca2+ channels (Murakami et al. 2012; Yaron et al. 2015).
Generation of ROS in mitochondria
Release of mtROS and mitochondrial DNA (mtDNA) from dysfunctional mitochondria are key upstream events that induce the assembly and activation of NLRP3 inflammasomes (Jin and Flavell 2010). ROS production induces dissociation of the thioredoxin-interacting protein (TXNIP) from thioredoxin-1(TRX1) in the cytoplasm and promotes its binding to NLRP3, thereby activating the NLRP3 inflammasome (Lane et al. 2013). At the same time, TXNIP may translocate into mitochondria and bind to thioredoxin-2 (TRX2), resulting in mitochondrial dysfunction (Lane et al. 2013). Oxidized mitochondrial DNA can directly activate the NLRP3 inflammasome by acting as a DAMP for NLRP3 activation (Lane et al. 2013; Chen et al. 2017; Zhang et al. 2010). Removal of ROS in macrophages with the ROS scavenger, N-acetylcysteine, reduced intracellular caspase-1 activation that in turn reduced production of the pro-inflammatory cytokine, IL-1β (Zhou et al. 2011). NLRP3 activation can also be inhibited by the nuclear factor erythroid 2-related factor 2 (NRF2) to limit ROS levels (Liu et al. 2017; Su et al. 2021). NRF2 may also attenuate NF-κB activation resulting in downregulation of the expression of multiple NLRP3 components thereby negatively regulating NLRP3 inflammasome activity (Su et al. 2021; Li et al. 2008).
Lysosomal disruption
Crystalline or granular substances enter cells through macrophage endocytosis resulting in lysosomal acidification and reduced stability of the phagolysosome membrane so that it ruptures and releases the particulates into the cytoplasm, with the net effect being NLRP3 inflammasome activation (Lima et al. 2013; Hornung et al. 2008). Although a role for lysosomal cathepsins in particulate-induced NLRP3 activation was implicated by work using cathepsin inhibitors, this was discounted by findings showing that genetic deletion of individual cathepsins had a minimal effect on NLRP3 activation which in turn suggested that the various cathepsins may have redundant roles in NLRP3 activation (Orlowski et al. 2015). As lysosomal damage in response to particulates activates both K+ efflux and Ca2+ flux, this implicates a convergence of these processes in many NLRP3 activation pathways (Muñoz-Planillo et al. 2013; Murakami et al. 2012; Katsnelson et al. 2016).
Metabolic changes
Although the enzyme, hexokinase, is well known to catalyze glucose phosphorylation, it may also have a role in NLRP3 inflammasome activation. For example, in bacterial infection, N-acetylglucosamine released from lysosomes, bound to hexokinase at the mitochondrial surface to induce its relocation to the cytosol (Wolf et al. 2016).This resulted in NLRP3 inflammasome activation independent of K+-efflux with mtDNA detected in the cytosol (Wolf et al. 2016). In other work, the NLRP3 inflammasome was activated by free fatty acids (FFAs) derived from dietary sources or by upregulation of FA synthesis (Wen et al. 2011; Moon et al. 2015). Conversely, the anti-inflammatory AMP-activated protein kinase, suppressed FA-induced inflammation by limiting ROS production and activating autophagy which led to inhibition of NLRP3 inflammasome activation (Li et al. 2009b). Metabolic changes such as fasting and caloric restriction also negatively regulate NLRP3 such as that which is mediated by the ketone body, β-hydroxybutyrate, which inhibited NLRP3 activation, suppressed caspase-1 activation and reduced IL-1β release by inhibiting K+ efflux (Youm et al. 2015).
Trans-Golgi disassembly
Trans-Golgi disassembly into vesicles termed the dispersed trans-Golgi network (dTGN), may be induced by a range of NLRP3 stimuli (Chen and Chen 2018). Phosphatidylinositol-4-phosphate is a negatively charged phospholipid on the dTGN that recruits NLRP3 through ionic bonding with the conserved polybasic region of the dispersed dTGN, resulting in NLRP3 aggregation, a prerequisite step for downstream ASC oligomerization and caspase-1 activation (Chen and Chen 2018). Observations that K+ efflux is essential for NLRP3 recruitment but not dTGN formation, suggest that K+ efflux-dependent and mitochondria-dependent NLRP3 activation may be separate pathways that converge on Golgi disassembly, but this requires additional investigation (Swanson et al. 2019).
Apart from the aforementioned pathways that may potentially activate the NLRP3 inflammasome and produce cellular dysfunction and inflammatory diseases, there are multiple mechanisms that may inhibit this process. These latter mechanisms include autophagy (Biasizzo and Kopitar-Jerala 2020), microRNA (miR) post-transcriptional regulation of NLRP3 (Tezcan et al. 2019), and silencing of the heat shock protein family (Zuo et al. 2018; Mi et al. 2021), to negatively regulate NLRP3 inflammasome activation and so provide adequate immune protection and avoid severe tissue damage to the host caused by harmful stimuli.
Nociceptive (pain) signaling pathway
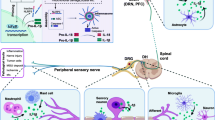
Dysregulation of the NLRP3 inflammasome can lead to excessive production of the pro-inflammatory cytokines, IL-1β and IL-18, leading to severe inflammation and/or a variety of diseases (Jin and Flavell 2010). The pro-inflammatory cytokines, IL-1β and IL-18 are secreted by macrophages and they can interact with their cognate receptors on nerve terminals to sensitize primary afferent sensory nerve fibers and induce pro-nociceptive signaling which is transduced into the dorsal horn of the spinal cord (Chen et al. 2014). This in turn may induce pronociceptive signaling in 2nd order neurons in the spinal cord that is propagated via the spinothalamic tract to higher order structures in the brain where it is interpreted as pain by the cerebral cortex (Doyle et al. 2019) (Fig. 3).

Nociceptive (Pain) signaling pathway
As shown in Fig. 3, key components of the nociceptive signaling (pain) pathway include peripheral nociceptors, first-order primary afferent sensory nerve fibers, the dorsal root ganglia that contain the cell bodies of primary sensory neurons, the dorsal horn of the spinal cord where the central terminals of primary afferent sensory nerve fibers form synapses with second order neurons, the spinothalamic tract and the brain.
The NLRP3 inflammasome is associated with chronic pain-related diseases
Neuropathic pain
Neuropathic pain is a major type of chronic pain characterized by spontaneous pain, allodynia and hyperalgesia (Zeilhofer et al. 2012; Bahari and Meftahi 2019). It may be caused by trauma and/or disease resulting in injury to peripheral nerves, posterior roots of the spinal cord, the spinal cord itself, and some central neurons. Neuroinflammatory responses contribute to the development of neuropathic pain following nerve injury with NLRP3 inflammasome activation contributing to these inflammatory responses (Abbaszadeh et al. 2018).
NLRP3 inflammasome activation in dorsal root ganglia (DRGs) from rodent models of neuropathic pain
Central neuropathic pain due to multiple sclerosis
Central neuropathic pain is a major complication of multiple sclerosis (MS) that affects up to 50% of patients with MS (Khan and Smith 2014). This type of pain is generally thought to be transduced by pathological changes in the brain and the dorsal horn of the spinal cord (Khan and Smith 2014). However, recent work in the myelin oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis (EAE) mouse model of MS-associated neuropathic pain has implicated an additional component involving activation of the complement system and the NLRP3 inflammasome in the lumbar dorsal root ganglia (DRGs) in the pathobiology of MS-associated neuropathic pain (Yousuf et al. 2019). In particular, there was transient activation of the complement system and prolonged activation of the NLRP3 inflammasome in the lumbar DRGs of EAE-mice that resulted in a small but significant increase in DRG levels of the pro-inflammatory cytokine, IL-1β, and marked hyper-excitability of medium-to-large-diameter Aβ nerve fibers that mediate mechanical hyperalgesia (Yousuf et al. 2019). In other work, chronic oral administration of the NLRP3 inhibitor, MCC950 in a relapsing–remitting EAE mouse model of MS-associated neuropathic pain progressively reversed neuropathic pain behaviour, further implicating a pathobiological role for NLRP3 inflammasome activation in MS-associated neuropathic pain (Khan et al. 2018).
Central post-stroke pain
Central post-stroke pain (CPSP) is defined as the neuropathic pain that arises either acutely or in the chronic phase of a cerebrovascular event and is a result of central lesions of the somatosensory nervous system (Liampas et al. 2020). CPSP has a prevalence of 11% with 31% of patients developing neuropathic pain symptoms within one month of stroke onset (Liampas et al. 2020). Ischemia/reperfusion injury (I/R) of the CNS after stroke, leads to cell necrosis or apoptosis, resulting in inflammation and an immune response (Li et al. 2009a). In a mouse model of thalamic hemorrhagic stroke, there was temporal development of pain behavior (mechanical and cold allodynia, heat hyperalgesia) in the hindpaw contralateral to the thalamic lesion, over a 14-day study period post-infarct induction (Huang et al. 2022). Additionally, there was decreased expression of miR-233, a miRNA known to negatively regulate the NLRP3 inflammasome, in the ipsilateral thalamus as early as 1-day post-infarct induction that persisted for the 14-day study duration (Huang et al. 2022). Concurrently, the NLRP3 inflammasome was activated and ipsilateral thalamic expression levels of caspase-1, ASC and NLRP3 were elevated by 1-day post-infarct which persisted until study completion on day 14 (Huang et al. 2022). Together, these data implicate a key role for NLRP3 inflammasome activation in the pathogenesis of post-stroke central neuropathic pain (Huang et al. 2022).
Chemotherapy induced neuropathic pain
In other work in a paclitaxel-induced rat model of chemotherapy induced peripheral neuropathy, there was a significant increase in NLRP3 inflammasome expression in CD68-labeled macrophages in the lumbar DRGs which was associated with ectopic firing of primary afferent sensory nerve fibers and the development of neuropathic pain behavior in these animals (Jia et al. 2017).
Radiculopathy
In a mouse model of radiculopathy, a neuropathic pain condition notoriously difficult to treat, there was temporal development of mechanical hyperalgesia in the bilateral hindpaws that was fully developed by day 3 and that persisted until at least day 15 of the model (Jin et al. 2017). At day 21 of the model, there was a marked increase in mRNA expression levels of multiple pro-inflammatory mediators including IL-1β, IL-6, TNFα and COX-2 with a fourfold decrease in the mRNA expression levels of the anti-oxidant enzyme, superoxide dismutase, SOD2 (Jin et al. 2017). Treatment of animals with the ‘free radical sponge’ fullerol, alleviated pain behavior and it suppressed otherwise increased mRNA expression levels of TNFα in the lumbar DRGs (Jin et al. 2017). In complementary work using cultured mouse lumbar DRG explants, incubation of explants with TNFα markedly increased the secretion of IL-1β that was suppressed by exposure to fullerol for 24 h (Jin et al. 2017). The increased secretion of IL-1β was underpinned by both increased expression of the NLRP3 inflammasome and caspase-1 and this was inhibited by fullerol (Jin et al. 2017). Together these findings suggest a role for NLRP3 inflammasome activation in the pathophysiology of radiculopathy, a type of neuropathic back and leg pain that is difficult to alleviate.
Lumbar disc herniation (LDH) is an important cause of radiculopathy (Zhang et al. 2017). In a rat model where autologous nucleus pulposus (NP) was implanted into one L5 DRG to simulate LDH, there was increased expression of NLRP3, ASC, Caspase-1, IL-1, IL-18 and other molecules in DRG neurons by one day after surgery and it peaked on day 7 post-implantation (Zhang et al. 2017). In the lumbar DRGs of the same animals (Zhang et al. 2017), there was also upregulated expression of calcitonin gene related peptide (CGRP) which is a pro-nociceptive neuropeptide that is a hallmark neurotransmitter released from small diameter C-fibers (Orita et al. 2013). Treatment of these animals with Bay11-7082, an inhibitor of both NF-kB activation and NLRP3 inflammasome activation, alleviated neuropathic pain behaviour thereby further implicating the NLRP3 inflammasome in the pathobiology of neuropathic pain (Zhang et al. 2017).
Neuropathic pain due to partial sciatic nerve injury in mice
In a mouse model of neuropathic pain induced by a partial sciatic nerve ligation (pSNL)-injury, there was a significant decrease in mRNA expression of miR-23a in the lumbar spinal cord (Pan et al. 2018). Conversely, overexpression of miR-23a in the spinal cord prevented pSNL-induced neuropathic pain whereas knockdown of miR-23a induced pain-like behaviour (Pan et al. 2018). In naïve mice, miR-23a knockdown increased spinal cord levels of thioredoxin-interacting protein (TXNIP) which was associated with induction of the NLRP3 inflammasome (Pan et al. 2018). In the spinal cord of pSNL-mice, miR-23a overexpression inhibited the increase of TXNIP and NLRP3 inflammasome activation and alleviated neuropathic pain behavior (Pan et al. 2018). Intrathecal injection of 681-siRNA for 3 consecutive days to knock down TXNIP expression, significantly alleviated pSNL-induced hyperalgesia and mechanical ectopic pain (Pan et al. 2018). Thus, miR-23a was confirmed to regulate neuropathic pain via the NLRP3/TXNIP inflammasome axis in pSNL-mice (Pan et al. 2018).
Chronic constriction injury of the sciatic nerve induced neuropathic pain
Many microRNAs (e.g. miR-145, miR-223, miR-23a, miR-183, miR-150) have been implicated in the pathogenesis of neuropathic pain (Ji et al. 2018; Shi et al. 2018; Xie et al. 2017). In work by others using the widely used unilateral chronic constriction injury (CCI) of the sciatic nerve mouse model of neuropathic pain, overexpression of Mir-34c in the lumbar spinal cord alleviated neuropathic pain behavior (Xu et al. 2019). Additionally, there was suppression of the activity of the NLRP3 inflammasome (decreased protein levels of NLRP3, ASC and caspase-1) and decreased inflammatory responses (downregulated production of the pro-inflammatory cytokines, TNFα, IL-1β and IL-18) in the lumbar spinal cord of these animals (Xu et al. 2019).
As noted in Sect. "Generation of ROS in mitochondria", the nuclear factor E2-related factor- 2 (Nrf2) signaling pathway can inhibit activation of the NLRP3 inflammasome by a mechanism involving nuclear translocation of Nrf2 (Liu et al. 2020). This had the same effect as the small molecule NLRP3 inhibitor, MCC950, in terms of alleviating neuropathic pain behavior in the CCI-rat model (Cohen and Mao 2014).
In other work in CCI-mice exhibiting mechanical hypersensitivity in the ipsilateral hindpaws, there was increased expression of connexin-43 hemichannels in lumbar spinal cord astrocytes and there was increased expression of key NLRP3 inflammasome components (NLRP3, ASC, caspase-1) in the lumbar spinal cord of these animals (Tonkin et al. 2018). Hindpaw hypersensitivity was significantly reduced in CCI-mice by intrathecal administration of the connexin-43 mimetic peptide, Peptide5, that blocks hemichannels in spinal cord astrocytes concurrent with a reduction to naïve levels of otherwise increased levels of NLRP3, its adaptor apoptosis-associated spec-like protein (ASC) and caspase-1 protein (Tonkin et al. 2018). Together these findings are consistent with that notion that pain relief evoked by Peptide 5 was transduced by specific inhibition of the NLRP3 inflammasome in the lumbar spinal cord of these mice (Tonkin et al. 2018).
In other work in the CCI-mouse model of peripheral neuropathic pain, intrathecal injection of divanillyl sulfone alleviated mechanical allodynia in the ipsilateral hindpaws (Shao et al. 2021). The mechanism likely involving induction of mitophagy in microglia to promote rapid clearance of reactive oxygen species and attenuate NLRP3 inflammasome activation in the ipsilateral spinal cord (Shao et al. 2021).
Painful diabetic neuropathy
Painful diabetic neuropathy (PDN) is a long term microvascular complication of poorly controlled diabetes that is often difficult to alleviate adequately (Fan and Gordon Smith 2022). PDN is the most common form of neuropathic pain globally as it affects up to 50% of patients with diabetes (Bril et al. 2011). Pain symptoms usually present with a "sock" and "glove"-like distribution and no single medication can prevent or completely reverse PDN (Javed et al. 2015). Although the exact pathogenesis of PDN is unclear, there was increased secretion of mRNA encoding NLRP3, ASC, IL-1β, and IL-18 in macrophages and peripheral blood monocytes of patients with type 2 diabetes mellitus (Lee et al. 2013). Additionally, there was increased expression of caspase-1, a key enzyme that causes apoptosis, in these cells from patients with type 2 diabetes (Lee et al. 2013). The secretion of the above related factors is mediated by the NLRP3 inflammasome (Lee et al. 2013). In addition, in a rat model of type 2 diabetes induced PDN, there was increased lumbar spinal cord expression of reactive oxygen species (ROS) as well as protein levels of NLRP3, TXNIP, caspase-1, interleukin 1-beta (IL-1β) and phosphorylated N-methyl-D-aspartic acid receptor subunit 2B (phospho-NR2B) (Wang et al. 2022). In the same rat model, once-daily treatment for 14-days with the ROS scavenger, N-tert-butyl-α-phenylnitrone (PBN) or TXNIP small interfering RNA, alleviated mechanical allodynia and thermal hyperalgesia in the bilateral hindpaws of these animals as well as decreasing protein levels of NLRP3, TXNIP, caspase-1, IL-1β, and phospho-NR2B (Wang et al. 2022). These findings together with complementary data from cultured microglia, implicate a role for ROS signaling through the TXNIP-NLRP3-NR2B pathway in the pathogenesis of PDN (Wang et al. 2022).
In summary, after nerve injury, there is induction of NLRP3 inflammasome activation and upregulated production of proinflammatory cytokines in the lumbar DRGs and in the lumbar spinal cord, which can lead to sensory neuron sensitization and the development of neuropathic pain. Based upon this knowledge, it is clear that new treatments that directly or indirectly inhibit NLRP3 inflammasome activation, have therapeutic potential as novel analgesic agents.
Inflammatory pain
Inflammatory pain is a common type of chronic pain that may be induced by inflammation associated with tissue damage due to trauma or bacterial infection (Abrahamsen et al. 2008). Inflammasomes regulate inflammation through the lysis of key cytokine precursors resulting in the secretion of mature pro-inflammatory cytokines (IL-1 and IL-18) that contribute to the development and maintenance of chronic inflammatory pain (Schlesinger 2014).
In the Complete Freund’s Adjuvant (CFA)-induced mouse model of chronic inflammatory pain in one hindpaw, there was increased expression of pro-inflammatory markers including NOX4, P-Jak2 / P-Stat3, and NLRP3 in the lumbar spinal cord (Yu et al. 2020). These changes were attenuated in CFA-mice administered muscone, the active ingredient of the Chinese medicine, musk, by intraperitoneal injection once-daily for 7-days (Yu et al. 2020). In addition, pain relief was evoked in the same animals in a dose-dependent manner (Yu et al. 2020), thereby implicating a role for NLRP3 activation in the pathobiology of chronic inflammatory pain.
In other work, intrathecal injection of the highly selective sphingosine-1-phosphate (S1P) receptor 1 subtype (S1PR1) agonist, SEW2871, evoked mechano-allodynia via activation of the NLRP3 inflammasome (increased expression of NLRP3, cleaved caspase 1 and mature IL-1β) in the lumbar spinal dorsal horn of rats (Doyle et al. 2019). These effects in rats were attenuated by treatment with S1PR1 antagonists or with the NLRP3 inflammasome inhibitor, MCC950 (Doyle et al. 2019). Additionally, intrathecal injection of SEW2871 in mice with astrocyte-specific deletions of S1pr1 did not evoke mechano-allodynia and the activation of cleaved caspase-1 was reduced (Doyle et al. 2019). Together, these findings showed that astrocyte-specific S1PR1 signaling is necessary for SIPR1 agonist-induced NLRP3 activation that underpins SIPR1 agonist induced mechano-allodynia in these animals (Doyle et al. 2019).
Gout
Gout is a sterile inflammatory disease with hyperuricemia, that is characterized by chronic monosodium urate (MSU) crystal deposition in various tissues (Goldberg et al. 2017). Gouty arthritis is one of the most common inflammatory pain disorders. When the serum urate concentration is higher than or equal to 0.42 mmol/L(7 mg/dL), it is considered clinically to be hyperuricemia (Dalbeth et al. 2021). Hyperuricemia is mainly related to decreased uricase activity (Kratzer et al. 2014), a gene mutation in the uric acid transporter (URAT1) (Tan et al. 2016). Symptoms of gout include joint pain, edema, redness and, in severe cases, disability (Merriman and Dalbeth 2011).
In 2006, Martinon et al. (Martinon et al. 2006) were the first to report that MSU crystals in the joints of patients with gout could activate NLRP3, thereby activating caspase-1 and promoting the maturation of IL-1β and IL-18. MSU crystals are a host-derived DAMP, that activates the NLRP3 inflammasome (Menu and Vince 2011). MSU crystals can cause multiple intracellular changes including mitochondrial injury (Nomura et al. 2015), increased xanthine oxidase (XO) activity (Bauernfeind et al. 2011), ROS production (Zhong et al. 2016), decreased intracellular ATP production (Nomura et al. 2015), inhibition of AMP-dependent protein kinase (AMPK) (Wang et al. 2016) and increased Nrf2 transcription (Jhang et al. 2015). The toll-like receptor (TLR) family of innate immune receptors are transmembrane receptors that bind extracellular ligands to trigger cellular activation and proliferation (Hsu et al. 2019). The innate immune components TLR-2, TLR-4, CD14 (TLR-4 ligand), NLRP3, ASC and caspase-1 are critical for the development of MSU crystal-induced inflammation (Duan et al. 2019; Giamarellos-Bourboulis et al. 2009; Sun and Zhang 2018).
In a mouse model of gouty arthritis involving MSU injection into the ankle joint, this induced ankle oedema, mechanical allodynia, neutrophil infiltration, oxidative stress, NLRP3 inflammasome activation and increased production of the pro-inflammatory cytokines, IL-1β and TNFα (Yin et al. 2020). Treatment of these mice with 4-doses of eucalyptol (anti-inflammatory oil contained in eucalyptus leaves) commencing at 1 h prior to model induction and continuing at intermittent intervals over 2-days, resulted in reduced ankle swelling and attenuation of mechanical allodynia in a manner mirroring mice treated similarly with the nonsteroidal anti-inflammatory drug, indomethacin (Yin et al. 2020). Both eucalyptol and indomethacin significantly reduced the otherwise upregulated mRNA expression of NLRP3, caspase-1, IL-1β and TRPV1 channels in ankle tissue from MSU-mice (Yin et al. 2020). Thus, eucalyptol inhibited MSU-induced activation of the NLRP3 inflammasome in the inflamed ankle joint tissues (Yin et al. 2020). In addition, for MSU-mice treated with the antioxidants, N-acetyl-L-cysteine or 2,2,6,6-tetramethylpiperidine 1-oxyl (Tempol) to reduce ROS generation and oxidative stress, there was a significant decrease in the otherwise upregulated expression of NLRP3, caspase 1, IL-1β and TRPV1 proteins in the ankle joint tissues (Yin et al. 2020). These findings support the notion that the antioxidants reduced NLRP3 inflammasome activation, IL-1β production and TRPV1 over-expression in ankle joint tissues of MSU-mice, in a manner that mimicked the effects of eucalyptol and indomethacin (Yin et al. 2020).β-hydroxybutyric acid, the most abundant ketone in vivo, inhibits activation of the NLRP3 inflammasome by reducing the priming and assembly steps, thereby reducing caspase 1-dependent secretion of IL-1β from neutrophils (Goldberg et al. 2017; Youm et al. 2015). Thus, β-hydroxybutyric acid, is an endogenous anti-inflammatory molecule with potential as a treatment for gout (Goldberg et al. 2017).
In summary, gouty arthritis is a debilitating chronic inflammatory arthritis caused by deposition of MSU crystals in the joints. MSU crystal-induced gouty flares are characterized by IL-1β-driven acute inflammation and intense pain and fever mediated by activation of the NLRP3 inflammasome in neutrophils to activate caspase-1 and increase the release of mature IL-1β and IL-18 in the inflamed joints (Martinon et al. 2006). In patients with gout, chronic deposition and presence of MSU crystals in the joints, facilitates on-going gouty flares underpinned by high systemic levels of NLRP3-derived IL-1β (Goldberg et al. 2017).
Fibromyalgia
Fibromyalgia (FM) is a clinical syndrome characterized by chronic widespread pain including headaches, pain or cramps in the lower abdomen, fatigue, unrefreshing sleep, cognitive and somatic symptoms and depression (Häuser et al. 2015). In various populations globally, the prevalence of fibromyalgia is in the range 2–4% with the ratio of women to men sufferers at 12:1 (Häuser et al. 2015). The exact pathophysiology of fibromyalgia is unclear but genetic, environmental, psychological and behavioral factors are implicated (Gupta et al. 2007; Kim et al. 2013).
The NLRP3 inflammasome has been implicated in the pathogenesis of FM (Cohen and Mao 2014). In blood mononuclear cells (BMCs) collected from patients with FM, mitochondrial dysfunction was accompanied by increased protein expression of NLRP3, caspase-1 activation and IL-1β expression (Cohen and Mao 2014). In these patients, there was also increased serum concentrations of the pro-inflammatory cytokines, IL-1β and IL-18 and decreased concentrations of co-enzyme Q10 (CoQ10) (Cohen and Mao 2014). CoQ10 deficiency induced by p-aminobenzoate treatment in blood mononuclear cells (BMCs) in mice, showed that there was NLRP3 inflammasome activation together with marked pain behavior in these mice (Cohen and Mao 2014). In a placebo-controlled clinical trial of oral CoQ10 in patients with FM, there was a reduction in NLRP3 inflammasome activation as well as the serum concentrations of IL-1β and IL-18 (Cohen and Mao 2014). Together, these findings implicate a role for NLRP3 activation and CoQ10 deficiency in the pathogenesis of FM and suggest that NLRP3 inflammasome inhibition may be a therapeutic opportunity for treating this disease (Cohen and Mao 2014).
In a rat model of reserpine induced fibromyalgia, intraperitoneal administration of the P2X7 purinergic receptor antagonist, Brilliant Blue G (BBG) at 50 mg/kg for 7-days, attenuated pain behavior and it prevented NLRP3 inflammasome activation and consequently inhibited the release of the pro-inflammatory cytokines IL-1β and IL-18 (D'Amico et al. 2021). Together these data suggest that inhibition of the P2X7 receptor to attenuate NLRP3 inflammasome activation may be a potential therapeutic approach for the treatment of fibromyalgia (D'Amico et al. 2021).
NLRP3 Inhibitors
Although multiple NLRP3 inhibitors have been reported in the past decade (Table 1), most have relatively low potency (uM) for inhibition of NLRP3 and so there is much room for improvement regarding discovery of ligands with nM inhibitory potency. Of the NLRP3 inhibitors listed in Table 1, only dapansutrile (OLT1177) has entered clinical trials in patients with chronic pain (gout) to date.
Conclusion
The NLRP3 inflammasome is a key component of the innate immune system that plays an essential role in the pathophysiology of various chronic inflammatory pain conditions as well as multiple types of central and peripheral neuropathic pain. Hence, targeting of the NLRP3 inflammasome may be an effective approach to address the large unmet medical need for a new generation of well-tolerated, safe and highly effective analgesic agents for the relief of chronic inflammatory pain and for alleviating neuropathic pain.
Data availability
Not applicable.
References
Abbaszadeh A, Darabi S, Hasanvand A, Amini-Khoei H, Abbasnezhad A, Choghakhori R, Aaliehpour A (2018) Minocycline through attenuation of oxidative stress and inflammatory response reduces the neuropathic pain in a rat model of chronic constriction injury. Iran J Basic Med Sci 21(2):138–144. https://doi.org/10.22038/ijbms.2017.24248.6053
Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN (2008) The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321(5889):702–705. https://doi.org/10.1126/science.1156916
Bahari Z, Meftahi GH (2019) Spinal α2-adrenoceptors and neuropathic pain modulation; therapeutic target. Br J Pharmacol 176(14):2366–2381. https://doi.org/10.1111/bph.14580
Bauernfeind F, Hornung V (2013) Of inflammasomes and pathogens–sensing of microbes by the inflammasome. EMBO Mol Med 5(6):814–826. https://doi.org/10.1002/emmm.201201771
Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187(2):613–617. https://doi.org/10.4049/jimmunol.1100613
Biasizzo M, Kopitar-Jerala N (2020) Interplay between NLRP3 inflammasome and autophagy. Front Immunol 11:591803. https://doi.org/10.3389/fimmu.2020.591803
Bril V, England J, Franklin GM, Backonja M, Cohen J, Del Toro D, Feldman E, Iverson DJ, Perkins B, Russell JW (2011) Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Muscle Nerve 3(4):345–352. https://doi.org/10.1002/mus.22092
Broz P, Monack DM (2011) Molecular mechanisms of inflammasome activation during microbial infections. Immunol Rev 243(1):174–190. https://doi.org/10.1111/j.1600-065X.2011.01041.x
Cao Y, Xu W, Huang Y, Zeng X (2020) Licochalcone B, a chalcone derivative from Glycyrrhiza inflata, as a multifunctional agent for the treatment of Alzheimer’s disease. Nat Prod Res 34(5):736–739. https://doi.org/10.1080/14786419.2018.1496429
Chen J, Chen ZJ (2018) PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564(7734):71–76. https://doi.org/10.1038/s41586-018-0761-3
Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR (2014) Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 137(Pt 8):2193–2209. https://doi.org/10.1093/brain/awu140
Chen W, Zhao M, Zhao S, Lu Q, Ni L, Zou C, Lu L, Xu X, Guan H, Zheng Z, Qiu Q (2017) Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflamm Res 66(2):157–166. https://doi.org/10.1007/s00011-016-1002-6
Cohen SP, Mao J (2014) Neuropathic pain: mechanisms and their clinical implications. BMJ 348:f7656. https://doi.org/10.1136/bmj.f7656
Cohen SP, Vase L, Hooten WM (2021) Chronic pain: an update on burden, best practices, and new advances. Lancet 397(10289):2082–2097. https://doi.org/10.1016/s0140-6736(21)00393-7
Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’neill, L. A. (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21(3):248–255. https://doi.org/10.1038/nm.3806
Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, Chitty JL, Fraser JA, Jennings MP, Robertson A, a. B. & Schroder, K. (2019) MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15(6):556–559. https://doi.org/10.1038/s41589-019-0277-7
Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, Debar L, Kerns R, Von Korff M, Porter L, Helmick C (2018) Prevalence of chronic pain and high-impact chronic pain among adults-United States, 2016. MMWR Morb Mortal Wkly Rep 67(36):1001–1006. https://doi.org/10.1558/mmwr.mm6736a2
Dalbeth N, Gosling AL, Gaffo A, Abhishek A (2021) Gout. Lancet 397(10287):1843–1855. https://doi.org/10.1016/s0140-6736(21)00569-9
D’amico R, Fusco R, Siracusa R, Impellizzeri D, Peritore AF, Gugliandolo E, Interdonato L, Sforza AM, Crupi R, Cuzzocrea S, Genovese T, Cordaro M, Di Paola R (2021) Inhibition of P2X7 purinergic receptor ameliorates fibromyalgia syndrome by suppressing NLRP3 pathway. Int J Mol Sci 22(12):6471. https://doi.org/10.3390/ijms22126471
Debono DJ, Hoeksema LJ, Hobbs RD (2013) Caring for patients with chronic pain: pearls and pitfalls. J Am Osteopath Assoc 113(8):620–627. https://doi.org/10.7556/jaoa.2013.023
Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, Mittal M, Hong Z, Kanneganti TD, Rehman J, Malik AB (2018) The TWIK2 potassium efflux channel in macrophages mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 49(1):56-65.e4. https://doi.org/10.1016/j.immuni.2018.04.032
Diatchenko L, Fillingim RB, Smith SB, Maixner W (2013) The phenotypic and genetic signatures of common musculoskeletal pain conditions. Nat Rev Rheumatol 9(6):340–350. https://doi.org/10.1038/nrrheum.2013.43
Domingo-Fernández R, Coll RC, Kearney J, Breit S, O’neill LAJ (2017) The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1β transcription and activate the NLRP3 inflammasome. J Biol Chem 292(29):12077–12087. https://doi.org/10.1074/jbc.M117.797126
Doyle TM, Chen Z, Durante M, Salvemini D (2019) Activation of sphingosine-1-phosphate receptor 1 in the spinal cord produces mechanohypersensitivity through the activation of inflammasome and IL-1β pathway. J Pain 20(8):956–964. https://doi.org/10.1016/j.jpain.2019.02.007
Duan L, Luo J, Fu Q, Shang K, Wei Y, Wang Y, Li Y, Chen J (2019) Decreased expression of CD14 in MSU-mediated inflammation may be associated with spontaneous remission of acute gout. J Immunol Res 2019:7143241. https://doi.org/10.1155/2019/7143241
Eccleston C, Cooper TE, Fisher E, Anderson B, Wilkinson NM (2017) Non-steroidal anti-inflammatory drugs (NSAIDs) for chronic non-cancer pain in children and adolescents. Cochrane Database Syst Rev 8(8):012537. https://doi.org/10.1002/14651858.CD012537.pub2
Economics, D. A. (2019). The cost of pain in Australia. Available: https://www2.deloitte.com/content/dam/Deloitte/au/Documents/Economics/deloitte-au-economics-cost-pain-australia-040419.pdf. Accessed Feb 20 2020.
Edwards RR, Dworkin RH, Sullivan MD, Turk DC, Wasan AD (2016) The role of psychosocial processes in the development and maintenance of chronic pain. J Pain 17(9 Suppl):T70-92. https://doi.org/10.1016/j.jpain.2016.01.001
Elliott AM, Smith BH, Hannaford PC, Smith WC, Chambers WA (2002) The course of chronic pain in the community: results of a 4-year follow-up study. Pain 99(1–2):299–307. https://doi.org/10.1016/s0304-3959(02)00138-0
El-Sharkawy LY, Brough D, Freeman S (2020) Inhibiting the NLRP3 Inflammasome. Molecules 25(23):5533. https://doi.org/10.3390/molecules25235533
Enthoven WT, Roelofs PD, Deyo RA, Van Tulder MW, Koes BW (2016) Non-steroidal anti-inflammatory drugs for chronic low back pain. Cochrane Database Syst Rev 2(2):Cd012087. https://doi.org/10.1002/14651858.Cd012087
Erdag E, Kucuk M, Aksoy U, Abacioglu N, Ozer A (2023) Docking study of ligands targeting NLRP3 inflammatory pathway for endodontic diseases. Chem Methodol 7(3):200–210. https://doi.org/10.2203/CHEMM.2022.367137.1623
Fan Q, Gordon Smith A (2022) Recent updates in the treatment of diabetic polyneuropathy. Fac Rev 11:30. https://doi.org/10.1270/r/11-30
Fayaz, A., Croft, P., Langford, R. M., Donaldson, L. J. & Jones, G. T. (2016). Prevalence of chronic pain in the UK: a systematic review and meta-analysis of population studies. BMJ Open, 6 (6). e010364. https://doi.org/10.1136/bmjopen-2015-010364.
Finnerup NB, Attal N, Haroutounian S, Mcnicol E, Baron R, Dworkin RH, Gilron I, Haanpää M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M (2015) Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 14(2):162–173. https://doi.org/10.1016/s1474-4422(14)70251-0
Franceschini A, Capece M, Chiozzi P, Falzoni S, Sanz JM, Sarti AC, Bonora M, Pinton P, Di Virgilio F (2015) The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J 29(6):2450–2461. https://doi.org/10.1096/fj.14-268714
Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10(3):241–247. https://doi.org/10.1038/ni.1703
Giamarellos-Bourboulis EJ, Mouktaroudi M, Bodar E, Van Der Ven J, Kullberg BJ, Netea MG, Van Der Meer JW (2009) Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1 beta by mononuclear cells through a caspase 1-mediated process. Ann Rheum Dis 68(2):273–278. https://doi.org/10.1136/ard.2007.082222
Goldberg EL, Asher JL, Molony RD, Shaw AC, Zeiss CJ, Wang C, Morozova-Roche LA, Herzog RI, Iwasaki A, Dixit VD (2017) β-Hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep 18(9):2077–2087. https://doi.org/10.1016/j.celrep.2017.02.004
Green JP, Yu S, Martín-Sánchez F, Pelegrin P, Lopez-Castejon G, Lawrence CB, Brough D (2018) Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci USA 115(40):E9371-e9380. https://doi.org/10.1073/pnas.1812744115
Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP (2010) NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 185(2):974–981. https://doi.org/10.4049/jimmunol.0904145
Gupta A, Silman AJ, Ray D, Morriss R, Dickens C, Macfarlane GJ, Chiu YH, Nicholl B, Mcbeth J (2007) The role of psychosocial factors in predicting the onset of chronic widespread pain: results from a prospective population-based study. Rheumatology (oxford) 46(4):666–671. https://doi.org/10.1093/rheumatology/kel363
Harker J, Reid KJ, Bekkering GE, Kellen E, Bala MM, Riemsma R, Worthy G, Misso K, Kleijnen J (2012) Epidemiology of chronic pain in denmark and sweden. Pain Res Treat. https://doi.org/10.1155/2012/371248
Häuser W, Ablin J, Fitzcharles MA, Littlejohn G, Luciano JV, Usui C, Walitt B (2015) Fibromyalgia. Nat Rev Dis Primers 1:15022. https://doi.org/10.1038/nrdp.2015.22
He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, Liu Q, Liang G, Deng X, Jiang W, Zhou R (2018) Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun 9(1):2550. https://doi.org/10.1038/s41467-018-04947-6
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9(8):847–856. https://doi.org/10.1038/ni.1631
Hsu HC, Hsieh CL, Wu SY, Lin YW (2019) Toll-like receptor 2 plays an essential role in electroacupuncture analgesia in a mouse model of inflammatory pain. Acupunct Med 37(6):356–364. https://doi.org/10.1136/acupmed-2017-011469
Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, Deng X, Liang G, Zhang H, Jiang W, Zhou R (2018) Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med 10(4):e8689. https://doi.org/10.1525/emmm.201708689
Huang T, Xiao Y, Zhang Y, Wang C, Chen X, Li Y, Ge Y, Gao J (2022) miR-223 ameliorates thalamus hemorrhage-induced central poststroke pain via targeting NLRP3 in a mouse model. Exp Ther Med 23(5):353. https://doi.org/10.3892/etm.2022.11280
Institute of Medicine Committee on Advancing Pain Research, C. & Education (2011). The National Academies Collection: Reports funded by National Institutes of Health. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington (DC).
Javed S, Petropoulos IN, Alam U, Malik RA (2015) Treatment of painful diabetic neuropathy. Ther Adv Chronic Dis 6(1):15–28. https://doi.org/10.1177/2040622314552071
Jayabalan N, Oronsky B, Cabrales P, Reid T, Caroen S, Johnson AM, Birch NA, O’sullivan JD, Gordon R (2023) A review of RRx-001: a late-stage multi-indication inhibitor of NLRP3 activation and chronic inflammation. Drugs. https://doi.org/10.1007/s40265-023-01838-z
Jhang JJ, Cheng YT, Ho CY, Yen GC (2015) Monosodium urate crystals trigger Nrf2- and heme oxygenase-1-dependent inflammation in THP-1 cells. Cell Mol Immunol 12(4):424–434. https://doi.org/10.1038/cmi.2014.65
Ji LJ, Shi J, Lu JM, Huang QM (2018) MiR-150 alleviates neuropathic pain via inhibiting toll-like receptor 5. J Cell Biochem 119(1):1017–1026. https://doi.org/10.1002/jcb.26269
Jia M, Wu C, Gao F, Xiang H, Sun N, Peng P, Li J, Yuan X, Li H, Meng X, Tian B, Shi J, Li M (2017) Activation of NLRP3 inflammasome in peripheral nerve contributes to paclitaxel-induced neuropathic pain. Mol Pain 13:1744806917719804. https://doi.org/10.1177/1744806917719804
Jiang Y, He L, Green J, Blevins H, Guo C, Patel SH, Halquist MS, Mcrae M, Venitz J, Wang XY, Zhang S (2019) Discovery of second-generation NLRP3 inflammasome inhibitors: design, synthesis, and biological characterization. J Med Chem 62(21):9718–9731. https://doi.org/10.1021/acs.jmedchem.9b01155
Jin C, Flavell RA (2010) Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol 30(5):628–631. https://doi.org/10.1007/s10875-010-9440-3
Jin L, Ding M, Oklopcic A, Aghdasi B, Xiao L, Li Z, Jevtovic-Todorovic V, Li X (2017) Nanoparticle fullerol alleviates radiculopathy via NLRP3 inflammasome and neuropeptides. Nanomedicine 13(6):2049–2059. https://doi.org/10.1016/j.nano.2017.03.015
Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, Meng R, Quong AA, Latz E, Scott CP, Alnemri ES (2010) Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J Biol Chem 285(13):9792–9802. https://doi.org/10.1074/jbc.M109.082305
Katsnelson MA, Rucker LG, Russo HM, Dubyak GR (2015) K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol 194(8):3937–3952. https://doi.org/10.4049/jimmunol.1402658
Katsnelson MA, Lozada-Soto KM, Russo HM, Miller BA, Dubyak GR (2016) NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am J Physiol Cell Physiol 311(1):C83-c100. https://doi.org/10.1152/ajpcell.00298.2015
Khan N, Smith MT (2014) Multiple sclerosis-induced neuropathic pain: pharmacological management and pathophysiological insights from rodent EAE models. Inflammopharmacology 22(1):1–22. https://doi.org/10.1007/s10787-013-0195-3
Khan N, Kuo A, Brockman DA, Cooper MA, Smith MT (2018) Pharmacological inhibition of the NLRP3 inflammasome as a potential target for multiple sclerosis induced central neuropathic pain. Inflammopharmacology 26(1):77–86. https://doi.org/10.1007/s10787-017-0401-9
Kim SK, Kim SH, Nah SS, Lee JH, Hong SJ, Kim HS, Lee HS, Kim HA, Joung CI, Bae J, Choe JY, Lee SS (2013) Association of guanosine triphosphate cyclohydrolase 1 gene polymorphisms with fibromyalgia syndrome in a Korean population. J Rheumatol 40(3):316–322. https://doi.org/10.3899/jrheum.120929
Kratzer JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, Ortlund EA, Johnson RJ, Gaucher EA (2014) Evolutionary history and metabolic insights of ancient mammalian uricases. Proc Natl Acad Sci USA 111(10):3763–3768. https://doi.org/10.1073/pnas.1320393111
Labianca R, Sarzi-Puttini P, Zuccaro SM, Cherubino P, Vellucci R, Fornasari D (2012) Adverse effects associated with non-opioid and opioid treatment in patients with chronic pain. Clin Drug Investig 32(Suppl 1):53–63. https://doi.org/10.2165/11630080-000000000-00000
Lane T, Flam B, Lockey R, Kolliputi N (2013) TXNIP shuttling: missing link between oxidative stress and inflammasome activation. Front Physiol 4:50. https://doi.org/10.3389/fphys.2013.00050
Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK (2013) Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62(1):194–204. https://doi.org/10.2337/db12-0420
Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, Kong AN (2008) Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol 76(11):1485–1489. https://doi.org/10.1016/j.bcp.2008.07.017
Li H, Ambade A, Re F (2009a) Cutting edge: Necrosis activates the NLRP3 inflammasome. J Immunol 183(3):1528–1532. https://doi.org/10.4049/jimmunol.0901080
Li XN, Song J, Zhang L, Lemaire SA, Hou X, Zhang C, Coselli JS, Chen L, Wang XL, Zhang Y, Shen YH (2009b) Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 58(10):2246–2257. https://doi.org/10.2337/db08-1512
Liampas A, Velidakis N, Georgiou T, Vadalouca A, Varrassi G, Hadjigeorgiou GM, Tsivgoulis G, Zis P (2020) Prevalence and management challenges in central post-stroke neuropathic pain: a systematic review and meta-analysis. Adv Ther 37(7):3278–3291. https://doi.org/10.1007/s12325-020-01388-w
Lima H Jr, Jacobson LS, Goldberg MF, Chandran K, Diaz-Griffero F, Lisanti MP, Brojatsch J (2013) Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle 12(12):1868–1878. https://doi.org/10.4161/cc.24903
Liu X, Zhang X, Ding Y, Zhou W, Tao L, Lu P, Wang Y, Hu R (2017) Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species-induced NLRP3 priming. Antioxid Redox Signal 26(1):28–43. https://doi.org/10.1089/ars.2015.6615
Liu P, Cheng J, Ma S, Zhou J (2020) Paeoniflorin attenuates chronic constriction injury-induced neuropathic pain by suppressing spinal NLRP3 inflammasome activation. Inflammopharmacology 28(6):1495–1508. https://doi.org/10.1007/s10787-020-00737-z
Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, Carta S, Tengesdal I, Nemkov T, D’alessandro A, Henry C, Jones GS, Goodrich SA, St Laurent JP, Jones TM, Scribner CL, Barrow RB, Altman RD, Skouras DB, Gattorno M, Grau V, Janciauskiene S, Rubartelli A, Joosten L, a. B. & Dinarello, C. A. (2018) OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A 115(7):E1530-e1539. https://doi.org/10.1073/pnas.1716095115
Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440(7081):237–241. https://doi.org/10.1038/nature04516
Mathur A, Hayward JA, Man SM (2018) Molecular mechanisms of inflammasome signaling. J Leukoc Biol 103(2):233–257. https://doi.org/10.1189/jlb.3MR0617-250R
Menu P, Vince JE (2011) The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol 166(1):1–15. https://doi.org/10.1111/j.1365-2249.2011.04440.x
Merriman TR, Dalbeth N (2011) The genetic basis of hyperuricaemia and gout. Joint Bone Spine 78(1):35–40. https://doi.org/10.1016/j.jbspin.2010.02.027
Mi J, Yang Y, Yao H, Huan Z, Xu C, Ren Z, Li W, Tang Y, Fu R, Ge X (2021) Inhibition of heat shock protein family A member 8 attenuates spinal cord ischemia-reperfusion injury via astrocyte NF-κB/NLRP3 inflammasome pathway : HSPA8 inhibition protects spinal ischemia-reperfusion injury. J Neuroinflammation 18(1):170. https://doi.org/10.1186/s12974-021-02220-0
Mills SEE, Nicolson KP, Smith BH (2019) Chronic pain: a review of its epidemiology and associated factors in population-based studies. Br J Anaesth 123(2):e273–e283. https://doi.org/10.1016/j.bja.2019.03.023
Moon JS, Lee S, Park MA, Siempos Ii, Haslip M, Lee PJ, Yun M, Kim CK, Howrylak J, Ryter SW, Nakahira K, Choi AM (2015) UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 125(2):665–680. https://doi.org/10.1172/jci78253
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38(6):1142–1153. https://doi.org/10.1016/j.immuni.2013.05.016
Murakami T, Ockinger J, Yu J, Byles V, Mccoll A, Hofer AM, Horng T (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 109(28):11282–11287. https://doi.org/10.1073/pnas.1117765109
Nomura J, So A, Tamura M, Busso N (2015) Intracellular ATP decrease mediates NLRP3 inflammasome activation upon nigericin and crystal stimulation. J Immunol 195(12):5718–5724. https://doi.org/10.4049/jimmunol.1402512
Orita S, Miyagi M, Kobori S, Gemba T, Ishikawa T, Inoue G, Toyone T, Aoki Y, Eguchi Y, Takahashi K, Ohtori S (2013) IκB kinase β inhibitor downregulates pain-related neuropeptide production in the sensory neurons innervating injured lumbar intervertebral discs in the dorsal root ganglia of rats. Spine J 13(3):284–288. https://doi.org/10.1016/j.spinee.2013.01.020
Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL (2015) Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J Immunol 195(4):1685–1697. https://doi.org/10.4049/jimmunol.1500509
Pan Z, Shan Q, Gu P, Wang XM, Tai LW, Sun M, Luo X, Sun L, Cheung CW (2018) miRNA-23a/CXCR4 regulates neuropathic pain via directly targeting TXNIP/NLRP3 inflammasome axis. J Neuroinflammation 15(1):29. https://doi.org/10.1186/s12974-018-1073-0
Raja SN, Carr DB, Cohen M, Finnerup NB, Flor H, Gibson S, Keefe FJ, Mogil JS, Ringkamp M, Sluka KA, Song XJ, Stevens B, Sullivan MD, Tutelman PR, Ushida T, Vader K (2020) The revised International Association for the Study of Pain definition of pain: concepts, challenges, and compromises. Pain 161(9):1976–1982. https://doi.org/10.1097/j.pain.0000000000001939
Reid KJ, Harker J, Bala MM, Truyers C, Kellen E, Bekkering GE, Kleijnen J (2011) Epidemiology of chronic non-cancer pain in Europe: narrative review of prevalence, pain treatments and pain impact. Curr Med Res Opin 27(2):449–462. https://doi.org/10.1185/03007995.2010.545813
Schlesinger N (2014) Anti-interleukin-1 therapy in the management of gout. Curr Rheumatol Rep 16(2):398. https://doi.org/10.1007/s11926-013-0398-z
Shah S, Mehta V (2012) Controversies and advances in non-steroidal anti-inflammatory drug (NSAID) analgesia in chronic pain management. Postgrad Med J 88(1036):73–78. https://doi.org/10.1136/postgradmedj-2011-130291
Shao S, Xu CB, Chen CJ, Shi GN, Guo QL, Zhou Y, Wei YZ, Wu L, Shi JG, Zhang TT (2021) Divanillyl sulfone suppresses NLRP3 inflammasome activation via inducing mitophagy to ameliorate chronic neuropathic pain in mice. J Neuroinflammation 18(1):142. https://doi.org/10.1186/s12974-021-02178-z
Shi J, Jiang K, Li Z (2018) MiR-145 ameliorates neuropathic pain via inhibiting inflammatory responses and mTOR signaling pathway by targeting Akt3 in a rat model. Neurosci Res 134:10–17. https://doi.org/10.1016/j.neures.2017.11.006
Shi Y, Wang H, Zheng M, Xu W, Yang Y, Shi F (2020) Ginsenoside Rg3 suppresses the NLRP3 inflammasome activation through inhibition of its assembly. FASEB J 34(1):208–221. https://doi.org/10.1096/fj.201901537R
Shi Y, Lv Q, Zheng M, Sun H, Shi F (2021) NLRP3 inflammasome inhibitor INF39 attenuated NLRP3 assembly in macrophages. Int Immunopharmacol 92(107358):1–11. https://doi.org/10.1016/j.intimp.2020.107358
Shim DW, Shin WY, Yu SH, Kim BH, Ye SK, Koppula S, Won HS, Kang TB, Lee KH (2017) BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci Rep 7(1):15020. https://doi.org/10.1038/s41598-017-15314-8
Su Y, Wang Y, Liu M, Chen H (2021) Hydrogen sulfide attenuates renal I/R-induced activation of the inflammatory response and apoptosis via regulating Nrf2-mediated NLRP3 signaling pathway inhibition. Mol Med Rep. https://doi.org/10.3892/mmr.2021.12157
Sun X, Zhang H (2018) miR-451 elevation relieves inflammatory pain by suppressing microglial activation-evoked inflammatory response via targeting TLR4. Cell Tissue Res 374(3):487–495. https://doi.org/10.1007/s00441-018-2898-7
Sutterwala FS, Haasken S, Cassel SL (2014) Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 1319(1):82–95. https://doi.org/10.1111/nyas.12458
Swanson KV, Deng M, Ting JP (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19(8):477–489. https://doi.org/10.1038/s41577-019-0165-0
Tan PK, Farrar JE, Gaucher EA, Miner JN (2016) Coevolution of URAT1 and uricase during primate evolution: implications for serum urate homeostasis and gout. Mol Biol Evol 33(9):2193–2200. https://doi.org/10.1093/molbev/msw116
Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, Cui J, Bai L, Wang J, Jiang W, Zhou R (2017) CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun 8(1):202. https://doi.org/10.1038/s41467-017-00227-x
Tang T, Gong T, Jiang W, Zhou R (2018) GPCRs in NLRP3 inflammasome activation, regulation, and therapeutics. Trends Pharmacol Sci 39(9):798–811. https://doi.org/10.1016/j.tips.2018.07.002
Tezcan G, Martynova EV, Gilazieva ZE, Mcintyre A, Rizvanov AA, Khaiboullina SF (2019) MicroRNA Post-transcriptional Regulation of the NLRP3 Inflammasome in immunopathologies. Front Pharmacol 10:451. https://doi.org/10.3389/fphar.2019.00451
Tonkin RS, Bowles C, Perera CJ, Keating BA, Makker PGS, Duffy SS, Lees JG, Tran C, Don AS, Fath T, Liu L, O’carroll SJ, Nicholson LFB, Green CR, Gorrie C, Moalem-Taylor G (2018) Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp Neurol 300:1–12. https://doi.org/10.1016/j.expneurol.2017.10.016
Vowles KE, Mcentee ML, Julnes PS, Frohe T, Ney JP, Van Der Goes DN (2015) Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 156(4):569–576. https://doi.org/10.1097/01.j.pain.0000460357.01998.f1
Wang Y, Viollet B, Terkeltaub R, Liu-Bryan R (2016) AMP-activated protein kinase suppresses urate crystal-induced inflammation and transduces colchicine effects in macrophages. Ann Rheum Dis 75(1):286–294. https://doi.org/10.1136/annrheumdis-2014-206074
Wang JW, Ye XY, Wei N, Wu SS, Zhang ZH, Luo GH, Li X, Li J, Cao H (2022) Reactive oxygen species contributes to type 2 diabetic neuropathic pain via the thioredoxin-interacting protein-NOD-like receptor protein 3- N -Methyl-D-aspartic acid receptor 2B pathway. Anesth Analg 135(4):865–876. https://doi.org/10.1213/ane.0000000000006117
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12(5):408–415. https://doi.org/10.1038/ni.2022
Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu NI, Coggeshall KM, Arditi M, Underhill DM (2016) Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166(3):624–636. https://doi.org/10.1016/j.cell.2016.05.076
Wu J, Fernandes-Alnemri T, Alnemri ES (2010) Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol 30(5):693–702. https://doi.org/10.1007/s10875-010-9425-2
Xie XJ, Ma LG, Xi K, Fan DM, Li JG, Zhang Q, Zhang W (2017) Effects of microRNA-223 on morphine analgesic tolerance by targeting NLRP3 in a rat model of neuropathic pain. Mol Pain 13:1744806917706582. https://doi.org/10.1177/1744806917706582
Xu L, Wang Q, Jiang W, Yu S, Zhang S (2019) MiR-34c ameliorates neuropathic pain by targeting NLRP3 in a mouse model of chronic constriction injury. Neuroscience 399:125–134. https://doi.org/10.1016/j.neuroscience.2018.12.030
Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, Tian Y, Glenn HL, Meldrum DR (2015) K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis 6(10):e1954. https://doi.org/10.1038/cddis.2015.277
Yin C, Liu B, Wang P, Li X, Li Y, Zheng X, Tai Y, Wang C, Liu B (2020) Eucalyptol alleviates inflammation and pain responses in a mouse model of gout arthritis. Br J Pharmacol 177(9):2042–2057. https://doi.org/10.1111/bph.14967
Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, Crawford PA, Biragyn A, Alnemri E, Dixit VD (2015) The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 21(3):263–269. https://doi.org/10.1038/nm.3804
Yousuf MS, Noh MC, Friedman TN, Zubkow K, Johnson JC, Tenorio G, Kurata HT, Smith PA, Kerr BJ (2019) Sensory neurons of the dorsal root ganglia become hyperexcitable in a T-Cell-mediated MOG-EAE model of multiple sclerosis. eNeuro. https://doi.org/10.1523/eneuro.0024-19.2019
Yu S, Zhao G, Han F, Liang W, Jiao Y, Li Z, Li L (2020) Muscone relieves inflammatory pain by inhibiting microglial activation-mediated inflammatory response via abrogation of the NOX4/JAK2-STAT3 pathway and NLRP3 inflammasome. Int Immunopharmacol 82:106355. https://doi.org/10.1016/j.intimp.2020.106355
Zahid A, Li B, Kombe AJK, Jin T, Tao J (2019) Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol 10(2538):1–10. https://doi.org/10.3389/fimmu.2019.02538
Zeilhofer HU, Benke D, Yevenes GE (2012) Chronic pain states: pharmacological strategies to restore diminished inhibitory spinal pain control. Annu Rev Pharmacol Toxicol 52:111–133. https://doi.org/10.1146/annurev-pharmtox-010611-134636
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464(7285):104–107. https://doi.org/10.1038/nature08780
Zhang A, Wang K, Ding L, Bao X, Wang X, Qiu X, Liu J (2017) Bay11-7082 attenuates neuropathic pain via inhibition of nuclear factor-kappa B and nucleotide-binding domain-like receptor protein 3 inflammasome activation in dorsal root ganglions in a rat model of lumbar disc herniation. J Pain Res 10:375–382. https://doi.org/10.2147/jpr.S119820
Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, Mcgeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M (2016) NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164(5):896–910. https://doi.org/10.1016/j.cell.2015.12.057
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225. https://doi.org/10.1038/nature09663
Zuo Y, Wang J, Liao F, Yan X, Li J, Huang L, Liu F (2018) Inhibition of heat shock protein 90 by 17-AAG reduces inflammation via P2X7 Receptor/NLRP3 inflammasome pathway and increases neurogenesis after subarachnoid hemorrhage in mice. Front Mol Neurosci 11:401. https://doi.org/10.3389/fnmol.2018.00401
Acknowledgements
The Centre for Integrated Preclinical Drug Development (CIPDD) in the School of Biomedical Sciences at The University of Queensland is supported financially by Therapeutic Innovation Australia (TIA). TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) programme. The authors acknowledge Queensland Government Smart State Research Facilities Programme for supporting the CIPDD research infrastructure.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The Centre for Integrated Preclinical Drug Development (CIPDD) in the School of Biomedical Sciences at The University of Queensland is supported financially by Therapeutic Innovation Australia (TIA). TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) programme. The authors acknowledge Queensland Government Smart State Research Facilities Programme for supporting the CIPDD research infrastructure.
Author information
Authors and Affiliations
Contributions
Both authors contributed to the conception of this review article. The first draft of the manuscript was written by CC and both authors commented on all manuscript drafts. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no relevant competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, C., Smith, M.T. The NLRP3 inflammasome: role in the pathobiology of chronic pain. Inflammopharmacol 31, 1589–1603 (2023). https://doi.org/10.1007/s10787-023-01235-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-023-01235-8