
Abstract
The “Thalidomide tragedy” is a landmark in the history of the pharmaceutical industry. Despite limited clinical trials, there is a continuous effort to investigate thalidomide as a drug for cancer and inflammatory diseases such as rheumatoid arthritis, lepromatous leprosy, and COVID-19. This review focuses on the possibilities of targeting inflammation by repurposing thalidomide for the treatment of idiopathic pulmonary fibrosis (IPF). Articles were searched from the Scopus database, sorted, and selected articles were reviewed. The content includes the proven mechanisms of action of thalidomide relevant to IPF. Inflammation, oxidative stress, and epigenetic mechanisms are major pathogenic factors in IPF. Transforming growth factor-β (TGF-β) is the major biomarker of IPF. Thalidomide is an effective anti-inflammatory drug in inhibiting TGF-β, interleukins (IL-6 and IL-1β), and tumour necrosis factor-α (TNF-α). Thalidomide binds cereblon, a process that is involved in the proposed mechanism in specific cancers such as breast cancer, colon cancer, multiple myeloma, and lung cancer. Cereblon is involved in activating AMP-activated protein kinase (AMPK)-TGF-β/Smad signalling, thereby attenuating fibrosis. The past few years have witnessed an improvement in the identification of biomarkers and diagnostic technologies in respiratory diseases, partly because of the COVID-19 pandemic. Hence, investment in clinical trials with a systematic plan can help repurpose thalidomide for pulmonary fibrosis.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease characterized by chronic fibrotic scarring of lung tissues, leading to a deterioration of lung function and respiratory failure. Disease progression ultimately leads to death within a few years. IPF is presumably caused by multiple causes such as smoking, drugs (E.g., bleomycin), inhalation of metallic particles, wood dust or silica, and exaggerated immune responses (Barratt et al. 2018). Environmental factors and epigenetic mechanisms such as DNA-methylation and histone modifications are major contributors to disease progression (Velagacherla et al. 2022). IPF is an abnormal wound healing process where micro-injuries to the alveolar epithelium do not heal appropriately (Richeldi et al. 2017). IPF is a chronic fibrotic disease and symptomatically overlaps with cryptogenic fibrosing alveolitis. In IPF, an altered extracellular matrix (ECM) replaces the healthy lung tissue, damaging alveolar integrity, which is reflected by decreased lung compliance, abnormal gas exchange, respiratory failure, and eventually, death. IPF histopathology reflects interstitial pneumonia with fibroblast foci as hallmark lesions (Martinez et al. 2017). IPF is caused by sustained micro-injuries to the alveolar epithelial tissue, combined with abnormal healing processes leading to the depletion of the basement membrane between the alveolar epithelium and the capillaries (Wuyts et al. 2020). IPF is most prevalent in South Korea, Canada, Poland, the United States, and Italy (Maher et al. 2021). The COVID-19 pandemic could have augmented the growing incidences of IPF (Tanni et al. 2021). The pathogenesis of post-COVID-19 pulmonary fibrosis involves TGF-β1, which facilitates the release of extracellular proteins, fibroblast proliferation, fibroblast migration and, myofibroblast conversion (Al-kuraishy et al. 2022). Furthermore, genetic susceptibility for IPF and COVID-19 seem to be similar, as confirmed by the fibrotic plasma biomarkers such as thrombospondin 2 (TSP2), glial-derived factor 15 (GDF15), insulin-like growth factor binding protein 7 (IGFBP7), and procollagen type III (PROC3) (Ackermann et al. 2022). Currently, the therapeutic management of IPF consists of symptomatic treatment and extensive palliative care, because nintedanib and pirfenidone are the only two drugs approved for the indication of IPF (Mudawi et al. 2021). In this review, we propose that thalidomide can mitigate inflammation in IPF, and can be repurposed either as monotherapy or in combination with nintedanib and pirfenidone.
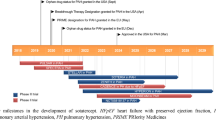
Thalidomide is a synthetic glutamic acid derivative α-(N-phthalimido)-glutarimide (Fig. 1), originally designed as a non-addictive and non-barbiturate sedative. (Kim and Scialli 2011). Thalidomide has one asymmetric carbon in the glutarimide ring, which acts as a chiral centre and gives rise to two enantiomers, R(+) and S(−). These enantiomers are capable of interconverting under appropriate biological conditions (Vargesson 2015). Being hailed as a ‘wonder drug’ at the time of its introduction, thalidomide was used as a quick and effective anti-emetic to treat morning sickness in pregnant women (Sharma and Kwatra 2016). It appeared to be non-toxic in rodent models. Hence, an LD50 value was not established, and it was assumed to be safe for humans (Cooper et al. 2010). Thalidomide received a lot of attention for being inexpensive and easily available without prescription. Thalidomide was widely marketed in European countries, Australia and Japan. It was first marketed in West Germany in 1957 under the trade name Contergan VR by Chemie Grunenthal, a German pharmaceutical company, and was later approved and distributed in 46 countries around the world under various brand names (Kumar et al. 2012). In 1960, around 14.6 tonnes of thalidomide were believed to be sold in Germany alone, making it the best-selling sedative in the market (Rehman et al. 2011). However, what came next was one of the most catastrophic man-made clinical disasters in history. There were multiple cases of limb malformations, birth defects and even death of thousands of babies born to mothers who consumed thalidomide during their early trimester of pregnancy (Kim and Scialli 2011). Thalidomide was withdrawn from the market in September 1961 when two scientists, McBride in Australia and Lenz in Germany, confirmed the association of thalidomide with congenital disabilities such as limb deformities, congenital cardiac, renal and other organ malformations (Mellin and Katzenstein 1962). Although thalidomide was swiftly withdrawn from the market, more than 10,000 infants suffered from congenital defects, with the survivors still suffering even today (Newbronner et al. 2019). However, the drug thalidomide is still an interesting molecule in drug discovery because of its anti-inflammatory and immunomodulatory properties and is now classified along with immunomodulatory imide drugs (IMiDs) such as lenalidomide, pomalidomide, apremilast, 3,6′-dithiopomalidomide, 3,6′-dithiothalidomide, N-adamantyl phthalimidine, and dithiophthalimide (Furihata et al. 2020). Some of these IMiDs are proposed for repurposing in neuropsychiatric and neurodegenerative disorders (Jung et al. 2021).

Structure of thalidomide showing its R( +) and S(-) enantiomers and its actions. The structure of thalidomide has asymmetric carbon (•); Both R( +) and S( −) enantiomers are equally active as they are interconvertible in the body
Therapeutic actions which enabled the re-entry of thalidomide
Due to the teratogenic effects, thalidomide was withdrawn late in November 1961 (Samel et al. 2019). However, the journey of thalidomide did not end there. In 1965, when thalidomide was administered as a sedative to patients with leprosy (Sheskin 1965), it was observed that along with its sedative action, thalidomide also reduced elevated body temperature, and stimulated the reabsorption of erythema nodosum-like skin lesions in patients with leprosy (Sheskin 1965). Thus, it was reintroduced into the market with extreme caution for the treatment of erythema nodosum leprosum, based on its immunomodulatory, anti-angiogenic, and anti-inflammatory actions. In the 1980s, thalidomide was given to patients with classic and definitive rheumatoid arthritis (Gutiérrez-Rodríguez 1984). In these patients, the erythrocyte sedimentation rate reportedly returned to normal within a few weeks, pain and joint inflammation disappeared, and the rheumatoid factor titers had significantly decreased (Gutierrez-Rodriguez et al. 1989). In 1986, a clinical study was conducted on the effect of thalidomide in 30 male patients suffering from Behçet’s disease, which is characterized by inflammation of the blood vessels in the body (Hamza 1986). The results demonstrated that thalidomide could be useful in treating clinical manifestations of Behçet’s disease. The side effects observed included skin rashes and somnolence, and were uncommon (Hamza 1986).
Thalidomide also selectively inhibited tumour necrosis factor-alpha (TNF-α) production in lipopolysaccharide-stimulated monocytes (Piura et al. 2013). However, there was no change in the levels of granulocytes / macrophage colony-stimulating factor, and cytokines [interleukin-6 (IL-6) and interleukin-1-beta (IL-1β)] produced by the monocytes. This could be due to increased degradation of TNF-α mRNA. This allowed the use of thalidomide in the treatment of severe inflammatory diseases (Majumder et al. 2012). Thalidomide has also been used in the treatment of wasting syndrome in HIV-AIDS patients (Reyes-Terán et al. 1996). In addition, the anti-TNF-α action of thalidomide was considerably helpful in treating tuberculous meningitis in children (van Toorn et al. 2021). An in vivo experiment in rabbits concluded that thalidomide is a potent inhibitor of angiogenesis (D’Amato et al. 1994). Among the putative angiogenesis inhibitors tested, thalidomide was the only drug that was effective orally suggesting that thalidomide is a feasible anticancer agent (Zhu et al. 2021). Thalidomide has also shown promising activity against various solid tumours such as breast cancer, renal cancer, Kaposi’s sarcoma, liver cancer, lung cancer, and several glioblastomas (Amare et al. 2021). US FDA authorized thalidomide for the treatment of erythema nodosum leprosum in 1998, and multiple myeloma in 2006 (Rehman et al. 2011). Its use in multiple myeloma was attributed to the immunomodulatory and anti-angiogenic activity which downregulates the expression of TNF-α and IL-6 by the stromal cells in the bone marrow, thereby inhibiting the proliferation of multiple myeloma cells (Holstein and McCarthy 2017). A recent compilation by Barbarossa et al. listed more than 130 analogues of thalidomide that were tested to treat cancer (Barbarossa et al. 2022). Efforts have now been made to strictly regulate the use of thalidomide to prevent potential harm from thalidomide. Several programmes have been developed to manage this. Risk Evaluation and Mitigation Strategies is a programme that controls the prescription of thalidomide (Brandenburg et al. 2017). The System for Thalidomide Education and Prescribing Safety (STEPS) is yet another initiative designed to supervise the distribution and use of thalidomide (Khalil et al. 2020).
Unfolding the mechanism of action of thalidomide for drug repurposing
Understanding the mechanism of teratogenicity is crucial for safer repurposing of thalidomide and its analogues for therapeutic applications. Soon after the horrifying thalidomide tragedy, research began on the teratogenicity of the drug. In 1979 (Blaschke et al. 1979), after thorough research on the thalidomide enantiomers, it was established that the (R)- and (S)- enantiomers possess varying biological characteristics, with the teratogenicity induced only by the (S)-enantiomer. No embryopathy was reported when the (R)- enantiomer was tested in animals (Tokunaga et al. 2018). There were several hypotheses proposed to explain the abnormal foetal development caused by thalidomide. One such theory was that thalidomide induces oxidative stress, thereby altering the NF-ĸB activity, causing impaired limb formation and other complications in embryos (Hansen et al. 2002). Another theory attributed the teratogenicity to the anti-angiogenic action of thalidomide, suggesting that there is a downregulation of growth factors and aberrant blood vessel formation at the limbs (D’Amato et al. 1994). However, these theories failed to elaborate on the targets of thalidomide and how these targets regulate its action (Asatsuma-Okumura et al. 2020).
In 2010, a breakthrough study identified that thalidomide binds to a protein called cereblon, linked to a crucial gene for mental retardation (Ito et al. 2010). Cereblon was recognized as the primary target for thalidomide. Cereblon is known to bind to another protein called damaged DNA binding protein 1 (DDB1) (Ito et al. 2011). Further, the DDB1 and Cul4 (cullin-4) configure an E3-ubiquitin ligase complex called Cullin-RING Ligase-4 (CRL4CRBN) (Chang and Keith Stewart 2011). This complex plays a vital role in the limb formation and expression of growth factors necessary for limb development (Ito and Handa 2012). Thus, it was concluded that the primary targets of thalidomide are (a) cereblon, which interacts directly with thalidomide, and (b) DDB1, which then interacts indirectly by its binding to cereblon (Ito et al. 2010). By binding to cereblon, thalidomide interferes with the normal functioning of the ligase complex and causes the degradation of certain neosubstrates, thus altering the expression of genes related to embryonic growth, leading to foetal abnormalities (Fig. 2) (Gao et al. 2020). Therefore, thalidomide and its analogues are now considered as cereblon-modulating drugs, especially for cancer treatment (Asatsuma-Okumura et al. 2019b). Further, Ito and Handa (2020) have explained the old and the current model of thalidomide mechanism of action to induce teratogenicity and its exploration in recent drug development (Ito and Handa 2020). Thalidomide downregulates fibroblast growth factor 8 (FGF8), thereby upregulating the apoptotic genes in the limbs (Kawamura et al. 2014). Therefore, the FGF8 downregulation could be another molecular mechanism that explains the abnormally short limbs in deformed infants exposed to thalidomide during embryonic development (Asatsuma-Okumura et al. 2019a). The cereblon protein is found primarily in the brain, particularly the cerebellum. Mutations in the cereblon gene is associated with mental retardation, compromising cognitive processes and memory (Choi et al. 2018). Apart from the brain, cereblon is largely present in the kidney, lung, liver, intestines, placenta, and prostate. Further, cereblon engages in the regulation of several other processes, such as the regulation of calcium and chloride channels (Kim et al. 2016). Cereblon also acts as a substrate receptor protein in the E3-ubiquitin ligase complex and determines the specificity of the substrate-binding to the complex. Hence, cereblon is pivotal in handling the degradation of many substrates (Liu et al. 2015). However, the recent study by Kowalski et al., also says that the cereblon mechanism vary highly between 14 species. Thalidomide does not have teratogenic effect in mice, while limb anomalies are absent in rats. In rabbits and sheep, thalidomide produces teratogenic effects, and the teratogenic characteristics in Rhesus monkeys are similar to that of humans (Kowalski et al. 2021).

Mechanism of thalidomide-induced teratogenicity. Thalidomide binds to cereblon (CRBN), which is a part of the E3-ubiquitin ligase complex with the damaged DNA binding protein 1 (DDB1), and cullin-4 (Cul4) forming Cullin-RING Ligase-4 (CRL4CRBN) complex. This leads to an interruption in the normal functioning of cereblon and recruitment of ‘neosubstrates’ such as SALL4 and p63 for degradation, initiating altered gene expression and subsequent limb malformations. Figures are created with created with BioRender.com
Etiopathogenesis and role of cereblon in pulmonary fibrosis
Despite extensive research over the past years, the pathogenesis of IPF remains unclear. Recent advances have linked the disease to inflammation and oxidative stress, which occurs in response to tissue injury. Generally, several endogenous and external factors contribute to tissue injury and damage the alveolar epithelium. Genetic factors such as polymorphisms of the surfactant protein or variations in the mucin 5B gene (MUC5B/Muc5b) (Michalski and Schwartz 2020), environmental factors such as inhalation of irritant dust particles, microbial infections, lifestyle habits such as cigarette smoking, are some of the attributable factors (Kärkkäinen et al. 2017; Denver et al. 2018). The aetiology of the most common lung fibrotic condition, IPF, is still not clearly understood. However, the genetic and epigenetic factors equally contribute to the etiopathogenesis of IPF (Velagacherla et al. 2022). The telomerase reverse transcriptase (TERT) gene is involved in more than 50% of IPF patients (van Batenburg et al. 2020). TERT is highly expressed in chronic smokers, which signifies the role of epigenetics (Diaz de Leon et al. 2010). What was earlier thought to be chronic inflammation is now being attributed to an aberrant wound healing in genetically susceptible individuals (Daccord and Maher 2016). It is now believed that there is dysregulation at any of the three processes: (i) injury (ii) inflammation (iii) repair, along with the overexpressed pro-fibrotic mediators such as TGF-β, and IL-1β. This leads to cellular and molecular modifications that develop fibrosis by the formation of myofibroblasts and deposition of ECM components such as collagen in the tissues (Wilson and Wynn 2009; Cinausero et al. 2017). Research has established that epigenetic factors and sustained injuries to the alveolar epithelial layer lead to the activation of alveolar epithelial cells (AECs). These activated AECs further engage in the attraction of inflammatory cells and promote fibrogenesis by activating fibroblasts. The fibroblasts differentiate into myofibroblasts and facilitate the process of fibrosis (Magnini et al. 2017) (Fig. 3).

Etiopathogenesis of pulmonary fibrosis. Repeated tissue injuries cause AECs to undergo apoptosis and re-epithelialization, causing the recruitment of inflammatory cells and profibrogenic mediators, and subsequent ECM accumulation, leading to fibrosis. ECM: Extracellular matrix, EMT: Epithelial mesenchymal transition. Figures are created with created with BioRender.com
Recent research on the implications of cereblon in IPF has opened up a new drug target (Kang et al. 2021). Cereblon modulates the energy metabolism by negatively regulating the activation of the AMP-activated protein kinase (AMPK)-signalling pathway (Lee et al. 2011). Thus, the role of cereblon-AMPK signalling pathway in IPF can be explored for drug discovery. In recent studies, AMPK activators such as metformin, have shown a protective effect against IPF by preventing the TGF-β/Smad signalling pathway and release of collagen, fibronectin, α-smooth muscle actin (α-SMA), and other fibrogenic factors (Rangarajan et al. 2018; Cheng et al. 2021; Wu et al. 2022). Therefore, activation of AMPK may inhibit the TGF-β-induced fibroblast proliferation (Chen et al. 2022). AMPK has also shown an inhibitory action on Forkhead box protein M1 (FOXM1), which is a crucial transcription factor in the process of cell proliferation. FOXM1 is now found to play a role in activating fibroblasts and their proliferation (Gu et al. 2021). The activation of AMPK also decreased the endoplasmic reticulum stress and unfolded protein response, both of which are driving factors for the development of fibrosis by inducing tissue injury in the alveolar epithelium (Choi et al. 2016; Burman et al. 2018). A computational investigation of a well-known AMPK activator DHFO, having structural features that are slightly different from thalidomide was reported to have better CRBN binding affinity than thalidomide (Nayek et al. 2022), Thus, the activation of the AMPK signalling pathway is crucial for protecting against IPF.
The overexpression of cereblon would lead to the inactivation of AMPK, and subsequent progression of IPF. Cereblon binds to the α-subunit of AMPK and reduces its overall catalytic activity, which suppresses the activation of AMPK, and worsens IPF (Kang et al. 2021). A recent study conducted by Kang et al. in 2021 has provided a strong basis for the association of cereblon in IPF by showing that the collagen and fibronectin were decreased in cereblon gene (CRBN) knockout mice. On the other hand, the TGF-β-induced phosphorylation and activation of Smad proteins, which occurs through CRBN activation, was decreased in the presence of an AMPK activator such as metformin (Kang et al. 2021). Moreover, higher expression of cereblon is also related to increased lung injury by oxidative stress, followed by inflammation. This can be subdued by repressing the expression of cereblon, which will downregulate the activation of the NF-ĸB pathway and its downstream mediators in the lungs (Yang et al. 2020). Hence, modulation or inhibition of cereblon by thalidomide and its analogues could be a novel approach to treating IPF (as depicted in Fig. 4).

Role of cereblon and actions of thalidomide in pulmonary fibrosis. Overexpression of cereblon (CRBN) leads to the downregulation of AMPK, which otherwise inhibits TGF-β-induced fibroblast proliferation, myofibroblast differentiation and epithelial-mesenchymal transition (EMT). Thus, CRBN inhibits the protective actions of AMPK against pulmonary fibrosis. By inhibiting CRBN thalidomide stops the progression of fibrosis. Figures are created with created with BioRender.com
Current treatment options for pulmonary fibrosis
Currently, there are two approved drugs for IPF available in the clinics, namely pirfenidone and nintedanib. They are well tolerated upon long-term treatment without any newer safety alerts (Cameli et al. 2020). Due to the poor understanding about pathogenesis of IPF, there have been several therapeutic failures over the past decades. This has left us with minimal treatment options for IPF. The earliest approach was to target the resulting inflammatory response using immunomodulators and anti-inflammatory drugs. Drugs targeting inflammatory and immune responses were recommended in the latest update of clinical practice guidelines on the treatment of IPF (Luppi et al. 2021). There are clinical trials conducted on drugs for the treatment of IPF that were later declared potentially harmful (Lota and Wells 2013). One such trial was based on the importance of the coagulation cascade in IPF (Kubo et al. 2005). The drug warfarin did not show any benefit to patients with progressive IPF, and moreover, warfarin treatment was associated with increased mortality in patients with IPF (Noth et al. 2012). Another drug named everolimus, a macrocyclic proliferation signal inhibitor with anti-fibroproliferative activity, was tested for its efficacy in IPF (Malouf et al. 2011). This trial involving 89 patients with IPF, (confirmed by surgical lung biopsy) concluded that everolimus was associated with rapid disease progression (Malouf et al. 2011). One of the widely used treatments for IPF was a combination of prednisone, azathioprine, and N-acetylcysteine. However, the safety of this drug combination was not established. After a 60 week treatment period and 32 weeks follow-up, this three-drug combination was conclusively implicated with an increased risk of hospitalization and death (Raghu et al. 2012). There are quite a few clinical trials of ineffective drugs which made a landmark contribution to IPF patient care. One such clinical trial was on sildenafil, a phosphodiesterase-5 inhibitor. The study on 180 patients treated with sildenafil did not meet its pre-defined significance (Zisman et al. 2010). However, some secondary outcomes, such as the degree of dyspnoea and quality of life, showed minor differences in favour of sildenafil. Another study on a combination of sildenafil and pirfenidone did not show any beneficial effect in preventing pulmonary hypertension associated with IPF (Behr et al. 2021). A prospective, randomized, double-blind, placebo-controlled, parallel-group, event-driven, morbidity–mortality clinical trial of bosentan, (an endothelin receptor antagonist) was conducted on 616 patients to investigate the benefit of modulating pulmonary hypertension associated with IPF (Giordano et al. 2010). However, bosentan failed to show any efficacy because there were no differences between treatment groups in health-related quality of life or dyspnoea (King et al. 2011).
Some of the investigational drugs under clinical development are TD139, PLN-74809, PLN-74809, TRK-250, PRM-151, and pamrevlumab (Glass et al. 2022). The investigational drug TD139 is a small molecule inhibitor of galectin-3 (Hirani et al. 2021). Integrins are known to activate TGF-β and have a role in pulmonary fibrosis (Sheppard 2008). Developmental endothelial locus-1 (Del-1) is an endogenous inhibitor of TGF-β activation (Kim et al. 2020). The newer molecule PLN-74809, a dual selective inhibitor of integrins αVβ1/αVβ6, is an emerging drug of the future (Decaris et al. 2021). PLN-74809 inhibits the deposition of collagen in lung tissue by blocking TGF-β pathway and inhibiting phosphorylation of Smad-3. It has shown efficacy in clinical trials (Decaris et al. 2021). TRK-250, produces siRNA in the body, and targets TGF-β mRNA is in clinical trial phase-1 (NCT03727802). PRM-151, a recombinant human serum amyloid P/pentraxin 2 (PTX2) (Raghu et al. 2019), is currently in phase-3 clinical trials (NCT04552899). The monoclonal antibody pamrevlumab, which targets connective tissue growth factor (CTGF) is currently in phase-2 clinical trial (Richeldi et al. 2020).
Drug targets for thalidomide in pulmonary fibrosis
Anti-fibrotic action
The potential use of thalidomide in interstitial pulmonary diseases has been debated since early 2000s (Ye 2006). The immunomodulatory action of thalidomide was seen as a desirable attribute with a potential to treat and manage debilitating diseases such as IPF. In addition, over recent years, the antiangiogenic action of thalidomide has also gained the attention of researchers. Thalidomide is considered helpful in fibrotic diseases mainly due to its immunomodulatory, antiangiogenic, antioxidant and anti-inflammatory actions (Ye 2006). Thalidomide is being proven as antifibrotic in bleomycin-induced mouse model of pulmonary fibrosis (Tabata et al. 2007). Inhibition of TGF-β is the major mechanism reported. Furthermore, thalidomide downregulates α-SMA in human foetal lung fibroblasts (Choe et al. 2010). In bleomycin-induced pulmonary fibrosis, thalidomide downregulates p-JNK and α-SMA (Liu et al. 2014). Dong and colleagues (2017) demonstrated that thalidomide inhibits pulmonary fibrosis both in vitro and in vivo. The same group has also demonstrated that the activity is due to regulation of the enzyme thioredoxin reductase (Dong et al. 2017a). A cell line-based study conducted on Human Embryonic Lung Fibroblasts (HELF) has established that thalidomide is capable of directly exerting an antifibrotic action by inhibiting the activity of connective tissue growth factor (CTGF). CTGF is responsible for cell transformation, angiogenesis, myofibroblast differentiation and even upregulated ECM synthesis in the fibroblasts (Wu et al. 2020). This study proved that it was safer to block CTGF using thalidomide, than to block TGF-β altogether to prevent the pro-fibrotic actions of TGF-β. Thalidomide has also shown antifibrotic activity in the kidneys of streptozotocin-induced diabetic rats via upregulating the phosphorylation of AMPKα and inhibiting the NF-ĸB and TGF-β1/Smad signalling pathways of inflammation (Zhang et al. 2018). Moreover, it has also prevented the epithelial-mesenchymal transformation (EMT) through TGF-β-induced Smad-independent pathways by suppressing the phosphorylation and activation of several downstream proteins such as ERK, p38, Akt, which ultimately reduce the expression of fibrotic factors such as collagen and α-SMA (Zhou et al. 2017). The process of EMT is also triggered by thalidomide, mediated by EGFL6/PAX6 axis (Tang et al. 2020). Thalidomide has proven to be helpful in diminishing the expression of fibronectin by preventing keloid formation, thus being beneficial in treating skin fibrosis (Liang et al. 2013). With this emerging body of evidence, thalidomide shows potential for use in preventing fibrotic progression in pulmonary fibrosis.
Immunomodulatory and anti-inflammatory actions
The pathological evolution of IPF is strongly regulated by the entry of inflammatory cytokines and inflammatory mediators, such as IL-6, IL-1β, TNF-α, and TGF-β, into the alveolar space (Akdis et al. 2016). Thalidomide exerts anti-inflammatory effect by selectively inhibiting the production of TNF-α and other cytokines such as IL-12p40, IL-18, and IL-8 produced by the alveoli macrophages upon stimulation (Sampaio et al. 1991; Ye 2006). Thalidomide shows immunomodulatory action by preventing the activation of immune cells such as Th1 cells and related cytokines (Horton and Hallowell 2012). Choe et al. reported that thalidomide successfully inhibits the ERK1/2 pathways of signalling, which are induced by TGF-β in bleomycin-induced pulmonary fibrosis in mice (Choe et al. 2010). TGF-β is an important marker of tissue fibrosis that regulates the production of ECM components such as fibronectin and collagen (Kim et al. 2018). Thalidomide inhibits the pathways downstream of TGF-β, such as p38/Smad signalling, ERK 1/2 signalling, and MAPK pathways, by preventing the phosphorylation and subsequent activation of the proteins involved in the pathways (Liang et al. 2013; Zhou et al. 2017). The recent findings on IMiDs such as lenalidomide and pomalidomide promote the recruitment of aromatase to cereblon, resulting in the degradation of aromatase by proteasomes in immune cells (Tochigi et al. 2020). El-Zahabi et al. 2020, have synthesized more than 40 thalidomide analogues, which were immunomodulators and ameliorated human TNF-α, caspase-8 (CASP8), human vascular endothelial growth factor (VEGF) and nuclear factor-kappa-BP65 (NF-κBP65) in human colon cancer (HCT-116) cells. Further, these derivatives have been shown to be immunomodulatory anticancer agents (El-Zahabi et al. 2020).
Antiangiogenic action
Since the anti-angiogenic action of thalidomide was established in 1994, thalidomide has been considered a potential drug candidate for treating several types of cancers and other metabolic disorders that require the inhibition of blood supply to tumours, or correct a pathologic angiogenic state (Behl et al. 2017). In addition, it is possible to extend this theory to the treatment of IPF. The role of angiogenesis in IPF has been constantly debated. It is still unclear how the aberrant neovascularization in the alveolar epithelium is related to the progression of IPF. However, nintedanib, a tyrosine kinase inhibitor, shows its antifibrotic action by targeting vascular endothelial growth factor receptors (VEGFR), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF)-receptors, all of which are involved in angiogenic signalling pathways (Ackermann et al. 2017). This indicates the possibility of using thalidomide as an anti-angiogenic agent in the treatment of IPF. A study conducted by Tabata et al. reported successful prevention of bleomycin-induced pulmonary fibrosis in mice by the anti-angiogenic action of thalidomide. It was observed that thalidomide, in addition to inhibiting angiogenesis in mice lung tissue, also reduced the expression of IL-6, TGF-β1, VEGF, Ang-1 Ang-2, and COL1A1 mRNA (Tabata et al. 2007). Thalidomide targets epidermal growth factor-like domain multiple 6 (EGFL6) and inhibits angiogenesis through EGFL6/PAX6 axis (Tang et al. 2020). The effect of thalidomide on EGFL6 is mediated through cereblon protein interaction with protease Lon N-terminal peptide, and this process is mediated by proteasome (Tang et al. 2020). Thalidomide and other IMiDs have reportedly reduced cell migration and adhesion by inhibiting the VEGF receptor, thereby reducing the capillary density on fibrotic lesions (Domingo et al. 2020), demonstrating that thalidomide could possibly be used as an anti-angiogenic for managing IPF.
Cereblon inhibitory action
CRBN has been established as the main target of thalidomide and its analogues (Ito et al. 2010). On binding to CRBN, thalidomide inhibits the activity of the E3-ubiquitin ligase of the multiple-protein complex CRBN-DDB1-Cul4 (Shortt et al. 2013). Cereblon is also involved in the pathogenesis of IPF (Kang et al. 2021). Thalidomide and other IMiDs such as lenalidomide and pomalidomide inhibit the action of cereblon and allow the expression of AMPK (Yang et al. 2021). Inhibition of AMPK is one of the mechanisms by which metformin inhibits proliferation and differentiation of human foetal lung fibroblast (Gu et al. 2021; Chen et al. 2022). AMPK modulation through cereblon shows a protective action and slows down the growth of fibrogenic proteins such as fibronectin, collagen α-SMA. Further, AMPK is down-regulated by the CRL4A-CRBN axis through the polyubiquitination of AMPKα isoforms (Kwon et al. 2019). Thus, cereblon inhibition could be the significant mechanism in mitigating fibrosis.
Antioxidant action
There is compelling evidence to implicate oxidative stress as one of the molecular mechanisms in IPF. Oxidative stress occurs from the imbalance between generation of reactive oxygen species (ROS) and the body’s ability to detoxify them (Todd et al. 2012; Estornut et al. 2022). Antioxidant enzymes (such as superoxide dismutase (SOD), catalase, glutathione peroxidase), and malondialdehyde (MDA), in the tissue homogenate are the markers of oxidative stress (Tsikas 2017). MDA accounts for lipid peroxidation or the extent of tissue damage occurring at the alveolar basement membrane (Tsikas 2017). The current standard drug pirfenidone demonstrated to have good antioxidant defence in human pulmonary artery smooth muscle cells (HPASMCs) (Fois et al. 2018). Further, pirfenidone and nintedanib exert beneficial effects on oxidative stress markers in humans (Fois et al. 2020; Li et al. 2022a; Wang and Qu 2022). In a study conducted by Dong et al. in 2017, treatment with thalidomide significantly increased SOD and normalized MDA and ROS in bleomycin-induced mouse model of pulmonary fibrosis. In another study, thalidomide significantly reduced MDA and ROS to near normal, and SOD was increased (Dong et al. 2017a). In IPF, the antioxidant defences are regulated significantly through the redox-sensitive transcription factor nuclear factor, erythroid-derived 2 (Nrf2) (Wang et al. 2022; Dong et al. 2022). The Nrf2 binds to specific antioxidant response elements (AREs), stimulates the antioxidant enzyme genes, and regulates their expression (Walters et al. 2008; Liu et al. 2019). An in vivo study conducted in a bleomycin-induced mice model demonstrated the abilities of pirfenidone to balance the antioxidant defence or regulate the oxidative stress through Nrf2/Bach1 equilibrium (Liu et al. 2017). Similarly, thalidomide attenuated radiation-induced pulmonary fibrosis through Nrf2-dependent downregulation of the TGF-β/Smad3 pathways (Bian et al. 2018). These studies suggest that thalidomide exerts its protective effect against pulmonary fibrosis by suppressing bleomycin-induced oxidative stress and the resulting damage to the membrane, thereby preventing inflammation.
Current status of preclinical and clinical studies
Over the past few years, there has been a lot of research on repurposing thalidomide in various disorders. However, its use is currently restricted to multiple myeloma and erythema nodosum leprosum. Table 1 represents 40 different studies with highly specific markers studied in IPF models. Despite abundant preclinical data on thalidomide acting against IPF, none have ventured into a clinical trial. Suboptimal confidence in thalidomide in a clinical setting could be attributed to teratogenic effects (Khalil et al. 2020). The multiple mechanisms of action of thalidomide in lung fibrosis urged us to produce this comprehensive report. Anti-inflammatory, immunomodulatory, and anti-angiogenic actions of thalidomide have been shown to be benefit management of aberrant inflammatory responses in IPF. The TNF-α-inhibitory and antifibrotic activity of thalidomide could help mitigate the harmful effects from the abnormal wound healing process during IPF. TGF-β inhibition halts the fibrosis process or progression of the disease (Ye and Hu 2021). See Table 1 for multiple mechanisms of action reported in preclinical and clinical studies, supporting the preventive effect of thalidomide in lung fibrosis.
Currently approved drugs for IPF are pirfenidone and nintedanib, with similar pros and cons. Pirfenidone is not preferred in case of comorbidities such as renal end-stage disease, because 80% of its clearance is via renal route. Similarly, nintedanib is not recommended for moderate to severe hepatic impairment because most of its metabolism and excretion occur within the hepatic system (Morrow et al. 2022). However, the preference for selection between pirfenidone and nintedanib is based on the patient’s preference on dosing, the adverse drug reactions (ADRs), cost, and insurance coverage (Morrow et al. 2022). Pirfenidone produces ADRs such as nausea, rash, abdominal pain, upper respiratory infection, diarrhoea, fatigue, headache, dyspepsia, dizziness, vomiting, anorexia, gastroesophageal reflux disease (GERD), sinusitis, insomnia, weight loss, and arthralgia (Galli et al. 2017). A combination of pirfenidone with thalidomide has the advantage of mitigating these ADRs. Along with anti-emetic activity, thalidomide has a sedative effect and improves sleep patterns in patients with lung fibrosis (Horton et al. 2012). A recent case study of an IPF patient treated with thalidomide for cough suppression showed an improvement in the patient condition, especially in tackling cough (Haraf et al. 2018). However, the efficacy of relieving or reversing fibrosis has not been tested in the case. Although thalidomide has been used for treating cough in IPF, it is only used when other drugs fail. The recently published case study is one such example (Haraf et al. 2018). Previously, a randomized study included only 24 patients with IPF and cough (Horton et al. 2012), and another study had only 11 patients having cough in IPF (Horton et al. 2008). The recent meta-analysis on thalidomide did not include data from patients with IPF (Xie et al. 2022). Thus, thalidomide has not yet undergone thorough clinical investigations owing to its teratogenic history. The preclinical effect of thalidomide is mainly mediated by inhibition of TFG-β, TNF-α, ILs and by antioxidant mechanisms. Significant advancement in diagnosis and prognosis of IPF, and availability of improved biomarkers for statistical comparison of therapeutic output, make clinical trials appropriate and timely (Chen et al. 2019b; Christe et al. 2019; Hochhegger et al. 2019; Tomassetti et al. 2020). Hence, based on the above highlights, we propose a prospective clinical investigation for thalidomide in the treatment of IPF.
Conclusion
There has been significant progress in thalidomide research, paving the way for its potential repurposing in inflammatory and neoplastic conditions. Thalidomide, which had once been revoked of its title of wonder drug, has managed to find a place in healthcare for the treatment of multiple myeloma and erythema nodosum leprosum. Despite the uncertainty regarding its potential in pulmonary fibrosis, a growing body of evidence substantiates the rationale of repurposing thalidomide for treating and slowing down the onset of fibrosis. The discovery of the thalidomide-binding protein, cereblon, was a breakthrough study that helped understand, to a certain extent, the mechanism of action of thalidomide at the molecular level. With recent information on the role of cereblon in the progression of IPF and newer biomarkers established for clinical trial assessments, thalidomide will have a promising role in preventing fibrosis, warranting future investigations.
Data availability
As this is a review manuscript, there is no data generated.
References
Ackermann M, Kamp JC, Werlein C et al (2022) The fatal trajectory of pulmonary COVID-19 is driven by lobular ischemia and fibrotic remodelling. EBioMedicine 85:104296. https://doi.org/10.1016/j.ebiom.2022.104296
Ackermann M, Kim YO, Wagner WL et al (2017) Effects of nintedanib on the microvascular architecture in a lung fibrosis model. Angiogenesis 20:359–372. https://doi.org/10.1007/s10456-017-9543-z
Akdis M, Aab A, Altunbulakli C et al (2016) Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J Allergy Clin Immunol 138:984–1010. https://doi.org/10.1016/j.jaci.2016.06.033
Al-kuraishy HM, Batiha GE-S, Faidah H et al (2022) Pirfenidone and post-Covid-19 pulmonary fibrosis: invoked again for realistic goals. Inflammopharmacology 30:2017–2026. https://doi.org/10.1007/s10787-022-01027-6
Amare GG, Meharie BG, Belayneh YM (2021) A drug repositioning success: the repositioned therapeutic applications and mechanisms of action of thalidomide. J Oncol Pharm Pract 27:673–678. https://doi.org/10.1177/1078155220975825
Amirshahrokhi K (2013) Anti-inflammatory effect of thalidomide in paraquat-induced pulmonary injury in mice. Int Immunopharmacol 17:210–215. https://doi.org/10.1016/j.intimp.2013.06.005
Amirshahrokhi K, Khalili A-R (2015) Thalidomide ameliorates cisplatin-induced nephrotoxicity by inhibiting renal inflammation in an experimental model. Inflammation 38:476–484. https://doi.org/10.1007/s10753-014-9953-7
Arai H, Furusu A, Nishino T et al (2011) Thalidomide prevents the progression of peritoneal fibrosis in mice. Acta Histochem Cytochem 44:51–60. https://doi.org/10.1267/ahc.10030
Asatsuma-Okumura T, Ando H, De Simone M et al (2019a) p63 is a cereblon substrate involved in thalidomide teratogenicity. Nat Chem Biol 15:1077–1084. https://doi.org/10.1038/s41589-019-0366-7
Asatsuma-Okumura T, Ito T, Handa H (2019b) Molecular mechanisms of cereblon-based drugs. Pharmacol Ther 202:132–139. https://doi.org/10.1016/j.pharmthera.2019.06.004
Asatsuma-Okumura T, Ito T, Handa H (2020) Molecular mechanisms of the teratogenic effects of thalidomide. Pharmaceuticals 13:95. https://doi.org/10.3390/ph13050095
Barbarossa A, Iacopetta D, Sinicropi MS et al (2022) Recent advances in the development of thalidomide-related compounds as anticancer drugs. Curr Med Chem 29:19–40. https://doi.org/10.2174/0929867328666210623143526
Barratt S, Creamer A, Hayton C, Chaudhuri N (2018) Idiopathic pulmonary fibrosis (IPF): an overview. J Clin Med 7:201. https://doi.org/10.3390/jcm7080201
Behl T, Kaur I, Goel H, Kotwani A (2017) Significance of the antiangiogenic mechanisms of thalidomide in the therapy of diabetic retinopathy. Vascul Pharmacol 92:6–15. https://doi.org/10.1016/J.VPH.2015.07.003
Behr J, Nathan SD, Wuyts WA et al (2021) Efficacy and safety of sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med 9:85–95. https://doi.org/10.1016/S2213-2600(20)30356-8
Bersani-Amado LE, Dantas JA, Damião MJ et al (2016) Involvement of cytokines in the modulation and progression of renal fibrosis induced by unilateral ureteral obstruction in C57BL/6 mice: effects of thalidomide and dexamethasone. Fundam Clin Pharmacol 30:35–46. https://doi.org/10.1111/fcp.12162
Bian C, Qin W-J, Zhang C-Y et al (2018) Thalidomide (THD) alleviates radiation induced lung fibrosis (RILF) via down-regulation of TGF-β/Smad3 signaling pathway in an Nrf2-dependent manner. Free Radic Biol Med 129:446–453. https://doi.org/10.1016/j.freeradbiomed.2018.10.423
Blaschke G, Kraft HP, Fickentscher K, Köhler F (1979) Chromatographic separation of racemic thalidomide and teratogenic activity of its enantiomers. Arzneimittel-Forschung/drug Res 29:1640–1642
Bobowski-Gerard M, Boulet C, Zummo FP et al (2022) Functional genomics uncovers the transcription factor BNC2 as required for myofibroblastic activation in fibrosis. Nat Commun 13:5324. https://doi.org/10.1038/s41467-022-33063-9
Brandenburg NA, Bwire R, Freeman J et al (2017) Effectiveness of risk evaluation and mitigation strategies (REMS) for lenalidomide and thalidomide: patient comprehension and knowledge retention. Drug Saf 40:333–341. https://doi.org/10.1007/s40264-016-0501-2
Burman A, Tanjore H, Blackwell TS (2018) Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol 68–69:355. https://doi.org/10.1016/J.MATBIO.2018.03.015
Cameli P, Refini RM, Bergantini L et al (2020) Long-term follow-up of patients with idiopathic pulmonary fibrosis treated with pirfenidone or nintedanib: a real-life comparison study. Front Mol Biosci. https://doi.org/10.3389/fmolb.2020.581828
Chang X-B, Keith Stewart A (2011) What is the functional role of the thalidomide binding protein cereblon? Int J Biochem Mol Biol 2:287–294
Chen C, Qi F, Shi K et al (2020) Thalidomide combined with low-dose short-term glucocorticoid in the treatment of critical Coronavirus Disease 2019. Clin Transl Med 10:e35. https://doi.org/10.1002/ctm2.35
Chen H, Xu H, Luo L et al (2019a) Thalidomide prevented and ameliorated pathogenesis of Crohn’s disease in mice via regulation of inflammatory response and fibrosis. Front Pharmacol. https://doi.org/10.3389/fphar.2019.01486
Chen L, Halai V, Leandru A, Wallis A (2019b) Interstitial lung disease: update on the role of computed tomography in the diagnosis of idiopathic pulmonary fibrosis. J Comput Assist Tomogr 43:898–905. https://doi.org/10.1097/RCT.0000000000000915
Chen Q, Wang Y, Sheng L, Huang Y (2022) Metformin suppresses proliferation and differentiation induced by BMP9 via AMPK signaling in human fetal lung fibroblast-1. Front Pharmacol. https://doi.org/10.3389/fphar.2022.984730
Cheng D, Xu Q, Wang Y et al (2021) Metformin attenuates silica-induced pulmonary fibrosis via AMPK signaling. J Transl Med 19:1–18. https://doi.org/10.1186/S12967-021-03036-5/FIGURES/9
Choe J-Y, Jung H-J, Park K-Y et al (2010) Anti-fibrotic effect of thalidomide through inhibiting TGF-β-induced ERK1/2 pathways in bleomycin-induced lung fibrosis in mice. Inflamm Res 59:177–188. https://doi.org/10.1007/s00011-009-0084-9
Choi SM, Jang AH, Kim H et al (2016) Metformin reduces bleomycin-induced pulmonary fibrosis in mice. J Korean Med Sci 31:1419–1425. https://doi.org/10.3346/JKMS.2016.31.9.1419
Choi T-Y, Lee S-H, Kim Y-J et al (2018) Cereblon maintains synaptic and cognitive function by regulating BK channel. J Neurosci 38:3571–3583. https://doi.org/10.1523/JNEUROSCI.2081-17.2018
Christe A, Peters AA, Drakopoulos D et al (2019) Computer-aided diagnosis of pulmonary fibrosis using deep learning and CT images. Invest Radiol 54:627–632. https://doi.org/10.1097/RLI.0000000000000574
Cinausero M, Aprile G, Ermacora P et al (2017) New frontiers in the pathobiology and treatment of cancer regimen-related mucosal injury. Front Pharmacol. https://doi.org/10.3389/fphar.2017.00354
Cooper CR, Poindexter C, Rohe B, Sikes RA (2010) Thalidomide and its analogues in prostate cancer therapy—a scientific update. Biochem (lond) 32:36–39. https://doi.org/10.1042/bio03205036
D’Amato RJ, Loughnan MS, Flynn E, Folkman J (1994) Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci 91:4082–4085. https://doi.org/10.1073/pnas.91.9.4082
Daccord C, Maher TM (2016) Recent advances in understanding idiopathic pulmonary fibrosis. F1000Research 5:1046. https://doi.org/10.12688/f1000research.8209.1
Decaris ML, Schaub JR, Chen C et al (2021) Dual inhibition of αvβ6 and αvβ1 reduces fibrogenesis in lung tissue explants from patients with IPF. Respir Res 22:265. https://doi.org/10.1186/s12931-021-01863-0
Deng M-Y, Ahmad KA, Han Q-Q et al (2021) Thalidomide alleviates neuropathic pain through microglial IL-10/β-endorphin signaling pathway. Biochem Pharmacol. https://doi.org/10.1016/j.bcp.2021.114727
Denver C, Eickelberg UO, Schwarz MI, Schwartz DA (2018) Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir Med 6:154–160. https://doi.org/10.1016/S2213-2600(18)30007-9
Diaz de Leon A, Cronkhite JT, Katzenstein A-LA, et al (2010) Telomere Lengths, Pulmonary Fibrosis and Telomerase (TERT) Mutations. PLoS One 5:e10680. https://doi.org/10.1371/journal.pone.0010680
Domingo S, Solé C, Moliné T et al (2020) Efficacy of thalidomide in discoid lupus erythematosus: insights into the molecular mechanisms. Dermatology 236:467–476. https://doi.org/10.1159/000508672
Dong X, Li X, Li M et al (2017a) Inhibitory effects of thalidomide on bleomycin-induced pulmonary fibrosis in rats via regulation of thioredoxin reductase and inflammations. Am J Transl Res 9:4390–4401
Dong X, Li X, Li M et al (2017b) Antiinflammation and antioxidant effects of thalidomide on pulmonary fibrosis in mice and human lung fibroblasts. Inflammation 40:1836–1846. https://doi.org/10.1007/s10753-017-0625-2
Dong Z, Yin EG, Yang M et al (2022) Role and mechanism of keap1/nrf2 signaling pathway in the regulation of autophagy in alleviating pulmonary fibrosis. Comput Intell Neurosci 2022:1–9. https://doi.org/10.1155/2022/3564871
El-Aarag B, Kasai T, Masuda J et al (2017) Anticancer effects of novel thalidomide analogs in A549 cells through inhibition of vascular endothelial growth factor and matrix metalloproteinase-2. Biomed Pharmacother 85:549–555. https://doi.org/10.1016/j.biopha.2016.11.063
El-Zahabi MA, Sakr H, El-Adl K et al (2020) Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg Chem 104:104218. https://doi.org/10.1016/j.bioorg.2020.104218
Estornut C, Milara J, Bayarri MA et al (2022) Targeting oxidative stress as a therapeutic approach for idiopathic pulmonary fibrosis. Front Pharmacol. https://doi.org/10.3389/fphar.2021.794997
Fois AG, Posadino AM, Giordo R et al (2018) Antioxidant activity mediates pirfenidone antifibrotic effects in human pulmonary vascular smooth muscle cells exposed to sera of idiopathic pulmonary fibrosis patients. Oxid Med Cell Longev. https://doi.org/10.1155/2018/2639081
Fois AG, Sotgiu E, Scano V et al (2020) Effects of pirfenidone and nintedanib on markers of systemic oxidative stress and inflammation in patients with idiopathic pulmonary fibrosis: a preliminary report. Antioxidants 9:1–15. https://doi.org/10.3390/antiox9111064
Furihata H, Yamanaka S, Honda T et al (2020) Structural bases of IMiD selectivity that emerges by 5-hydroxythalidomide. Nat Commun. https://doi.org/10.1038/s41467-020-18488-4
Galli JA, Pandya A, Vega-Olivo M et al (2017) Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: tolerability and adverse drug reactions. Respirology 22:1171–1178. https://doi.org/10.1111/resp.13024
Gao S, Wang S, Fan R, Hu J (2020) Recent advances in the molecular mechanism of thalidomide teratogenicity. Biomed Pharmacother 127:110114. https://doi.org/10.1016/j.biopha.2020.110114
Giordano N, Puccetti L, Papakostas P et al (2010) Bosentan treatment for Raynauds phenomenon and skin fibrosis in patients with systemic sclerosis and pulmonary arterial hypertension: an open-label, observational, retrospective study. Int J Immunopathol Pharmacol 23:1185–1194. https://doi.org/10.1177/039463201002300422
Glass DS, Grossfeld D, Renna HA et al (2022) Idiopathic pulmonary fibrosis: current and future treatment. Clin Respir J. https://doi.org/10.1111/crj.13466
Gu X, Han YY, Yang CY et al (2021) Activated AMPK by metformin protects against fibroblast proliferation during pulmonary fibrosis by suppressing FOXM1. Pharmacol Res 173:105844. https://doi.org/10.1016/J.PHRS.2021.105844
Gutiérrez-Rodríguez O (1984) Thalidomide a promising new treatment for rheumatoid arthritis. Arthritis Rheum 27:1118–1121. https://doi.org/10.1002/art.1780271006
Gutierrez-Rodriguez O, Starusta-Bacal P, Gutierrez-Montes O (1989) Treatment of refractory rheumatoid arthritis - the thalidomide experience. J Rheumatol 16:158–163
Hamza MH (1986) Treatment of Behçet’s disease with thalidomide. Clin Rheumatol 5:365–371. https://doi.org/10.1007/BF02054255
Hansen JM, Gong S-G, Philbert M, Harris C (2002) Misregulation of gene expression in the redox-sensitive NF-?b-dependent limb outgrowth pathway by thalidomide. Dev Dyn 225:186–194. https://doi.org/10.1002/dvdy.10150
Haraf R, Flora AS, Assaly R (2018) Thalidomide as a cough suppressant in idiopathic pulmonary fibrosis. Am J Ther 25:E687–E688. https://doi.org/10.1097/MJT.0000000000000695
Hirani N, MacKinnon AC, Nicol L et al (2021) Target inhibition of galectin-3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. Eur Respir J. https://doi.org/10.1183/13993003.02559-2020
Hochhegger B, Marchiori E, Zanon M et al (2019) Imaging in idiopathic pulmonary fibrosis: diagnosis and mimics. Clinics. https://doi.org/10.6061/clinics/2019/e225
Holstein SA, McCarthy PL (2017) Immunomodulatory drugs in multiple myeloma: mechanisms of action and clinical experience. Drugs 77:505–520. https://doi.org/10.1007/s40265-017-0689-1
Horton MR, Danoff SK, Lechtzin N (2008) Thalidomide inhibits the intractable cough of idiopathic pulmonary fibrosis. Thorax 63:749. https://doi.org/10.1136/thx.2008.098699
Horton MR, Hallowell RW (2012) Revisiting thalidomide: fighting with caution against idiopathic pulmonary fibrosis. Drugs Today 48:661–671. https://doi.org/10.1358/dot.2012.48.10.1855760
Horton MR, Santopietro V, Mathew L et al (2012) Thalidomide for the treatment of cough in idiopathic pulmonary fibrosis: a randomized trial. Ann Intern Med 157:398–406. https://doi.org/10.7326/0003-4819-157-6-201209180-00003
Hosseini-Chegeni A, Jazaeri F, Yousefi-Ahmadipour A et al (2019) Thalidomide attenuates the hyporesponsiveness of isolated atria to chronotropic stimulation in BDL rats: The involvement of TNF-α, IL-6 inhibition, and SOCS1 activation. Iran J Basic Med Sci 22:1259–1266
Hsu VM, Denton CP, Domsic RT et al (2018) Pomalidomide in patients with interstitial lung disease due to systemic sclerosis: a phase II, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. J Rheumatol 45:405–410. https://doi.org/10.3899/jrheum.161040
Ito T, Ando H, Handa H (2011) Teratogenic effects of thalidomide: molecular mechanisms. Cell Mol Life Sci 68:1569–1579. https://doi.org/10.1007/s00018-010-0619-9
Ito T, Ando H, Suzuki T et al (2010) Identification of a primary target of thalidomide teratogenicity. Science 80(327):1345–1350. https://doi.org/10.1126/science.1177319
Ito T, Handa H (2012) Deciphering the mystery of thalidomide teratogenicity. Congenit Anom (Kyoto) 52:1–7. https://doi.org/10.1111/j.1741-4520.2011.00351.x
Ito T, Handa H (2020) Molecular mechanisms of thalidomide and its derivatives. Proc Japan Acad Ser B Phys Biol Sci 96:189–203. https://doi.org/10.2183/PJAB.96.016
Jung YJ, Tweedie D, Scerba MT et al (2021) Repurposing immunomodulatory imide drugs (IMiDs) in neuropsychiatric and neurodegenerative disorders. Front Neurosci. https://doi.org/10.3389/fnins.2021.656921
Kang HJ, Lee KJ, Woo J et al (2021) Cereblon contributes to the development of pulmonary fibrosis via inactivation of adenosine monophosphate-activated protein kinase α1. Exp Mol Med 53:885–893. https://doi.org/10.1038/s12276-021-00619-6
Kang Y, Zhang C, He Y et al (2022) Thalidomide attenuates skin lesions and inflammation in rosacea-like mice induced by long-term exposure of LL-37. Drug Des Devel Ther 16:4127–4138. https://doi.org/10.2147/DDDT.S393122
Kärkkäinen M, Kettunen H-P, Nurmi H et al (2017) Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir Res 18:160. https://doi.org/10.1186/s12931-017-0642-6
Kawamura Y, Yamashita T, Yamauchi T et al (2014) Effects of thalidomide on Fgf8, Bmp4 and Hoxa11 expression in the limb bud in Kbl: JW rabbit embryos. Congenit Anom (kyoto) 54:54–62. https://doi.org/10.1111/cga.12046
Khalil A, Kamar A, Nemer G (2020) Thalidomide-revisited: are COVID-19 patients going to be the latest victims of yet another theoretical drug-repurposing? Front Immunol 11:1248. https://doi.org/10.3389/FIMMU.2020.01248/BIBTEX
Khan MI, Momeny M, Ostadhadi S et al (2018) Thalidomide attenuates development of morphine dependence in mice by inhibiting PI3K/Akt and nitric oxide signaling pathways. Prog Neuro-Psychopharmacol Biol Psychiatry 82:39–48. https://doi.org/10.1016/j.pnpbp.2017.12.002
Khan MI, Ostadhadi S, Mumtaz F et al (2017) Thalidomide attenuates the development and expression of antinociceptive tolerance to μ-opioid agonist morphine through L-arginine-iNOS and nitric oxide pathway. Biomed Pharmacother 85:493–502. https://doi.org/10.1016/j.biopha.2016.11.056
Kim D-Y, Lee S-H, Fu Y et al (2020) Del-1 an endogenous inhibitor of TGF-β activation attenuates fibrosis. Front Immunol. https://doi.org/10.3389/fimmu.2020.00068
Kim HK, Ko TH, Nyamaa B et al (2016) Cereblon in health and disease. Pflügers Arch - Eur J Physiol 468:1299–1309. https://doi.org/10.1007/s00424-016-1854-1
Kim JH, Scialli AR (2011) Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol Sci 122:1–6. https://doi.org/10.1093/toxsci/kfr088
Kim KK, Sheppard D, Chapman HA (2018) TGF-β1 signaling and tissue fibrosis. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a022293
King TE, Brown KK, Raghu G et al (2011) BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 184:92–99. https://doi.org/10.1164/rccm.201011-1874OC
Kowalski TW, Caldas-Garcia GB, Gomes JDA et al (2021) Comparative genomics identifies putative interspecies mechanisms underlying crbn-sall4-linked thalidomide embryopathy. Front Genet. https://doi.org/10.3389/fgene.2021.680217
Kubo H, Nakayama K, Yanai M et al (2005) Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest 128:1475–1482. https://doi.org/10.1378/chest.128.3.1475
Kumar N, Sharma U, Singh C, Singh B (2012) Thalidomide: chemistry, therapeutic potential and oxidative stress induced teratogenicity. Curr Top Med Chem 12:1436–1455. https://doi.org/10.2174/156802612801784407
Kwon E, Li X, Deng Y et al (2019) AMPK is down-regulated by the CRL4A-CRBN axis through the polyubiquitination of AMPKα isoforms. FASEB J 33:6539–6550. https://doi.org/10.1096/fj.201801766RRR
Lee KM, Jo S, Kim H et al (2011) Functional modulation of AMP-activated protein kinase by cereblon. Biochim Biophys Acta Mol Cell Res 1813:448–455. https://doi.org/10.1016/J.BBAMCR.2011.01.005
Li D, Zhang X-W, Jiang X-Q et al (2015) Protective effects of thalidomide on pulmonary injuries in a rat model of paraquat intoxication. J Inflamm (united Kingdom). https://doi.org/10.1186/s12950-015-0093-0
Li L, Chen Y, Shi C (2022a) Nintedanib ameliorates oxidized low-density lipoprotein induced inflammation and cellular senescence in vascular endothelial cells. Bioengineered 13:6196–6207. https://doi.org/10.1080/21655979.2022.2036913
Li Y, Cai W, Jin F et al (2022b) Thalidomide alleviates pulmonary fibrosis induced by silica in mice by inhibiting ER stress and the TLR4-NF-κB pathway. Int J Mol Sci. https://doi.org/10.3390/ijms23105656
Liang CJ, Yen YH, Hung LY et al (2013) Thalidomide inhibits fibronectin production in TGF-β1-treated normal and keloid fibroblasts via inhibition of the p38/Smad3 pathway. Biochem Pharmacol 85:1594–1602. https://doi.org/10.1016/J.BCP.2013.02.038
Liu JH, Wu HH, Zhao YK et al (2018) Thalidomide improves psoriasis-like lesions and inhibits cutaneous vegf expression without alteration of microvessel density in imiquimod-induced psoriatic mouse model. Curr Vasc Pharmacol 16:510–521. https://doi.org/10.2174/1570161115666171004123428
Liu Q, Gao Y, Ci X (2019) Role of Nrf2 and its activators in respiratory diseases. Oxid Med Cell Longev 2019:1–17. https://doi.org/10.1155/2019/7090534
Liu X, Qian L, Nan H et al (2014) Function of the transforming growth factor-β1/c-Jun N-terminal kinase signaling pathway in the action of thalidomide on a rat model of pulmonary fibrosis. Exp Ther Med 7:669–674. https://doi.org/10.3892/etm.2013.1457
Liu Y, Huang X, He X et al (2015) A novel effect of thalidomide and its analogs: Suppression of cereblon ubiquitination enhances ubiquitin ligase function. FASEB J 29:4829–4839. https://doi.org/10.1096/FJ.15-274050/-/DC1
Liu Y, Lu F, Kang L et al (2017) Pirfenidone attenuates bleomycin-induced pulmonary fibrosis in mice by regulating Nrf2/Bach1 equilibrium. BMC Pulm Med. https://doi.org/10.1186/s12890-017-0405-7
Lota HK, Wells AU (2013) The evolving pharmacotherapy of pulmonary fibrosis. Expert Opin Pharmacother 14:79–89. https://doi.org/10.1517/14656566.2013.758250
Lu Y, Zhao C, Lei L et al (2020) Effects of thalidomide on Th17, Treg cells and TGF-β1/Smad3 pathway in a mouse model of systemic sclerosis. Int J Rheum Dis 23:406–419. https://doi.org/10.1111/1756-185X.13769
Luppi F, Kalluri M, Faverio P et al (2021) Idiopathic pulmonary fibrosis beyond the lung: understanding disease mechanisms to improve diagnosis and management. Respir Res 22:109. https://doi.org/10.1186/s12931-021-01711-1
Magnini D, Montemurro G, Iovene B et al (2017) Idiopathic pulmonary fibrosis: molecular endotypes of fibrosis stratifying existing and emerging therapies. Respiration 93:379–395. https://doi.org/10.1159/000475780
Maher TM, Bendstrup E, Dron L et al (2021) Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res 22:197. https://doi.org/10.1186/s12931-021-01791-z
Majumder S, Rama Chaitanya Sreedhara S, Banerjee S, Chatterjee S (2012) TNF α signaling beholds thalidomide saga: a review of mechanistic role of TNF-α signaling under thalidomide. Curr Top Med Chem 12:1456–1467. https://doi.org/10.2174/156802612801784443
Malouf MA, Hopkins P, Snell G, Glanville A (2011) An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology 16:776–783. https://doi.org/10.1111/j.1440-1843.2011.01955.x
Martinez FJ, Collard HR, Pardo A et al (2017) (2017) Idiopathic pulmonary fibrosis. Nat Rev Dis Prim 31(3):1–19. https://doi.org/10.1038/nrdp.2017.74
Mellin GW, Katzenstein M (1962) The saga of thalidomide - neuropathy to embryopathy, with case reports of congenital anomalies. N Engl J Med 267:1238–1244. https://doi.org/10.1056/NEJM196212132672407
Michalski JE, Schwartz DA (2020) Genetic risk factors for idiopathic pulmonary fibrosis: Insights into immunopathogenesis. J Inflamm Res 13:1305–1318. https://doi.org/10.2147/JIR.S280958
Miyazato K, Tahara H, Hayakawa Y (2020) Antimetastatic effects of thalidomide by inducing the functional maturation of peripheral natural killer cells. Cancer Sci 111:2770–2778. https://doi.org/10.1111/cas.14538
Morrow LE, Hilleman D, Malesker MA (2022) Management of patients with fibrosing interstitial lung diseases. Am J Heal Pharm 79:129–139. https://doi.org/10.1093/ajhp/zxab375
Mudawi D, Heyes K, Hastings R et al (2021) An update on interstitial lung disease. Br J Hosp Med 82:1–14. https://doi.org/10.12968/hmed.2020.0556
Nayek U, Basheer Ahamed SI, Mansoor Hussain UH et al (2022) Computational investigations of indanedione and indanone derivatives in drug discovery: indanone derivatives inhibits cereblon, an E3 ubiquitin ligase component. Comput Biol Chem. https://doi.org/10.1016/j.compbiolchem.2022.107776
Nematbakhsh M, Rafieyan S, Mirkheshti N et al (2009) Thalidomide reduces the level of nitric oxide in Bleomycin induced pulmonary fibrosis model. Clin Exp Med Lett 50:75–76
Newbronner E, Glendinning C, Atkin K, Wadman R (2019) The health and quality of life of Thalidomide survivors as they age—evidence from a UK survey. PLoS ONE. https://doi.org/10.1371/journal.pone.0210222
Noth I, Anstrom KJ, Calvert SB et al (2012) A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. https://doi.org/10.1164/rccm.201202-0314OC
Piura B, Medina L, Rabinovich A et al (2013) Thalidomide distinctly affected TNF-α, IL-6 and MMP secretion by an ovarian cancer cell line (SKOV-3) and primary ovarian cancer cells. Eur Cytokine Netw 24:122–129. https://doi.org/10.1684/ecn.2013.0342
Raghu G, Anstrom KJ, King TE Jr et al (2012) Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 366:1968–1977. https://doi.org/10.1056/NEJMoa1113354
Raghu G, van den Blink B, Hamblin MJ et al (2019) Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: an open-label extension study. Lancet Respir Med 7:657–664. https://doi.org/10.1016/S2213-2600(19)30172-9
Rangarajan S, Bone NB, Zmijewska AA et al (2018) Metformin reverses established lung fibrosis in a bleomycin model. Nat Med 24:1121. https://doi.org/10.1038/S41591-018-0087-6
Rehman W, Arfons LM, Lazarus HM (2011) The rise, fall and subsequent triumph of thalidomide: lessons learned in drug development. Ther Adv Hematol 2:291. https://doi.org/10.1177/2040620711413165
Reyes-Terán G, Sierra-Madero JG, Martínez Del Cerro V et al (1996) Effects of thalidomide on HIV-associated wasting syndrome: a randomized, double-blind, placebo-controlled clinical trial. AIDS 10:1501–1507. https://doi.org/10.1097/00002030-199611000-00007
Rezaie MJ, Rostamzadeh A, Keshavarz G et al (2019) Effect of Thalidomide on Cox-2 expression in bleomycin-induced pulmonary fibrosis in mice. Biomed Res Ther 6:2974–2982. https://doi.org/10.15419/bmrat.v6i1.518
Richeldi L, Collard HR, Jones MG (2017) Idiopathic pulmonary fibrosis. Lancet 389:1941–1952. https://doi.org/10.1016/S0140-6736(17)30866-8
Richeldi L, Fernández Pérez ER, Costabel U et al (2020) Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir Med 8:25–33. https://doi.org/10.1016/S2213-2600(19)30262-0
Samel C, Albus C, Nippert I et al (2019) Life situation of women impaired by Thalidomide embryopathy in North Rhine-Westphalia—a comparative analysis of a recent cross-sectional study with earlier data. BMC Womens Health 19:51. https://doi.org/10.1186/s12905-019-0745-y
Sampaio EP, Sarno EN, Galilly R et al (1991) Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med 173:699–703. https://doi.org/10.1084/jem.173.3.699
Sharma D, Kwatra SG (2016) Thalidomide for the treatment of chronic refractory pruritus. J Am Acad Dermatol 74:363–369. https://doi.org/10.1016/j.jaad.2015.09.039
Sheppard D (2008) The role of integrins in pulmonary fibrosis. Eur Respir Rev 17:157–162. https://doi.org/10.1183/09059180.00010909
Sheskin J (1965) Thalidomide in the treatment of lepra reactions. Clin Pharmacol Ther 6:303–306. https://doi.org/10.1002/cpt196563303
Shortt J, Hsu AK (2013) Johnstone RW (2013) Thalidomide-analogue biology: immunological, molecular and epigenetic targets in cancer therapy. Oncogene 3236(32):4191–4202. https://doi.org/10.1038/onc.2012.599
Tabata C, Tabata R, Kadokawa Y et al (2007) Thalidomide prevents bleomycin-induced pulmonary fibrosis in mice. J Immunol 179:708–714. https://doi.org/10.4049/jimmunol.179.1.708
Talaat R, El-Sayed W, Agwa HS et al (2015) Anti-inflammatory effect of thalidomide dithiocarbamate and dithioate analogs. Chem Biol Interact 238:74–81. https://doi.org/10.1016/j.cbi.2015.05.017
Tang C-T, Zhang Q-W, Wu S et al (2020) Thalidomide targets EGFL6 to inhibit EGFL6/PAX6 axis-driven angiogenesis in small bowel vascular malformation. Cell Mol Life Sci 77:5207–5221. https://doi.org/10.1007/s00018-020-03465-3
Tang KW, Hsu WL, Chen CR et al (2021) Discovery of triazolyl thalidomide derivatives as anti-fibrosis agents. New J Chem 45:3589–3599. https://doi.org/10.1039/D0NJ03139A
Tanni SE, Fabro AT, de Albuquerque A et al (2021) Pulmonary fibrosis secondary to COVID-19: a narrative review. Expert Rev Respir Med 15:791–803. https://doi.org/10.1080/17476348.2021.1916472
Tochigi T, Miyamoto T, Hatakeyama K et al (2020) Aromatase is a novel neosubstrate of cereblon responsible for immunomodulatory drug-induced thrombocytopenia. Blood 135:2146–2158. https://doi.org/10.1182/blood.2019003749
Todd NW, Luzina IG, Atamas SP (2012) Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 5:11. https://doi.org/10.1186/1755-1536-5-11
Tokunaga E, Yamamoto T, Ito E, Shibata N (2018) Understanding the thalidomide chirality in biological processes by the self-disproportionation of enantiomers. Sci Rep 8:17131. https://doi.org/10.1038/s41598-018-35457-6
Tomassetti S, Ravaglia C, Wells AU et al (2020) Prognostic value of transbronchial lung cryobiopsy for the multidisciplinary diagnosis of idiopathic pulmonary fibrosis: a retrospective validation study. Lancet Respir Med 8:786–794. https://doi.org/10.1016/S2213-2600(20)30122-3
Tsikas D (2017) Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: analytical and biological challenges. Anal Biochem 524:13–30. https://doi.org/10.1016/J.AB.2016.10.021
van Batenburg AA, Kazemier KM, van Oosterhout MFM et al (2020) From organ to cell: Multi-level telomere length assessment in patients with idiopathic pulmonary fibrosis. PLoS ONE 15:e0226785. https://doi.org/10.1371/journal.pone.0226785
van Toorn R, Zaharie S-D, Seddon JA et al (2021) The use of thalidomide to treat children with tuberculosis meningitis: a review. Tuberculosis. https://doi.org/10.1016/j.tube.2021.102125
Vargesson N (2015) Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res Part C Embryo Today Rev 105:140–156. https://doi.org/10.1002/bdrc.21096
Velagacherla V, Mehta CH, Nayak Y, Nayak UY (2022) Molecular pathways and role of epigenetics in the idiopathic pulmonary fibrosis. Life Sci 291:120283. https://doi.org/10.1016/j.lfs.2021.120283
Walters DM, Cho H-Y, Kleeberger SR (2008) Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: a potential role for Nrf2. Antioxidants Redox Signal 10:321–332. https://doi.org/10.1089/ars.2007.1901
Wang C, Qu L (2022) The anti-fibrotic agent nintedanib protects chondrocytes against tumor necrosis factor-ɑ (TNF-ɑ)-induced extracellular matrix degradation. Bioengineered 13:5318–5329. https://doi.org/10.1080/21655979.2022.2036899
Wang L, Wang S, Xue A et al (2021) Thalidomide inhibits angiogenesis via downregulation of VEGF and angiopoietin-2 in Crohn’s Disease. Inflammation 44:795–807. https://doi.org/10.1007/s10753-020-01378-8
Wang Y, Wei J, Deng H et al (2022) The role of Nrf2 in pulmonary fibrosis: molecular mechanisms and treatment approaches. Antioxidants. https://doi.org/10.3390/antiox11091685
Wilson M, Wynn T (2009) Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol 2:103. https://doi.org/10.1038/MI.2008.85
Wu X, Xiao X, Chen X et al (2022) Effectiveness and mechanism of metformin in animal models of pulmonary fibrosis: a preclinical systematic review and meta-analysis. Front Pharmacol. https://doi.org/10.3389/fphar.2022.948101
Wu Y, Liu L, Zhang J et al (2020) Thalidomide inhibits the gene promoter of connective tissue growth factor in human embryonic lung fibroblasts. Ann Palliat Med 9:2516–2523. https://doi.org/10.21037/apm-19-398
Wuyts WA, Wijsenbeek M, Bondue B et al (2020) Idiopathic pulmonary fibrosis: best practice in monitoring and managing a relentless fibrotic disease. Respiration 99:73–82. https://doi.org/10.1159/000504763
Xie J, Zhang C, Li S et al (2022) Efficacy and safety of thalidomide as a pre-medication of chemotherapy-induced nausea and vomiting (CINV) following highly emetogenic chemotherapy (hec): a systematic review and meta-analysis. Front Oncol. https://doi.org/10.3389/fonc.2021.818839
Yang H, Song Z, Hong D (2020) CRBN knockdown mitigates lipopolysaccharide-induced acute lung injury by suppression of oxidative stress and endoplasmic reticulum (ER) stress associated NF-κB signaling. Biomed Pharmacother 123:109761. https://doi.org/10.1016/J.BIOPHA.2019.109761
Yang S-J, Jeon S, Baek JW et al (2021) Regulation of AMPK activity by CRBN is independent of the thalidomide-CRL4CRBN protein degradation axis. Pharmaceuticals. https://doi.org/10.3390/ph14060512
Ye Q (2006) Thalidomide reduces IL-18, IL-8 and TNF- release from alveolar macrophages in interstitial lung disease. Eur Respir J 28:824–831. https://doi.org/10.1183/09031936.06.00131505
Ye Z, Hu Y (2021) TGF-β1: Gentlemanly orchestrator in idiopathic pulmonary fibrosis (review). Int J Mol Med. https://doi.org/10.3892/ijmm.2021.4965
Zhang H, Yang Y, Wang Y et al (2018) Renal-protective effect of thalidomide in streptozotocin-induced diabetic rats through anti-inflammatory pathway. Drug Des Devel Ther 12:89. https://doi.org/10.2147/DDDT.S149298
Zhang L, Yang W-L (2012) Effect of thalidomide on the expressions of IL-6, TNF-α and TGF-β 1 in BALF of elder patients with idiopathic pulmonary fibrosis. J xi’an Jiaotong Univ (medical) Sci. 33:622–625
Zhao L, Xiao K, Wang H et al (2009) Thalidomide has a therapeutic effect on interstitial lung fibrosis: evidence from in vitro and in vivo studies. Clin Exp Immunol 157:310–315. https://doi.org/10.1111/j.1365-2249.2009.03962.x
Zheng F, Zhu J, Zhang W et al (2021) Thal protects against paraquat-induced lung injury through a microRNA-141/HDAC6/IκBα-NF-κB axis in rat and cell models. Basic Clin Pharmacol Toxicol 128:334–347. https://doi.org/10.1111/bcpt.13505
Zhou XL, Xu P, Chen HH et al (2017) (2017) Thalidomide inhibits TGF-β1-induced epithelial to mesenchymal transition in alveolar epithelial cells via smad-dependent and smad-independent signaling pathways. Sci Reports 71(7):1–10. https://doi.org/10.1038/s41598-017-15239-2
Zhu H, Shi X, Ju D et al (2014) Anti-Inflammatory effect of thalidomide on h1n1 influenza virus-induced pulmonary injury in mice. Inflammation 37:2091–2098. https://doi.org/10.1007/s10753-014-9943-9
Zhu N, Wang L, Guo H et al (2021) Thalidomide suppresses angiogenesis through the signal transducer and activator of transcription 3/SP4 signaling pathway in the peritoneal membrane. Front Physiol. https://doi.org/10.3389/fphys.2021.712147
Zisman DA, Schwarz M, Anstrom KJ et al (2010) A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med 363:620–628. https://doi.org/10.1056/NEJMoa1002110
Acknowledgements
The authors thank BioRender software used in creating the figures and visualisations. The authors express their gratitude to M K Unnikrishnan, Professor, NGSM Institute of Pharmaceutical Sciences, Nitte University, for mentoring, revising and helping to correct the scientific writing.
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal. There was no funding support for this work.
Author information
Authors and Affiliations
Contributions
CRediT authorship contribution statement. NND: Conceptualization, Methodology, Visualisation, Data Curation, Writing—Original Draft. VA: Methodology, Data collections, Visualisation, Writing—Original Draft. KB: Formal analysis, Writing—Review and Editing. SM: Methodology, Formal analysis. BHB: Methodology, Formal analysis. YN: Conceptualization, Resources, Data Curation, Visualisation, Writing—Review and Editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence in publishing this paper.
Ethics approval
There was no human or animal studies involved.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dsouza, N.N., Alampady, V., Baby, K. et al. Thalidomide interaction with inflammation in idiopathic pulmonary fibrosis. Inflammopharmacol 31, 1167–1182 (2023). https://doi.org/10.1007/s10787-023-01193-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-023-01193-1