
Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is a progressive debilitating lung disease with considerable morbidity. Heterogeneity in epidemiologic studies means the full impact of the disease is unclear.
Methods
A targeted literature search for population-based, observational studies reporting incidence and/or prevalence of IPF from January 2009 to April 2020 was conducted. Identified studies were aggregated by country. For countries with multiple publications, a weighted average was determined. Incidence and prevalence data were adjusted for between-study differences where possible. The final model included adjusted estimates of incidence and prevalence per 10,000 of the population with 95% confidence intervals. As prevalence estimates vary depending on the definitions used, estimates were based on a specific case definition of IPF.
Results
Overall, 22 studies covering 12 countries met the inclusion criteria, with 15 reporting incidence and 18 reporting prevalence estimates. The adjusted incidence estimates (per 10,000 of the population) ranged from 0.35 to 1.30 in Asia–Pacific countries, 0.09 to 0.49 in Europe, and 0.75 to 0.93 in North America. Unadjusted and adjusted incidence estimates were consistent. The adjusted prevalence estimates ranged from 0.57 to 4.51 in Asia–Pacific countries, 0.33 to 2.51 in Europe, and 2.40 to 2.98 in North America. South Korea had the highest incidence and prevalence estimates. When prevalence estimates were compared to country-specific rare disease thresholds, IPF met the definition of a rare disease in all countries except South Korea. There were notable geographic gaps for IPF epidemiologic data.
Conclusions
Due to differences in study methodologies, there is worldwide variability in the reported incidence and prevalence of IPF. Based on the countries included in our analysis, we estimated the adjusted incidence and prevalence of IPF to be in the range of 0.09–1.30 and 0.33–4.51 per 10,000 persons, respectively. According to these prevalence estimates, IPF remains a rare disease. For consistency, future epidemiologic studies of IPF should take age, sex, smoking status, and the specificity of case definitions into consideration.
Similar content being viewed by others

Background
Idiopathic pulmonary fibrosis (IPF) is a rare chronic progressive disease of unknown etiology that affects both physical and emotional well-being [1,2,3]. It is characterized by irreversible loss of lung function due to fibrosis, which manifests as symptoms of increasing cough and dyspnea and impaired quality of life [2,3,4,5,6]. Lung transplantation is limited to a minority of patients and patients primarily rely on antifibrotic therapy plus several supportive/palliative treatments. Despite recent advances, current IPF therapies only slow disease progression and prognosis is poor, with a median survival of 2–3 years if left untreated [7]. Accordingly, reliance on healthcare services is considerable, contributing to a marked socioeconomic burden of disease [8, 9].
Epidemiology estimates of IPF are derived using various data sources. For those using claims databases, it is important to differentiate between specific versus non-specific case definitions of IPF, as estimates can vary drastically depending on the definitions used [10,11,12,13]. A specific case definition is obtained from an accurate diagnosis of IPF, which requires observation of clinical characteristics as well as confirmation of specific pulmonary patterns via high-resolution chest imaging and sometimes lung biopsy [1]. However, some patients are diagnosed with IPF without precise diagnostic procedures and as such can only be considered under a broad (non-specific) case definition.
Single studies describing the epidemiology of IPF can also be misleading if age, sex, and other risk factors are not taken into consideration [1, 10]. The mean age of IPF patients is around 65–70 years, with incidence increasing with age [14,15,16]. Globally, patient numbers are rising, which may be attributed to, among other causes, an aging population, a higher degree of disease awareness and improved diagnostic tools [17,18,19]. Furthermore, IPF affects males more than females [10], and risk factors such as smoking [20, 21], metal/wood dust inhalation [22], and genetic factors [23, 24] are frequently recorded as being associated with development of IPF.
Overall, owing to diagnostic challenges, updated diagnostic criteria, and differences in study methodologies there is substantial heterogeneity between studies providing estimated epidemiology data in IPF [1, 10], impacting the understanding of global disease burden. Indeed, a detailed knowledge of the incidence and prevalence of IPF provides additional disease understanding that is crucial for therapeutic and healthcare system planning, particularly when considering the socioeconomic burden of the disease. By re-evaluating the published literature, this study sought to produce adjusted incidence and prevalence for IPF by country.
Methods
This was a targeted literature review to identify studies estimating epidemiologic measures of IPF published between 2009 and 2020. Statistical modeling was applied to the epidemiologic estimates obtained from the identified studies to provide adjusted incidence and prevalence data.
Study design and data processing
The PubMed and EMBASE databases were searched for population based, observational studies from January 2009 to January 2019 using a search strategy derived from the following PICO (population, intervention, comparison, outcome) formulation: (i) patients with IPF (no restriction on case definitions); (ii) any intervention; (iii) any comparator; (iv) with outcomes including quantitative measures of IPF incidence (authors’ definition) and IPF prevalence (authors’ definition) (Additional file 1: Table S1). EMBASE was also searched to identify congress abstracts from 2014 to 2019, and supplementary gray literature searches were performed. We conducted a secondary supplemental search utilizing the same search terms between January 2019 and April 2020. No publications which met the threshold for inclusion in our analysis were identified through this supplementary search. Identified studies were aggregated at country-level and estimates further categorized based on the case definition (“specific” [i.e. narrow] or “broad”) used to identify patients with IPF. Studies were classified by two individuals in a blinded manner with adjudication by a third person where opinions differed with regards to the classification of the IPF identification. Collectively, studies utilizing broad classification criteria tended toward a generalized search of pertinent medical records for diagnostic classification according to the International Classification of Diseases (ICD) or a related coding system, without any additional diagnostic steps being undertaken. Studies reporting specific classifications typically required confirmatory imaging and/or pathology in addition to the ICD code classification or required review by medically trained staff.
Statistical analysis
Incidence and prevalence data were adjusted to fit a negative binomial general linear model developed under a fixed-effects framework, using a study population offset parameter to adjust for population size of each study. An initial “full model” included age, sex, study year, diagnostic criteria, study region/country, and population size; any covariates in the model that were not significant at an alpha-level of 0.05 were removed (except age and sex, which were included in all models). In instances where data on age or proportion of male patients were not directly provided, appropriate estimates for a given study population were used or a value was imputed using the average of all the other studies. The outcome variable in the model was the total number of IPF cases, whether for incidence or prevalence. For countries with multiple publications, a weighted average was determined using the underlying study population number as the weighting coefficient. The final model included adjusted estimates of incidence and prevalence per 10,000 of the population with 95% confidence intervals. Model-associated adjustments for prevalence estimates are provided in Additional file 1: Table S2. For prevalence estimates, a sensitivity analysis was performed using broad IPF case definitions.
Prevalence estimates were compared to country-specific rare disease thresholds [25,26,27,28,29,30,31]. For countries where a threshold of cases, as opposed to a prevalence, is utilized, the prevalence estimates were multiplied by the countries 2020 United Nations population estimate [32] to determine a total number of estimated cases.
Results
Study selection
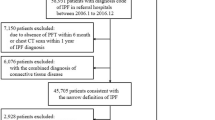
Following the removal of duplicate articles, the literature search yielded 3188 hits (Fig. 1). The abstracts of these publications were reviewed and the full-text versions of 294 manuscripts were examined against the PICO criteria for selection (Additional file 1: Table S1). Of the 74 articles that met the criteria, 22 provided incidence and/or prevalence, IPF case identification descriptions, and details on the underlying patient populations. The included studies were classified as “specific” or “broad” according to how IPF patients were identified [11,12,13, 33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50] (Additional file 1: Table S3). Of those studies reporting a specific IPF case definition, 15 reported incidence estimates and underlying population details [11,12,13, 33, 35, 37, 38, 40, 41, 43, 44, 46, 47, 51,52,53] (Additional file 1: Table S4) and 18 described prevalence estimates and underlying population details [11,12,13, 33, 35, 37,38,39,40,41,42,43,44,45,46,47, 50, 52] (Additional file 1: Table S5). Of the 15 studies reporting incidence estimates, five used primary databases including medical charts and other direct sources and 10 used secondary research databases including claims data. In addition, eight studies reported incidence per population and seven reported incidence per patient-year. For prevalence estimates, eight studies used primary databases and 10 used secondary databases. In total, the studies covered 12 countries and corresponded to review of 229,611,497 patient records globally.

An overview of articles reviewed and study flow. *Some studies included both a broad and a specific case definition. IPF idiopathic pulmonary fibrosis, PICO population, intervention, comparison, outcome
Estimated incidence
The adjusted incidence estimates (per 10,000 of the population) for each country ranged from 0.35 to 1.30 in Asia–Pacific countries, 0.09 to 0.49 in Europe, and 0.75 to 0.93 in North America (Table 1). Overall, unadjusted and adjusted incidence estimates were similar. Both age and country were identified as statistically significant variables within the model. There are clear epidemiologic knowledge gaps in substantial geographic regions including Africa, South America, South Asia, and the Middle East (Fig. 2a).

Global heat maps of adjusted IPF incidence (a) and prevalence (b) for included studies with specific IPF definitions. IPF idiopathic pulmonary fibrosis
Estimated prevalence
The adjusted prevalence estimates (per 10,000 of the population) for each country ranged from 0.57 to 4.51 in Asia–Pacific countries, 0.33 to 2.51 in Europe, and 2.40 to 2.98 in North America (Table 2). Overall, unadjusted and adjusted prevalence estimates were similar. Notable exceptions were South Korea and the United States, both of which demonstrated higher adjusted prevalence when compared to unadjusted estimates (4.51 vs. 3.70 and 2.40 vs. 1.37, respectively). Conversely, the United Kingdom demonstrated a modestly reduced adjusted versus unadjusted IPF prevalence (0.78 vs. 1.16). The adjusted prevalence estimates (per 10,000 of the population) from the sensitivity analysis (using broad IPF case definitions) for each country ranged from 0.79 to 5.67 (Table 3).
South Korea was the only country where the threshold for rare disease status (< 20,000 cases [26]) was exceeded by the adjusted prevalence estimate (4.51/10,000, equating to approximately 23,136 patients [assuming a population of 51.3 million] [32]), although the unadjusted estimate was within the rare disease criteria (3.70/10,000, equating to approximately 18,981 patients) (Table 2). Within the sensitivity analysis using the broader definitions of IPF, IPF prevalence estimates still met rare disease thresholds although the upper confidence interval exceeded the threshold in all cases (Table 3).
Both age and country were identified as statistically significant variables within the model. Each year increase in average age was associated with a 6.2% increase in IPF prevalence over the unadjusted estimate. Geographic evidence gaps for prevalence were similar to those observed for incidence (Fig. 2b).
Discussion
To our knowledge, this is the first targeted literature review including a model for adjusted analyses of IPF incidence and prevalence. Of the countries analyzed, estimates of the adjusted incidence of IPF are in the range of 0.09 to 1.30 per 10,000 persons globally. Overall, the countries with the highest incidence of IPF are South Korea, Canada, and the United States. Fewer countries were available to evaluate when compared with the prevalence model.
Based on the countries included in our analysis, estimates of the adjusted prevalence of IPF are in the range of 0.33 to 4.51 per 10,000 persons globally. Because most studies had similar proportions of male patients and age distributions, the IPF estimates remained relatively unchanged between unadjusted and adjusted prevalence. Overall, the countries with the highest apparent prevalence of IPF include South Korea, Canada, Poland, the United States, and Italy, although the extent to which variations reflect true differences in prevalence rather than methodologic differences is open to question.
In all but one country (South Korea), IPF would be classified as a rare disease according to national guidelines. South Korea utilizes very stringent criteria for defining rare disease status of < 20,000 cases (an estimated prevalence of < 3.91/10,000 persons based on a population of 51.3 million). This is somewhat lower than the 5/10,000 threshold used by most European countries. Regardless, for South Korea, the mean adjusted prevalence is the highest of the countries evaluated and around a third greater than the country with the next highest adjusted prevalence (Canada). This difference may be due to an overestimation of cases due to the study populations (elderly with a high proportion of male patients), the definitions used in the South Korean studies, or due to genetic or environmental factors. For example, in 2011, an increase in lung injuries was observed in South Korea due to humidifier disinfectant use [54]. South Korea has also experienced high levels of particulate matter air pollution [55], which might be associated with the incidence of IPF [56].
Broadly, trends were consistent between the incidence and prevalence models. However, compared to other countries Taiwan ranked differently for incidence and prevalence. Taiwan had the fifth highest incidence of IPF (out of nine countries), yet in the prevalence model it was the second lowest behind Greece (out of 12 countries). The reason for this is unclear, as in both cases Taiwan was only subject to mild alterations in point-estimates for incidence and prevalence. The large Taiwan study showed evidence of a continual shift to greater IPF burden across the study period (1997–2007), and it is conceivable that there is simply a lag between the increased incidence observed and the associated prevalence [43]. However, the study also indicated that the median time from diagnosis to death was 0.7 years based on specific IPF case definitions, compared with 3.47 years in a comparable study from the United States [11, 43]. The shorter survival time recorded in Taiwan, which may have been partly due to delayed diagnosis of IPF and less access to specific IPF treatments at the time of the study [43, 57], could account for the lower observed prevalence.
Overall, the primary prevalence analyses were comparable with the sensitivity analyses. When the broader IPF definition was used to identify patients, the estimates of IPF prevalence increased compared with the specific definition. The broader definition can result in a considerably larger number of patients falsely being classified as having IPF. In the study from the United States by Raghu et al., the broad case subgroup enrolled approximately 60% more patients than the specific case subgroup [46]. Indeed, Strongman and colleagues noted a nearly threefold difference in IPF prevalence in the UK when utilizing a broad versus a specific IPF case definition [13]. In our study, when utilizing broad case definitions, the inference is similar to the principal findings, that there is substantial between-country heterogeneity.
This study has some limitations. A relatively small number of studies are included with high heterogeneity between them including differences in case definitions, type of database analyzed, and timing of data collection. For example, data were collected earlier for some countries (such as Greece [52]) and may provide an underestimate of incidence and prevalence as diagnostic criteria, assessments and use of a multidisciplinary team approach to diagnosis and care have evolved over time [58]. However, the coding for IPF has not altered in line with changes to the guidelines. As such, we do not anticipate that changes in the way we diagnose IPF have had a major impact on incidence and prevalence data. Of note, any potential impact of changes in diagnostic approach on IPF epidemiology are likely compounded by reported increases in the incidence of IPF over time [59]. Further to this, during the development of our model, we assessed whether publication year was a significant variable and found it not predictive of IPF incidence or prevalence (either positively or negatively).
Our analysis also has limited geographic spread, with economically similar countries represented. In some countries, such as Germany, the healthcare system does not easily allow for structured data analysis [60]. In others, particularly low- or middle-income countries, few epidemiologic data are available, possibly due to reduced access to diagnostic tools and healthcare professionals with the expertise needed to provide an accurate diagnosis. Of the included studies, limited data were provided on covariates that could have been informative had they been available for analysis. For example, smoking status is a well-known risk factor associated with IPF prevalence [20, 21], but was not available for integration into our model. Other hard-to-quantify parameters, such as exposure to environmental hazards or overall healthcare system capacity, may also be influential features. For incidence, the development of a robust model was challenging, as data can be reported as a function of observed patient time (typically per patient-years) or as a function of the population observed. An adjustment was made to allow for the studies to be combined, and as such our results should be considered exploratory and in the context of the prevalence results. Finally, we note that the quality of data in the included studies may impact the validity of the study findings; however, due to the correlation between coding systems and diagnostic reliability, the impact is unlikely to be extensive [13, 59].
Conclusions
Reported IPF incidence and prevalence are variable worldwide, even with statistical adjustment made where possible for between-study differences. Based on the countries included in our analysis, the adjusted incidence and prevalence of IPF are estimated to be in the range of 0.09–1.30 and 0.33–4.51 per 10,000 persons, respectively. According to these prevalence estimates, IPF remains a rare disease. Future epidemiologic studies of IPF should take age, sex, other risk factors, and the specificity of case definitions into consideration to better characterize the IPF patient population.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its additional information files.
Abbreviations
- ALAT:
-
Latin American Thoracic Society
- ATS:
-
American Thoracic Society
- CI:
-
Confidence interval
- CT:
-
Computed tomography
- ERS:
-
European Respiratory Society
- FVC:
-
Forced vital capacity
- HRCT:
-
High-resolution computed tomography
- ICD:
-
International Classification of Diseases
- IIP:
-
Idiopathic interstitial pneumonia
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- JRS:
-
Japanese Respiratory Society
- NA:
-
Not applicable
- NHI:
-
National Health Insurance
- NHIS:
-
National Health Insurance Service
- NR:
-
Not reported
- PICO:
-
Population, intervention, comparison, outcome
- RID:
-
Rare intractable diseases
- UIP:
-
Usual interstitial pneumonia
References
American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277–304.
Quinn C, Wisse A, Manns ST. Clinical course and management of idiopathic pulmonary fibrosis. Multidiscip Respir Med. 2019;14:35.
Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074.
van Manen MJ, Geelhoed JJ, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2017;11:157–69.
Kreuter M, Swigris J, Pittrow D, Geier S, Klotsche J, Prasse A, et al. The clinical course of idiopathic pulmonary fibrosis and its association to quality of life over time: longitudinal data from the INSIGHTS-IPF registry. Respir Res. 2019;20:59.
Kreuter M, Swigris J, Pittrow D, Geier S, Klotsche J, Prasse A, et al. Health related quality of life in patients with idiopathic pulmonary fibrosis in clinical practice: insights-IPF registry. Respir Res. 2017;18:139.
Sharif R. Overview of idiopathic pulmonary fibrosis (IPF) and evidence-based guidelines. Am J Manag Care. 2017;23(11 Suppl):S176–82.
Diamantopoulos A, Wright E, Vlahopoulou K, Cornic L, Schoof N, Maher TM. The burden of illness of idiopathic pulmonary fibrosis: a comprehensive evidence review. Pharmacoeconomics. 2018;36:779–807.
Yu YF, Wu N, Chuang CC, Wang R, Pan X, Benjamin NN, et al. Patterns and economic burden of hospitalizations and exacerbations among patients diagnosed with idiopathic pulmonary fibrosis. J Manag Care Spec Pharm. 2016;22:414–23.
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. AJRCCM. 2006;174:810–6.
Fernández Pérez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 2010;137:129–37.
Tarride JE, Hopkins RB, Burke N, Guertin JR, O’Reilly D, Fell CD, et al. Clinical and economic burden of idiopathic pulmonary fibrosis in Quebec, Canada. Clinicoecon Outcomes Res. 2018;10:127–37.
Strongman H, Kausar I, Maher TM. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv Ther. 2018;35:724–36.
Jo HE, Randhawa S, Corte TJ, Moodley Y. Idiopathic pulmonary fibrosis and the elderly: diagnosis and management considerations. Drugs Aging. 2016;33:321–34.
Collard HR. The age of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:771–2.
Fernandez-Fabrellas E, Molina-Molina M, Soriano JB, Portal JAR, Ancochea J, Valenzuela C, et al. Demographic and clinical profile of idiopathic pulmonary fibrosis patients in Spain: the SEPAR National Registry. Respir Res. 2019;20:127.
World Health Organization. Global health and aging. 2011. https://www.who.int/ageing/publications/global_health.pdf. Accessed 10 Jan 2021.
Hoyer N, Prior TS, Bendstrup E, Wilcke T, Shaker SB. Risk factors for diagnostic delay in idiopathic pulmonary fibrosis. Respir Res. 2019;20:103.
Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46:795–806.
Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc. 2006;3:293–8.
Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005;172:1146–52.
Miyake Y, Sasaki S, Yokoyama T, Chida K, Azuma A, Suda T, et al. Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Ann Occup Hyg. 2005;49:259–65.
Checa M, Ruiz V, Montaño M, Velázquez-Cruz R, Selman M, Pardo A. MMP-1 polymorphisms and the risk of idiopathic pulmonary fibrosis. Hum Genet. 2008;124:465–72.
Rosas IO, Ren P, Avila NA, Chow CK, Franks TJ, Travis WD, et al. Early interstitial lung disease in familial pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:698–705.
Ministry of Health Labour and Welfare. Overview of orphan drug/medical device designation system. 2009. https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/orphan_drug.html. Accessed 21 Jan 2021.
Wong-Rieger D, Claxton W, Vines R, Padilla C, Tsang KP, Hickinbotham L. An Asia Pacific alliance for rare diseases. Patient. 2015;8:11–7.
Khosla N, Valdez R. A compilation of national plans, policies and government actions for rare diseases in 23 countries. Intractable Rare Dis Res. 2018;7:213–22.
RD-Action. State of the art of rare disease—activities in EU member states and other European countries: Denmark report. 2017. http://www.rd-action.eu/wp-content/uploads/2017/10/Denmark-Report-06.10.2017.pdf. Accessed 21 Jan 2021.
European Commission. Regulation (EC) no 141/2000 of the European Parliament and of the Council. 1999. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf. Accessed 21 Jan 2021.
The Canadian Agency for Drugs and Technologies in Health. Drugs for rare diseases: evolving trends in regulatory and health technology assessment perspectives. 2016. https://www.cadth.ca/sites/default/files/pdf/ES0300_Rare_Disease_Drugs_e.pdf. Accessed 21 Jan 2021.
US Food and Drug Administration. Developing products for rare diseases and conditions. 2018. https://www.fda.gov/industry/developing-products-rare-diseases-conditions. Accessed 21 Jan 2021.
United Nations Population Fund. World population dashboard. 2020. https://www.unfpa.org/data/world-population-dashboard. Accessed 29 Jan 2021.
Agabiti N, Porretta MA, Bauleo L, Coppola A, Sergiacomi G, Fusco A, et al. Idiopathic pulmonary fibrosis (IPF) incidence and prevalence in Italy. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31:191–7.
Bartley K, Levine A, Arnheim-Dahlstrom L, Ferrara G, Kirchgaessler K, Linder R, et al. Description of a national pulmonary fibrosis cohort in Sweden. Thorax. 2017;72(Suppl 3):A164–5 (Abstract P49).
Duchemann B, Annesi-Maesano I, Jacobe de Naurois C, Sanyal S, Brillet PY, Brauner M, et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur Respir J. 2017;50:1602419.
Esposito DB, Lanes S, Donneyong M, Holick CN, Lasky JA, Lederer D, et al. Idiopathic pulmonary fibrosis in United States automated claims. Incidence, prevalence, and algorithm validation. Am J Respir Crit Care Med. 2015;192:1200–7.
Harari S, Madotto F, Caminati A, Conti S, Cesana G. Epidemiology of idiopathic pulmonary fibrosis in northern Italy. PLoS ONE. 2016;11:e0147072.
Hopkins RB, Burke N, Fell C, Dion G, Kolb M. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur Respir J. 2016;48:187–95.
Hyldgaard C, Hilberg O, Muller A, Bendstrup E. A cohort study of interstitial lung diseases in central Denmark. Respir Med. 2014;108:793–9.
Kaunisto J, Kelloniemi K, Sutinen E, Hodgson U, Piilonen A, Kaarteenaho R, et al. Re-evaluation of diagnostic parameters is crucial for obtaining accurate data on idiopathic pulmonary fibrosis. BMC Pulm Med. 2015;15:92.
Kim SW, Myong JP, Yoon HK, Koo JW, Kwon SS, Kim YH. Health care burden and medical resource utilisation of idiopathic pulmonary fibrosis in Korea. Int J Tuberc Lung Dis. 2017;21:230–5.
Kondoh S, Chiba H, Nishikiori H, Umeda Y, Kuronuma K, Otsuka M, et al. Validation of the Japanese disease severity classification and the GAP model in Japanese patients with idiopathic pulmonary fibrosis. Respir Investig. 2016;54:327–33.
Lai CC, Wang CY, Lu HM, Chen L, Teng NC, Yan YH, et al. Idiopathic pulmonary fibrosis in Taiwan—a population-based study. Respir Med. 2012;106:1566–74.
Lee HE, Myong JP, Kim HR, Rhee CK, Yoon HK, Koo JW. Incidence and prevalence of idiopathic interstitial pneumonia and idiopathic pulmonary fibrosis in Korea. Int J Tuberc Lung Dis. 2016;20:978–84.
Natsuizaka M, Chiba H, Kuronuma K, Otsuka M, Kudo K, Mori M, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med. 2014;190:773–9.
Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J. 2016;48:179–86.
Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med. 2014;2:566–72.
Raimundo K, Chang E, Broder MS, Alexander K, Zazzali J, Swigris JJ. Clinical and economic burden of idiopathic pulmonary fibrosis: a retrospective cohort study. BMC Pulm Med. 2016;16:2.
Snell N, Strachan D, Hubbard R, Gibson J, Maher T, Jarrold I. Epidemiology of idiopathic pulmonary fibrosis in the UK: findings from the British Lung Foundation’s ‘Respiratory Health of the Nation’ project [Abstract P272]. Thorax. 2016;71(Suppl 3):A236.
Szafrański W. Interstitial lung diseases among patients hospitalized in the department of respiratory medicine in radom district hospital during the years 2000–2009. Pneumonol Alergol Pol. 2012;80:523–32.
Han SH, Mok YJ, Jee SH, Danoff SK. Incidence and mortality of idiopathic pulmonary fibrosis in South Korea. Am J Respir Crit Care Med. 2013;201:A1460.
Karakatsani A, Papakosta D, Rapti A, Antoniou KM, Dimadi M, Markopoulou A, et al. Epidemiology of interstitial lung diseases in Greece. Respir Med. 2009;103:1122–9.
Ley B, Urbania T, Husson G, Vittinghoff E, Brush DR, Eisner MD, et al. Code-based diagnostic algorithms for idiopathic pulmonary fibrosis. Case validation and improvement. Ann Am Thorac Soc. 2017;14:880–7.
Kim HJ, Lee M-S, Hong S-B, Huh JW, Do K-H, Jang SJ, et al. A cluster of lung injury cases associated with home humidifier use: an epidemiological investigation. Thorax. 2014;69:703–8.
IQAir. Air quality in South Korea. 2021. https://www.iqair.com/us/south-korea. Accessed 22 Feb 2021.
Conti S, Harari S, Caminati A, Zanobetti A, Schwartz Joel D, Bertazzi PA, et al. The association between air pollution and the incidence of idiopathic pulmonary fibrosis in Northern Italy. Eur Respir J. 2018;51:1700397.
Yang SN, Perng DW, Ko HK, Chang YL, Hsu CC, Huang HY, et al. Epidemiologic analysis of Taiwanese patients with idiopathic pulmonary fibrosis. Healthcare (Basel). 2020;8:580.
Wells AU. Managing diagnostic procedures in idiopathic pulmonary fibrosis. Eur Respir Rev. 2013;22:158–62.
Navaratnam V, Fleming KM, West J, Smith CJP, Jenkins RG, Fogarty A, et al. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax. 2011;66:462–7.
Neubauer S, Kreis K, Klora M, Zeidler J. Access, use, and challenges of claims data analyses in Germany. Eur J Health Econ. 2017;18:533–6.
Acknowledgements
Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copyediting, fact checking and referencing) was provided by Louise Niven D.Phil, CMPP (Aspire Scientific Ltd, Bollington, UK) and funded by Galapagos NV (Mechelen, Belgium).
Funding
This study was funded by Galapagos NV (Mechelen, Belgium).
Author information
Authors and Affiliations
Contributions
TMM and GS contributed to the study design and data analysis or interpretation; EB contributed to data collection; LD contributed to data collection and data analysis or interpretation; JMK, HP and MK contributed to data analysis or interpretation; all authors reviewed and critically revised the manuscript, approved the final draft, and are accountable for the accuracy and integrity of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
TMM, via his institution, has received industry-academic funding from GlaxoSmithKline and UCB, and has received consultancy or speaker fees from Apellis, AstraZeneca, Bayer, Blade Therapeutics, Boehringer Ingelheim, Bristol Myers Squibb, Galapagos, GlaxoSmithKline, Indalo, Novartis, Pliant, ProMetic, Respivant, Roche, Samumed, and UCB outside the submitted work. EB reports grants and personal fees from Galapagos during the conduct of the study, and from AstraZeneca, Boehringer Ingelheim and Roche outside the submitted work. LD and GS are employees of Cytel, which received funding from Galapagos NV for the current work. JL and JMK are employees of and warrant holders at Galapagos NV. HP is an employee of Immensity Consulting, Inc. which received funding from Galapagos NV for the current work. MK reports grants and personal fees from Galapagos during the conduct of the study, and grants and personal fees from Boehringer Ingelheim and Roche outside the submitted work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
PICO search criteria. Table S2. Model-associated adjustments for prevalence estimates adjusted per country. Table S3. Included studies and associated IPF categories. Table S4. List of studies for IPF incidence estimates. Table S5. List of studies for IPF prevalence estimates (primary analysis).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Maher, T.M., Bendstrup, E., Dron, L. et al. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res 22, 197 (2021). https://doi.org/10.1186/s12931-021-01791-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-021-01791-z