
Abstract
The ecological interactions parasitic insects have with their hosts may contribute to their prodigious diversity, which is unrivaled among animals. Many insects assumed to be polyphagous generalists have been shown to consist of several differentiated races, each occupying a different host-niche. The sunflower maggot fly, Strauzia longipennis, has long been thought to consist of two or more races due to its substantial intra-specific morphological variation. Here, we use nuclear and mitochondrial markers to test the hypothesis that S. longipennis is a complex of two or more partially reproductively isolated races. We collected S. longipennis flies as pupae from roots of Jerusalem artichoke (Helianthus tuberosus) and as adults swept from leaves of mature H. tuberosus across the breadth of a field season. Flies were scored for morphological variety (typica or vittigera), mitochondrial haplotype (A or B) and a panel of 176 AFLP loci. Bayesian clustering and neighbor-joining phylogenetic analyses of AFLP data supported the existence of at least three, possibly four, genetic races of Strauzia (clusters I, II, III, and V), as well as a small number of putative interracial hybrids (cluster IV). Clusters I and III each consisted of flies of both morphological varieties and both haplotype groups, while flies in cluster II were all of variety typica and all but one was of mitochondrial haplotype B. Flies in cluster II were also collected only as adults on H. tuberosus and not among flies reared from pupae collected from H. tuberosus roots, suggesting that they use a different plant as their larval host. Mean capture date was significantly different between flies of each genetic race, indicating that partial allochronic isolation may be one contemporary barrier to gene flow between races. Evidence that mitochondrial genomes and morphological traits have moved between lineages implies a model of speciation-with-gene-flow for S. longipennis races.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phytophagous (plant-eating) insects are tremendously diverse—constituting perhaps as much as one third of all animal species (Strong et al. 1984)—and their speciation has been tied to extrinsic reproductive barriers arising during changes in host plant use (Bush 1969; Wood and Keese 1990; Berlocher 1998; Nason et al. 2002; Funk et al. 2011). Many insects find mates on or near the female’s oviposition substrate (the host plant), so emergent preferences for novel hosts may often translate into assortative mating and behaviorally isolate different host-associated populations (e.g., Emelianov et al. 2001; Berlocher and Feder 2002). The result is the formation of partially isolated races, presumed to be an early step along the so-called “speciation continuum” (Mallet 2008). Genetically differentiated races within morphologically defined species have also been found to coexist on a single plant species, particularly among taxa that have recently undergone adaptive radiations (Marsteller et al. 2009; Stireman et al. 2012). Further, recent empirical and theoretical work demonstrating that populations can diverge even in the face of effective migration (Nosil 2008; Nosil and Feder 2012), as well as new information about the permeability of genomes to between-lineage gene flow (Turner et al. 1999; Michel et al. 2010; Gompert et al. 2010) both suggest that persistent hybridization between closely related lineages may occur without leading to lineage fusion or breakdown of reproductive barriers. Such studies imply that ecology plays a central role in insect diversity, but many questions remain as to the interplay between gene flow and host plant mediated selection during the early stages of speciation.
The sunflower maggot, Strauzia longipennis (Wiedemann) (Diptera: Tephritidae) may be an ideal subject for investigating the dynamics of speciation and gene flow among phytophagous insects because it is thought to consist of multiple genetic races at various stages of divergence (Lisowski 1979; Axen et al. 2010). Most flies in genus Strauzia are host-specialists; they oviposit and undergo larval development on a single species of plant. Adult phenology across the genus is variable and corresponds to the time of the year that each species’ host plant matures (Stoltzfus 1988). S. longipennis is an exception; it has been described as using at least 5 different hosts: three species in genus Helianthus (sunflowers) and two other plants in the family Asteraceae, Smallanthus uvedalia and Ageratina altissima (Foote et al. 1993). Several other genera of Tephritidae are known to have undergone race formation and speciation through host association (Bush 1969; Abrahamson and Weis 1997), so the uniquely broad host range of S. longipennis is an indication that it could be a conglomerate of cryptic lineages.
Strauzia longipennis is also uniquely variable in its morphology when compared with congeners, a situation that has caused long-standing taxonomic difficulty (Foote et al. 1993; Steyskal 1986). The fly was initially described as a single species with as many as seven different morphological ‘varieties,’ for which many characters gradually transition from one variety into another (Loew 1873). Subsequent taxonomic revisions attempted to split S. longipennis into two and later into seven species (Lisowski 1985; Steyskal 1986; Stoltzfus 1988), but no characters reliably distinguished any one of the postulated species, so taxonomic authorities have resisted each suggested split (Foote et al. 1993). Currently, as many as six “unofficial” varieties make up the S. longipennis complex as defined by Foote et al. (1993). Of these, three morphological varieties have been named as associates of species of Helianthus though none has been shown to be exclusive to any one host species. Strauzia longipennis var. longitudinalis is found only in the Eastern United States. The other two, S. longipennis var. typica (hereafter ‘typica’) and S. longipennis var. vittigera (hereafter ‘vittigera’) co-occur sympatrically in the Midwest and are the focus of this study. Vittigera flies are defined by two symmetrical lines of dark pigment on their dorsal thorax, while typica flies have relatively unpigmented thoraxes (Fig. 1). Flies with thoracic pigmentation intermediate between vittigera and typica are also occasionally collected (Axen et al. 2010).

The two primary thoracic banding patterns found among S. longipennis flies: S. longipennis variety typica, left; S. longipennis variety vittigera, right
Attempts to resolve Strauzia phylogeny using genetic markers have proved similarly frustrating. A study of electrophoretic variants (allozymes) among flies collected from several different plant hosts and sites in Illinois resolved six distinct clusters defined by fixed or strong allele frequency differences (Lisowski 1979), two of which correspond to the now-named species S. arculata and S. intermedia (attacking Helianthus grosseserratus and Rudbeckia laciniata, respectively) (Steyskal 1986). Four clusters, however, overlapped broadly in host plant use and morphology, and electrophoretic intermediates between clusters were found, calling into question the extent of their reproductive isolation (Lisowski 1979). More recently, mitochondrial data collected from adult Strauzia captured on H. tuberosus revealed two major haplotype groups, leading to the tentative conclusion that two incipient species had been identified (Axen et al. 2010). However, again morphology did not perfectly correspond to haplotype: several different combinations of wing and thoracic characters were common among flies with both haplotypes.
Our goal in this study was to determine whether S. longipennis is a complex of two or more partially reproductively isolated races and whether extrinsic barriers to gene flow are associated with reproductive isolation. We had four specific aims. First, we used both mitochondrial and nuclear markers to determine whether S. longipennis collected from H. tuberosus consists of multiple cryptic species or races, as suggested by Lisowski (1979). Second, we asked whether races differed in their thoracic banding patterns (vittigera vs. typica). Third, we used collection dates of individual flies to determine whether cryptic lineages were allochronically isolated from one another. Fourth, we compared populations of wild caught adult flies with fly pupae extracted from H. tuberosus tissue to determine whether all fly lineages captured as adults on a plant actually use that plant as their larval host. We show that S. longipennis is a complex of at least three genetic races at various stages of divergence and that these races are partially allochronically isolated from one another. Neither mitochondrial haplotype group identity nor morphological variety was 100 % diagnostic for distinguishing between individuals from different races, providing support for historical and/or contemporary introgression between races.
Materials and methods
Study organisms
As with many temperate tephritid flies, Strauzia have just a single generation per year, emerging as adults in the spring or summer. Adult male Strauzia occupy positions on the underside of host plant leaves and aggressively defend territories (Stoltzfus 1988). Females move between plants and successful mating may last anywhere from a few minutes to several hours. Females may mate with multiple males over the course of their lifetime (Westdal and Barrett 1962). Once mated, females then oviposit into the host plant stem; a single lab-reared female can lay as many as 31 eggs in a day (Westdal and Barrett 1962). Larvae feed on the pith of the plant and pupate either in the root crown or in the soil, depending on the fly species (Brink 1922; Stoltzfus 1988). All Strauzia overwinter as pupae.
Strauzia longipennis (Wiedemann) pupae were dissected from tubers of Helianthus tuberosus growing in Mount Vernon, IA, USA (41°55.358N, 91°25.101W) and Cedar Rapids, IA (41°57.292N, 91°34.733W) in April and May of 2011. Tubers representing plants from the 2010 growing season were extracted from soil and split with a sharp knife to reveal puparia enclosed within. Most plants contained a single Strauzia puparium, but two puparia were occasionally found in the same plant. Pupae were held in moist vermiculite at room temperature (approximately 18 °C) until flies eclosed. No exit scars were found on plant stalks, and no pupae were found in soil around plants, supporting a previous observation that Strauzia associated with H. tuberosus overwinter exclusively in roots and not in the soil (Lisowski 1979).
Adult Strauzia flies were caught off of leaves of Helianthus tuberosus from May 23 through July 11, 2011 at the same two sites described above and at a third site in Iowa City, IA (41°38.453N, 91°34.138W). All sites were between 14 and 38 km of one another, as multiple independent sampling sites from a relatively small geographic area ensures that genetic differences will be minimally influenced by geographic distance. Two other sunflowers previously recorded as hosts to S. longipennis, Helianthus annuus and H. strumosus, were present at all sites, but were relatively uncommon and flies were not sampled from these plants. Fly collections were attempted every 2 days, for approximately 1–2 h at each site. The date and site of capture, thoracic banding pattern (Fig. 1; vittigera, typica, or intermediate; see Axen et al. (2010) for details of morphological classification), and sex of each fly was recorded. Flies were frozen and held at −80 °C in 95 % EtOH for use in genetic work.
DNA extraction and mtDNA haplotyping
Whole genomic DNA was extracted from all flies (both those collected as adults and those reared from root-extracted pupae) using DNeasy Blood & Tissue kits (Qiagen Sciences, Maryland, USA). A 648 bp segment of the mitochondrial COI gene was PCR-amplified using the primers LepF1 & LepR1 (Smith et al. 2008) and the following cycling parameters: 94 °C for 3 min, followed by 35 cycles of 94 °C for 30 s, 46 °C for 1 min, and 72 °C for 2 min, with a final extension of 72 °C for 4 min. Reactions were cleaned using Shrimp Alkaline Phosphatase (USB, Swampscott, MA) and Exonuclease I (New England Biolabs, Ipswich, MD) and cycle sequencing was performed in both forward and reverse directions on an ABI 3730 DNA Analyzer using BigDye 3.1 (Applied Biosystems, Foster City, CA) sequencing chemistry. Forward and reverse reads were used to create consensus sequences for each individual and consensi were then automatically aligned using the program SeqMan (DNASTAR, Inc., Madison, WI). TCS 1.21 (Clement et al. 2000) was used to construct a haplotype network using statistical parsimony and a 95 % connection limit.
AFLP genotyping and phylogenetic inferences
Fly DNA extracts were scored for nuclear genomic variation using an AFLP protocol following Egan et al. (2008). Briefly, genomic DNA samples from the same flies as above were digested using the restriction enzymes EcoRI and MseI (New England Biolabs, Ipswich, MA) and site-specific adaptors were ligated onto cut sites. Samples were then PCR-amplified using a pair of preselective primers [Eco + C (5′-GACTGCGTACCAATTCC-3′) and Mse + C (5′-GATGAGTCCTGAGTAAC-3′)], and then amplified a second time in three independent reactions using a fluorescently labeled forward primer Eco + CAG (5′-GACTGCGTACCAATTCCAG-3′) and each of three different reverse primers: Mse + CAA (5′-GATGAGTCCTGAGTAACAA-3′), Mse + CTG (5′-GATGAGTCCTGAGTAACTG-3′) and Mse + CGA (5′-GATGAGTCCTGAGTAACGA-3′). Selective amplification reactions were genotyped on an ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA) and fragment analysis was performed using GeneMarker 2.2.0 (Softgenetics, State College, PA). A panel of loci sized between 100 and 450 bp was first called automatically by GeneMarker and then all loci were inspected visually. Loci with consistently weak signals were dropped from the panel. All individuals were selectively amplified and genotyped twice and individuals with inconsistent signals were not included in the analysis. In total, 176 polymorphic loci were identified and scored for 91 individual flies.
AFLP-SURV (Vekemans 2002) was used to generate 10,000 bootstrapped Nei’s D (Lynch and Milligan 1994) distance matrices which were then converted into distance networks using the programs NEIGHBOR and CONSENSE in PHYLIP 3.6 (Felsenstein 1989). Pairwise Fst was also calculated between clusters identified by distance networks using AFLP-SURV and ARLEQUIN 3.5.1.3 (Excoffier and Lischer 2010). Bayesian clustering analysis of AFLP loci was performed using the program STRUCTURE (Pritchard et al. 2000) for each of K = 2–11 with a burn-in of 500,000 and 750,000 Markov Chain Monte Carlo replications under a model of admixture and correlated allele frequencies. Two methods were used to evaluate patterns of genetic structuring resulting from STRUCTURE analyses. First, we used the K at which log-likelihood values (Ln(K)) were at their highest, as in the methods of Falush et al. (2003) and other authors (Michel et al. 2007). Second, we used the statistic ΔK of Evanno et al. (2005), shown in their simulations to produce the most consistent estimate of the true K under conditions of moderate to strong structure. Both ΔK and Ln(K) are ad hoc statistics, each which has been shown to perform well against different types of simulated data (Pritchard et al. 2000; Evanno et al. 2005).
Results
Collections
Almost every tuber inspected in this study contained a S. longipennis pupa. A total of 33 flies were reared from 49 pupae dissected from tubers, and 30 of these were variety vittigera (19♂, 11♀). The remaining three adult flies, all females, had a morphology intermediate between variety typica and variety vittigera. No variety typica flies were reared from pupal collections. An additional 106 adult flies were caught on or near H. tuberosus plants across the duration of the summer. Of these, 35 were variety vittigera (25♂, 10♀), 63 were variety typica (51♂, 12♀), and 8 were of intermediate morphology (6♂, 2♀). Adult flies of both morphological varieties were collected at all three sites in this study. No other species of Strauzia other than S. longipennis was reared from or captured on H. tuberosus.
mtDNA
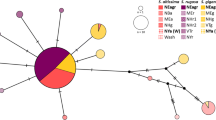
Two major haplotype groups were resolved across all flies for which sequence was obtained (n = 98; Fig. 2). These corresponded to the haplotype groups found by Axen et al. (2010). Each haplotype group was comprised of one primary haplotype found in the majority of individual flies in each group, and then a number of less common haplotypes that differed from the primary haplotype by between one and seven base pair substitutions. The two primary haplotypes from each group differed from each another at 18 of 658 bp. One group, hereafter haplotype group A, was composed of 38 flies of vittigera morphology (19♂, 19♀), nine of typica morphology (3♂, 6♀), and two flies with an intermediate morphology (2♂). The second group, hereafter haplotype group B, was composed of 39 flies of typica morphology (21♂, 18♀), eight of vittigera morphology (5♂, 3♀), and two flies of intermediate morphology (2♂). Both haplotype groups were obtained from flies at all three collection sites, and geographic location was not significantly associated with haplotype (Fisher’s Exact Test; df = 2, P = 0.07).

Parsimony network for a 658 bp segment of mitochondrial cytochrome oxidase I (COI) in 98 Strauzia longipennis individuals. Each connection = 1 base substitution. Sizes of polygons correspond to the number of flies having a particular haplotype. Small circles indicate haplotypes found in a single individual fly. Colors correspond to morphological IDs: black = S. longipennis variety vittigera, white = S. longipennis variety typica; gray = intermediate morphology. Numbers inside of shapes designate the number of flies of a particular morphology with that haplotype. “A” and “B” delineate the two major haplotype groups. The haplotype in group B marked with a asterisks denotes two individual flies with a unique haplotype that may be distinct from other lineages based on AFLP data (see Fig. 3, cluster V)
AFLP data
Analyses of AFLP data were consistent with the existence of genetic races. The neighbor-joining network resolved three well-supported clusters of flies collectively corresponding to the majority of individuals collected (clusters I, II, and III; Fig. 3). Two additional clusters (clusters IV and V) consisted of five flies and two flies, respectively. Cluster I (n = 40 flies; 24♂, 16♀) was more strongly differentiated from the other clusters: 16 of 176 loci (9.1 %) were amplified exclusively in cluster I (i.e., never in clusters II or III), seven loci (4.0 %) were exclusive to clusters II and III, and four loci (2.3 %) were ‘fixed’ between cluster I and clusters II and II. Clusters II (25 flies; 13♂, 2♀) and III (19 flies; 11♂, 8♀) were less strongly differentiated from one another: no fixed differences were found at any AFLP loci, but 10 loci were absent in either cluster II or cluster III and found at frequencies >0.15 in the other cluster. Pairwise FST between clusters I, II, and III ranged from 0.08 to 0.34 (Table 1).

Neighbor-joining network of Nei genetic distances generated for 91 individual S. longipennis flies and based on 176 polymorphic AFLP loci. Branches in black denote flies with vittigera morphology, gray lines denote flies with typica morphology, and dashed lines denote flies with an intermediate morphology (Fig. 1b). Closed circles are flies with a group A mitochondrial haplotype, while open circles are flies with a group B haplotype (refer to Fig. 2). Numbers at branch nodes denote bootstrap support (10,000 bootstraps). “T” denotes flies dissected and then reared from tubers of Helianthus tuberosus, while flies without a “T” were all collected as adults on H. tuberosus plants
Log likelihood values generated from STRUCTURE analyses of AFLP data best supported K = 2 or 4 populations, depending on the method of evaluation used. The ΔK of Evanno et al. (2005), which performs well in identifying the true number of clusters under conditions of moderate to strong genetic structure, showed a modal maximum at a K = 2. For a K = 2 (Fig. 4), flies in cluster I had strong proportional membership with one simulated population (0.990), and flies in clusters II and III to the second population (0.995 and 0.991, respectively). Cluster IV and V flies were of intermediate membership (0.535 and 0.834 proportional assignment to the second population, respectively). Evaluation of unmodified Ln(K) values showed a maximum likelihood value at K = 4, before values decreased and then fluctuated widely, and so we also evaluated this as a potential reflection of “true” genetic structure. For a K = 4, most flies were placed into one of three populations, corresponding strongly to the clusters I, II, and III resolved by the phylogenetic analysis (Fig. 4). Cluster IV flies were grouped into their own fourth population, while cluster V flies were not strongly allied with any single population.

Bar plots of STRUCTURE analyses (admixture with correlated allele frequencies) for 91 individual S. longipennis flies assigned to predefined groups according to the five clusters resolved from the NJ distance network (Fig. 3). a The ΔK method of Evanno et al. (2005) best supported a K = 2, while b methods of Falush et al. (2003), which rely on identifying the value of K with the highest log likelihood value, best supported a K = 4. Both values of K identify structure also resolved by the distance network, and both indicate the existence of cryptic genetic races within S. longipennis
Patterns of morphology and mitochondrial haplotypes were different between genetic clusters. Cluster I contained flies of both haplotypes (24 Haplotype A, 13 Haplotype B, 3 unscored) and of both morphologies (10 typica, 26 vittigera, 4 intermediate). No association between haplotype and morphology was found among individual flies in cluster I (Fisher’s Exact Test; df = 1, P = 0.08). Cluster II was composed exclusively of flies of typica morphology (n = 25), and all but one fly had the group B haplotype. Conversely, 14 of 19 flies in cluster III were of vittigera morphology (4 variety typica, 1 intermediate) and 14 of 17 flies scored for mitochondrial sequence had a group A haplotype (3 group B; 2 unscored). As with cluster I, no association between haplotype and morphology was found for cluster III (FET; df = 1, P = 0.12).
Cluster IV (5 flies; 3♂, 2♀) was rooted at a position intermediate between cluster I and clusters II and III (Fig. 3). Two cluster IV flies had typica morphology, and three had vittigera morphology. Two of the vittigera morphology flies were tuber-extracted, while the other three flies were captured as adults. Four cluster IV flies were scored for COI, and both haplotypes groups were represented (Fig. 3). Moreover, two of the four AFLP loci with fixed differences between cluster I and clusters II and III were amplified at intermediate frequencies among the cluster IV flies, and cluster IV flies were almost perfectly divided in their population assignment for a K = 2 (Fig. 4). These data point towards cluster IV representing naturally occurring F1 hybrids between cluster I and flies from cluster II and/or III.
Cluster V consisted of just two female flies and was rooted closely to cluster II (Fig. 3). However, we tentatively distinguish it from other clusters due to an intermediate STRUCTURE assignment in both evaluated values of K (Fig. 4), and because both cluster V flies shared a group B haplotype found in no other flies in this study (Fig. 2). Both cluster V flies (2♀) were of variety typica morphology and were collected on the final day of the sampling period.
Adult flies from all three major genetic clusters were captured at all three collection sites. Flies reared from H. tuberosus tuber-extracted pupae were only from clusters I (8♂, 7♀) and III (5♂, 3♀). No cluster II flies were reared from pupae collected from H. tuberosus tubers.
Allochrony between genetically differentiated S. longipennis
Although the range of capture dates for the three primary genetic clusters of S. longipennis overlapped, mean date of capture in the field was significantly different between clusters (Fig. 5; ANOVA: F = 41.08, df = 2, P < 0.0001). Cluster III was captured earliest in the year (mean capture date = June 9th), followed by cluster I (June 29th), then cluster II (July 6th). The three putative F1 hybrids (Cluster IV) captured as adults were all caught on the same day (June 30th) at the Cedar Rapids, IA site. Both cluster V flies were collected in Cedar Rapids, IA on the final day of the sampling period (July 11th).

Field collection dates of adult S. longipennis flies from genetic clusters resolved through analyses of 176 AFLP loci (Figs. 3, 4). Grey bars denote standard errors around mean collection dates for each cluster. Figures following each category label show the primary thoracic banding patterns found among flies in each cluster. Lowercase letters denote significant differences in mean eclosion dates between clusters
Discussion
Strauzia longipennis is a complex of three or more genetically differentiated races
A scan of the S. longipennis nuclear genome using AFLPs resolved at least three genetically differentiated clusters of flies (Figs. 3, 4). Cluster I differs most radically from clusters II and III: several fixed differences between these clusters imply that gene flow is greatly reduced between these groups, at least in some regions of the genome. We therefore consider cluster I to be a distinct genetic race. The fixed differences between cluster I flies and other clusters might even warrant elevation to species status, if one were to abide by the genomic cluster species concept (GCSC), which defines species as “…a distinguishable group of individuals that has few or no intermediates when in contact with other such clusters.” (Mallet 1995). However, the GCSC in generally criticized for having no set threshold for the tightness of clustering or for the number of differences required to diagnose species (Coyne and Orr 2004) and application of the GCSC to complexes of closely related organisms in various stages of divergence has failed to identify a threshold for species status (e.g., Berlocher 1999), so we will maintain the term ‘race’ until further evidence warrants a change. We do note that the fixed allelic differences combined with contemporary allochronic isolation (see below) suggest extremely limited effective gene flow into or out of this cluster.
Clusters II and III were more closely related to one another than either was to cluster I, but support for their independence was also strong. No fixed allelic differences were found between AFLP loci in clusters II and III, but several alleles were found at high frequency in one cluster and were entirely absent in the other. All but one cluster II fly (1/25) had a group B mitochondrial haplotype, while cluster III flies had primarily group A haplotypes (14/17). Based on this evidence, we propose that clusters II and III are also genetic races.
Ecological data suggest that a plant other than H. tuberosus is likely the primary host for flies in cluster II. None of the S. longipennis reared from pupae extracted from tubers of H. tuberosus were from cluster II, and adults of cluster II captured on H. tuberosus were almost exclusively male (23 of 25). Males in genus Strauzia tend to be territorial, staking out leaves and waiting for females to approach (Stoltzfus 1988). That few cluster II females were captured suggests that cluster II males may simply be making errors in host plant choice, while females have more strict fidelity. Alternatively, cluster II flies may oviposit on H. tuberosus but not complete development (note that 16 of 49 pupae dissected from tubers did not emerge as adult flies). Additional support for an alternative host for cluster II males comes from taxonomic work associating flies of typica morphology with domesticated H. annuus (Lisowski 1985; Stoltzfus 1988), a plant species found at all three collections sites. Lisowski’s (1979) allozyme study also identified a genetic cluster of flies reared from H. strumosus and H. annuus, but with no apparent H. tuberosus association. Future work should involve sampling of Strauzia from other sympatric Helianthus species, including H. annuus, to determine the natal host for cluster II and to allow for comparative study of host environments.
Putative F1 hybrids were also found in nature. The five flies that comprise Cluster IV were of intermediate assignment for a K = 2 in our STRUCTURE analysis, apparent hybrids between cluster I flies and flies in clusters II and III. All AFLP loci fixed between cluster I and clusters II and III were found at intermediate proportions among cluster IV flies, indicating a mixed ancestry. Cluster IV flies also had a mixture of both morphologies and both mtDNA haplotype groups. That fixed differences persist between cluster I and clusters II and III suggests that even though F1 hybrids exist, certain regions of the genome are robust to introgression, while other regions may be more porous (e.g., Turner et al. 1999; Nosil 2008; Michel et al. 2010). A finer scale, sequence-based marker system may be necessary to evaluate the status of hybrids and their contribution to gene flow in this system (e.g. RAD-tags: Gompert et al. 2010).
The two flies in Cluster V may represent still another reproductively isolated group. Phylogenetic analysis grouped cluster V as sister to cluster II, both clusters share a common morphology, and results from STRUCTURE simulations suggest they may be natural F2 backcross flies. However, there was strong support for the monophyly of these two individuals (Fig. 3), and that the two cluster V flies share a unique haplotype found in no other flies in our study (Fig. 2, asterisk) suggests that they may be distinct from other flies collected. Both cluster V flies were collected on the final day of sampling, and so may represent a fourth, late summer, race that was incompletely sampled in this study.
Genetically differentiated Strauzia fly populations are partially allochronically isolated
Significant differences in the date of capture for adult flies in each cluster suggest that allochrony in adult emergence may be a premating reproductive barrier between fly species and races. Flies live an average of 13.5 ± 7.8 (SE) days under ideal lab conditions (n = 16; AAF, unpublished data), so differences of up to 28 days between clusters in their peak incidence in the field depress the propensity for gene flow between demes. Allochronic differentiation is common among phytophagous insects occupying different ecological niches: differences in diapause length and eclosion timing partially or completely isolate races and species of Diptera (Filchak et al. 1999; Joy and Crespi 2007), Hymenoptera (Forbes et al. 2009), and Lepidoptera (Scriber and Ording 2005; Santos et al. 2007), among others (Abbot & Withgott, 2010). Temporal differences are typically correlated with shifts to new hosts with divergent life histories (although see Scriber and Ording 2005). Here, as at least two of three Strauzia races are using H. tuberosus as their larval host (see below), and as these races occur as adults at different times during the year, we hypothesize that cluster I and cluster III flies colonized H. tuberosus independently, and that they currently occupy different temporal niches on the same host plant. Females of cluster III, found earlier in the season, likely oviposit into H. tuberosus earlier than cluster I females. As the plant grows and adds tissue during the course of the season, temporal staggering of ovipositon events may allow for both taxa to coexist.
Allochronic differences between genetic races of S. longipennis do not necessarily reflect that allochrony was an important pre-mating barrier at the time of initial divergence, nor that it is the only contemporary barrier to gene flow. For instance, many phytophagous insect races also differ in their host discrimination behaviors, which, if mating occurs on or near different hosts, can translate into assortative mating and prezygotic isolation (Funk 1998; Craig et al. 2001; Linn et al. 2003; Matsubayashi et al. 2011). Divergent host performance characters may also isolate demes, wherein feeding or development on a non-natal host plant results in a reduction in fitness (Via 1991; Brazner and Etges 1993; de Jong et al. 2007). Hybrids between insects adapted to different host plants may also be less fit on all hosts than either parental race, a form of extrinsic postzygotic barrier (Dambroski et al. 2005). Finally, intrinsic pre- or postzygotic barriers can isolate taxa, including hybrid sterility and hybrid inviability (Hollocher and Wu 1996), or incompatibility resulting from cytoplasmically inherited endosymbionts (Werren 1997). We must therefore expect that allochronic isolation between S. longipennis races is not the only current barrier to gene flow in this system.
Introgression of mitochondria and morphology: evidence for speciation-with-gene-flow
We propose that the two mitochondrial haplotype groups and the two morphological varieties described by Axen et al. (2010) reflect relatively ancient genetic structure, now obscured due to introgression between lineages. Mitochondrial haplotype groups A and B differ at 18 of the total 658 base pairs, which under the assumption of a standard insect molecular clock, dates their most recent common ancestral sequence to between 0.77 and 1.19 Mya (Brower 1994; Papadopoulou et al. 2010). These do not, however, correspond to differences between contemporary lineages as defined by the more powerful AFLP scan. No genetic cluster identified using nuclear data had just a single haplotype group, and the most divergent lineage, Cluster I, possessed a particularly heterogeneous mixture of both haplotype groups. The most parsimonious explanation for such patterns is that mitochondrial genomes have been captured by non-ancestral lineages (e.g., Llopart et al. 2005). Likewise, the two divergent morphological types (vittigera and typica) also appear to have moved between lineages, given that two of the three races contained a mix of both vittigera and typica morphologies. An alternative explanation might be that morphological variants have evolved in parallel in different lineages, but this seems a more convoluted explanation for these patterns.
Taken together, patterns of genetic differentiation and morphological variation suggest a model of speciation-with-gene-flow for S. longipennis races (Fig. 6), wherein movement of genetic material between demes has occurred without a breakdown of genetically differentiated clusters. Speciation-with-gene-flow models are increasingly relevant to emerging animal systems, especially insects, and promise to offer much new information about the genomics of speciation. Such models strongly imply that complete reproductive isolation is not necessary for genetic divergence, and that divergent lineages may experience even relatively high rates of gene flow without consequent lineage fusion (Nosil 2008). To date, such patterns have been elegantly documented in a range of phytophagous insects, including Anopheles mosquitoes (Turner et al. 1999; White et al. 2010), Acyrthosiphon pea aphids (Via and West 2008), Neochlamisus leaf beetles (Funk et al. 2011), and Rhagoletis fruit flies (Schwarz et al. 2005; Michel et al. 2010), and early indications are that reduced recombination in some number of genomic regions “protect” important allelic combinations. The study of organisms at various stages along the so-called “speciation continuum” (Hendry et al. 2009), ultimately allows for a better understanding of how reproductive isolating barriers arise and are maintained.

A hypothesis for speciation and gene flow among three morphologically cryptic races of Strauzia collected from H. tuberosus. First, after an ancestral split between cluster I and the ancestor of clusters II and III, each lineage may have seized or evolved a unique haplotype and/or morphology. Subsequent gene flow between lineages may have resulted in reciprocal capture of both mitochondrial haplotypes and morphological variation, resulting in their imperfect contemporary relationship with actual species limits. Divergence between clusters II and III probably represents a more recent repeat of the above: again, two lineages diverge and begin to sort mitochondria and morphology but continue to experience gene flow without lineage fusion
References
Abbot P, Withgott JH (2010) Phylogenetic and molecular evidence for allochronic speciation in gall-forming aphids (Pemphigus). Evolution 58:539–553
Abrahamson WG, Weis AE (1997) Evolutionary ecology across three trophic levels. Princeton University Press, Princeton
Axen HJ, Harrison JL, Gammons JR, Mcnish IG, Blythe LD, Condon MA (2010) Incipient speciation in Strauzia longipennis (Diptera: Tephritidae): two sympatric mitochondrial DNA lineages in Eastern Iowa. Ann Entomol Soc Am 103:11–19
Berlocher SH (1998) Can sympatric speciation via host or habitat shift be proven from phylogenetic and biogeographic evidence? In: Howard DJ, Berlocher SH (eds) Endless forms: species and speciation. Oxford University Press, Oxford, pp 99–113
Berlocher SH (1999) Host race or species? Allozyme characterization of the ‘flowering dogwood fly’, a member of the Rhagoletis pomonella complex. Heredity 83:652–662
Berlocher SH, Feder JL (2002) Sympatric speciation in phytophagous insect: moving beyond controversy? Annu Rev Entomol 47:773–815
Brazner JC, Etges WJ (1993) Pre-mating isolation is determined by larval rearing substrates in cactophilic Drosophila mojavensis. 2. Effects of larval substrates on time to copulation, mate choice and mating propensity. Evol Ecol 7:605–624
Brink JE (1922) The sunflower maggot (Straussia longipennis Wied.). Ann Rep Entomol Soc Ont 53:72–74
Brower AV (1994) Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc Natl Acad Sci USA 91:6491–6495
Bush GL (1969) Sympatric host race formation and speciation in frugivorous flies of the genus Rhagoletis (Diptera, Tephritidae). Evolution 23:237–251
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Coyne JA, Orr HA (2004) Speciation. Sinauer, Sunderland
Craig TP, Horner JD, Itami JK (2001) Genetics, experience and host plant preference in Eurosta solidaginis: implications for host shifts and speciation. Evolution 55:773–782
Dambroski HR, Linn C Jr, Berlocher SH, Forbes AA, Roelofs W, Feder JL (2005) The genetic basis for fruit odor discrimination in Rhagoletis flies and its significance for sympatric host shifts. Evolution 59:1953–1964
de Jong PW, Breuker CJ, de Vos H, Vermeer KMCA, Oku K, Verbaarschot P, Nielsen JK, Brakefield PM (2007) Genetic differentiation between resistance phenotypes in the phytophagous flea beetle, Phyllotreta nemorum. J Insect Sci 9:1–8
Egan SP, Nosil P, Funk DJ (2008) Selection and genomic differentiation during ecological speciation: isolating the contributions of host association via a comparative genome scan of Neochlamisus bebbianae leaf beetles. Evolution 62:1162–1181
Emelianov I, Dres M, Baltensweiler W, Mallet J (2001) Host-induced assortative mating in host races of the larch budmoth. Evolution 55:2002–2010
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Felsenstein J (1989) PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 5:164–166
Filchak KE, Feder JL, Roethele JB, Stolz U (1999) A field test for host-plant dependent selection on larvae of the apple maggot fly, Rhagoletis pomonella. Evolution 53:187–200
Foote RH, Blanc FL, Norrbom AL (1993) Handbook of the fruit flies (Diptera: Tephritidae) of America north of Mexico. Cornell University Press, Ithaca
Forbes AA, Powell THQ, Stelinski LL, Smith JJ, Feder JL (2009) Sequential sympatric speciation across trophic levels. Science 323:776–779
Funk DJ (1998) Isolating a role for natural selection in speciation: host adaptation and sexual isolation in Neochlamisus bebbianae leaf beetles. Evolution 52:1744–1759
Funk DJ, Egan SP, Nosil P (2011) Isolation by adaptation in Neochlamisus leaf beetles: host-related selection promotes neutral genomic divergence. Mol Ecol 20:4671–4682
Gompert Z, Forister ML, Fordyce JA, Nice CC, Williamson RJ, Buerkle CA (2010) Bayesian analysis of molecular variance in pyrosequences quantifies population genetic structure across the genome of Lycaeides butterflies. Mol Ecol 19:2455–2473
Hendry AP, Bolnick DI, Berner D, Peichel CL (2009) Along the speciation continuum in stickleback. J Fish Biol 75:2000–2036
Hollocher H, Wu CI (1996) The genetics of reproductive isolation in the Drosophila simulans clade: X vs autosomal effects and male vs female effects. Genetics 143:1243–1255
Joy JB, Crespi BJ (2007) Adaptive radiation of gall-inducing insects within a single host-plant species. Evolution 61:784–795
Linn C Jr, Feder JL, Nojima S, Dambroski HR, Berlocher SH, Roelofs W (2003) Host fruit odor discrimination and sympatric race formation in the apple maggot. PNAS 100:11490–11493
Lisowski EA (1979) Biochemical systematics of the genus Strauzia (Diptera: Tephritidae). University of Illinois, Thesis
Lisowski EA (1985) Taxonomy and biology of Strauzia (Diptera: Tephritidae) in Illinois. University of Illinois, Dissertation
Llopart A, Lachaise D, Coyne JA (2005) Multilocus analysis of introgression between two sympatric sister species of Drosophila: Drosophila yakuba and D. santomea. Genetics 171:197–210
Loew H (1873) Monographs of the Diptera of North America, Part III. Misc Coll Smithsonian Inst 256:1–351
Lynch M, Milligan BG (1994) Analysis of population genetic structure with RAPD markers. Mol Ecol 3:91–99
Mallet J (1995) A species definition for the modern synthesis. Trends Ecol Evol 10:294–299
Mallet J (2008) Hybridization, ecological races and the nature of species: empirical evidence for the ease of speciation. Philos Trans R Soc Lond B 363:2971–2986
Marsteller S, Adams DC, Collyer ML, Condon M (2009) Six cryptic species on a single species of host plant: morphometric evidence for possible reproductive character displacement. Ecol Entomol 34:66–73
Matsubayashi K, Kahono S, Katakura H (2011) Divergent host plant specialization as the critical driving force in speciation between populations of a phytophagous ladybird beetle. J Evol Biol 24:1421–1432
Michel AP, Rull J, Aluja M, Feder JL (2007) The genetic structure of hawthorn-infesting Rhagoletis pomonella populations in Mexico: implications for sympatric host race formation. Mol Ecol 16:2867–2878
Michel AP, Sim S, Powell THQ, Taylor MS, Nosil P, Feder JL (2010) Widespread genomic divergence during sympatric speciation. Proc Natl Acad Sci USA 107:9724–9729
Nason JD, Heard SB, Williams FR (2002) Host-associated genetic differentiation in the goldenrod elliptical-gall moth, Gnorimoschema gallaesolidaginis (Lepidoptera: Gelechiidae). Evolution 56:1475–1488
Nosil P (2008) Speciation with gene flow could be common. Mol Ecol 17:2103–2106
Nosil P, Feder JL (2012) Genomic divergence during speciation: causes and consequences. Phil Trans R Soc B Biol Sci 367:332–342
Papadopoulou A, Anastasiou I, Vogler AP (2010) Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Mol Biol Evol 27:1659–1672
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Santos H, Rousselet J, Magnoux E, Paiva M-R, Branco M, Kerdelhué C (2007) Genetic isolation through time: allochronic differentiation of a phenologically atypical population of the pine processionary moth. Proc R Soc Lond Ser B Biol Sci 274:935–941
Schwarz D, Matta BM, Shakir-Botteri NL, McPheron BA (2005) Host shift to an invasive plant triggers rapid animal hybrid speciation. Nature 436:546–549
Scriber JM, Ording GJ (2005) Ecological speciation without host plant specialization; possible origins of a recently described cryptic Papilio species. Entomol Exp Appl 115:247–263
Smith MA, Rodriguez JJ, Whitfield JB, Deans AR, Janzen DH, Hallwachs W, Hebert PDN (2008) Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology, and collections. Proc Natl Acad Sci USA 105:12359–12364
Steyskal GC (1986) Taxonomy of the adults of the genus Strauzia Robineau-Desvoidy (Diptera, Tephritidae). Insecta Mundi 1:101–117
Stireman JO, Devlin H, Abbot P (2012) Rampant host- and defensive phenotype-associated diversification in a goldenrod gall midge. J Evol Biol 25:1991–2004
Stoltzfus WB (1988) The Taxonomy and Biology of Strauzia (Diptera: Tephritidae). J Iowa Acad Sci 95:117–126
Strong DR, Lawton JA, Southwood TRE (1984) Insects on plants: community patterns and mechanisms. Blackwell Scientific Publications, Oxford
Turner TL, Hahn MW, Nuzhdin SV (1999) Genomic islands of speciation in Anopheles gambiae. PLoS Biol 3:e285
Vekemans X (2002) AFLP-SURV 1.0: a program for genetic diversity analysis with AFLP (and RAPD) population data. Laboratoire de Génétique et Ecologie Végetale
Via S (1991) Specialized host plant performance of pea aphid clones is not altered by experience. Ecology 72:1420–1427
Via S, West J (2008) The genetic mosaic suggests a new role for hitchhiking in ecological speciation. Mol Ecol 17:4334–4345
Werren JH (1997) Wolbachia and speciation. In: Howard DJ, Berlocher SH (eds) Endless forms: species and speciation. Oxford University Press, Oxford, pp 245–260
Westdal P, Barrett C (1962) Injury by the sunflower maggot, Strauzia longipennis (Wied.) (Diptera: Trypetidae), to sunflowers in Manitoba. Can J Plant Sci 42:11–14
White BJ, Cheng C, Simard F, Costantini C, Besansky NJ (2010) Genetic association of physically unlinked islands of genomic divergence between incipient species of Anopheles gambiae. Mol Ecol 19:925–939
Wood TK, Keese M (1990) Host-plant-induced assortative mating in Enchenopa treehoppers. Evolution 44:619–628
Acknowledgments
The authors would like to thank Andy Roth, Gaby Hamerlinck, Steve Hendrix, Amanda Nelson, and Katie Weldon for collection assistance. Heather Axen provided illustrations of Strauzia thoracic morphology. Stewart Berlocher offered unique insight into the history of Strauzia research. Raul Medina and two anonymous reviewers provided helpful comments on an earlier draft of this manuscript. Special thanks to Gery Hehman and the Carver Center for Genomics for sequencing and genotyping support. Part of this research was supported by NSF DEB-0949361 to M.A.C.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Forbes, A.A., Kelly, P.H., Middleton, K.A. et al. Genetically differentiated races and speciation-with-gene-flow in the sunflower maggot, Strauzia longipennis . Evol Ecol 27, 1017–1032 (2013). https://doi.org/10.1007/s10682-012-9622-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10682-012-9622-y