
Abstract
Quinoa (Chenopodium quinoa Willd.) is recognized as an important crop to improve global food security. It has gained international recognition because of the nutritional value of its seeds and its broad agronomic resilience. Although several studies have attempted to characterize the genetic diversity of quinoa, none have focused on evaluating germplasm from Ecuador; the latter considered a relevant subcenter of diversity for the species. In this study, 84 accessions representing the species’ cultivated range in the Ecuadorian Andes were characterized using 15 species-specific SSR markers. The extent of allelic richness (196 alleles) and genetic heterozygosity (HE = 0.71) detected for these accessions demonstrate that Ecuadorian quinoa is highly diverse. Phenetic analyzes structured Ecuadorian germplasm into 3 subgroups; each containing genotypes from all surveyed provinces. Average expected heterozygosity was high for all 3 subgroups (0.53 ≤ HE ≤ 0.72), and Nei-pairwise comparisons showed significant genetic divergence among them (0.31 ≤ NeiDST ≤ 0.84). The lack of a clear geographic pattern in the genetic structure of Ecuadorian quinoa led us to believe that the 3 reported subgroups constitute independent genetic lineages representing ancestral landrace populations which have been disseminated throughout Ecuador via informal seed networks. Nevertheless, a Wilcoxon test showed that at least one subgroup had been subject to intensive inbreeding and selection; and possibly corresponds to the local commercial variety INIAP-Tunkahuan. Our results show that ancestral quinoa diversity in Ecuador has prevailed despite the introduction of commercial varieties, and should be preserved for future use in breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Quinoa (Chenopodium quinoa Willd.) is an annual species of the Amarantheceae endemic to South America. In recent years it has gained international attention as a highly nutritious food source. The crop produces gluten-free seeds with a high protein content, an excellent balance of essential amino-acids, and an optimal composition of fibers, lipids, vitamins and minerals (Koziol 1992; Vega-Gálvez et al. 2010; Ruiz et al. 2014). Quinoa is also recognized as a promising crop for global food security. The species can thrive in a wide range of agroecosystems, including nutrient-deficient, arid and saline environments (Jacobsen et al. 2003). Its agronomic resilience offers unexplored avenues for the production of nutritious food in marginal lands unsuitable for conventional agriculture (Jacobsen et al. 2003; Ruiz et al. 2014).
Despite its potential, quinoa remains an underutilized agricultural species and significant breeding efforts are necessary to transform the crop into an international commodity (Zurita-Silva et al. 2014; Bazile et al. 2016). In this regard, genetic diversity studies can provide an assessment of the conservation status of this valuable agricultural resource to enable the identification of new sources of allelic variation that can be exploited to increase the crop’s productivity and geographic range. Quinoa originated nearly 7000 years ago in the Andean Altiplano following the domestication of an ancestral tetraploid (Jarvis et al. 2017). Subsequently, the species underwent a millenary process of selection and diversification (led by generations of farmers) which culminated in its geographic expansion throughout western South America (Bazile et al. 2013). Today, quinoa is associated with 5 major ecotypes (Altiplano, Salar, Yunga, Valley and Lowland); each one highly adapted to specific environments, yet highly rich in genetic variation (Bazile et al. 2013, 2016). Over the past two decades, several attempts have been made at characterizing the species’ diversity across its endemic range using a variety of molecular marker technologies (Mason et al. 2005; Christensen et al. 2007; Fuentes et al. 2009, 2012; Jarvis et al. 2008, 2017; Tártara et al. 2012; Zhang et al. 2017). These studies suggest that quinoa can be classified into two distinct germplasm pools: the Andean highland group (with its center of diversity in the Altiplanos of Bolivia and Peru) and the lowland diversity group (with its center of diversification in the coastal regions of central and southern Chile) (Christensen et al. 2007; Fuentes et al. 2009; Jarvis et al. 2017).
For long, Ecuador has been considered a subcenter of diversification for the species (Tapia et al. 2015; Bazile et al. 2016), but germplasm from the region has been largely unrepresented in molecular studies. The first evidence of cultivation of quinoa in Ecuador dates back to approximately 300–500 BC (Yugcha 1996), and historical records indicate that the crop had been cultivated in the central highlands of Ecuador well before the Spanish conquest of South America (Peralta and Mazón 2015). Molecular evidence suggests that Ecuadorian landraces originate from the highland diversity pools of Peru and Bolivia (Christensen et al. 2007) and have been classified as Valley ecotypes (Gandarillas et al. 1989; Peralta and Mazón 2015). Notwithstanding, when compared with germplasm from Peru and Bolivia, Ecuadorian landraces display unique, defining characteristics; they tend to mature later, grow taller and have a higher potential for biomass accumulation and grain yield (Gandarillas et al. 1989; Tapia et al. 2015). Agronomic and morphological evaluations also suggest that Ecuadorian germplasm harbors broad genetic variation (Gandarillas et al. 1989; Tapia et al. 2015). Gandarillas et al. (1989) could classify Ecuadorian quinoa into six different races; all of them distinct to Peruvian and Bolivian ecotypes, and each one showing a significant degree of within-group diversity. Certainly, Ecuadorian quinoa diversity could be a relevant source of novel and unique adaptive characteristics for breeding programs and should be explored further (Christensen et al. 2007; Bazile et al. 2016). Thus, the main objective of this study was to assess the extent of genetic diversity of cultivated quinoa from the Ecuadorean Andes in order to understand its conservation status and future potential for breeding.
Methods
Germplasm sampling and collection
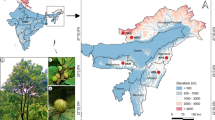
A total of 84 quinoa accessions were collected from 7 provinces across the Ecuadorian highlands (Fig. 1, Supplementary Table 1). At present, quinoa is cultivated in Ecuador by small scale farmers (< ~ 0.3 Ha) primarily in the central and northern provinces of Chimborazo, Cotopaxi and Imbabura, and to a lesser extent in the remaining Andean provinces (Fig. 1) (Peralta and Mazón 2015). Sampling was conducted at localities where quinoa is produced primarily as a staple for own-consumption. For each accession, 500 g of farmer’s seed were purchased directly at cultivation sites, or in local markets. Farmers were interrogated into the origin of their germplasm to avoid collection of the widely disseminated variety, INIAP-Tunkahuan, which was released in the early 90s and remains the only commercial variety available in Ecuadorian markets (Peralta and Mazón 2015). Estimates indicate that over 60% of the production of quinoa in Ecuador is associated to INIAP-Tunkahuan (Peralta and Mazón 2015). The latter was also included in our analyzes and was purchased at the Ecuadorian National Institute for Agricultural Research (INIAP) located in Quito, Ecuador. Furthermore, 5 quinoa varieties (Atlas, Pasto, Riobamba, Carmen, Red Carina) developed at Wageningen University and Research (Wageningen, The Netherlands) were included in this study to investigate the genetic relationship of Ecuadorian germplasm relative to genetic variants from other regions of the Andes. The 5 varieties (henceforth referred to as Dutch varieties) were derived through the systematic crossing of specific genotypes adapted to the climatic and day-length conditions of North-Western Europe, followed by pedigree selection (Jacobsen 2015). The genetic background of these lines is known to be primarily lowland Chilean (Van Loo, Personal Communication, 2018). Thus, even though they represent a reduced sample of the genetic diversity of lowland germplasm, we expected them to highly contrast with Ecuadorian quinoa given evidence showing the discrete segregation of the highland and lowland diversity groups (Christensen et al. 2007; Fuentes et al. 2009; Jarvis et al. 2017).

Geographical map of Ecuador indicating C. quinoa collection-sites as established by geo-referenced coordinates
Molecular characterization using SSR markers
Collected seeds were germinated in the greenhouse of the Plant Biotechnology Laboratory of Universidad San Francisco de Quito (Cumbayá, Ecuador). Per genotype, 5 seeds were sown in trays with regular potting soil. Following germination, young leaves were collected to isolate total genomic DNA using the CTAB protocol described by Cota-Sánchez et al. (2006).
Fifteen species-specific SSR markers were used for the characterization of the complete panel of 84 Ecuadorian quinoa accessions and 5 Dutch quinoa varieties. These markers were selected from a set of approximately 400 SSR markers previously reported and validated by Mason et al. (2005) and Jarvis et al. (2008). Selected markers were chosen for collectively displaying the highest number of alleles per locus (> 8) and highest of levels heterozygosity (HE > 0.7) in their respective studies, as well as for showing clear amplification signals in Ecuadorian germplasm (Data not shown). Table 1 summarizes primer sequences, SSR motifs, annealing temperatures and expected allele size ranges for all selected markers. It is important to highlight that primer design included the addition of 15 extra nucleotides (GCCTCCCTCGCGCCA) to the original sequence of the forward sequence to enable multiplexing and analysis by capillary electrophoresis as described by Blacket et al. (2012). Briefly, genomic DNA (20 ng) was PCR amplified in a 10 μl reaction containing 50 mM KCl, 20 mM Tris-HCl (pH 8.4), 2.5 mM MgCl2, 0.15 µM Forward Primer, 0.5 µM Reverse Primer, 0.2 µM mM dNTPs, 0.5 µM fluorescent tagged universal primer and 1 U Platinum Taq Polymerase (Life Technologies, Carlsbad, California). PCR amplification conditions consisted of 35 cycles of a 30 s denaturation at 94 °C, annealing of 90 s at 59–63 °C (depending on the primer pair employed, Table 1), and extension at 72 °C for 60 s. All reactions were performed in a T-Personal Series Thermocycler (Biometra, Gottingen, Germany). Amplified products were finally sent to Macrogen (Seoul, Korea) for analysis by capillary electrophoresis on a 3730XL ABI sequencer (ABI Systems, California).
Data analyzes
A total of 89 genotypes were used for downstream data analyzes. Allele size determination and SSR marker scoring were performed using the GeneMarker software package (Softgenetics, Pennsylvania). Standard genetic diversity indices, including allele number per locus (Na), expected heterozygosity (HE), Nei genetic distances (NeiDST) and allelic frequencies were calculated using the R-based adegent 2.0.0 statistical genetic package (Jombart 2008). Principle co-ordinates analyzes were also performed using adegent 2.0.0. Bayesian population structure analyzes were conducted using STRUCTURE (Pritchard et al. 2000) assuming an admixture model, evaluating between 1 and 7 subpopulations (K) through a total of 300,000 MCMC iterations and burn in of 100,000 iterations, with 5 replicates per K value. COANCESTRY (Wang 2011) was used to estimate inbreeding coefficients in the collected germplasm. Analysis were conducted using a threat value of Si = 1 to calculate Wang coefficients of inbreeding (WI) among identified sub-populations. Finally, BOTTLENECK (Piry et al. 1999) was used to estimate the possibility of genetic bottlenecks in identified sub-populations according to Wilcoxon indices.
Results
Genetic diversity indices
A collection of 84 quinoa genotypes from the Ecuadorian Andes were characterized using 15 genomic SSR markers. These markers proved highly informative and produced 196 alleles in the collection, with a range of 5 to 28 alleles per locus (mean = 13) (Table 2). Five quinoa varieties of Dutch origin were also included in the analysis and produced a total of 37 alleles. From these, only 9 alleles (21%) were common with variants detected in Ecuadorian germplasm.
Overall, a large proportion (~ 60%) of alleles discovered in Ecuadorian germplasm were observed in less than 5% of evaluated genotypes. Common alleles (i.e., those with allelic frequencies over 25%) were observed for only 10 SSR markers, and on average 55 (~ 65%) of the 84 evaluated genotypes shared a common allele at any of these loci. As expected for a predominately autogamous species, genetic heterogeneity was low but not uncommon; on average; 16% of all evaluated genotypes were heterozygous at any given SSR locus. Residual levels of allelic heterogeneity at individual loci could be explained by unintended outcrossing (i.e., with other genotypes and/or wild relatives) during seed maintenance (González Marín 2009). The degree of cross-pollination for this species has been estimated to range between 8 and 10% (Christensen et al. 2007).
Expected heterozygosity (HE) for the evaluated markers ranged from 0.40 to 0.93, with a mean of 0.71 (Table 2). These results suggest a high degree of genetic diversity for Ecuadorian quinoa. Prior assessments of the genetic diversity of the species using multi-origin germplasm have reported HE values ranging from 0.57 to 0.82 (Mason et al. 2005; Christensen et al. 2007; Jarvis et al. 2008; Fuentes et al. 2009, 2012; Tártara et al. 2012). Differences in HE between studies have been ascribed to the size and geographic composition of the evaluated germplasm, as well as the choice of SSR markers used for analysis. In general, higher HE was observed in studies analyzing germplasm collections spanning broad geographic ranges (Christensen et al. 2007; Fuentes et al. 2009). HE for Ecuadorian quinoa was high despite the restricted sampling area of this study (i.e., relative to the geographic span of cultivated quinoa in the Andes). These findings must be handled with caution, however, since the exclusive use of only highly polymorphic SSR markers may have inflated reported HE (Christensen et al. 2007; Fuentes et al. 2012; Tártara et al. 2012).
Population structure
In this study, we found evidence of population structure in cultivated quinoa from Ecuador. Phenetic analysis of 84 genotypes and 5 varieties of Dutch origin using STRUCTURE (Pritchard et al. 2000) clearly separated the collection into 4 subgroups according to Evanno delta K values (Fig. 2). Ecuadorian germplasm was structured into 3 distinct clusters (labeled A, B and C; Fig. 2); none of which included Dutch genotypes These clusters were highly heterogeneous in geographic composition and contained individuals from most, or all surveyed provinces. Correspondingly, a mantel test among clusters did not generate a significant correlation between genetic and geographic distances (r2 = 0.00023; p = 0.82). We therefore discarded the possibility of geographic patterning for the genetic diversity for Ecuadorian germplasm.

Population structure of 84 C. quinoa accessions with an optimal model of K = 4. Subgroups A, B and C are exclusively composed of Ecuadorian accessions. Subgroup D is composed by 5 varieties of Dutch origin
Population structure was investigated further with a principal coordinates analysis (PCoA). A two-dimension biplot using the first two coordinates (PCo1 vs. PCo2) accounted for approximately 45% (PCo1 = 34.4%; PCo2 = 10.4%) of the genetic diversity observed in the panel (Fig. 3a). Ecuadorian germplasm structured into 3 reasonably delimited clusters which closely reflected the grouping patterns revealed by STRUCTURE analysis (Fig. 2). Notwithstanding, in the PCoA biplot group A and group C shared a considerable region of overlapping genotypes. A careful examination of these overlapping genotypes revealed that these also presented a high degree of genetic admixture in the Bayesian clustering analysis. Unexpectedly, the 5 quinoa varieties of Dutch origin could not be resolved from Ecuadorian germplasm and clustered in group C. However, when a third dimension (PCo3 = 5.55%) was added to the analysis, Dutch materials could separate from Ecuadorian germplasm (Fig. 3b, Fig. 3c). This result was more coherent with the fact that Dutch varieties showed limited allelic correspondence with Ecuadorian germplasm. The addition of a third coordinate also revealed alternative relationship patterns between groups A, B and C (Fig. 3b, c). For instance, in the PCo1 vs. PCo3 biplot, groups A and B overlapped completely to form a major new group (Fig. 3c) These alternative clustering patterns show that Ecuadorian germplasm shows varying degrees of genetic relatedness for specific sets of SSR markers groups.

Population structure of 84 C. quinoa accessions based on multivariate PCoA analysis under different dimensions: a. PCo1 vs. PCo2; b. PCo1 vs. PCo3; and c. PCo2 vs. PCo3. Subgroups A, B and C are exclusively composed of Ecuadorian accessions. Subgroup D is composed by 5 varieties of Dutch origin. Color schemes to represent groups are based on the identities revealed with STRUCTURE analysis
Patterns of genetic variation in cultivated quinoa from Ecuador
Genetic diversity values, pairwise Nei coefficients, average number of alleles per locus, and unique alleles found between the 3 main population subgroups identified by clustering analyzes (i.e., STRUCTURE, PCoA) are presented in Table 3. In general, mean expected heterozygosity over 15 SSR markers was high for all 3 groups, with the highest diversity index found for group C (HE = 0.72) and the lowest for group B (HE = 0.53). Accordingly, pairwise comparisons of Nei’s coefficient demonstrated that the 3 groups diverged significantly from each other, albeit at different levels (Table 3). Thus, while groups B and C exhibited a high degree of genetic divergence (NeiDST = 0.84), groups A and B appeared more related (NeiDST = 0.31) (Table 3). These results are consistent with clustering patterns obtained via PCoA analyzes (Fig. 2a), where group C shows a greater degree of separation from groups A and B along the first dimension (PCo-1 = 35%; which accounts for the greatest degree of genetic differentiation in the collection), while groups A and B form an almost contiguous group along the first coordinate which is primarily resolved by the second dimension (PCo-2 = 12%).
Analysis of allelic richness per cluster shows that group B has the highest percentage of rare alleles (46%) detected in the collection, while group C exhibits the lowest (13%). However, group C presents the highest number of private alleles, an indication of greater allelic diversity. Finally, COANCESTRY analyzes (Wang 2011) showed a negative Wang Inbreeding index (WI = −0.186) for the entire collection, which indicates that on average all analyzed genotypes show a low degree of inbreeding amongst them. Notwithstanding, when individual groups were analyzed, cluster B presented a positive inbreeding coefficient value (WI = 0.066), indicative of a high degree of alleles which share identity by descent. These results were further confirmed by population bottleneck analyzes which showed a positive and significant (p < 0.001) bottleneck effect for subgroup B. None of the other subgroups presented any evidence for demographic bottlenecks, thus suggesting that these clusters have a lower degree of inbreeding and a higher degree of genotypic heterogeneity.
Discussion
Genetic diversity of cultivated quinoa in Ecuador
In this study, we have demonstrated that cultivated quinoa from Ecuador is highly diverse. The degree of allelic richness (Na = 196, mean = 13alleles/locus) and genetic heterozygosity (HE = 0.71) detected for 84 accessions using species-specific SSR markers was comparable with the vast allelic diversity reported for Argentinian, Bolivian, Chilean and Peruvian germplasm (Mason et al. 2005; Christensen et al. 2007; Jarvis et al. 2008; Fuentes et al. 2009, 2012; Tártara et al. 2012). Furthermore, our molecular results agree with phenotypic studies showing the extensive breadth of variation of Ecuadorian quinoa for morphological, agronomic and bromatological characters (Gandarillas et al. 1989; Tapia et al. 2015).
As expected, cultivated quinoa from Ecuador shared a limited proportion of its allelic diversity with 5 quinoa varieties bred in the Netherlands, and known to have been derived from lowland Chilean material. Molecular evidence suggests that Ecuadorian germplasm originates from the species’ highland diversity group; the latter has been repeatedly shown to diverge significantly from lowland Chilean genetic diversity (Christensen et al. 2007; Fuentes et al. 2012, 2009; Jarvis et al. 2017). We suspect that the coincidence of 9 alleles between Ecuadorian and Dutch germplasm can be explained by the fact that the evaluated varieties include, albeit to a limited extent, Ecuadorian diversity in their genetic constitution (Van Loo, Personal Communication, 2018). However, it remains unclear whether these 9 alleles are unique to Ecuadorian germplasm, or whether they can also be found in the northern- or southern-highland (Bolivia, Chilean Highlands) diversity gene pools. For all evaluated markers, detected alleles fell within the expected size-range as reported in literature (Mason et al. 2005; Christensen et al. 2007; Jarvis et al. 2008; Fuentes et al. 2009), but it was impossible to assess whether unique alleles were discovered in Ecuadorian germplasm (i.e., relative to Chilean, Bolivian and Peruvian gene pools) as previous studies did not disclose individual allele-size information. Future molecular assessments of Ecuadorian quinoa diversity should include a wider range of samples from the highland and lowland diversity groups, as well as local, wild Chenopodium germplasm. The latter could prove particularly relevant. Fertility assessments, phenetic analyzes and molecular data have demonstrated that interspecific gene flow occurs naturally between quinoa and its weedy relatives (Wilson 1980, 1990; González Marín 2009; Bazile et al. 2013). It has been suggested that historical patterns of crop-weed natural hybridization and differentiation, accompanied by human-mediated selection processes, were central to the diversification of distinct, locally-adapted landraces (Wilson 1990). The collection and characterization of weedy Chenopodium species could therefore shed light into the evolutionary origin and molecular distinctiveness of Ecuadorian quinoa diversity, and also offer novel sources of allelic variation for crop improvement.
Population structure in Ecuadorian quinoa
A key objective of this study was to investigate patterns of population structure in Ecuadorian quinoa. Our analyzes demonstrate that modern cultivated germplasm can be resolved into 3 distinct clusters showing varying degrees of genetic exchange among them (Figs. 1, 2). Each cluster was highly heterogeneous in geographic composition and included genotypes representing the species’ continuous range of cultivation throughout the Ecuadorian highlands. Ultimately, the lack of a clear geographic pattern in the genetic structure of Ecuadorian quinoa led us to believe that reported clusters constitute independent genetic lineages representing either ancestral landrace populations or modern bred varieties which have been disseminated throughout the territory via informal seed networks. Historically, the flow of landraces and local seed varieties in the Andes has been governed by the complex (and often poorly understood) dynamics of informal seed networks connecting farmers over different altitudes and across latitudes (Zimmerer 2003; Almekinders et al. 2007; Bazile et al. 2013).
Following the Spanish conquest of South America, quinoa became a marginal crop primarily consumed by indigenous communities as part of their cultural identity. In these communities, the tradition of cultivating quinoa was passed down from generation to generation, along with germplasm carefully preserved and curated via selection (Fuentes et al. 2009). Today, many autochthonous farmers still conserve their “family landraces” as part of their intangible heritage, and prefer to cultivate their landrace collections over commercial varieties (Peralta and Mazón 2015). In our analyzes, group A and group C displayed high levels of allelic diversity (Table 3) and genotypic heterogeneity (i.e., COANCESTRY tests show that individuals within these clusters tend to be outbred, and consequently have a low degree of genetic relatedness). It therefore seems likely that each group constitutes a unique ancestral lineage representative of the species’ extant diversity originally introduced into Ecuador. We must highlight, however, that while these groups were genetically divergent (NeiDST = 0.62), they also displayed a certain degree of genetic exchange among them. This was especially evident in PCoA biplot analyzes (Fig. 3a, b) where both groups shared regions with overlapping genotypes. Two possibilities come into play. The first one explains that Ecuadorian diversity originates from a specific gene pool (e.g. founder effect) which was differentiated into an array of locally adapted landraces through a process of domestication led by generations of farmers, as previously suggested by Christiansen et al. (2007). At least phenotypically, Ecuadorian germplasm expresses a certain degree of uniformity (i.e., hence their denomination as “valley ecotypes”). When compared in the field with germplasm from Peru and Bolivia, Ecuadorian germplasm displays unique defining characteristics; these tend to mature later, grow taller and have a higher potential for biomass accumulation and grain yield than their counterparts from Peru and Bolivia (Gandarillas et al. 1989; Tapia et al. 2015). However, given that groups A and C have unique and defining patterns of allelic diversity (data not shown), a second possibility would explain that each lineage derives from divergent allelic pools within the northern-highland diversity group. Under this hypothesis, a broader degree of diversity was introduced into the region, but this diversity was likely subject to a process of partial homogenization which favored the fixation of specific alleles necessary for adaptation to Ecuadorian latitudes (Jellen et al. 2015; Peralta and Mazón 2015).
In contrast to groups A and C, group B appears to be exclusively composed by genotypes corresponding to a commercial variety. To develop this theory, we investigated the genetic membership of the commercial variety INIAP-Tunkahuan, which clustered inside group B (Fig. 2). In principle, the subgroup’s high degree of expected heterozygosity (HE = 0.56) and allelic richness (Na = 96) would indicate a high degree of within-population genetic diversity. Notwithstanding, group B was also characterized by having the highest incidence of rare alleles (~ 46%) and the lowest level of genetic variability among the 3 detected population subgroups (Table 3). In fact, COANCESTRY and Bottleneck analyzes revealed that individuals from group B were highly related to each other, and that the gene pool had been subject to an intensive process of allelic selection and fixation. INIAP-Tunkahuan was released in the early 90s as part of a governmentally funded initiative to boost the productivity and consumption of quinoa nationwide, and its production has been promoted ever since via the commercialization of certified seed and subsidized contract-farming (Peralta and Mazón 2015). However, since its introduction to low-income farmers, INIAP-Tunkahuan has also been traded in farmer seed-exchange networks without official denomination. Moreover, it seems likely that during its dissemination in informal seed-networks, the genetic homogeneity of the variety has been lost either by intentional or unintentional outcrossing with cultivated or wild populations (González Marín 2009).
Implications for conservation and breeding
While this study reveals that cultivated germplasm harbors a vast degree of allelic diversity, it also hints at an ongoing process of genetic erosion. From 84 genotypes representing the species’ cultivation range in the country, 32 (~ 40% of the evaluated germplasm) belonged to population subgroup B; which we believe is exclusively composed of INIAP-Tunkahuan genotypes showing varying degrees of genetic admixture (i.e., possibly resulting from intended or unintended outcrossing with wild populations and wild relatives). During sampling, farmers were interrogated into the origin of their genetic material to avoid collection of INIAP-Tunkahuan, and to procure sampling of the species’ extant original genetic diversity in the region. Nevertheless, it is now clear that the INIAP-Tunkahuan has been introduced into informal seed networks and that the variety’s high-performance has inadvertently led to its wide-adoption in the market place.
With the strengthening of local and international quinoa markets, pressure exists to homogenize quinoa seed in order to respond to commercial quality demands. Under these conditions, farmers are expected to lean towards the production of certified varieties designed to meet market demands over local landraces (i.e., as has been the case for INIAP-Tunkahuan in formal and informal seed-exchange networks). In parallel, the decline of traditional agricultural landscapes due to population migration could be one of the reasons that have led to a further loss of the species’ diversity preserved by indigenous farming communities (Fuentes et al. 2012; Peralta and Mazón 2015). This loss of biodiversity poses a threat to future breeding programs of the species which will rely on its diversity to further exploit agronomic resilience and nutritional quality (Jarvis et al. 2017).
Ultimately, genetic diversity studies are essential tools for the conservation and promotion of agricultural biodiversity. With our research, we have provided a preliminary assessment of the conservation status of quinoa in the highlands of Ecuador; the latter considered a subcenter of domestication for the species. Our study shows that important measures need to be taken to preserve the species’ genetic variation carefully preserved by local indigenous farming communities.
Change history
25 November 2019
Due to an unfortunate error of miscommunication, two of the co-authors of this manuscript were omitted from the original publication. The correct representation of the authors and their affiliations are listed here and should be treated as definitive.
References
Almekinders CJ, Thiele G, Danial DL (2007) Can cultivars from participatory plant breeding improve seed provision to small-scale farmers? Euphytica 153:363–372
Bazile D, Fuentes F, Mujica A (2013) Historical perspectives and domestication. In: Bhargava A, Srivastava S (eds) Quinoa: botany, production and uses. CABI, Wallingford, pp 16–35
Bazile D, Jacobsen SE, Verniau A (2016) The global expansion of quinoa: trends and limits. Front Plant Sci 7:622
Blacket MJ, Robin C, Good RT, Lee SF, Miller AD (2012) Universal primers for fluorescent labelling of PCR fragments—an efficient and cost-effective approach to genotyping by fluorescence. Mol Ecol Resour 12(3):456–463
Christensen S et al (2007) Assessment of genetic diversity in the USDA and CIP-FAO international nursery collections of quinoa (Chenopodium quinoa Willd.) using microsatellite markers. Plant Genet Resour 5:82–95
Cota-Sánchez JH, Remarchuk K, Ubayasena K (2006) Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol Biol Rep 24(2):161
Fuentes F, Martinez E, Hinrichsen P, Jellen Maughan P (2009) Assessment of genetic diversity patterns in Chilean quinoa (Chenopodium quinoa Willd.) germplasm using multiplex fluorescent microsatellite markers. Conserv Genet 10:369–377
Fuentes F, Bazile D, Bhargava A, Martinez EA (2012) Implications of farmers’ seed exchanges for on-farm conservation of quinoa, as revealed by its genetic diversity in Chile. J Agric Sci 150:702–716
Gandarillas S, Nieto C, Castillo T (1989) Razas de quinua en Ecuador. INIAP, Quito, pp 1–16
González Marín S (2009) Estudio de flujo de genes en quinua (Chenopodium quinoa w.) en campo de agricultores mediante el uso de marcadores microsatélites. ESPE, Sangolquí
Jacobsen SE (2015) Adaptation and scope for quinoa in northern latitudes of Europe. In: Bazile D, et al. (eds) State of the art report of Quinoa in the World in 2013. FAO and CIRAD, Rome, pp 436–446
Jacobsen SE, Mujica A, Jensen CR (2003) The resistance of quinoa (Chenopodium quinoa Willd.) to adverse abiotic factors. Food Rev Int 19(1–2):99-109
Jarvis D et al (2008) Simple sequence repeat marker development and genetic mapping in quinoa (Chenopodium quinoa Willd.). J Genet 87:39–51
Jarvis D et al (2017) The genome of Chenopodium quinoa. Nature 542:307
Jellen EN, Maughan PJ, Fuentes F, Kolano B (2015) Botany, phylogeny and evolution. In: Bazile D, et al. (eds) State of the art report of Quinoa in the World in 2013. FAO and CIRAD, Rome, pp 12–23
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405
Koziol MJ (1992) Chemical composition and nutritional evaluation of quinoa (Chenopodium quinoa Willd.). J Food Compos Anal 5(1): 35-68
Mason S et al (2005) Development and use of microsatellite markers for germplasm characterization in quinoa (Chenopodium quinoa Willd.). Crop Sci 45:1618–1630
Peralta E, Mazón N (2015) Quinoa in Ecuador. In: Bazile D, et al. (eds) State of the art report of Quinoa in the World in 2013. FAO and CIRAD, Rome, pp 388–400
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genet 155(2):945–959
Ruiz KB et al (2014) Quinoa biodiversity and sustainability for food security under climate change: a review. Agron Sustain Dev 34(2):349–359
Tapia C, Peralta E, Mazón N (2015) Colecciones núcleo de quinua (Chenopodium quinoa Willd.) del Banco de Germoplasma del INIAP: Ecuador. Revista Cientifica AXIOMA 2:5–9
Tártara SC, Manifesto MM, Bramardi SJ, Bertero HD (2012) Genetic structure in cultivated quinoa (Chenopodium quinoa Willd.), a reflection of landscape structure in Northwest Argentina. Conserv Genet 13:1027–1038
Vega-Gálvez A, Miranda M, Vergara J, Uribe E, Puente L, Martínez EA (2010) Nutrition facts and functional potential of quinoa (Chenopodium quinoa Willd.), an ancient Andean grain: a review. J Sci Food Agric 90(15): 2541- 2547
Wang J (2011) COANCESTRY: a program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol Ecol Resourc 11(1):141–145
Wilson HD (1980) Artificial hybridization among species of Chenopodium sect. Chenopodium. Syst Bot 5:253–263
Wilson HD (1990) Quinoa and relatives (Chenopodium sect. Chenopodium subsect. Cellulata). Econ Bot 44:92–110
Yugcha T (1996) Zonificación potencial del cultivo de la quinua en el callejón interandino del Ecuador. MAG, Quito, p 10
Zhang T et al (2017) Development of novel InDel markers and genetic diversity in Chenopodium quinoa through whole-genome re-sequencing. BMC Genomics 18(1):685
Zimmerer KS (2003) Geographies of seed networks for food plants (potato, ulloco) and approaches to agrobiodiversity conservation in the Andean countries. Soc Natur Resour 16(7):583–601
Zurita-Silva A, Fuentes F, Zamora P, Jacobsen SE, Schwember AR (2014) Breeding quinoa (Chenopodium quinoa Willd.): potential and perspectives. Mol Breed 34(1): 13-30
Acknowledgements
This research was funded with a Chancellor's Grant (2015) from Universidad San Francisco de Quito USFQ (Quito-Ecuador). Germplasm acess and research permit was granted by the Ministry of Environment of Ecuador (MAE-DNB-CM-2016-0044). The authors would like to acknowledge the technical assistance offered by researchers at the Plant Biotechnology Laboratory (COCIBA, USFQ), as well as Dr. Leonardo Zurita for his assistance with georeferenced mapping. The authors would also like to acknowledge Dr. Robert van Loo (Wageningen University and Research) for provision of 5 Dutch quinoa varieties and his valuable insights on the manuscript, as well as Dr. Gerard van der Linden (Wageningen University and Research) and Viviana Jaramillo (Wageningen University and Research) for supporting our efforts to search and collect quinoa germplasm throughout the Andes of Ecuador.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Salazar, J., de Lourdes Torres, M., Gutierrez, B. et al. Molecular characterization of Ecuadorian quinoa (Chenopodium quinoa Willd.) diversity: implications for conservation and breeding. Euphytica 215, 60 (2019). https://doi.org/10.1007/s10681-019-2371-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-019-2371-z