
Abstract
Habitat loss and fragmentation are critical threats to biodiversity. Consequent decreases in population size and connectivity can impact genetic diversity and, thus, future adaptability and resilience to environmental change. Understanding landscape patterns of genetic diversity, including patterns of adaptive variation, can assist in developing conservation strategies that maximise population persistence and adaptability in the face of environmental change. Using a reduced-representation genomic approach, we investigated genetic diversity, structure, and adaptive variation across an aridity gradient in the woodland forb Arthropodium fimbriatum. Moderate levels of genetic diversity (HS = 0.14–0.23) were found in all 13 sampled provenances. Inbreeding varied among provenances (FIS = 0.08–0.42) but was not associated with estimated population size. Four genetic clusters were identified, including one highly differentiated cluster. Higher pairwise FST (0.23–0.42) between the three provenances of this cluster and the remaining 10 provenances (pairwise FST between 10 provenances 0.02–0.32) suggested two highly divergent lineages or potentially a cryptic species. After excluding the three highly differentiated populations, outlier and genotype-environment association analysis identified 275 putatively adaptive loci suggesting genomic signatures of climate adaptation in A. fimbriatum is primarily associated with changes in aridity. Combined, these results suggest that all provenances have conservation value, contributing to the maintenance of genetic diversity and adaptive variation in this species. The uncovering of a potential cryptic taxon highlights the power of genomics approaches in conservation genetics and the importance of understanding the role of landscape variation shaping genetic variation to effectively define conservation management units in an era of rapid biodiversity decline.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Habitat loss and fragmentation are among the leading threats facing species and their populations (Young et al. 1996; Honnay and Jacquemyn 2007; Leimu et al. 2010; Aguilar et al. 2019). Such landscape changes can be further compounded by the ongoing pressure of climate change that is already affecting species and their ecosystems (Parmesan 2006; Scheffers et al. 2016). These threats can impair the evolutionary potential of species through the random loss of genetic diversity and the adverse effect of inbreeding. For example, climate stress potentially intensifies the negative effects of inbreeding depression (Nickolas et al. 2019). To avoid extinction, populations need to track their climate niche through migration or adapt via genetic changes, or persist in the altered environment by individuals adjusting their phenotype (plasticity) (Aitken et al. 2008; Hoffmann and Sgró 2011). Given their sessile nature and often slow dispersal rates, range shifts in plants are unlikely to keep pace with ongoing climate change (Corlett and Westcott 2013), and may be further limited in fragmented environments by a lack of suitable habitat to move into (Leimu et al. 2010). In altered, rapidly changing environments, in situ adaptation will likely be an important mechanism that will enhance survival of many plant species (Jump and Peñuelas 2005).
Maintaining high genetic diversity within populations will be important for facilitating adaptation and resilience to biotic and abiotic stressors (Jump et al. 2009; Sgrò et al. 2011; Kardos et al. 2021). While it has been argued that there exists a poor relationship between genetic diversity and a species’ risk of extinction (Teixeira and Huber 2021), evidence suggests that high genetic diversity is associated with greater fitness (Reed and Frankham 2003; DeWoody et al. 2021) and minimises small population effects—such as genetic drift and inbreeding—that can decrease adaptive potential (Kardos et al. 2021). For example, seagrass populations with higher genetic diversity had faster and greater recovery following an extreme heat event (Reusch et al. 2005).
Beyond levels of genetic diversity, adaptation to local environments across a species’ range can create distinct population-level genetic differences. Local adaptation to the home-site environment is common in both plants and animals (Leimu and Fischer 2008; Hereford 2009). Indeed, species often show adaptation to abiotic factors—e.g. soils (Selby and Willis 2018), temperature and precipitation (Fournier-Level et al. 2011; McLean et al. 2014; Prober et al. 2016) and biotic factors (e.g. disease resistance (Hamilton et al. 2013) and animal browsing (O’Reilly-Wapstra et al. 2004)). Local adaptive variation could enhance future adaptability of populations by introducing genotypes already adapted to new conditions, either by natural gene flow between populations (Sgrò et al. 2011) or by active management [e.g. assisted gene flow strategies; Prober et al. 2015; Aitken and Bemmels 2016)]. Conversely, gene flow may introduce maladapted variation or result in outbreeding depression, especially between populations adapted to contrasting environments, highly divergent lineages or even cryptic species (Hufford and Mazer 2003). For conservation, understanding landscape patterns of genetic diversity, population differentiation, and the extent and drivers of local adaptation, including identifying the key environmental drivers of adaptation, will assist in developing and implementing in situ and ex situ strategies that maximise population persistence in the face of climate change, habitat loss and fragmentation.
Landscape genomics has become a valuable approach for assessing patterns of genetic diversity and adaptive variation. In addition to explaining neutral patterns of diversity, landscape genomics is also revealing cryptic lineages (Steane et al. 2015), potential signatures of adaptative genomic variation and associated environmental drivers of such variation (e.g. Yeaman et al. 2016; Guerrero et al. 2018), including extensions to exploring genomic vulnerability to future climate change (e.g. Rellstab et al. 2016; Jordan et al. 2017; Dauphin et al. 2020). A large number of studies to date have focused on woody species, for example, trees such as eucalypts (e.g. Steane et al. 2014; Butler et al. 2022), oaks (e.g. Gugger et al. 2021), poplars (e.g. Zhang et al. 2019) and conifers (e.g. Yeaman et al. 2016; Lotterhos et al. 2018). In contrast, landscape genomic studies for herbaceous species have focused predominately on the model plant Arabidopsis thaliana (Fournier-Level et al. 2011; Hancock et al. 2011) and related species (Franks et al. 2016; Rellstab et al. 2017; Lobréaux and Miquel 2020; Walden et al. 2020), with fewer studies on non-model herbaceous species (e.g. Manel et al. 2012; Roda et al. 2017).
The temperate eucalypt woodlands of south-east Australia are target ecosystems for conservation and restoration. A 200-year legacy of land use change and fragmentation has resulted in a significant decline in many woodland understorey species in these ecosystems (Prober and Thiele 1995), and a suite of eucalypt woodlands are listed as Threatened Ecological Communities under Federal legislation. Of particular concern is the loss of ground-layer species in these ecosystems owing to weed invasion, nutrification, and livestock grazing (Prober and Thiele 2005). Re-creating this important structural layer is a priority not only to conserve these plant species, but to facilitate functional connectivity and habitat for dependent native animals (Prober and Thiele 2004; Harrison et al. 2022). The trees of these ecosystems have been the focus of genetic and genomic studies (Prober and Brown 1994; Gauli et al. 2014; Jordan et al. 2017; Supple et al. 2018; Murray et al. 2019), but there is little known about the genetic and adaptive variation of associated herbaceous forb species (though see Prober et al. 1998). Furthermore, genomic studies may also identify cryptic taxon (e.g. Binks et al. 2021) that would need to be considered on their own for conservation and restoration.
The current study uses a landscape genomics approach to explore the role of environmental features in shaping genetic diversity and adaptive variation in the woodland forb species, Arthropodium fimbriatum R.Br (nodding chocolate-lily). This species is an ecologically important component of the ground-layer flora of temperate grassy eucalypt woodlands (Prober and Thiele 2005). Arthropodium fimbriatum is a geophyte, forming underground storage tubers to survive the hot summers. This tuber also formed a significant ‘root staple’ food source in traditional diets of local indigenous peoples (Gott 2008). Though no data are available, A. fimbriatum is presumed to be insect pollinated, with limited seed dispersal given seeds have no dispersal structures. This species is currently under taxonomic revision (J. Conran pers. comm.) and its boundaries may be refined in the future. Effective conservation and restoration of this species therefore requires an understanding of genetic variation, including adaptive variation and potential cryptic lineages to guide management decisions. We undertook this study with the aim of generating the first population genomic information for this ecologically and culturally important species. The study aimed to provide baseline knowledge to guide conservation and restoration strategies under future climates. We investigated (i) genetic diversity within and variation (differentiation) among provenances of A. fimbriatum in central New South Wales, including the presence of any cryptic lineages, and (ii) genomic signatures of climate adaptation and key climate drivers of adaptive variation.
Methods
Study area
Arthropodium fimbriatum was sampled from 13 sites (hereafter referred to as ‘provenances’) in central New South Wales, Australia (Fig. 1). This area was selected because: (1) the grassy eucalypt woodlands that predominate in this area are of conservation significance owing to widespread clearing and decline of component understorey plant species and fauna; (2) it captured a substantial proportion of the core temperature range of the species—mean annual temperature across distribution = mean of 14.9 °C ± 1.4 standard deviation (based on all A. fimbriatum occurrence data and mean annual temperature data from Atlas of Living Australia [www.ala.org.au, accessed: 30th June 2022]), (3) relatively shallow elevation gradients (range 161–614 m) enabled us to minimise autocorrelation between climatic variables (mean annual rainfall [Moran I = 0.29, P = 0.041], mean annual temperature [Moran I = 0.34, P = 0.024]) and spatial location; and (4) rainfall seasonality was similar across all sites (range 7–15%), despite a latitudinal cline from increasingly summer- to winter-dominant rainfall patterns (Moran I = 0.39, P = 0.015). Note that the taxonomy of A. fimbriatum across its broader distribution is currently under review (J. Conran pers. comm.). In this study we used the current taxonomic treatment of A. fimbriatum which also included A. sp. Albury (NSW FloraOnline 2022).

Map of the 13 sampled provenances of Arthropodium fimbriatum across New South Wales, Australia. Provenance abbreviations (see Table 1) are shown next to the coloured points, where unique colours correspond to the genetic clusters based on the genomics PCA (red = cluster 1, yellow = cluster 2, green = cluster 3, blue = cluster 4; see Fig. 2). Colour gradient indicates the first climate principal component (climPC1) representing aridity, with blue being cooler, wetter areas and red being warmer, drier areas. Inset map of Australia shows the location of the study region (red box) relative to the recorded occurrences of A. fimbriatum (points) obtained from the Atlas of Living Australia (www.ala.org.au, accessed: 20th Dec 2021)
Despite only a portion of the range of A. fimbriatum being represented, the sampled area covers climate gradients informative for conservation in this species. In particular, sampled sites spanned a mean annual temperature range of 3.7 °C (13.4–17.1 °C; mean period: 1979–2013), a range exceeding projected increases in annual mean maximum temperature under a high emissions scenario by 2050 (+ 1.5 to + 3 °C, RCP 8.5) and in the range of projected increases under a high emissions scenario by 2090 (+ 3 to + 4.5 °C, RCP 8.5; Prober et al. 2017). Rainfall projections for the region are more variable, with changes of − 25 to + 5% in mean annual rainfall; our rainfall gradient ranged from 450 to 746 mm (mean period: 1979–2013).
Provenances and sample collection
For each provenance, seed was collected from 4 to 10 individual plants (Table 1, Fig. 1). Of the sampled provenances, most were from cemeteries (n = 10), which are important refuges for declining woodland plant species in fragmented agricultural landscapes due to being relatively undisturbed and protected from livestock grazing (Prober and Thiele 1995). The remaining three sites were two travelling stock reserves and a roadside woodland remnant patch. The number of individuals within a provenance was estimated visually, based on population area and density, and classified into three broad size classes of ‘small’ (approximately < 2000 plants), ‘medium’ (approximately 2000 to 20,000) and ‘large’ (approximately > 20,000).
DNA extraction, DArTseq genotyping and SNP filtering
Seed from the 13 provenances was grown by Greening Australia (Canberra) in 2016. Leaf samples were taken from one randomly chosen adult plant per maternal seed collection, to minimise the chance of familiar relationships between samples, and dried on silica gel (4–10 samples per provenance; Table 1). Total genomic DNA was extracted using E.Z.N.A.® Plant DNA DS Kit (Omega Bio-tek, USA), following the manufacturer’s protocol. Approximately 1 µg of high molecular weight genomic DNA per sample was sent to Diversity Arrays Technologies Pty Ltd (Canberra, Australia) for DArTseq genotyping (see Supplementary Methods for details). In brief, DArTseq is a reduced-representation genomic approach, generating SNP data by combining DArT genomic complexity reduction using restriction enzymes with next generation sequencing (Kilian et al. 2012). For A. fimbriatum, PstI and MseI restriction enzymes were used for complexity reduction (see Supplementary Methods). Individual SNP genotypes were called by Diversity Arrays Technology using their proprietary bioinformatics pipeline (see Supplementary Methods).
A total of 96 samples were genotyped using DArTseq, including seven technical replicates (Table 1). Technical replicates were used to check reproducibility of genotypes (see below) and then removed from further analyses, leaving 90 samples in the final data set.
All data filtering and analyses were performed in R version 4.1.0 (R Core Team 2018). A total of 225,339 SNPs were identified across the 90 samples. For analysis, the SNP dataset was filtered based on DArTseq metadata to retain only SNPs with a call rate greater than 0.95, repeatability greater than 0.99, and minor allele frequency greater than or equal to 0.05. Only one SNP per DArTseq fragment was retained by filtering out ‘secondaries’ (i.e., multiple SNPs present on the same DArTseq fragment). All filtering was performed using the R package dartR (Gruber et al. 2018) and yielded a final dataset of 8760 high quality SNPs for analysis for all 13 provenances (hereafter ‘full dataset’) and 7189 SNPs for 10 provenances when the highly differentiated cluster of provenances was excluded (hereafter ‘10 provenance dataset’; see “Results”).
Following filtering, all loci for both datasets had less than 5% missing data. Linkage disequilibrium, as tested by correlations between genotypes for all pairs of SNPs using the ‘cor’ function in R, was low for both datasets, with > 98% of pairwise comparisons having r2 ≤ 0.2. Deviation from Hardy–Weinberg Equilibrium were tested for each locus within each population using the ‘gl.hwe.pop’ function in dartR. All loci were retained as less than 2.3% of loci within any given population were out of HWE (average = 1.4%) and less than 6 (of 13) populations were out of HWE for any locus (average = 0.18). Genotypes were compared between the two samples in each of the 7 pairs of technical replicates for the ‘full dataset’ of 8,760 SNPs. Similarity (same scored genotype) ranged from 90 to 98%, with an average similarity of 96% (Supplementary Table S1).
Genetic diversity and population genetic structure analyses
Provenance-level genetic diversity metrics and overall differentiation among provenance (FST) were estimated using the ‘basic.stats’ function of the R package hierfstat (v 0.04-22; Goudet and Jombart 2015) using the ‘full dataset’. Mean and standard deviation (s.d.) for gene diversity (HS) and observed heterozygosity (Ho) were calculated from provenance-level per-locus estimates. Unbiased gene diversity (UHS) as calculated as 2n/(2n−1)*HS, where n is the population sample size. Unbiased gene diversity was calculated per locus for each provenance, and the mean and standard deviation then calculated per provenance. Overall Wright’s inbreeding coefficient (FIS) per provenance was then calculated as 1 − Ho/HS as per hierfstat. To determine whether FIS differed significantly from zero, bootstrapped 95% confidence intervals were estimated by running 10,000 bootstraps using the ‘boot.ppfis’ function in hierfstat. The percentage of polymorphic loci (P) was determined from provenance-level per-locus allele frequencies. A Spearman’s rank correlation was used to test if provenance-level genetic diversity estimates were correlated with sample size or estimated population sizes (small, medium or large. See Table 1), using the ‘cor.test’ function (method = ‘spearman’) in R.
Contemporary effective population size (Ne) was calculated for each of the four genetic clusters identified (see below) using the linkage disequilibrium method (Hill 1981; Waples 2006; Waples and Do 2010) with all alleles included, and jackknife estimates of confidence intervals (Jones et al. 2016), implemented in NeEstimator V2.1 (Do et al. 2014).
Differentiation between provenances was assessed by calculating pairwise FST values using the R package StAMPP (v1.6.1; ‘stamppFst’ function; Pembleton et al. 2013) using the ‘full dataset’. To investigate whether provenance differentiation was associated with geographic distance (i.e. isolation by distance—IBD), a Mantel test was executed in the R package vegan (v 2.5-6; Oksanen et al. 2019) using genetic differentiation measured as FST/(1−FST) and ln(geographic distance + 1) following Rousset (1997). Pairwise geographic distances between provenance were calculated using the function ‘rdist.earth’ of R package fields (v 10.3; (Nychka et al. 2017)). Significance of the Mantel correlation was tested using 999 permutations.
The potential influence of missing data, the minor allele frequency (MAF) threshold, and differences in sample sizes within populations were checked separately by recalculating genetic diversity metrics (HS, UHS, HO, FIS and P) and pairwise FST with three different subsets of the ‘full dataset’—(1) using only loci without any missing data (2,260 loci), (2) using loci with a MAF ≥ 0.1 (4898 loci), and (3) rarefaction of each population to the lowest sample size (n = 4) through random resampling. Random resampling was performed 100 times and the mean and standard deviation across resamples calculated. Correlations between metric estimates using these data subsets and the ‘full dataset’ were calculated using the ‘cor’ function in R. Results were deemed robust to missing data, minor allele frequency and within-population sample size, with absolute values being similar (Supplementary Tables S2 and S3) and highly correlated to results from the ‘full dataset’ (r > 0.99 for all measures of HS, UHS, HO, P, FIS and pairwise FST, except for FIS with no missing data (r = 0.91) and rarefied P (r = 0.93). Hence, all analyses presented are those undertaken using the original filtering parameters and within-population sample sizes (‘full dataset’ and ‘10 provenance dataset’).
Two approaches were used to investigate population genomic structure across the samples. First, a multivariate approach was used by undertaking a Principal Component Analysis (PCA) using the ‘glPCA’ function of adegenet. Second, the number of ancestral, genetic clusters (K) was determined using a sparse nonnegative matrix factorisation algorithm (sNMF), implemented using the ‘snmf’ function of the R package LEA (v 3.4.0; Frichot and François 2015). The sNMF model was run for values of K between 1 and 13, where 13 assumes each provenance is a unique ancestral gene pool. Each value of K was replicated 15 times using default parameters. The optimum number of clusters was estimated using the cross-entropy criterion following Frichot et al. (2014), where the suggested number of clusters corresponds to the lowest cross-entropy value. Individual proportion memberships—the least-square estimate of ancestry proportions (Q-matrix)—were plotted for the optimum value of K, using the run of K that yielded the lowest cross entropy score. Two additional values of K, with similarly low cross-entropy values, were also plotted and explored. PCA and sNMF analyses were performed on both the ‘full dataset’ and ‘10 provenance dataset’, the latter having ‘cluster 1’ provenances removed (see “Results”), and therefore only testing values of K from 1 to 10 (10 being maximum number of provenances).
Genomic signatures of climate adaptation
Correlations between local climate and provenance-level SNP allele frequencies were explored to investigate evidence of climate adaptation across the sampled range. The three provenances from genetic ‘cluster 1’ (Figs. 1, 2; see “Results”) were excluded from this analysis given their strong differentiation to reduce the probability of putatively adaptive SNPs been driven by outlier populations. All climate association analyses were therefore performed on the ‘10 provenance dataset’.
Climate data for each provenance were acquired from CHELSA (Karger et al. 2017; http://chelsa-climate.org/ accessed 07/10/2019) for the standard 19 BIOCLIM variables (Xu and Hutchinson 2012) representing eleven temperature and eight precipitation variables averaged across the period between 1976 and 2013. To reduce the complexity of the climate data to the main, independent climate gradients that transgressed the sampled provenances, the climate variation was summarised by a Principal Component Analysis (PCA) using the R package FactorMineR (v 2.4; Lê et al. 2008). Principal components (PC) with an eigenvalue greater than one were retained and indicated that single PC axis accounted for more variation than a single variable alone. This resulted in the first three PCs that explained 93% of the climate variation among provenances (climPC1-3; Supplementary Table S4) being retained. These PC axes were then ‘characterised’ using the Pearson correlation coefficient between the original climate variables and each PC axis (Supplementary Table S4) following Costa e Silva et al. (2018).
Genomic signatures of adaptation and relationship to climate for the 10 provenances were explored using two approaches—an outlier approach and a multivariate, genotype-environment association (GEA) analysis. For both analyses, missing SNP data were imputed for the 10 provenances using the ‘impute’ function of LEA, which draws on population structure identified by ‘snmf’ to inform imputation. Missing genotypes were imputed using the ‘mode’ method and informed by the ‘snmf’ run of the optimal K value for the ‘10 provenance dataset’ with the lowest cross entropy score (K = 3; see “Results”). The SNPs were further filtered to those where both alleles were present in at least 3 of the 10 provenances (prior to imputation) and thus could be more confidently associated with environmental differences rather than population structure, drift or other processes (n = 6706).
Signatures of adaptation with no a priori assumptions of environmental drivers, were investigated using the R package pcadapt (v 4.3.3 Privé et al. 2020). pcadapt is an individual-based genome scan approach that compares SNPs to K principal components (pcadapt-PCs) representing population structure. Outlier, putatively adaptive SNPs were identified as those whose relationship with the K pcadapt-PCs (population structure) differed from most SNPs, suggesting a difference from neutral patterns and therefore potential selection acting upon this SNP (Luu et al. 2017). By not including a priori variables, this approach allows for the identification of putatively adaptive SNPs that may be associated with environmental variables not included in the genotype-environment association analysis (see below). For analysis, eight pcadapt-PCs were retained to account for population structure based on an initial run and exploration of the screeplot and the pcadapt-PC plots, showing population structure represented in pcadapt-PC 1 to 8 (Supplementary Fig. S1). To control for false discovery rates, q-values were calculated from p-values using the qvalue package (v 2.24.0; Storey et al. 2021) in R. Outliers which we declared as putatively adaptive loci were those with a q < 0.001 (FDR 0.1%).
A redundancy analysis (RDA) was used to explore genomic signatures of adaptation through a multivariate, genotype-environment association (GEA) approach. Partial RDA was performed with vegan using default settings, to assess genetic variation (individual imputed genotypes; response data) explained by climate (explanatory variables; climPC1-3), conditional on population structure (first two PCs from population structure analysis; see above). Correcting for population structure can reduce power and affect error rates if there are low levels of genetic differentiation among populations (Forester et al. 2018). However, as there was a moderate level of genetic differentiation among the 10 provenances (see “Results”), we accounted for population structure in the GEA analysis. Significance of the partial RDA model and RDA axes was assessed using the ‘anova.cca’ function with default parameters (999 permutations). Outliers—putative climate-adapted SNPs were identified as those with scores greater than 3 standard deviations from the RDA axis mean.
For outlier SNPs identified by both analyses (i.e. strong adaptive candidates), Pearson’s correlations between provenance-level allele frequencies and climate variables (PC) were calculated using the ‘cor’ function in R to determine the climate variable with which each SNP was most strongly associated. Population-level allele frequencies for the outlier SNPs were calculated using the ‘basic.stats’ function for the 10 provenance dataset.
The potential adaptability of the 10 provenances was explored by calculating the percentage of polymorphic loci within a population. Putatively adaptive loci that are fixed (monomorphic) have no diversity for selection to act upon and therefore have no capacity to contribute to in situ provenance adaptation. Conversely, polymorphic loci have variation upon which selection could act and thus contribute to future adaptation if selection gradients change. Therefore, a high percentage of polymorphic adaptive loci within a provenance was interpreted as suggestive of higher adaptive potential. The percentage of polymorphic ‘adaptive’ loci per provenance was calculated for (i) all loci in the 10 provenance dataset, (ii) the loci identified by each individual analysis (pcadapt, RDA), and (iii) the subset of common loci identified by both analyses.
Results
Genetic diversity and population structure
Genetic diversity within populations, as measured by gene diversity (HS), varied from 0.14 to 0.23, with unbiased gene diversity estimates being slightly higher but showing similar relative patterns between provenances (Table 1). Observed heterozygosity (Ho) was lower in all populations, ranging from 0.11 to 0.20. Reflecting lower Ho than HS, all populations showed evidence of inbreeding (FIS), which ranged from 0.08 to 0.23 and were all significantly different from zero based on bootstrapped 95% confidence intervals (Table 1). Only approximately half of the SNPs were polymorphic in any given provenance—the percentage of polymorphic loci ranged from 33 to 67% (Table 1). Levels of genetic diversity and inbreeding were not correlated to either sample size or estimated population size (p > 0.05), with some small populations having genetic diversity estimates similar to those of large populations (Table 1).
There was a moderate level of differentiation among the 13 sampled provenances, with overall FST = 0.25 and pairwise FST ranging between 0.02 and 0.42 (Supplementary Table S3). High levels of FST were driven primarily by three highly differentiated provenances—Canowindra, Stockinbingal, and Wallendbeen. Pairwise FST values of these three provenances when compared to the other ten ranged from 0.23 to 0.42. In contrast, pairwise FST values among the other ten provenances were slightly lower, ranging from 0.02 to 0.32 (see below; Supplementary Table S3). Overall FST for the 10 provenances excluding the three highly differentiated provenances was 0.18. There was evidence of isolation by distance (IBD) across the sampled range but only when the three highly differentiated provenances were removed (full dataset: Mantel r = 0.14, p = 0.129; 10 provenance dataset: r = 0.55, p = 0.002; Supplementary Fig. S2).
Both the PCA and sNMF analyses provided evidence of population genetic structure across the sampled range of A. fimbriatum. As expected based on the pairwise FST results, the PCA showed Canowindra, Stockinbingal and Wallendbeen to be highly differentiated from the other 10 sampled populations, forming a separate genetic cluster (hereafter ‘Cluster 1’; Fig. 2a). The PCA also revealed three additional genetic clusters (Fig. 2a), and these remained even after excluding cluster 1 (Supplementary Fig. S3). The presence of four genetic clusters in the full dataset was confirmed with the sNMF analysis (Fig. 2b). Cross-entropy criterion analysis suggested K = 4 or 5 as the optimal number of genetic clusters (Supplementary Fig. S4a). For K = 4, the clustering matched that in the PCA, with Baldry, Ungarie and Monteagle showing strong signals of admixture between clusters. As K increased, Euchareena and Geurie (K = 5) and Stockingbingal (K = 6) formed separate clusters (Supplementary Fig. S5). Similar results were found when cluster 1 populations were removed, with the optimal number of clusters becoming K = 3 or 4 (Supplementary Figs. S4b and S5).

Population structure of Arthropodium fimbriatum using the ‘full dataset’ (8760 SNPs) a Principal Components Analysis (PCA). Four genetic clusters were identified and are indicated by the ellipses that surround the 13 sampled provenances (different coloured points). See Fig. 1 for geographic locations of (colour-coded) genetic clusters. b Plot of sNMF cluster membership for the optimal value of K = 4 ancestral populations (see Supplementary Fig. S5 for K = 5 and K = 6). Each vertical bar represents an individual, where the same colour corresponds to shared ancestral affinities. The most highly genetically differentiated provenances (Cluster 1; Can [Canowindra], Wal [Wallendbeen] and Sto [Stockinbingal]) are shown in red. See Table 1 for abbreviations of provenance names
Contemporary effective population size was less than 100 for all genetic clusters except cluster 2. Cluster 2 had the highest estimate of Ne = 145.9 (95% Confidence Interval (CI) 106.5–168.2), followed by cluster 1, the highly differentiated cluster, (Ne = 64.0, 95% CI 38.4–121.6) and cluster 4 (Ne = 63.4, 95% CI 18.6–21.5), with cluster 3 having the lowest estimate of Ne = 36.9 (95% CI 60.6–136.4).
Genomic signatures of climate adaptation
Evidence suggesting the presence of genomic adaptation was found in A. fimbriatum. Excluding the highly differentiated ‘Cluster 1’, a total of 275 SNPs across the 10 populations were identified as putatively under selection. The environment-naïve approach of pcadapt identified 147 outlier SNPs (Supplementary Fig. S6). Further to this, the multivariate partial RDA found environment explained a small but significant portion of genetic variation (adjusted r2 = 0.04, model p = 0.001), with 142 unique outlier SNPs identified across the three RDA axes (Table 2). Fourteen outliers were common to both methods. Of these, the majority were most strongly associated with climPC1, representing aridity (10 SNPs, |r|= 0.33–0.80). Three SNPs were most strongly associated with climPC2 which represented continentality (|r|= 0.23–0.52) and one SNP with climPC3 which represented rainfall seasonality (|r|= 0.20).
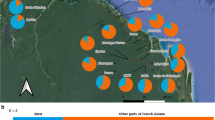
The percentage of putatively adaptive loci that were polymorphic, and therefore the proportion of putatively adaptive loci that had variation upon which selection may act and contribute to in situ adaptation, differed between provenances and analysis method (Fig. 3). Most putatively adaptive loci identified by pcadapt were not polymorphic within provenances (27–57% polymorphic adaptive loci), suggesting little adaptive variation for selection. Conversely, most (> 70%) putatively adaptive loci identified by RDA were polymorphic within provenances, suggesting a large amount of variation for selection and adaptation in all provenances. Of the putatively adaptive loci identified by both methods, 33–73% were polymorphic within a provenance. Comparing the shared loci between provenances, Marrar, Merryville and Monteagle had the highest amount of putatively adaptive variation (higher percentage of polymorphic loci, > 65%), while Baldry, Euchareena had the lowest (< 40%).

Proportion of loci that are monomorphic (grey) or polymorphic (white) within each of the 10 provenances of A. fimbriatum based on a all 7,189 SNPs (‘10 provenance dataset’), b outlier SNPs identified by pcadapt, c partial RDA and d SNPs found by both pcadapt and partial RDA. Pie charts for each of the 10 provenances are overlaid on the first climate principal component (climPC1) representing aridity
Discussion
This is one of the first studies to identify potential genomic signals of adaptation to climate in an herbaceous woodland forb of conservation interest in Australia. This study found evidence of genetic diversity maintained within provenances despite detection of inbreeding and notable habitat fragmentation. High levels of population differentiation among provenances of A. fimbriatum and structure were detected across central New South Wales (NSW). Of note was the potential presence of a cryptic lineage (cluster 1) that was highly differentiated from the other ancestral lineages (clusters 2–4). Furthermore, after excluding the potential cryptic lineage (‘Cluster 1’), provenances of the other ancestral lineages showed potential genomic signatures of selection to climate, particularly aridity, suggesting evidence for local adaptation across this range. Together, these results suggest remaining populations of A. fimbriatum have high conservation value despite fragmentation, since they have maintained genetic diversity and potentially important adaptive variation that may enhance adaptability and conservation actions.
Genetic diversity and structure
Provenances of A. fimbriatum in this study appear to have maintained moderate levels of genetic diversity, despite habitat loss and fragmentation across the sampled region. Absolute genetic diversity estimates vary between marker types (Broadhurst et al. 2017; Fischer et al. 2017), limiting direct comparisons of diversity levels among studies. Given SNP-based estimates of genetic diversity are lower than hypervariable markers such as microsatellites (Fischer et al. 2017), genetic diversity in A. fimbriatum appears slightly below the mean diversity found in other Australian herbaceous species (mean expected heterozygosity of 0.22 and 0.63 for allozymes and microsatellites respectively) as well as short-lived perennials globally (0.12–0.13 based on allozymes; references within Broadhurst et al. 2017), though similar to SNP-based expected heterozygosity estimates in Arabidopsis halleri (Fischer et al. 2017). While habitat loss may be expected to reduce genetic diversity, due to decreases in population size and gene flow (Young et al. 1996; Aguilar et al. 2008), this is not always the case. Moderate levels of genetic diversity, despite fragmentation, have been found in other herbaceous species (gene diversity 0.18–0.312, AFLPs; Honnay et al. 2006; Reisch et al. 2017) including in south-eastern Australia (expected heterozygosity 0.13–0.28, allozymes; Young et al. 1999). For example, small populations of Microseris lanceolata, a herbaceous perennial of eucalypt woodlands across south-eastern Australia, displayed genetic diversity similar to that of large populations, as measured by number of alleles (allozymes; Prober et al. 1998). Similarly, moderate diversity has also been maintained in fragmented populations of the south-eastern Australian grassland herbaceous perennial Ptilotus macrocephalus (microsatellite expected heterozygosity 0.41–0.54; Ahrens and James 2016).
Species in remnant patches of vegetation may have the potential to maintain high levels of genetic diversity or show a lag in the expression of the negative impacts of habitat fragmentation, as suggested by a relationship between genetic diversity and historic, rather than contemporary, landscape. Genetic diversity was related to historic (1954) habitat patch area, not current area or population size (2011) for Campanula rotundifolia in Sweden (Plue et al. 2017). Genetic diversity in several calcareous grassland species in Germany was related to distance to nearest grassland in 1830, prior to fragmentation, not current habitat area or population size (Reisch et al. 2017). With multiple generations since clearing commenced ca. 180 years ago in NSW (Prober and Thiele 1993), our findings suggest A. fimbriatum has retained modest genetic diversity, potentially by maintaining moderate population sizes despite habitat loss. Even in very small habitat remnants of less than 1 ha, A. fimbriatum populations can generally contain more than 500 individuals and could be potentially buffered from the effects of genetic drift. However, caution should be exercised when interpreting these results. First, changes in genetic diversity may take time to appear (Aguilar et al. 2008). Second, estimates of contemporary effective population size for each genetic cluster (genetic population) were small, suggesting these provenances may be at risk of future genetic diversity loss through inbreeding or genetic drift, and subsequent decreases in population fitness and adaptability. While Ne estimates for three of the four genetic clusters was greater than 50, the size suggested for avoiding short-term effects of inbreeding (Jamieson and Allendorf 2012), all Ne estimates were much lower than the suggested Ne > 500 (Jamieson and Allendorf 2012) or > 1000 (Frankham et al. 2014) necessary to support adaptation and long term evolutionary potential. Focus should therefore remain on maintaining population sizes and diversity within provenances of A. fimbriatum to maintain adaptability within these provenances.
Of interest are the low diversity measures, signaled by low percentage of polymorphic loci, in the provenances of cluster 1 (Canowindra, Stockingbingal and Wallendbeen). The studied progeny came from large and relatively intact populations that are unlikely to have gone through a genetic bottleneck. What is driving this lower diversity requires further investigation, though one possibility may be explained in part by the discovery of a cryptic lineage (see below) and the possibility of reproductive barriers with the other lineage represented by the 10 provenances.
Significant population structure suggests distinct genetic differences among provenances of A. fimbriatum across central NSW. Population differentiation was generally higher than the mean of other Australian herbaceous species—FST = 0.25 and 0.18 for 13 and 10 provenances respectively of A. fimbriatum versus a mean FST of 0.11 for Australian herbaceous species generally based on allozymes and microsatellites (Broadhurst et al. 2017). The spatial arrangement of genetic clusters of A. fimbriatum could indicate IBD-style gene flow between geographically close provenances or geographic differences in selective pressures resulting in spatially distinct adaptative genetic differences (see next section).
In contrast to moderate differentiation between three of the four identified genetic clusters, one cluster (cluster 1) was highly differentiated, suggesting a potentially cryptic lineage. While such divergence among provenances could arise due to differences in ploidy levels, as observed in other Australian herbaceous species (Brown and Young 2000; Broadhurst et al. 2012), there was no evidence that any of the provenances in this study were polyploids (D. Steane, unpub. data, 2018). An alternative hypothesis is that the level of differentiation is due to isolation by environment. While this was not specifically tested, there were no obvious differences in climate or soils of the home-site environments between this cluster and the other provenances. For example, the Wallendbeen provenance grows on rich volcanic soil in gently hilly terrain in White Box eucalypt (Eucalyptus albens) woodlands, while the Stockinbingal and Canowindra provenances occur on flatter plains, on red brown soils typical of Grey Box eucalypt (E. microcarpa) woodlands. However, we can not dismiss the possibility that provenances of cluster 1 originated from other parts of the species’ distribution and were translocated by First Peoples of Australia as they moved across the landscape. Movement of A. fimbriatum seed may explain genetic similarities between provenances within this cluster despite the geographic distance between them. A broader genetic analysis of provenances across the distribution of A. fimbriatum is warranted and may help determine the genetic origins of the cluster 1 provenances or whether they represent a cryptic lineage of this species. Indeed, the A. fimbriatum species complex is currently undergoing taxonomic review and several more taxa have been proposed (J. Conran, personal communication).
Of additional note was the Monteagle provenance (belonging to cluster 4) situated geographically close to provenances comprising cluster 1 but remaining highly differentiated from this cluster. This could not be convicingly explained by differences in the home-site environment or drift. With the evidence at hand, the most plausible explaination is either the presence of a potential cryptic lineage or movement of cluster 1 samples into the region from other areas of the species distribution, as discussed above. Further sampling of more provenances across this region, and the broader distribution, would help to disentangle the full geographic distribution of the distinct genetic clusters and lineages.
Genomic signatures of climate adaptation
This study identified putative signals of genomic adaptation to home-site climate variation in A. fimbriatum, particularly in response to differences in aridity. These signals were identified across the 10 populations (clusters 2–4) excluding the potential cryptic lineage. Evidence suggestive of local adaptation to climate has been identified in other herbaceous species, through common garden and reciprocal transplant trials (e.g., Leinonen et al. 2009; Pickup et al. 2012) as well as genotype-environment association analyses (Fournier-Level et al. 2011; Hancock et al. 2011; Lasky et al. 2012). Beyond the model species Arabidopsis thaliana, and close relatives (e.g., Rellstab et al. 2017; Lobréaux and Miquel 2020), studies of genomic signatures of adaptation in herbaceous species have received less attention than forest trees. In an early study of genomic adaptation in non-model plants, Manel et al (2012) found evidence of genomic signatures of adaptation, primarily associated with temperature and precipitation variables, in alpine species, including herbaceous species. Isolation by environment was identified in a microsatellite study of Ptilotus macrocephalus across south-eastern Australia, suggesting significant genetic variation associated with both temperature and precipitation (Ahrens & James 2016). While the present study focused on a component of the species’ gene pool, although representative of the species’ core climate envelope, broadening the scope to across the native range of A. fimbriatum may uncover further associations between climate and genomic variation beyond putative adaptation to aridity.
In situ adaptation will likely be an important mechanism for this species to persist into the future given both projected changes in temperature and precipitation and, thus, aridity across this region (IPCC 2021) and altered land use limiting dispersal. The identification of polymorphic genomic variants associated with aridity suggest there may already be adaptive variation related to aridity within the sampled range of A. fimbriatum, that could aid future adaptation. Altered land use, however, may restrict long-distance gene flow and limit the geographic spread of this adaptive genomic variation, leaving provenances reliant on adaptation from standing genetic variation. Moderate genetic diversity overall, as well as variation in putatively adaptive loci, suggest that all provenances within this region have some variation upon which selection could act. However, the large number of loci fixed for putatively adaptive provenance-specific alleles suggests that assisted gene-flow within ancestral lineages may provide an avenue by which to increase the genetic diversity available for selection and, therefore, improve the adaptive capacity of provenances in the face of climate change. Further studies to better characterise adaptive variation across A. fimbriatum and within individual provenances, including increasing the number of sampled provenances and individuals within each provenance to gain more accurate estimates of allele frequencies, would assist in understanding the amount of standing variation available, and thus potential adaptability of provenances.
Conservation and restoration
Understanding landscape patterns of genetic diversity, including adaptive diversity and differentiation, can help inform biodiversity conservation decisions (Hoffmann et al. 2015), including seed sourcing for restoration plantings under climate change (Prober et al. 2015). Despite the lack of a reference genome for A. fimbriatum, the set of loci identified in this study and their associated climatic variables may be considered as a generalised genomic signature of possible climate adaptation, describing landscape patterns of genetic variation that may be important for adaptation to climate and allowing inferences about important climatic drivers of this variation. The potential presence of adaptive variation among populations suggests there may exist important genomic differences that could be exploited to enhance the resilience and adaptability of current populations as well as future restoration plantings. Furthermore, these results suggest that even small provenances of A. fimbriatum have reasonable levels of genetic diversity and, thus, conservation value. Maintaining current diversity, including putative adaptive diversity will be important for conservation and restoration of this species.
The highly divergent cryptic lineage found in this study requires further investigation. Cryptic lineages have been observed in other Australian species (Broadhurst et al. 2012; Steane et al. 2015; Binks et al. 2021), and could have significant implications for the intentional movement of material across the landscape for conservation or restoration. For example, while the inclusion of highly divergent lineages in mixed provenance restoration plantings may introduce hybrid vigour to a population in the short term, it may perversely result in outbreeding depression (i.e. reduced vigour) in later generations (Edmands 2007; Hoffmann et al. 2020), although this may be less common than initially thought (Frankham et al. 2011). Furthermore, the adaptive variation identified in the other provenances may not be applicable to this cryptic lineage, which may have experienced different selective pressures and adaptive responses. Gaining a deeper understanding of potential cryptic variation in A. fimbriatum and how adaptive variation within the different lineages may vary will be critical to determine how this might influence conservation and restoration decisions. This study helps provide information towards understand the taxonomy of this species.
Conclusion
Provenances of the woodland forb, Arthropodium fimbriatum, contain moderate levels of genetic diversity and putative adaptive variation that could be harnessed in conservation and restoration plantings to aid adaptability and create resilient revegetation in the face of climate change. However, the evidence of a cryptic lineage warrants consideration. We caution mixing provenances from different clusters for use in conservation and restoration until the taxonomy has been reviewed and further note that patterns of adaptive genetic variation within this lineage may differ from those identified in the other lineages in this study. Further, population genetic studies across a broader distribution of A. fimbriatum would help untangle the identity of the cryptic lineage as well as identify the range of adaptive variation available for use in restoration.
Data availability
The SNP data used in this study are available via the University of Tasmania’s Research Data Portal, https://doi.org/10.25959/a493-6p26.
References
Aguilar R, Quesada M, Ashworth L et al (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188. https://doi.org/10.1111/j.1365-294X.2008.03971.x
Aguilar R, Cristobal-Perez EJ, Balvino-Olvera FJ et al (2019) Habitat fragmentation reduces plant progeny quality: a global synthesis. Ecol Lett 22:1163–1173
Ahrens CW, James EA (2016) Regional genetic structure and environmental variables influence our conservation approach for feather heads (Ptilotus macrocephalus). J Hered 107:238–247. https://doi.org/10.1093/jhered/esw009
Aitken SN, Bemmels JB (2016) Time to get moving: assisted gene flow of forest trees. Evol Appl 9:271–290. https://doi.org/10.1111/eva.12293
Aitken SN, Yeaman S, Holliday JA et al (2008) Adaptation, migration or extirpation: climate change outcomes for tree populations. Evol Appl 1:95–111. https://doi.org/10.1111/j.1752-4571.2007.00013.x
Binks RM, Steane DA, Byrne M (2021) Genomic divergence in sympatry indicates strong reproductive barriers and cryptic species within Eucalyptus salubris. Ecol Evol 11:5096–5110. https://doi.org/10.1002/ece3.7403
Broadhurst LM, Murray BG, Forrester R, Young AG (2012) Cryptic genetic variability in Swainsona sericea (A. Lee) H. Eichler (Fabaceae): lessons for restoration. Aust J Bot 60:429–438. https://doi.org/10.1071/BT12026
Broadhurst L, Breed M, Lowe A et al (2017) Genetic diversity and structure of the Australian flora. Divers Distrib 23:41–52. https://doi.org/10.1111/ddi.12505
Brown AHD, Young AG (2000) Genetic diversity in tetraploid populations of the endangered daisy Rutidosis leptorrhynchoides and implications for its conservation. Heredity 85:122–129. https://doi.org/10.1046/j.1365-2540.2000.00742.x
Butler JB, Harrison PA, Vaillancourt RE et al (2022) Climate adaptation, drought susceptibility, and genomic-informed predictions of future climate refugia for the Australian forest tree Eucalyptus globulus. Forests 13:575
Corlett RT, Westcott DA (2013) Will plant movements keep up with climate change? Trends Ecol Evol 28:482–488. https://doi.org/10.1016/j.tree.2013.04.003
Costa e Silva J, Harrison P, Wiltshire R, Potts B (2018) Evidence that divergent selection shapes a developmental cline in a forest tree species complex. Ann Bot 122:181–194.https://doi.org/10.1093/aob/mcy064
Dauphin B, Rellstab C, Schmid M et al (2020) Genomic vulnerability to rapid climate warming in a tree species with a long generation time. Glob Change Biol 27:1181–1195. https://doi.org/10.1111/gcb.15469
DeWoody JA, Harder AM, Mathur S, Willoughby JR (2021) The long-standing significance of genetic diversity in conservation. Mol Ecol 30:4147–4154. https://doi.org/10.1111/mec.16051
Do C, Waples RS, Peel D et al (2014) NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Edmands S (2007) Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol Ecol 16:463–475
Fischer MC, Rellstab C, Leuzinger M et al (2017) Estimating genomic diversity and population differentiation—an empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genomics 18:1–15. https://doi.org/10.1186/s12864-016-3459-7
Forester BR, Lasky JR, Wagner HH, Urban DL (2018) Comparing methods for detecting multilocus adaptation with multivariate genotype–environment associations. Mol Ecol 27:2215–2233. https://doi.org/10.1111/mec.14584
Fournier-Level A, Korte A, Cooper MD et al (2011) A map of local adaptation in Arabidopsis thaliana. Science 334:86–89. https://doi.org/10.1126/science.1209271
Frankham R, Ballou JD, Eldridge MDB et al (2011) Predicting the probability of outbreeding depression. Conserv Biol 25:465–475. https://doi.org/10.1111/j.1523-1739.2011.01662.x
Frankham R, Bradshaw CJA, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol Conserv 170:56–63. https://doi.org/10.1016/j.biocon.2013.12.036
Franks SJ, Kane NC, O’Hara NB et al (2016) Rapid genome-wide evolution in Brassica rapa populations following drought revealed by sequencing of ancestral and descendant gene pools. Mol Ecol 25:3622–3631. https://doi.org/10.1111/mec.13615
Frichot E, François O (2015) LEA: an R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929. https://doi.org/10.1111/2041-210X.12382
Frichot E, Mathieu F, Trouillon T et al (2014) Fast and efficient estimation of individual ancestry coefficients. Genetics 196:973–983. https://doi.org/10.1534/genetics.113.160572
Gauli A, Steane DA, Vaillancourt RE, Potts BM (2014) Molecular genetic diversity and population structure in Eucalyptus pauciflora subsp. pauciflora (Myrtaceae) on the island of Tasmania. Aust J Bot 62:175–188. https://doi.org/10.1071/BT14036
Gott B (2008) Indigenous use of plants in south-eastern Australia. Telopea 12:215–226
Goudet J, Jombart T (2015) hierfstat: Estimation and tests of hierarchical F-statistics. R package version 0.04-22
Gruber B, Unmack PJ, Berry OF, Georges A (2018) Dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Resour 18:691–699
Guerrero J, Andrello M, Burgarella C, Manel S (2018) Soil environment is a key driver of adaptation in Medicago truncatula: new insights from landscape genomics. New Phytol 219:378–390. https://doi.org/10.1111/nph.15171
Gugger PF, Fitz-Gibbon ST, Albarrán-Lara A et al (2021) Landscape genomics of Quercus lobata reveals genes involved in local climate adaptation at multiple spatial scales. Mol Ecol 30:406–423. https://doi.org/10.1111/mec.15731
Hamilton MG, Williams DR, Tilyard PA et al (2013) A latitudinal cline in disease resistance of a host tree. Heredity 110:372–379
Hancock AM, Brachi B, Faure N et al (2011) Adaptation to climate across the Arabidopsis thaliana genome. Science 334:83–86. https://doi.org/10.1126/science.1209244
Harrison P, Davidson N, Bailey T et al (2022) A decade of restoring a temperate woodland: lessons learned and future directions. Ecol Manag Restor
Hereford J (2009) A quantitative survey of local adaptation and fitness trade-offs. Am Nat 173:579–588. https://doi.org/10.1086/597611
Hill W (1981) Estimation of effective population size from data on linkage disequilibrium. Genet Res Camb 38:209–216
Hoffmann AA, Sgró CM (2011) Climate change and evolutionary adaptation. Nature 470:479–485. https://doi.org/10.1038/nature09670
Hoffmann A, Griffin P, Dillon S et al (2015) A framework for incorporating evolutionary genomics into biodiversity conservation and management. Clim Chang Responses 2:1. https://doi.org/10.1186/s40665-014-0009-x
Hoffmann AA, Miller AD, Weeks AR (2020) Genetic mixing for population management: from genetic rescue to provenancing. Evol Appl 1–19. https://doi.org/10.1111/eva.13154
Honnay O, Jacquemyn H (2007) Susceptibility of common and rare plant species to the genetic consequences of habitat fragmentation. Conserv Biol 21:823–831
Honnay O, Coart E, Butaye J et al (2006) Low impact of present and historical landscape configuration on the genetics of fragmented Anthyllis vulneraria populations. Biol Conserv 127:411–419. https://doi.org/10.1016/j.biocon.2005.09.006
Hufford KM, Mazer SJ (2003) Plant ecotypes: genetic differentiation in the age of ecological restoration. Trends Ecol Evol 18:147–155. https://doi.org/10.1016/S0169-5347(03)00002-8
IPCC (2021) Summary for policymakers. In: Masson-Delmotte V, Zhai P, Pirani A et al (eds) Climate change 2021: the physical science basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change
Jamieson IG, Allendorf FW (2012) How does the 50/500 rule apply to MVPs? Trends Ecol Evol 27:578–584. https://doi.org/10.1016/j.tree.2012.07.001
Jones AT, Ovenden JR, Wang Y-G (2016) Improved confidence intervals for the linkage disequilibrium method for estimating effective population size. Heredity 117:217–223
Jordan R, Hoffmann AA, Dillon SK, Prober SM (2017) Evidence of genomic adaptation to climate in Eucalyptus microcarpa: implications for adaptive potential to projected climate change. Mol Ecol 26:6002–6020. https://doi.org/10.1111/mec.14341
Jump AS, Peñuelas J (2005) Running to stand still: adaptation and the response of plants to rapid climate change. Ecol Lett 8:1010–1020. https://doi.org/10.1111/j.1461-0248.2005.00796.x
Jump AS, Marchant R, Peñuelas J (2009) Environmental change and the option value of genetic diversity. Trends Plant Sci 14:51–58. https://doi.org/10.1016/j.tplants.2008.10.002
Kardos M, Armstrong E, Fitzpatrick S et al (2021) The crucial role of genome-wide genetic variation in conservation. Proc Natl Acad Sci 118:e2104642118. https://doi.org/10.1073/pnas.2104642118
Karger DN, Conrad O, Bohner J et al (2017) Climatologies at high resolution for the earth’s land surface areas. Sci Data 4:170122
Kilian A, Wenzl P, Huttner E, et al (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. In: Data production and analysis in population genomics. Humana Press, Totowa, NJ, pp 67–89
Lasky JR, Des Marais DL, McKay JK et al (2012) Characterizing genomic variation of Arabidopsis thaliana: the roles of geography and climate. Mol Ecol 21:5512–5529. https://doi.org/10.1111/j.1365-294X.2012.05709.x
Lê S, Josse J, Husson F (2008) FactoMineR: an R package for multivariate analysis. J Stat Softw 25:1–18
Leimu R, Fischer M (2008) A meta-analysis of local adaptation in plants. PLoS ONE 3:1–8. https://doi.org/10.1371/journal.pone.0004010
Leimu R, Vergeer P, Angeloni F, Ouborg NJ (2010) Habitat fragmentation, climate change, and inbreeding in plants. Ann N Y Acad Sci 1195:84–98. https://doi.org/10.1111/j.1749-6632.2010.05450.x
Leinonen PH, Sandring S, Quilot B et al (2009) Local adaptation in european populations of Arabidopsis lyrata (Brassicaceae). Am J Bot 96:1129–1137. https://doi.org/10.3732/ajb.0800080
Lobréaux S, Miquel C (2020) Identification of Arabis alpina genomic regions associated with climatic variables along an elevation gradient through whole genome scan. Genomics 112:729–735. https://doi.org/10.1016/j.ygeno.2019.05.008
Lotterhos KE, Yeaman S, Degner J et al (2018) Modularity of genes involved in local adaptation to climate despite physical linkage. Genome Biol 19:1–24. https://doi.org/10.1186/s13059-018-1545-7
Luu K, Bazin E, Blum MGB (2017) pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol Ecol Resour 17:67–77. https://doi.org/10.1111/1755-0998.12592
Manel S, Gugerli F, Thuiller W et al (2012) Broad-scale adaptive genetic variation in alpine plants is driven by temperature and precipitation. Mol Ecol 21:3729–3738. https://doi.org/10.1111/j.1365-294X.2012.05656.x
McLean EH, Prober SM, Stock WD et al (2014) Plasticity of functional traits varies clinally along a rainfall gradient in Eucalyptus tricarpa. Plant Cell Environ 37:1440–1451. https://doi.org/10.1111/pce.12251
Murray KD, Janes JK, Jones A et al (2019) Landscape drivers of genomic diversity and divergence in woodland Eucalyptus. Mol Ecol 28:5232–5247. https://doi.org/10.1111/mec.15287
Nickolas H, Harrison PA, Tilyard P et al (2019) Inbreeding depression and differential maladaptation shape the fitness trajectory of two co-occurring Eucalyptus species. Ann For Sci 76:10
Nychka D, Furrer R, Paige J, Sain S (2017) fields: tools for spatial data. R Package Vers 10:3. https://doi.org/10.5065/D6W957CT
O’Reilly-Wapstra J, McArthur C, Potts BM (2004) Linking plant genotype, plant defensive chemistry and mammal browsing in a Eucalyptus species. Funct Ecol 18:677–684
Oksanen J, Blanchet FG, Friendly M et al (2019) vegan: community ecology package. R package version 2.5-6. https://cran.r-project.org/package=vegan
Parmesan C (2006) Ecological and evolutionary responses to recent climate change. Annu Rev Ecol Evol Syst 37:637–669. https://doi.org/10.1146/annurev.ecolsys.37.091305.110100
Pembleton L, Cogan N, Forster J (2013) StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol Ecol Resour 13:946–959
Pickup M, Field DL, Rowell DM, Young AG (2012) Predicting local adaptation in fragmented plant populations: implications for restoration genetics. Evol Appl 5:913–924. https://doi.org/10.1111/j.1752-4571.2012.00284.x
Plue J, Vandepitte K, Honnay O, Cousins SAO (2017) Does the seed bank contribute to the build-up of a genetic extinction debt in the grassland perennial Campanula rotundifolia? Ann Bot 120:373–385. https://doi.org/10.1093/aob/mcx057
Privé F, Luu K, Vilhjálmsson BJ et al (2020) Performing highly efficient genome scans for local adaptation with R Package pcadapt version 4. Mol Biol Evol 37:2153–2154. https://doi.org/10.1093/molbev/msaa053
Prober SM, Brown AHD (1994) Conservation of the grassy White Box woodlands: population genetics and fragmentation of Eucalyptus albens. Conserv Biol 8:1003–1013
Prober S, Thiele K (1993) The ecology and genetics of remnant grass White Box woodlands in relation to their conservation. Vic Nat 110:30–36
Prober SM, Thiele KR (1995) Conservation of the grassy white box woodlands: relative contributions of size and disturbance to floristic composition and diversity of remnants. Aust J Bot 43:349–366
Prober SM, Thiele KR (2004) Floristic patterns along an east–west gradient in grassy box woodlands of Central New South Wales. Cunninghamia 8:306–325
Prober SM, Thiele KR (2005) Restoring Australia’s temperate grasslands and grassy woodlands: integrating function and diversity. Ecol Manag Restor 6:16–27. https://doi.org/10.1111/j.1442-8903.2005.00215.x
Prober SM, Spindler DLH, Brown AHD (1998) Conservation of the grassy white box woodlands: effects of remnant population size on genetic diversity in the allotetraploid herb Microseris lanceolata. Conserv Biol 12:1279–1290. https://doi.org/10.1111/j.1523-1739.1998.97100.x
Prober SM, Byrne M, McLean EH et al (2015) Climate-adjusted provenancing: a strategy for climate-resilient ecological restoration. Front Ecol Evol 3:65. https://doi.org/10.3389/fevo.2015.00065
Prober SM, Potts BM, Bailey T et al (2016) Climate adaptation and ecological restoration in eucalypts. Proc R Soc Victoria 128:40–53. https://doi.org/10.1071/RS16004
Prober SM, Colloff MJ, Abel N et al (2017) Informing climate adaptation pathways in multi-use woodland landscapes using the values-rules-knowledge framework. Agric Ecosyst Environ 241:39–53. https://doi.org/10.1016/j.agee.2017.02.021
R Core Team (2018) R: a language and environment for statistical computing. R Found. Stat. Comput, Vienna
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Reisch C, Schmidkonz S, Meier K et al (2017) Genetic diversity of calcareous grassland plant species depends on historical landscape configuration. BMC Ecol 17:1–13. https://doi.org/10.1186/s12898-017-0129-9
Rellstab C, Zoller S, Walthert L et al (2016) Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Mol Ecol 25:5907–5924. https://doi.org/10.1111/mec.13889
Rellstab C, Fischer MC, Zoller S et al (2017) Local adaptation (mostly) remains local: reassessing environmental associations of climate-related candidate SNPs in Arabidopsis halleri. Heredity 118:193–201. https://doi.org/10.1038/hdy.2016.82
Reusch TBH, Ehlers A, Hammerli A, Worm B (2005) Ecosystem recovery after climatic extremes enhanced by genotypic diversity. Proc Natl Acad Sci 102:2826–2831. https://doi.org/10.1073/pnas.0500008102
Roda F, Walter GM, Nipper R, Ortiz-Barrientos D (2017) Genomic clustering of adaptive loci during parallel evolution of an Australian wildflower. Mol Ecol 26:3687–3699. https://doi.org/10.1111/mec.14150
Rousset F (1997) Genetic differentiation. Genetics 145:1219–1228
Scheffers BR, De Meester L, Bridge TCL et al (2016) The broad footprint of climate change from genes to biomes to people. Science 354:aaf7671. https://doi.org/10.1126/science.aaf7671
Selby JP, Willis JH (2018) Major QTL controls adaptation to serpentine soils in Mimulus guttatus. Mol Ecol 27:5073–5087. https://doi.org/10.1111/mec.14922
Sgrò CM, Lowe AJ, Hoffmann AA (2011) Building evolutionary resilience for conserving biodiversity under climate change. Evol Appl 4:326–337. https://doi.org/10.1111/j.1752-4571.2010.00157.x
Steane DA, Potts BM, McLean E et al (2014) Genome-wide scans detect adaptation to aridity in a widespread forest tree species. Mol Ecol 23:2500–2513. https://doi.org/10.1111/mec.12751
Steane DA, Potts BM, McLean E et al (2015) Genome-wide scans reveal cryptic population structure in a dry-adapted eucalypt. Tree Genet Genomes 11:1–14. https://doi.org/10.1007/s11295-015-0864-z
Storey JD, Bass AJ, Dabney A (2021) qvalue: Q-value estimation for false discovery rate control. R package version 2.24.0. http://github.com/jdstorey/qvalue
Supple MA, Bragg JG, Broadhurst LM et al (2018) Landscape genomic prediction for restoration of a Eucalyptus foundation species under climate change. elife 7:e31835. https://doi.org/10.1101/200352
Teixeira JC, Huber CD (2021) The inflated significance of neutral genetic diversity in conservation genetics. Proc Natl Acad Sci 118:1–10. https://doi.org/10.1073/pnas.2015096118
Walden N, Lucek K, Willi Y (2020) Lineage-specific adaptation to climate involves flowering time in North American Arabidopsis lyrata. Mol Ecol 29:1436–1451. https://doi.org/10.1111/mec.15338
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Xu T, Hutchinson M (2012) New developments in the ANUCLIM bioclimatic modelling package. Int Congr Environ Model Softw 212
Yeaman S, Hodgins KA, Lotterhos KE et al (2016) Convergent local adaptation to climate in distantly related conifers. Science 353:23–26
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Young AG, Brown AHD, Zich FA (1999) Genetic structure of fragmented populations of the endangered daisy Rutidosis leptorrhynchoides. Conserv Biol 13:256–265. https://doi.org/10.1046/j.1523-1739.1999.013002256.x
Zhang M, Suren H, Holliday JA (2019) Phenotypic and genomic local adaptation across latitude and altitude in Populus trichocarpa. Genome Biol Evol 11:2256–2272
Acknowledgements
This study was supported through a Research Fellowship jointly funded by CSIRO and the University of Tasmania (DS) and by the Australian Department of Agriculture, Water and Environment’s Biodiversity Knowledge project series. PAH was supported by the Australian Research Council Industrial Transformation Training Centre for Forest Value (IC150100004). We thank four reviewers for their considered feedback that helped improve this manuscript.
Funding
Open access funding provided by CSIRO Library Services. This study was supported through a Research Fellowship jointly funded by CSIRO and the University of Tasmania (DS) and by the Australian Department of Agriculture, Water and Environment’s Biodiversity Knowledge project series. PAH was supported by the Australian Research Council Industrial Transformation Training Centre for Forest Value (IC150100004).
Author information
Authors and Affiliations
Contributions
SMP conceived the study and all authors contributed to the study design. Fieldwork was conducted by SMP. Laboratory experimental work was conducted by MP. Analysis was performed by RJ, MP and PAH with all authors contributing to interpretation of results. RJ and MP wrote the first draft of the manuscript with all authors reviewing and contributing to the final manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jordan, R., Price, M., Harrison, P.A. et al. Landscape genomics reveals signals of climate adaptation and a cryptic lineage in Arthropodium fimbriatum. Conserv Genet 24, 473–487 (2023). https://doi.org/10.1007/s10592-023-01514-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01514-5