
Abstract
Amphibian populations world-wide are threatened by declines and extinctions mainly due to habitat loss and fragmentation. Habitat fragmentation threatens the yellow-bellied toad Bombina variegata in the northern and western regions of its distribution where it is strictly protected. We studied the genetic structure and diversity of populations at three geographical scales using microsatellite loci to detect potential threats for population persistence. At the local scale, we sampled four neighbouring localities at 1–2.6 km distance to detect effects of short-term (decades) fragmentation on connectivity. At the regional scale, five additional localities in the mountains of the Westerwald (Rhineland-Palatinate, Germany) were studied at up to 50.1 km distance to analyse genetic diversity and population structure. At the continental scale, we included data from regions in the northern distribution with fragmented populations (Hesse and Lower Saxony, Germany) and more continuous populations in the South (Alsace, France; Geneva, Switzerland; Trentino, Italy) to evaluate variation of genetic diversity. At the local scale, short-term fragmentation caused significant genetic differentiation between breeding assemblages only 1.4 km apart from each other. At the regional scale, we found notable genetic distance among localities. At the continental scale, we identified Alsace, Trentino and Geneva in the South as regions with low genetic structuring and high allelic richness, and the northern remaining regions in Germany as deeply structured with reduced allelic richness. We suggest that reduced genetic diversity and habitat fragmentation in northern regions makes these populations particularly vulnerable to decline. In conclusion, informed conservation management of B. variegata should focus on measures maintaining or improving connectivity among neighbouring populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Worldwide declines affect increasing numbers of amphibian species (Stuart et al. 2004). Besides the main cause habitat loss, environmental contaminants, UV-B irradiation, diseases, invasive species, exploitation and climate change contribute to the observed declines (Beebee and Griffiths 2005; Hof et al. 2011). Reduced genetic variation may further increase vulnerability of amphibian populations and enhance the risk of decline (Allentoft and O’Brien 2010; Chen et al. 2012). Isolation of populations and bottleneck effects, great intraspecific variation of genetic diversity and distinct effective population sizes were observed among amphibian populations (Funk et al. 2005; Monsen and Blouin 2004; Razpet et al. 2016). In European amphibians, continental-scale decrease of genetic diversity is not only caused by anthropogenic habitat deterioration, but also associated with recolonization histories following the last glaciation (Dufresnes and Perrin 2015). Indeed, current distributions of European taxa are often explained by northern expansions from Pleistocene refuge areas, (i.e. from the three Mediterranean peninsulas Iberia, Italy and the Balkans, or the Caucasus). These expansions were frequently accompanied by reductions in genetic diversity (Zeisset and Beebee 2008). Many successful recolonizations originated in the Balkan refuge area, possibly because barriers to northern migration were weaker than barriers on the way from the Alps or Pyrenees (Zeisset and Beebee 2008).
The European yellow-bellied toad Bombina variegata exemplifies a postglacial recolonization history (Gollmann and Gollmann 2012). The species is currently listed “least concern” according to the IUCN red list of endangered species, but as the population trend is decreasing, it is protected under the EU Habitats Directive 92/43/EE and considered endangered in many European countries (Kuzmin et al. 2009). At its northern range limit in Germany, many yellow-bellied toad populations have gone extinct mainly due to habitat loss, while others have declined during the past decades (Gollmann and Gollmann 2012; Veith 1996). As extant populations are often fragmented because of a low dispersal capacity (e.g., Gollmann and Gollmann 2012; Jehle and Sinsch 2007) and habitat fragmentation (Veith 1996), B. variegata is presently considered “critically endangered” in Germany (Laufer et al. 2020; Schlüpmann et al. 2011).
In target species of conservation interest, patterns of genetic diversity and the impact of fragmentation on connectivity among populations are commonly assessed by means of molecular tools (Balloux and Lugon-Moulin 2002; Holderegger et al. 2019). As microsatellites are sensitive markers for the detection of gene flow and thus genetic structuring due to high mutation rates, they are often used as a surrogate measure for landscape connectivity (Baguette et al. 2013). Here we explore genetic diversity and connectivity among populations of yellow-bellied toads at the local, regional, and continental scale. At the local and regional scale, we used microsatellite analyses to test for the effects of short- and long-term habitat fragmentation on isolation and genetic drift to evaluate implications for conservation. We complemented our data on the continental scale with published microsatellite data for B. variegata populations throughout Europe. In the following we explain our study objectives for the three geographic scales in detail:
-
1.
At the local scale, we asked whether the appearance of dispersal barriers in a former panmictic continuous population caused interruption of gene flow and genetic structuring among breeding assemblages.
-
2.
At the regional scale, we expected considerable population structure owing to the expansion of road and rail networks during the last century.
-
3.
At the continental scale, we compared genetic diversity and connectivity as well as isolation by distance effects between more northern and more southern populations and expected less fragmentation and higher diversity in the southern populations in line with phylogeographic patterns.
Materials and methods
Field study: sampling area and procedure
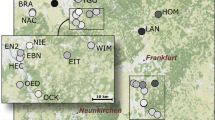
We sampled yellow-bellied toads B. variegata from nine study localities in Rhineland-Palatinate, Germany (Fig. 1; Table 1). At the local scale (approx. 2.5 km² survey area; Fig. 1a), we sampled four neighbouring study sites (1–4, Table 1) located at the former military training area Schmidtenhöhe near Koblenz (Hantzschmann and Sinsch 2019; Hantzschmann et al. 2019). About 40 years ago, the toads at these localities formed a single panmictic population considered as the largest B. variegata population in Rhineland-Palatinate (Veith 1996). At the regional scale (approx. 1000 km² survey area; Fig. 1b), we sampled toads at another five localities (5–9, Table 1) inhabiting clay and loamy sand pits in the Westerwald region of Rhineland-Palatinate. Geographical distances between localities are given as line-of-sight distance between the centres of breeding pond groups. The pairwise geographical distances range from 1.0 to 50.1 km (Table 2).

Geographical distribution of study sites. Local scale with short-term fragmentation. a Former military training area Schmidtenhöhe. Regional scale with long-term fragmentation. B Northern Rhineland-Palatinate (localities 1–4: Schmidtenhöhe; 5: Mogendorf; 6: Ruppach-Goldhausen; 7: Elkenroth; 8: Galgenkopf; 9: Bad Hönningen). Continental scale (C): populations in Lower Saxony (green); Hesse (blue); Rhineland-Palatinate (yellow); Alsace (red); Geneva (purple); Trentino (turquoise). See Table 1 for coordinates. Maps were created using data by the Naturschutzverwaltung Rheinland-Pfalz, Geobasisdata, Kataster- und Vermessungsverwaltung Rheinland-Pfalz, and GADM online platform (https://gadm.org/data.html)
We hand-captured and toe-clipped 16–30 individuals per locality, obtaining 182 samples for microsatellite analyses (Table 1). We sampled toads from localities 1–4 in May and July 2017, localities 5–8 in July 2018 and locality 9 in June 2019. We avoided replicate sampling by checking individuals for clipped toes and comparing the unique ventral pattern with the photographs of previously collected toads.
DNA extraction and genotyping
We genotyped the toads using 10 colour-labelled microsatellite markers originally developed for Bombina bombina (Stuckas and Tiedemann 2006; Hauswaldt et al. 2007). DNA was extracted from phalange tissue (toe clips) using a Qiagen blood and tissue kit. DNA was then amplified in a locus-specific polymerase chain reaction (PCR) on a MultiGene OptiMax (Labnet International) in 10 µl volumes containing 4.55–5.55 µl dH2O, 1 µl Buffer (Bioline), 5 pmol (1.0 µl) Primer, 0.3–0.4 µl My-Taq-Polymerase (Bioline) and 1–2 µl undiluted DNA. PCR conditions followed the protocol of Weihmann et al. (2009) using reduced elongation (3 min) and adapted annealing temperatures (Primer 1A and 8A: 59,5 °C; 5F and B14: 56 °C; 9H: 48 °C; 10F: 68 °C; 12F: 58 °C; B13: 41 °C; F2: 54 °C; F22: 51 °C). For the B13 locus, we used a different procedure: 10 µl volume containing 3.45–6.45 µl dH2O, 2 µl HF-Buffer (Thermo Fisher), 0.2 µl dNTPs, 0.5 µl MgCl2, 0.1 µl Phusion High Fidelity-Polymerase (Thermo Fisher), 5 pmol (1.0 µl) per primer and 1.0–2 0.0 µl undiluted DNA denaturated at 98 °C, followed by 35 cycles at 98 °C (10 s), 41 °C (30 s), 72 °C (30 s) and a final elongation at 72 °C (8 min). Fragment lengths were analysed by Eurofins Genomics, Germany, at an ABI 3130 XL sequencing machine and allele sizes were determined using the software GeneMapper 5.
Local and regional scale population genetic analyses
We checked the resulting fragment lengths using the software Microchecker 2.2.3 for null alleles (Van Oosterhout et al. 2004). Genetic diversity was examined by calculating allele number and private alleles with corresponding frequencies in GeneAlEx (Peakall and Smouse 2012). Furthermore, allelic richness (AR), deviations from Hardy-Weinberg equilibrium using Fisher’s exact test, and inbreeding coefficients (FIS) with corresponding confidence intervals were calculated using the package diveRsity 1.9.9 in R (Keenan et al. 2013). Linkage disequilibrium between loci (10,000 permutations), an analysis of molecular variance (AMOVA), observed (Ho) and expected (He) heterozygosity with corresponding standard deviation were calculated using the software Arlequin 3.5.2.2 (Excoffier and Lischer 2010). We examined population connectivity through an analysis of genetic isolation by geographic distance (IBD) transforming the raw FST values (formula: FST/(1-FST); Vacher and Ursenbacher 2014) and applying a Mantel test in GeneAlEx (Peakall and Smouse 2012). In addition, pairwise genetic difference between populations (weighed FST-statistics according to Weir and Cockerham 1984; Michalakis and Excoffier 1996) were calculated using the software Arlequin 3.5.2.2 (Excoffier and Lischer 2010). Genetic drift due to bottleneck effects was examined using the software Bottleneck 1.2.02 (Piry et al. 1999) with default settings. As it is unknown under which mutation models the microsatellite loci evolved, we compared the Wilcoxon’s sign rank test that is based on heterozygosity excess for (1) the stepwise mutation model (SMM) and (2) the two-phase model (TPM) with the mode shift test that uses allele frequency distribution (Cornuet and Luikart 1996, Luikart et al. 1998). We performed a cluster analysis to identify the number of genetic clusters in our data and a Discriminant Analysis of Principal Components (DAPC) to describe diversity between populations using the package adegenet in R (Jombart 2008). The population structure on the regional scale was alternatively investigated estimating the most probable number of genetic clusters in Structure 2.3 (Pritchard et al. 2009) using the admixture model without prior information on sample population. The evaluated number of clusters (k) was set from 1 to 10 with 20 runs per k after a burn-in period of 100,000 followed by 500,000 iterations. We used the Evanno method (∆k; Evanno et al. 2005) in Structure Harvester (Earl and vonHoldt 2012) to evaluate the most appropriate number of genetic clusters k. Corresponding charts were obtained using CLUMPAK (Kopelman et al. 2015). We used the R package tess3r (Caye et al. 2018) to visualize the spatial population structure for the most appropriate number of genetic clusters. Gene flow was detected and visualised applying divMigrate-online (Sundqvist et al. 2016). We used the Nm migration statistic and default settings for the analysis of relative migration at the local scale, while statistically significant directional migration was detected applying 1000 bootstraps at the regional scale.
Continental scale population genetic analyses
For the continental-scale analysis (Fig. 1c), we retrieved published microsatellite data throughout the western range of B. variegata. This included regions at the northern range margin in Germany (Lower Saxony: n = 150, 11 localities, the same 10 microsatellite loci, Weihmann et al. 2009; Weihmann et al. 2019; Hesse: n = 281, 20 localities, the same 10 microsatellite loci, Guicking et al. 2017), urbanised areas in Geneva, Switzerland (n = 305, 12 localities, 5 of the same loci, Tournier 2017), forested areas in Alsace, France (n = 290, 10 localities, 6 of the same loci, Vacher and Ursenbacher 2014) and different areas in Trento, Italy (n = 200, 9 localities, 9 of the same loci, Cornetti 2013; Cornetti et al. 2016), representing the southernmost region in this dataset. As raw data were not available for every region, we refrained from applying the same analyses we used for our data at the local and regional scale. Instead, we compared the published data sets on the six regions in respect to genetic diversity, i.e. AR and He, and genetic differentiation among populations, i.e. FST. We used an analysis of variance (ANOVA) to assess continental genetic diversity. If ANOVA detected significant differences among the regions, we applied a multiple regression analysis to test for the impact of latitude, longitude and altitude to detect continental clines on the target variables. Within each region, the isolation by distance (IBD) pattern was modelled by calculating the linear relationship between geographical distance and transformed raw FST-value (Fst/[1-Fst], Vacher and Ursenbacher 2014). Slopes of regression lines as a measure of intensity of isolation among regional populations were compared using the 95% confidence intervals. Statistical procedures were performed using the program package Statgraphics Centurion version 18.1.01. The significance level was set at alpha = 0.05.
Results
Eight of the ten loci studied were polymorphic in the Westerwald region (Table 3). The MicroChecker analysis did not provide evidence for null alleles or large allelic dropout in any of the localities. Consequently, all sampled individuals were included in the analyses. We detected 40 alleles including eight private alleles (Tables 3 and 4). The average number of alleles per locus was 4.2 (range: 1–8; Table 3). Linkage disequilibrium (LD) was found in six out of nine localities (1, 5–9), but as the LD was neither consistent over loci nor localities, we assumed that genotypes were independent among loci analogous to Guicking et al. (2017). We did not detect deviations from Hardy-Weinberg equilibrium (Fisher’s exact test; p > 0.05) or evidence for significant inbreeding at any of the localities (Table 4).
Local scale (Schmidtenhöhe): Genetic diversity, population structure and migration
The expected heterozygosity He was similar at the four localities of the Schmidtenhöhe (range: 0.43–0.45; Table 4). Toads at locality 3 had the lowest number of alleles and lowest AR and showed evidence for a bottleneck event (one-tailed Wilcoxon’s test for two-phase model: p < 0.05, positive mode shift test; Table 4). Toads at locality 2 had the highest number of alleles and the highest AR (Table 4). Private alleles were found in low frequency at locality 1 (Table 4). Most of the genetic variation (96%) was explained by variation within localities, only 4% were attributable to variation among localities (AMOVA with weighted FST statistics over all loci: p < 0.001). Breeding assemblages that were at least 1.4 km distant from each other showed a corresponding genetic differentiation (AMOVA with weighted FST statistics over all loci: p < 0.05; Table 2). Toads from localities 1 and 2 at about 1 km distance did not show significant genetic differentiation. This is in line with the results of the analysis for migration indicating that gene flow still exists among the four localities (Fig. 2A). The highest gene flow was detected among the neighbouring localities 1 and 2, whereas migration to the most distant locality 3 seems to be limited. Directional migration was not statistically significant.

a Gene flow visualized through relative migration among populations of the local scale. b Directional migration at the regional scale using 1000 bootstraps
Regional scale (Westerwald, Rhineland-Palatinate): genetic diversity, population structure and migration
The expected heterozygosity (He) at the nine study localities varied in a broader range than at the local scale (range: 0.37–0.48; Table 4). Locality 7 had the lowest He, localities 6 and 8 the highest. Locality 6 showed the highest AR (Table 4). Private alleles were found in localities 6, 8, and 9 in high frequencies (range: 0.16–0.19; Table 4). We found evidence for a recent bottleneck in toads at localities 6 and 9 (Table 4). Most of the genetic variation (83.9%) was explained by variation within populations, whereas 16.1% of the variation was attributable to variation among populations (AMOVA with weighted FST statistics over all loci: p < 0.001). We found significant genetic differentiation between locality pairs covering a distance range of 1.4–50.1 km (Table 2) with a significant global FST value of 0.16 (AMOVA with weighted FST statistics over all loci: p < 0.001). The analysis of IBD showed a significant positive correlation between genetic and geographic distance (Mantel test: p < 0.05, R2 = 0.45; IBD = 0.06 + 0.0055*Geographical distance [km]; Fig. 3). The most likely population structure using the Evanno method suggested three different genetic clusters of B. variegata in the Westerwald (Fig. 4a). Localities 1–4 were assigned to cluster 1, localities 6 and 9 to cluster 2 and localities 7 and 8 to cluster 3; Toads from locality 5 were of admixed origin (Fig. 4b). This is in line with the tess3r graphical output displaying the spatial population structure for k = 3 (Fig. 4c). The Discriminant Analysis of Principal Components yielded a differentiated, finer scaled pattern of the genetic structure of B. variegata, as the BIC values decrease with the number of clusters (Suppl. Figure 1). The graphical output of the DAPC analysis however, jointly clustered toads at geographically close localities (Suppl. Figure 2). Statistically significant directional migration was detected for localities 6 and 9 (Fig. 2b).

Isolation by distance: regression of genetic distance (Fst/(1-Fst)) on geographic distance in the Westerwald region

a Delta K plot representing the most probable number (K = 3) of genetic groups in the Westerwald region; using the Evanno method. b Results of the structure analysis of study localities in the Westerwald region representing three (K = 3) genetic clusters. C Spatial geographic population structure of the localities at the regional scale for k = 3
Continental scale (Europe): genetic diversity and isolation by distance
Allelic richness of B. variegata differed significantly among study regions in Germany, France and Italy (ANOVA: F4,52 = 13.99, P < 0.0001; Table 5). Specifically, AR was significantly lower in Germany than that in France and Italy (Multiple group comparison: P < 0.05). Variation of latitude, longitude, and altitude of localities studied accounted for 41.8% of variance in AR (Multiple regression model: AR = 15.04–0.22*Latitude-0.11*Longitude–0.00069*Altitude; F3,52=13.48, P < 0.0001). Specifically, AR decreased significantly from south to north and from east to west. In contrast, He was similar in five out of six regions studied (ANOVA: F5,64=3.32, P = 0.0104; Table 5). The only significant deviation was detected between the Geneva and Alsace regions (Multiple group comparison: P < 0.05). Latitude, longitude, and altitude of localities did not account for a significant amount of variance in He (Multiple regression analysis: F3,64=1.62, P = 0.1937). The genetic differentiation between pairs of localities within the six regions studied, i.e. global FST and slopes of IBD linear regression analyses, differed considerably between the southern and the northern regions (Table 6). Isolation by distances up to 150 km was low in Trentino (Italy) and Alsace (France), whereas it was up to four times larger in a distance range of 20–70 km in the German and Swiss regions. There were exceptions from the rule in all regions, but the greatest genetic differentiation between locality pairs was detected in the neighbouring regions Hesse and Westerwald (Rhineland-Palatinate) at the northern range limit of B. variegata. Despite of the regional scatter of pairwise genetic differentiation, IBD regression models differed significantly with respect to slopes between Westerwald (Rhineland-Palatinate) on one side and Alsace, Geneva and Trentino on the other (Table 6).
Discussion
In this study we measured genetic population structure, connectivity and genetic diversity at three geographical scales in the endangered yellow-bellied toad by applying population genetic tools. At the local scale (Schmidtenhöhe) we found that a former panmictic population shows indications of increasing fragmentation despite low distances among breeding assemblages. At the regional scale (Westerwald) we found evidence for the existence of significant population structure, and at the continental scale we found that population fragmentation is more severe and genetic diversity reduced in the northern study regions as compared to more southern study regions.
In the following we discuss the observed genetic diversity and population structure of yellow-bellied toads in the context of local, regional and continental scale and evaluate the predictions on genetic structuring in view of the short-term and long-term isolation of populations in the Westerwald region.
Local scale
The small-scale system of breeding assemblages studied at the Schmidtenhöhe mirrors a 30–40 years’ history of habitat fragmentation, as toad dispersal has been impeded through succession that emerged rapidly after the withdrawal of tanks (Hantzschmann and Sinsch 2019). As predicted, this short-term fragmentation caused low, but significant, differentiation among three of the four study localities. This is in line with a similar genetic structuring of B. variegata inhabiting the Geneva region in which structuring was attributed to habitat fragmentation by urbanisation (Tournier 2017).
The ability of individuals to reach a neighbouring conspecific population, i.e. movement capacity, depends in part on density-dependent motivation to disperse, landscape resistance, and life expectancy (Cayuela et al. 2016; Hantzschmann et al. 2019; Sinsch 2014; Stevens et al. 2006). Dry open grasslands without water bodies pose a high landscape resistance for movements of Bufo bufo, Rana dalmatina and Lissotriton vulgaris (Jeliazkov et al. 2019) and the same may apply to B. variegata. CMR surveys at the Schmidtenhöhe did not provide evidence for recent among-locality migrations of yellow-bellied toads even though distances are smaller than observed lifetime dispersal distances of the species (1.2–4.5 km; Gollmann and Gollmann 2005; Jehle and Sinsch 2007; Plytycz and Bigaj 1984). This suggests that dry open grasslands without water bodies may constitute effective dispersal barriers for these toads (Hantzschmann and Sinsch 2019). However, gene flow still exists among the four localities, but decreases with increasing distance. This is in line with observations on the migratory behaviour of B. variegata (Hartel 2008), as the number of individuals reaching neighbouring breeding sites decreased with increasing distance between ponds. The FST values are not significant between the two nearest sites (1 and 2, distance: 1 km) and some infrequent and therefore undetected migration might still occur over distances up to 1 km. These numbers are in accordance with annual migratory ranges of B. variegata (20–732 m) measured in capture-mark-recapture studies (Abbühl and Durrer 1996; Beshkov and Jameson 1980; Hantzschmann and Sinsch 2019; Hartel 2008; Jacob et al. 2009; Jordan 2012). We assume that dispersal in the Schmidtenhöhe populations is limited by the absence of stepping stones, e.g. ponds. In line with our predictions, we conclude that in small-sized yellow-bellied toad populations, habitat fragmentation through succession leads to genetic structuring within a few decades.
Regional scale
Modelling genetic structure of the Westerwald region yielded different results depending on the statistical approach applied. The Evanno method (STRUCTURE: Bayesian iterative algorithm; Porras-Hurtado et al. 2013) suggests the presence of three genetic clusters. One cluster included geographically distant localities (6 and 9; Table 1). This assignment is compatible with unauthorized translocation of individuals or animal-mediated egg dispersal between those sites. The latter however has not been reported for yellow-bellied toads. Instead, the directional migration to localities 6 and 9 (Fig. 2B) indicates a founder effect for colonized regions. This is in line with the potential bottleneck detected for these localities and may explain the STRUCTURE clustering as well. In contrast, the DAPC method (multivariate approach; Jombart et al. 2010) yielded a finer scaled pattern in which the grouping of localities to clusters was consistent with their spatial arrangement (Suppl. Figure 2). We suggest that these discrepancies result from the different statistical approaches and conclude that there are at minimum three genetic clusters that may be sub-structured.
Amphibians are strongly affected by habitat fragmentation and subsequent isolation of local populations that promote bottlenecks as well as high inbreeding levels and thus reduced fitness (Andersen et al. 2004; Angelone 2010; Apodaca et al. 2012; Ficetola and De Bernardi 2004). The pronounced IBD in the Westerwald populations (steep regression line, Table 6) indicates an ancestry from a common metapopulation that has been recently fragmented and isolated. The low levels of gene flow emphasize the interrupted exchange of individuals in this region as well. The high frequency of private alleles indicates genetic drift effects due to the isolation of populations. Drift effects may be strengthened by the presumably small population size and the short reproductive lifespan in the investigated populations that decreases an individual´s chance to contribute to the local gene pool (Cayuela et al. 2019; Hantzschmann and Sinsch 2019). Landscape fragmentation in Hesse is considered the main cause of isolation among B. variegata populations (Guicking et al. 2017). This is probably true for the Westerwald region as well because during the 19th century the road and rail network expanded parallel to increased clay mining (Schenk 1993). Clay mines provided secondary habitats for yellow-bellied toads, but the expansion of transport network contributed to landscape fragmentation. We suggest that this habitat fragmentation promoted the isolation of toad populations and resulted in the recent patchy distribution. Furthermore, many stepping-stone populations between the extant populations have disappeared during the past decades (Veith 1996). In conclusion, we consider the notable genetic structuring of the Westerwald populations as the result of long-term, i.e. about 100 years, habitat fragmentation and isolation.
Continental scale
The phylogeography of some European amphibians is characterised by genetic diversity gradients, e.g. declining diversity from south to north and east to west, caused by postglacial recolonization from glacial refuge areas (e.g. the European tree frog Hyla arborea, Dufresnes et al. 2013; the natterjack toad Epidalea calamita, Allentoft et al. 2009; Rowe et al. 1998, 2006; Beebee and Rowe 2000; the spadefoot toad Pelobates fuscus, Eggert et al. 2006; the Italian agile frog Rana latastei, Garner et al. 2004, the common frog Rana temporaria, Palo et al. 2004). As predicted, we observed a decrease of genetic diversity (AR) from south to north and from east to west for B. variegata as well. Allelic richness is a genetic diversity measure that is more sensitive to founder events followed by expansions than heterozygosity (Greenbaum et al. 2014). Thus, we consider demographic processes following the postglacial dispersal to account for the lower genetic diversity at the northern range margin in B. variegata (Fijarczyk et al. 2011; Rowe et al. 2006). Reduced genetic diversity may additionally be a consequence of small effective population size resulting from habitat fragmentation (Andersen et al. 2004; Arens et al. 2006; Hitchings and Beebee 1997; Marsh et al. 2008; Noël et al. 2006; Reh and Seitz 1990; Tournier 2017; Willi et al. 2006; Zancolli et al. 2014). Consequently, we attribute the marked IBD pattern among populations in the German regions to reduced gene flow caused by landscape fragmentation and demographic processes, whereas the low genetic structuring in Alsace, Geneva and Trentino suggests a still intact connectivity among populations.
Implications for conservation management
As the conservation status of post-glacial European amphibian populations worsen with increasing distances from the species’ glacial refugia, reduced genetic diversity may affect resilience towards current threats such as habitat fragmentation (Dufresnes and Perrin 2015). Our study suggests that postglacial colonisation processes and habitat fragmentation are the main causes of pronounced genetic structuring and reduced AR of B. variegata populations in German regions. As a reestablishment of connectivity between currently isolated populations over distances larger than 5 km is visionary because of current land use and low dispersal capacity of this species, conservation measures should focus on the preservation of remaining populations by improving habitats. In the case of small, isolated and genetically impoverished populations, translocation may improve adaptability to changing environmental conditions (Frankham et al. 2017). However, translocation of specimens, even to populations following a bottleneck, should be considered with caution, as local populations may be adapted to distinct habitat features and pathogens may be spread unintentionally (Orizaola et al. 2010; Taft and Roff 2012; Verhoeven et al. 2011). Improving connectivity between local populations seems to be the crucial factor to mitigate genetic structuring and to increase resilience towards the variation of environmental factors (Frankham et al. 1999; Schön et al. 2011). Small-scale groups of isolated local breeding assemblages (example: Schmidtenhöhe) should be transformed to an interacting metapopulation system by stepping-stone ponds less than 1 km apart to enable movements of some individuals between neighbouring populations. Thus, informed conservation management of B. variegata populations could consist in the creation of small-scale metapopulations by offering satellite habitats around isolated extant breeding groups, for colonization. These metapopulation systems would still be isolated from each other, but more resilient to local extinction. On a regional scale, the still existing connectivity among populations such as those in Alsace and Trentino should be conserved to avoid the negative effects of habitat fragmentation currently present in the three German and threatening the persistence of yellow-bellied toads.
References
Abbühl R, Durrer H (1996) Habitatpräferenz und Migrationsverhalten bei der Gelbbauchunke (Bombina variegata variegata) in einer seminatürlichen Versuchsanlage. Salamandra 32:23–30
Allentoft M, O’Brien J (2010) Global Amphibian declines, loss of genetic diversity and fitness. Rev Divers 2:47–71. https://doi.org/10.3390/d2010047
Allentoft ME, Siegismund HR, Briggs L, Andersen LW (2009) Microsatellite analysis of the natterjack toad (Bufo calamita) in Denmark: populations are islands in a fragmented landscape. Conserv Gen 10:15–28. https://doi.org/10.1007/s10592-008-9510-8
Andersen LW, Fog K, Damgaard C (2004) Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea). Proc R Soc Lond B: 271:1293–1302. https://doi.org/10.1098/rspb.2004.2720
Angelone S (2010) Are differences in fitness traits related to genetic clusters? An empirical test on the European tree frog. Biol Conserv 143:471–478. https://doi.org/10.1016/j.biocon.2009.11.014
Apodaca JJ, Rissler LJ, Godwin JC (2012) Population structure and gene flow in a heavily disturbed habitat: implications for the management of the imperilled Red Hills salamander (Phaeognathus hubrichti). Conserv Gen 13:913–923. https://doi.org/10.1007/s10592-012-0340-3
Arens P, Bugter R, van’t Westende W, Zollinger R, Stronks J, Vos CC, Smulders MJM (2006) Microsatellite variation and population structure of a recovering Tree frog (Hyla arborea L.) metapopulation. Conserv Gen 7:825–835. https://doi.org/10.1007/s10592-005-9112-7
Baguette M, Blanchet S, Legrand D, Stevens VM, Turlure C (2013) Individual dispersal, landscape connectivity and ecological networks. Biol Rev 88:310–326. https://doi.org/10.1111/brv.12000
Balloux F, Lugon-Moulin N (2002) The estimation of population differentiation with microsatellite markers. Mol Ecol 11:155–165. https://doi.org/10.1046/j.0962-1083.2001.01436.x
Beebee TJC, Griffiths RA (2005) The amphibian decline crisis: a watershed for conservation biology? Biol Conserv 125:271–285. https://doi.org/10.1016/j.biocon.2005.04.009
Beebee TJC, Rowe G (2000) Microsatellite analysis of natterjack toad Bufo calamita Laurenti populations: consequences of dispersal from a Pleistocene refugium. Biol J Linnean Soc 69:367–381. https://doi.org/10.1006/bijl.1999.0368
Beshkov VA, Jameson DL (1980) Movement and abundance of the yellow-bellied toad Bombina variegata. Herpetologica 36:365–370
Caye K, Jay F, Michel O, François O (2018) Fast inference of individual admixture coefficients using geographic data. Ann Appl Stat 12:586–608. https://doi.org/10.1214/17-AOAS1106
Cayuela H, Boualit L, Arsovski D, Bonnaire E, Pichenot J, Bellec A, Miaud C, Léna JP, Joly P, Besnard A (2016) Does habitat unpredictability promote the evolution of a colonizer syndrome in amphibian metapopulations? Ecology 97:2658–2670. https://doi.org/10.1002/ecy.1489
Cayuela H, Cruickshank SS, Brandt H, Ozgul A, Schmidt BR (2019) Habitat-driven life history variation in an amphibian metapopulation. Oikos 128:1265–1276. https://doi.org/10.1111/oik.06286
Chen SY, Zhang YJ, Wang XL, Sun JY, Xue Y, Zhang P, Zhou H, Qu LH (2012) Extremely Low genetic diversity indicating the endangered status of Ranodon sibiricus (Amphibia: Caudata) and implications for phylogeography. Plos One 7(3):e33378. https://doi.org/10.1371/journal.pone.0033378
Cornetti L (2013) Conservation genetics of the yellow-bellied toad (Bombina variegata) and the common lizard (Zootoca vivipara) in the Italian Alps. PhD-Thesis, University of Ferrara
Cornetti L, Benazzo A, Hoban S, Vernesi C, Bertorelle G (2016) Ancient, but not recent, population declines have had a genetic impact on alpine yellow-bellied toad populations, suggesting potential for complete recovery. Conserv Genet 17:727–743. https://doi.org/10.1007/s10592-016-0818-5
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Dufresnes C, Perrin N (2015) Effect of biogeographic history on population vulnerability in European amphibians. Conserv Biol 29:1235–1241. https://doi.org/10.1111/cobi.12490
Dufresnes C, Wassef J, Ghali K, Brelsford A, Stöck M, Lymberakis P, Crnobrnja-Isailovic J, Perrin N (2013) Conservation phylogeography: does historical diversity contribute to regional vulnerability in European tree frogs (Hyla arborea)? Mol Ecol 22:5669–5684. https://doi.org/10.1111/mec.12513
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eggert C, Cogălniceanu D, Veith M, Dzukic G, Taberlet P (2006) The declining Spadefoot toad, Pelobates fuscus (Pelobatidae): paleo and recent environmental changes as a major influence on current population structure and status. Conserv Genet 7:185–195. https://doi.org/10.1007/s10592-006-9124-y
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver.3.5: a new series of programs to perfom population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Ficetola GF, De Bernardi F (2004) Amphibians in a human-dominated landscape: the community structure is related to habitat features and isolation. Biol Conserv 119:219–230. https://doi.org/10.1016/j.biocon.2003.11.004
Fijarczyk A, Nadachowska K, Hofman S, Litvinchuk SN, Babik W, Stuglik M, Gollmann G, Choleva L, Cogălniceanu D, Vukov T, Dzukic G, Szymura JM (2011) Nuclear and mitochondrial phylogeography of the European fire-bellied toads Bombina bombina and Bombina variegata supports their independent histories. Mol Ecol 20:3381–3398. https://doi.org/10.1111/j.1365-294X.2011.05175.x
Frankham R, Ballou JD, Ralls K, Eldridge MD, Dudash MR, Fenster CB, Lacy RC, Sunnucks P (2017) Genetic management of fragmented animal and plant populations. Oxford University Press, Oxford
Frankham R, Lees K, Montgomery ME, England PR, Lowe EH, Briscoe DA (1999) Do population size bottlenecks reduce evolutionary potential? Anim Conserv 2:255–260. https://doi.org/10.1111/j.1469-1795.1999.tb00071.x
Frei M, Csencsics D, Brodbeck S, Schweizer E, Bühler C, Gugerli F, Bolliger J (2016) Combining landscape genetics, radio-tracking and long-term monitoring to derive management implications for natterjack toads (Epidalea calamita) in agricultural landscapes. J Nat Conserv 32:22–34. https://doi.org/10.1016/j.jnc.2016.04.002
Funk WC, Blouin MS, Corn PS, Maxell BA, Pilliod DS, Amish S, Allendorf FW (2005) Population structure of Columbia spotted frogs (Rana luteiventris) is strongly affected by the landscape. Mol Ecol 14:483–496. https://doi.org/10.1111/j.1365-294X.2005.02426.x
Garner TW, Pearman PB, Angelone S (2004) Genetic diversity across a vertebrate species’ range: a test of the central-peripheral hypothesis. Mol Ecol 13:1047–1053. https://doi.org/10.1111/j.1365-294X.2004.02119.x
Gollmann G, Gollmann B (2005) Postmetamorphic growth and movements in yellow-bellied toads, Bombina variegata: approaching life-path analysis. Herpetologia Petropolitana 12:143–145
Gollmann B, Gollmann G (2012) Die Gelbbauchunke. Laurenti, Bielefeld
Greenbaum G, Templeton AR, Zarmi Y, Bar-David S (2014) Allelic richness following population founding events – A stochastic modeling framework incorporating gene flow and genetic drift. PLoS One 9(12):e115203. https://doi.org/10.1371/journal.pone.01152031
Guicking D, Finke L, Wittich M, Pfeiffer I, Veith M, Geske C, Braukmann U, Weising K, Neubeck C (2017) Conservation genetics of Bombina v. variegata (Anura: Bombinatoridae) in northern Hesse. Germany Salamandra 53:201–211
Hantzschmann AM, Sinsch U (2019) Struktur und Dynamik von Gelbbauchunken-Populationen (Bombina variegata) im Westerwald- Konsequenzen für das regionale Artenschutz-Management. Z Feldherpet 26:218–235
Hantzschmann AM, Gollmann B, Gollmann G, Sinsch U (2019) The fast–slow continuum of longevity among yellow-bellied toad populations (Bombina variegata): intrinsic and extrinsic drivers of variation. PeerJ 7:e8233. https://doi.org/10.7717/peerj.8233
Hartel T (2008) Movement activity in a Bombina variegata population from a deciduous forested landscape. North-West J Zool 4:79–90
Hauswaldt JS, Schröder C, Tiedemann R (2007) Nine new tetranucleotide microsatellite markers for the fire-bellied toad (Bombina bombina). Mol Ecol Notes 7:49–52. https://doi.org/10.1111/j.1471-8286.2006.01516.x
Hitchings SP, Beebee TJC (1997) Genetic substructuring as a result of barriers to gene flow in urban Rana temporaria (common frog) populations: implications for biodiversity conservation. Heredity 79:117–127
Hof C, Araujo MB, Jetz W, Rahbek C (2011) Additive threats from pathogens, climate and land-use change for global amphibian diversity. Nature 480(7378):516–519. https://doi.org/10.1038/nature10650
Holderegger R, Balkenhol N, Bolliger J, Engler JO, Gugerli F, Hochkirch A, Nowak C, Segelbacher G, Widmer A, Zachos FE (2019) Conservation genetics: Linking science with practice. Mol Ecol 28:3848–3856. https://doi.org/10.1111/mec.15202
Jacob A, Scheel B, Buschmann H (2009) Raumnutzung in einer Metapopulation der Gelbbauchunke (Bombina variegata) an ihrer nördlichen Verbreitungsgrenze. Z Feldherpet 16:85–102
Jehle R, Sinsch U (2007) Wanderleistung und Orientierung von Amphibien: eine Übersicht. Z Feldherpet 14:137–256
Jeliazkov A, Lorrilliere R, Besnard A, Garnier J, Silvestre M, Chiron F (2019) Cross-scale effects of structural and functional connectivity in pond networks on amphibian distribution in agricultural landscapes. Freshwater Biol 64:997–1014. https://doi.org/10.1111/fwb.13281
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94. https://doi.org/10.1186/1471-2156-11-94
Jordan M (2012) Untersuchung von Populationsgrösse und Dispersionsverhalten der Gelbbauchunken-Population im Talkessel Schwyz-Ingenbohl. Bachelor thesis, Zürcher Hochschule für Angewandte Wissenschaften. Downloaded on 6 February 2020 at http://www.karch.ch/karch/de/home/aktivitaten/forschungsprojekte/dissertationen-und-masterarbeite.html
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Kuzmin S, Denoël M, Anthony B, Andreone F, Schmidt B, Ogrodowczyk A, Ogielska M, Vogrin M, Cogalniceanu D, Kovács T, Kiss I, Puky M, Vörös J, Tarkhnishvili D, Ananjeva N (2009) Bombina variegata. The IUCN Red List of Threatened Species 2009:e.T54451Ahttps://doi.org/10.2305/IUCN.UK.2009.RLTS.T54451A11148290.en. Downloaded on 27 April 2020
Laufer H, Höppner C, Nöllert A (2020) Gelbbauchunke (Bombina variegata). In: Rote-Liste-Gremium Amphibien und Reptilien: Rote Liste und Gesamtartenliste der Amphibien (Amphibia) Deutschlands. Naturschutz Biologische Vielfalt 170(4):38–39
Luikart G, Allendorf FW, Cornuet J-M, Sherwin WB (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89:238–247. https://doi.org/10.1093/jhered/89.3.238
Marsh DM, Page RB, Hanlon TJ, Corritone R, Little EC, Seifert DE, Cabe PR (2008) Effects of roads on patterns of genetic differentiation in red-backed salamanders, Plethodon cinereus. Conserv Gen 9:603–613. https://doi.org/10.1007/s10592-007-9377-0
Michalakis Y, Excoffier L (1996) A generic estimation of population subdivision using distances between alleles with special reference to microsatellite loci. Genetics 142:1061–1064
Monsen KJ, Blouin MS (2004) Extreme isolation by distance in a montane frog Rana cascadae. Conserv Gen 5:827–835. https://doi.org/10.1007/s10592-004-1981-z
Noël S, Ouellet M, Galois P, Lapointe FJ (2006) Impact of urban fragmentation on the genetic structure of the eastern red-backed salamander. Conserv Gen 8:599–606. https://doi.org/10.1007/s10592-006-9202-1
Orizaola G, Quintela M, Laurila A (2010) Climatic adaptation in an isolated and genetically impoverished amphibian population. Ecography 33:730–737. https://doi.org/10.1111/j.1600-0587.2009.06033.x
Palo JU, O´Hara RB, Laugen AT, Laurila A, Primmer CR, Merilä J (2003) Latitudinal divergence of common frog (Rana temporaria) life history traits by natural selection: evidence from a comparison of molecular and quantitative genetic data. Mol Ecol 12:1963–1978. https://doi.org/10.1046/j.1365-294X.2003.01865.x
Palo JU, Schmeller DS, Laurila A, Primmer CR, Kuzmin SL, Merilä J (2004) High degree of population subdivision in a widespread amphibian. Mol Ecol 13:2631–2644. https://doi.org/10.1111/j.1365-294X.2004.02269.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Piry S, Luikart G, Cornuet JM (1999) Bottleneck: A programm for detecting recent effective population size reductions from allele data frequencies. Downloaded on 10 December 2019 at http://www1.montpellier.inra.fr/CBGP/software/Bottleneck/bottleneck.html
Plytycz B, Bigaj J (1984) Preliminary studies on the Growth and Movements of the Yellow-bellied Toad, Bombina variegata (Anura: Discoglossidae). Amphibia-Reptilia 5:81–86
Porras-Hurtado L, Ruiz Y, Santos C, Phillips C, Carracedo Á, Lareu MV (2013) An overview of STRUCTURE: applications, parameter settings, and supporting software. Front Genet 4:98. https://doi.org/10.3389/fgene.2013.00098
Pritchard J, Wen X, Falush D (2009) Documentation for structure software: Version 2.3
QGIS Development Team (2019) QGIS Geographic Information System. Open Source Geospatial Foundation Projekt. http://qgis.osgeo.org
Razpet A, Šunje E, Kalamujić B, Tulić U, Pojskić N, Stanković D, Krizmanić I, Marić S (2016) Genetic differentiation and population dynamics of Alpine salamanders (Salamandra atra, Laurenti 1768) in Southeastern Alps and Dinarides. Herpetol J 26:109–119
Reh W, Seitz A (1990) The influence of land use on the genetic structure of populations of the common frog Rana temporaria. Biol Conserv 54:239–249
Rowe G, Beebee TJC, Burke T (1998) Phylogeography of the natterjack toad Bufo calamita in Britain: genetic differentiation of native and translocated populations. Mol Ecol 7:751–760. https://doi.org/10.1046/j.1365-294x.1998.00387.x
Rowe G, Harris J, Beebee TJC (2006) Lusitania revisited: A phylogeographic analysis of the natterjack toad Bufo calamita across its entire biogeographical range. Mol Phyl Evol 39:335–346. https://doi.org/10.1016/j.ympev.2005.08.021
Schenk G (1993) Der Westerwälder Tonbergbau von 1950–1988 - Ein Beitrag zur Geschichte der Grundstoffindustrie im Nachkriegsdeutschland. PHD thesis, Universität-Gesamthochschule Siegen
Schlüpmann M, Bußmann M, Hachtel M, Haese U (2011) Gelbbauchunke - Bombina variegata In: Arbeitskreis Amphibien und Reptilien Nordrhein-Westfalen (ed) Handbuch der Amphibien und Reptilien Nordrhein-Westfalens. Laurenti, Bielefeld, pp 507–542
Schön I, Raepsaet A, Goddeeris B, Bauwens D, Mergeay J, Vanoverbeke J, Martens K (2011) High genetic diversity but limited gene flow in Flemish populations of the crested newt, Triturus cristatus. Belg J Zool 141:3–13
Sinsch U (2014) Movement ecology of amphibians: From individual migratory behaviour to spatially structured populations in heterogeneous landscapes. Can J Zool 92:491–502. https://doi.org/10.1139/cjz-2013-0028
Stevens VM, Verkenne C, Vandewoestijne S, Wesselingh RA, Baguette M (2006) Gene flow and functional connectivity in the natterjack toad. Mol Ecol 15:2333–2344. https://doi.org/10.1111/j.1365-294X.2006.02936.x
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL, Waller RW (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306(5702):1783–1786. https://doi.org/10.1126/science.1103538
Stuckas H, Tiedemann R (2006) Eight new microsatellite loci for the critically endangered fire-bellied toad Bombina bombina and their cross-species applicability among anurans. MolEcol Notes 6:150–152. https://doi.org/10.1111/j.1471-8286.2005.01171.x
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D (2016) Directional genetic differentiation and relative migration. Ecology Evolution 6:3461–3475. https://doi.org/10.1002/ece3.2096
Taft HR, Roff DA (2012) Do bottlenecks increase additive genetic variance? Conserv Gen 13:333–342. https://doi.org/10.1007/s10592-011-0285-y
Tournier E (2017) Endangered species conservation in peri-urban habitats. PHD thesis, Universita degli studi di Padova
Vacher JP, Ursenbacher S (2014) Structuration et caractérisation génétiques des populations de sonneur à ventre jaune Bombina variegata en Alsace. Technical report. Downloaded on 10 December 2019 at http://www.bufo-alsace.org/wp-content/uploads/2015/07/rapport_bombina_alsace-1.pdf
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Veith M (1996) Verbreitung und Gefährdung der Gelbbauchunke, Bombina v. variegata (Linnaeus, 1758), in Rheinland-Pfalz. Naturschutzreport 11:177–186
Verhoeven KJF, Macel M, Wolfe LM, Biere A (2011) Population admixture, biological invasions and the balance between local adaption and inbreeding depression. Proc R Soc B (2011) 278:2–8. https://doi.org/10.1098/rspb.2010.1272
Vörös J, Alcobendas M, Martinez-Solano I, Garcia-Paris M (2006) Evolution of Bombina bombina and Bombina variegata (Anura: Discoglossidae) in the Carpathian Basin: A history of repeated mt-DNA introgression across species. Mol Phylogenetics Evol 38:705–718. https://doi.org/10.1016/j.ympev.2005.08.010
Waraniak JM, Fisher JDL, Purcell K, Mushet DM, Stockwell CA (2019) Landscape genetics reveal broad and fine-scale population structure due to landscape features and climate history in the northern leopard frog (Rana pipiens) in North Dakota. Ecol Evol 9:1041–1060. https://doi.org/10.1002/ece3.4745
Weihmann F, Podloucky R, Hauswaldt S, Pröhl H (2009) Naturschutzgenetische Untersuchungen von Populationen der Gelbbauchunke (Bombina v. variegata) im südlichen Niedersachsen. Z Feldherpet 16:183–200
Weihmann F, Weihmann S, Weihmann T (2019) Conservation genetic analysis of a Central-European range-margin population of the yellow-bellied toad (Bombina v. variegata). Conserv Gen 20:557–569. https://doi.org/10.1007/s10592-019-01156-6
Weir BS, Cockerham CC (1984) Estimating F-Statistics for the Analysis of Population Structure. Evolution 38:1358–1370
Willi Y, Van Buskirk J, Hoffmann AA (2006) Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst 37:433–458. https://doi.org/10.1146/annurev.ecolsys.37.091305.110145
Zancolli G, Rödel MO, Steffan-Dewenter I, Storfer A (2014) Comparative landscape genetics of two river frog species occurring at different elevations on Mount Kilimanjaro. Mol Ecol 23:4989–5002. https://doi.org/10.1111/mec.12921
Zeisset I, BeebeeTJC (2008) Amphibian phylogeography: a model for understanding historical aspects of species distributions. Heredity 101:109–119. https://doi.org/10.1038/hdy.2008.30
Acknowledgements
We thank the Heinrich Böll Stiftung for the PhD grant (A.M. Hantzschmann). The authors are grateful to the Struktur- und Genehmigungsdirektion Nord; Koblenz; Germany; for funding and cooperation during the project; and for issuing permissions. The support of S. Wöhle; D. Trense and B. Nilow-Lange during the laboratory work and data analysis is greatly acknowledged. The authors also thank the Keramikmuseum Westerwald and A. Zeischka-Kenzler for support and providing literature. The manuscript benefitted greatly from the comments of two anonymous reviewers.
Funding
Open Access funding enabled and organized by Projekt DEAL..
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10592_2020_1320_MOESM1_ESM.tif
Supplementary file1 Suppl. Figure 1. Bayesian Information Criterion (BIC) values for different genetic clusters obtained through the find.clusters command for all retained Principal Components (n = 30) using the R package adegenet (DOC 17 kb)
10592_2020_1320_MOESM2_ESM.tif
Supplementary file2 Suppl. Figure 2. Results of the DAPC (discriminant analysis of principal components) on localities in the Westerwald region (DOC 9416 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hantzschmann, A.M., Sinsch, U., Göttlicher, C. et al. Conservation genetics of yellow-bellied toads (Bombina variegata): a matter of geographical scale and isolation. Conserv Genet 22, 83–96 (2021). https://doi.org/10.1007/s10592-020-01320-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-020-01320-3