
Abstract
Molecular methods can play a crucial role in species management and conservation. Despite the usefulness of genetic approaches, they are often not explicitly included as part of species recovery plans and conservation practises. The Natterjack toad (Epidalea calamita) is regionally Red-Listed as Endangered in Ireland. The species is declining and is now present at just seven sites within a highly restricted range. This study used 13 highly polymorphic microsatellite markers to analyse the population genetic diversity and structure. Genetic diversity was high with expected heterozygosity between 0.55 and 0.61 and allelic richness between 4.77 and 5.92. Effective population sizes were small (Ne < 100 individuals), but not abnormal for pond breeding amphibians. However, there was no evidence of historical or contemporary genetic bottlenecks or high levels of inbreeding. We identified a positive relationship between Ne and breeding pond surface area, suggesting that environmental factors are a key determinant of population size. Significant genetic structuring was detected throughout the species’ range, and we identified four genetic entities that should be considered in the species’ conservation strategies. Management should focus on preventing further population declines and future loss of genetic diversity overall and within genetic entities while maintaining adequate local effective population size through site-specific protection, human-mediated translocations and head-start programs. The apparent high levels of genetic variation give hope for the conservation of Ireland’s rarest amphibian if appropriately protected and managed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Given the current global ‘amphibian crisis’ (IUCN 2020), comprehensive understanding of declining and threatened species’ genetic structure is essential for effective conservation strategies and population management (Emel and Storfer 2012). Small population size promotes loss of rare alleles, increased homozygosity and accumulation of detrimental recessive alleles (Westemeier et al. 1998; Madsen et al. 1999), resulting in local declines and elevated extinction risk (Frankel and Soule 1981; Shaffer 1990; Hedrick 2001; Emel and Storfer 2012). Amphibians are particularly at risk of reduced genetic diversity due to their sensitivity to environmental change, low dispersal capability, and strong natal philopatry (Beebee and Griffiths 2005; Beebee 2005; Kraaijeveld-Smit et al. 2005).
Low genetic variability in amphibians has been linked to an increased presence of physical abnormalities (Hitchings and Beebee 1988) and negative impacts on oxygen consumption (Mitton et al. 1986), immune response (O’Brien and Evermann 1988; Altizer et al. 2003; Cabido et al. 2010, 2011), ability to compete for resources (Rowe and Beebee 2005), clutch size (McAlpine 1993), hatching success (Blaustein et al. 1994) and growth rates (Rowe and Beebee 2004). For example, loss of genetic diversity in the Italian agile frog (Rana latastei) has been related to higher susceptibility to Ranavirus (Pearman and Garner 2005). Such findings are a concern given the major role disease plays in global amphibian declines (Daszak et al. 2003; Skerratt et al. 2007; Wake and Vredenburg, 2008). Thus, omitting genetics from design of species management plans may lead to inappropriate allocation of resources, ineffective conservation strategies, and further population declines (Frankham 2003; Cushman 2006; Noel et al. 2007).
Species genetic management is often overlooked in conservation programmes due to lack of resources or expertise (Taylor et al. 2017). Understanding of the genetic structure of endangered populations can inform a targeted management approach. Genetic analysis permits assignment of individuals to genetic clusters independent of the sampling location and identifies discrete units for management (Moritz 1994; Palsbøll et al. 2007; Schwartz et al. 2007). Below the species-level, conservation priorities focus on intraspecific diversity for evolutionary plasticity and adaptability in response to environmental change (Shaffer et al. 2015). For instance, genetic analysis of the Australian agamid lizard (Diporiphora nobbi) identified several Evolutionary Significant Units (ESUs), emphasising the importance of local populations of a widespread species in harbouring intraspecific genetic diversity (Driscoll and Hardy 2005). Conversely, where genetic diversity is critically low, conservation managers may need to actively consider genetic rescue by deliberately maximising assisted gene flow between distinct units (Beauclerc et al. 2010; Whiteley et al. 2015). An isolated population of adders (Vipera berus) in Sweden dramatically increased in number after the introduction of new genes to avoid severe inbreeding averting local extirpation (Madsen et al. 1999, 2004). Thus, the approach taken to genetic management is species-, population- and context-dependent.
Effective population size (Ne) plays a crucial role in predicting a population’s extinction risk and is often more valuable than estimating absolute census size (Beebee 2005). It is defined as the size of an idealized population that meets Hardy–Weinberg assumptions showing the same rate of loss of genetic diversity as the target population (Wright 1931). Effective population size can be derived from genetic data with no additional life-history information (Schwartz et al. 1998). Furthermore, molecular markers allow historical patterns of population decline or expansion to be inferred, informing contemporary management. There are many factors that can influence Ne, including census size, sex ratio and past declines, breeding strategy, habitat carrying capacity and connectivity between populations (Waples and Gaggiotti 2006; Mills 2007). Wang et al. (2011) found that suitability of breeding habitats can contribute to variation in Ne among populations of the California tiger salamander (Ambystoma californiense). Thus, understanding the relationship between demographic parameters, key habitat features, and effective population size is essential for a comprehensive conservation approach.
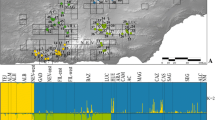
Our study assesses the impact of small population size and recent declines on the genetic structure of the Natterjack toad (Epidalea calamita), to inform species conservation management. In Ireland, the species is at its northwesternmost range edge margin (Gasc et al. 1997) and is highly range restricted, occurring in seven isolated breeding sites on the coast of Co. Kerry (Fig. 1; Reyne et al. 2021a). One of those breeding sites (Caherdaniel) was established in the 1990s as a result of introduction of individuals from the Magharees to increase the species geographic range. Regardless of conservation efforts, the species is in decline and is regionally Red-Listed as Endangered in Ireland (King et al. 2011). The Natterjack appears native to Ireland and, despite reporting an apparent bottleneck, May and Beebee (2008) suggested its genetic diversity was “reassuringly high” despite a lack of gene flow from other populations. However, low levels of polymorphism in the selected microsatellite markers may have contributed to their low reported allelic richness (1.86–2.89). Since then, the Natterjack toad population in Ireland has continued to decline, with > 90% loss of spawning at some breeding sites (Reyne et al. 2019, 2021a). In an effort to improve habitat availability, over 100 new breeding ponds were created as part of an agri-environment scheme since 2008 by the National Parks and Wildlife Service (NPWS). NPWS now manages a ‘head-start and translocation’ programme through which eggs strings and tadpoles are collected annually, raised in captivity and metamorph toadlets released back into the wild, supplementing existing populations as part of assisted migration and translocation to newly created ponds. Since 2016, 13 translocations have been performed to aid colonisation of artificial ponds. As yet, there are no data available to assess the head-start and translocation program success, as breeding will occur 4 to 5 years post release when toads have reached sexual maturity and return to the ponds for breeding (Beebee 1979). A primary goal of ex situ conservation is to preserve maximum intraspecific genetic variability to ensure long-term survival (Pelletier et al. 2009). However, inbreeding is a major problem in many ex situ breeding programs (Witzenberger and Hochkirch 2011; Santymire et al. 2019; Phillips et al. 2020) with success often determined by the appropriate selection of founders from source populations (Tzika et al. 2009). Selecting individuals and populations for ex situ breeding and translocations should, therefore, be informed by empirical genetic data ensuring provenance and/or appropriate management of genetic lineages (Witzenberger and Hochkirch 2011; IUCN/SSC 2013). Hence, the specific objectives of our study were to: (i) provide genetic characterisation of each breeding site, (ii) reconstruct parentage of offspring samples, (iii) estimate the effective and census population sizes, (iv) evaluate the impact of pond characteristics on effective population size, (v) detect any genetic bottleneck effect(s) due to historical or recent declines, (vi) quantify genetic differentiation among breeding sites, (vii) designated discrete conservation genetic entities, if appropriate, and (viii) provide management recommendations.

Map showing the location of the Natterjack toad breeding ponds in Co Kerry, Ireland. Colours represent different breeding sites and numbers represent number of breeding ponds per site
Methods
Field surveys and sample collection
We conducted fieldwork in 2017 during the Natterjack toad’s breeding season (April–July). Breeding ponds were visited every 7 to 10 days throughout the duration of the breeding season. We recorded the total number of egg strings in each pond and measured the size (surface area) of each breeding site. For full details of sampling methods see Reyne et al. (2019). We collected minimum 30 DNA samples per breeding site from different, well-developed Natterjack toad egg strings (embryos with clearly defined head and tail) within the same pond or whole tadpoles from different breeding ponds to avoid analysing full siblings (see Fig. 1 for the location of the breeding sites and number of breeding ponds/areas per site). We did not sample any egg strings or tadpoles that were part of the head-start and translocation program i.e., translocated between sites. Samples were preserved in 100% ethanol at ambient temperature until extraction.
Genotyping and data validation
Genomic DNA was extracted following a high salt extraction protocol (Supporting Protocol S1). Thirteen highly polymorphic fluorescently labelled microsatellite markers were amplified (Table 1). PCR reactions were performed in two multiplexes, and forward primers were labelled with 6-FAM™, NED™ and VIC™ fluorescent dye (Applied Biosystems, Integrated DNA Technologies). Multiplex PCR reactions had a total volume of 10 µl, and contained 1 µl of genomic DNA, 5 µl Type-it Multiplex PCR Master Mix (Qiagen) and primer mix of labelled forward and reverse with 0.1–0.3 µM final concentrations. PCR conditions were: an initial denaturation of 95 °C for 15 min; followed by 35 cycles of 94 °C for 30 s, annealing temperature (Ta) of 55 °C or 58 °C for 90 s for multiplex one and two respectively, and 72 °C for 60 s, with a final extension at 60 °C for 30 min. Samples were randomized across breeding sites during the genotyping analysis to avoid bias and negative controls were used throughout. PCR products were diluted ten times with ddH2O, and 1 µl diluted PCR product, was added to 8.95 µl Hi-Di™ Formamide (Thermo Scientific) and 0.05 µl GeneScan™ 600LIZ ladder (Applied Biosystems). Fragment analysis was performed on ABI 3730xl Genetic Analyzer (Applied Biosystems).
Alleles were scored with GeneMarker® V1.8. We rounded all genotype calls to an even or odd number based on the known motif of respective locus. Differences between assigned and actual allele size were between 0.1 and 1 bp. To calculate potential genotyping errors (Bonin et al. 2004; Pompanon et al. 2005), 28 samples (approx. 10%) were selected randomly using random number function in Microsoft Excel (2016), genotyped three times, and genotyping error rates (allelic dropout and false allele) were calculated using PEDANT v.1.0 software (Johnson and Haydon 2007). To calculate error rates from three repeated genotypes, we compared each repeat and averaged the error estimates, as recommended by Johnson (2007).
Genetic diversity
To quantify genetic diversity, we estimated allelic richness (AR), observed heterozygosity (HO), expected heterozygosity (HE) and inbreeding coefficient (FIT) for each locus and breeding site using FSTAT version 2.9.4 (Goudet 1995) and Genepop 4.2 (Raymond and Rousset 1995). We also calculated rarefied private allelic richness (AP) in HP-rare (Kalinowski 2004, 2005). Compliance with Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) was tested among loci. Statistical significance was adjusted for multiple comparisons using strict Bonferroni correction.
Polygyny
Polygyny is a common anuran breeding strategy. While multiple paternity of egg clutches as a result of polyandrous mating or clutch piracy have been observed in several amphibian species (e.g., VietIs et al. 2004; Knopp and Merilä 2009; Byrne and Roberts 2012), it is unlikely for the Natterjack toad due to species segregated-pair breeding behaviour (May et al. 2011). Polygyny was estimated following Ficetola et al. (2010). We used the computer software COLONY (Jones and Wang 2010) to estimate half-sibling groups among the sampled eggs and tadpoles. We assumed that all eggs and tadpoles had different mothers based on the sample collection method (i.e., samples were collected from different egg strings or different breeding ponds), but that one male could fertilise more than one egg string. The reconstructed sibships were used to assess the number of egg strings fertilized by each male. We identified all half-siblings and calculated the degree of polygyny at a breeding site level as the average number of egg strings fertilized by reproductive males. Eggs and tadpoles without half-siblings were treated as the offspring of males that fertilized only one egg string. Variance in reproductive success at a breeding site level was calculated as the variance in the number of egg strings fertilized by breeding males. We estimated Spearman’s rank correlations to investigate relationships among polygyny, variance in reproductive success and number of egg strings. All statistical analyses in the study were performed in R 4.0.0. using the stats package (R Core Team 2019) unless stated overwise.
Population measures
We used egg string counts to monitor population size, a widely used method by previous Natterjack toad surveys in Great Britain (Smith and Skelcher 2019) and Ireland (Bécart et al. 2007; Sweeney et al. 2013; Reyne et al. 2019). Females typically deposit one egg string per year (Buckley and Beebee 2004); thus the number of egg strings can be used as a proxy for the female breeding population size. We plotted the cumulative number of egg strings at each pond visit for each population and the asymptote was calculated using a generalized additive model (GAM) which allowed 95% confidence intervals to be associated with counts. The lower confidence limit (LCL) accounted for potential double counting and the upper confidence limit (UCL) accounted for egg strings potentially being missed.
We estimated effective population size (Ne) using LDNe 1.0 software (Waples and Do 2008). Any bias caused by a small sample size is corrected by implementing the bias-correction method of Waples (2006). We ran the model with three different critical values (Pcrit) of allele frequency. Pcrit was set at 0.05, 0.02 and 0.01 which means that all alleles with frequency lower than the critical value were excluded from analysis (Waples 2006). We used linear regression to evaluate the role of number of breeding females in explaining Ne.
Breeding area
Total available area to breed at each breeding site was calculated as a sum of pond surface areas where breeding activity was recorded. We performed linear regressions between available breeding surface area and each of the following response variables: effective population size (Ne), mean observed heterozygosity (HO), mean allelic richness (AR) and polygyny to examine the relationship between breeding pond characteristics and genetic diversity.
Bottlenecks
BOTTLENECK v.1.2.0.2 was used to identify that recently had undergone a reduction in effective population size through testing for significant deviations from the mutation-drift equilibrium. The program was run under two mutational models for microsatellite data: two-phased (TPM) and stepwise mutation (SMM). The TPM model was run with 95% single-step mutations, 5% multiple-step mutations and variance among multiple steps of 12, as recommended by Piry et al. (1999). A Wilcoxon single-rank test was used to test for heterozygosity excess. Significant results of heterozygosity excess indicate evidence of a recent reduction in the effective population size (Piry et al. 1999). We also performed a two-way ANOVA between mean observed heterozygosity and mean allelic richness across markers and breeding sites to compare genetic diversity between stable and declining populations.
Population genetic structure
We performed discriminant analysis of principal components (DAPC), a multivariate analysis which is free from HWE assumptions and shows greater resolution of population structure when levels of genetic differentiation are low (Jombart et al. 2010). We used snapclust clustering algorithm to identify optimal number of genetic clusters (K) and assign individuals to panmictic populations (Beugin et al. 2018). The optimal number of clusters to retain in the analysis was identified using the Akaike Information Criterion (Akaike 1998) and the Kullback Information Criterion (Cavanaugh 1999) where lower values indicate better fit. Analyses were carried out in the adegent package in R following Jombat (2008) and Beugin et al. (2018).
FST (Weir and Cockerham 1984) was used to investigate patterns of differentiation among populations and as an indirect measurement of historical gene flow. We tested for significance across these comparisons using 10,000 permutations. Statistical significance was adjusted for multiple comparisons using strict Bonferroni correction. Analysis was done using R package hierfstat (Goudet 2005) and FSTAT v2.9.4 (Goudet 1995). Genetic structure was also assessed using analysis of molecular variance (AMOVA, Excoffier et al. 1992) which calculates the proportion of the total genetic variance originating within and between breeding sites. Permutation tests were performed at three hierarchical levels: among breeding sites, among individuals within breeding sites and within individuals. Analyses were conducted in Arlequin v3.5.2.2. (Excoffier and Lischer 2010).
Results
In total, 316 Natterjack toad samples were collected from all breeding sites in Ireland and successfully genotyped. Sample sizes varied between 32 and 58 individuals (Table 2). All 13 microsatellite loci amplified successfully. Genotyping error rate varied among loci ranging from 0 to 0.10 for allelic dropout and from 0 to 0.03 for false alleles (Table S1). All microsatellite markers were polymorphic with allelic richness ranging from 4.77 to 5.69 (Table 2). Mean observed and expected heterozygosity per locus were 0.54 and 0.59 respectively (Table S2). LD analysis performed 78 pairwise comparisons between loci, identifying 28 linked pairs (36%, Table S3). At a population level, Glenbeigh had the lowest (0.49) and Caherdaniel the highest (0.59) observed heterozygosity (Table 2). The mean inbreeding coefficient across all populations was low (ranged 0.04–0.16; Table 2). The breeding sites and overall, the Natterjack toad population in Ireland were not in HWE (p = 0.01).
The number of egg strings fertilized by individual males was between 1 and 8. The degree of polygyny at a population level varied between 2.13 and 3.73 (Table 2). Variance of male breeding success differed strongly among populations and ranged from 1.98 to 13.22 (Table 2). There was no significant relationship between polygyny, variance of breeding success and number of egg strings i.e., breeding females (p > 0.05).
In total, we recorded 1457 egg strings. There was a significant sigmoidal accumulation of egg strings for all populations resulting in narrow 95% confidence intervals (Fig. S1; Table 3). The narrow 95% confidence intervals for most populations are indicative of reasonably precise estimates. Based on the number of egg strings, the most productive population was at the Magharees sand dune and the least productive at Inch (Table 3). Estimates of Ne were low, ranging from 19 at Caherdaniel to 519 at Yganavan but typically < 100 individuals at most sites (Table 3). There was no significant relationship between the number of breeding females and Ne regardless of the critical value of allele frequencies used to calculate Ne (p > 0.05).
Linear regression suggested no relationship between available breeding surface area and mean allelic richness AR (Fdf=1,5 = 1.658, p = 0.254, R2 = 0.249), mean observed heterozygosity HO (Fdf=1,5 = 0.383, p = 0.563, R2 = 0.071) or degree of polygyny (Fdf=1,5 = 3.037, p = 0.142, R2 = 0.378). There was a positive relationship between breeding surface area and Ne when the critical value of allele frequency was 0.05 and 0.01 (Ne 0.05: Fdf=1,5 = 136.195, p < 0.001, R2 = 0.965; Ne 0.01: Fdf=1,5 = 37.430, p = 0.007, R2 = 0.882) but the relationship was less clear with an allele frequency of 0.02 (Ne 0.02: Fdf=1,5 = 4.921, p = 0.077, R2 = 0.496).
BOTTLENECK analysis suggested significant deviations from mutational drift equilibrium for some breeding sites (Table 4). However, there is no evidence for significant heterozygosity excess at any breeding site. In addition, there was no evidence of mode-shift in allele frequencies as all distributions were L-shaped suggesting no recent reduction in the effective population size. Additionally, a two-way ANOVA showed no significant difference in the allelic richness (Fdf=6,84 = 0.594, p = 0.735) and observed heterozygosity (Fdf=6,84 = 0.490, p = 0.814) among breeding sites.
Cluster analysis suggested an optimal number of K = 5 using AIC and K = 4 using KIC (Fig. S2). KIC approach indicated a better fit (sharp decrease in the values followed by a sharp increase), hence the number of clusters retained in the analysis was four. DAPC indicated that the Magharees differed from the rest of the breeding sites with no overlap (Fig. 2A). The Caherdaniel was also distinct from all other breeding sites, although it had similar DAPC axis 1 scores (which carried most of the observed variation) to the Magharees breeding site from which it was derived. The Castlemaine Harbour sites (Glenbeigh, Yganavan, Inch, Dooks and Roscullen) clustered together but re-analysis after the exclusion of the Magharees and Caherdaniel suggested Roscullen was distinct from the rest whilst Glenbeigh had minimal overlap (Fig. 2B). Snapclust analysis revealed similar patterns where Magharees and Caherdaniel breeding sites were different from the rest of the breeding aggregations; however it failed to clearly differentiate among the Castlemaine Harbour breeding sites (Fig. S3). Based on our analysis, we identified four genetic entities: (1) Magharees, (2) Caherdaniel, (3) Roscullen and (4) the remainder of the Castlemaine Harbour sites i.e., Dooks, Glenbeigh, Yganavan and Inch.

Discriminant analysis of principal component (DAPC) of Natterjack toad populations. Ordination plots shows the first two principal components of the DAPC for A all seven breeding sites and B separate for the five Castlemaine Harbour sites. Dots on the DAPC graphs present individuals. Labels are placed at the centre of dispersion for each population. Eigen values suggest that the first two components explain the biggest genetic variation in the dataset. Marginal density graphs show distribution of data for DAPC values
AMOVA analysis suggested most molecular variation (96.43%) was attributed to within individuals while variation between breeding sites accounted for 4.58% (Table 5). AMOVA detected significant (p < 0.001) differentiation among sites. The pairwise FST values suggested higher levels of gene flow between Glenbeigh, Yganavan, Dooks and Inch with FST ranging between 0.016 and 0.020 (Table 6). Higher FST values (> 0.05) were observed between Magharees and the rest of the populations apart from Caherdaniel.
Discussion
The regionally Red-Listed Irish Natterjack toad population, whilst having undergone substantial declines in census population size (Reyne et al. 2021a), exhibited no evidence of genetic bottlenecks or inbreeding with relatively high genetic diversity (allelic richness and heterozygosity), despite low effective population sizes. Analysis of population structure suggested four distinct genetic entities that should be considered in species conservation programmes.
All seven Natterjack toad breeding sites surveyed in Ireland were polymorphic at the 13 microsatellite loci. Expected heterozygosity (0.55–0.61) and observed heterozygosity (0.49–0.59) were higher than previous estimates for the same populations in Ireland and higher than most estimates for Natterjack toad populations throughout Europe (Table S4). However, estimates of heterozygosity and allelic richness are not directly comparable due to the use of a different set of microsatellite markers. It is also important to emphasis, that neutral and selected genes can provide different information regarding species genetic diversity and adaptative capacity. In this study, we used microsatellite markers consequently focusing only on the putatively neutral genetic diversity. However, it is increasingly recognised that adaptive genetic diversity is a more important indicator of species ability to adapt to changing environments and population persistence in the wild. Hence, shifts towards genome-wide single nucleotide polymorphisms (SNPs) or a combined approach can help develop a more functional perspective of the genetic diversity (Mable 2019).
Pond breeding amphibians are predisposed to lower levels of genetic variation compared to other taxa resulting from high amplitude fluctuations in population size (Alford and Richards 1999; Newman and Squire 2001). For example, the Natterjack toad typically breeds in ephemeral ponds where breeding success can depend on stochastic climatic variation i.e., wet warm years (Beebee and Griffiths 2000; Baker et al. 2011; Smith and Skelcher 2019; Reyne et al. 2021b). However, small, declining amphibian populations have been shown to retain high genetic diversity, for example, the black toad (Bufo exsul, Wang 2009), the critically endangered Montseny brook newt (Calotrina arnoli, Valbuena-Ureña et al. 2017) and the Hula painted frog (Latonia nigriventer, Perl et al. 2018), considered to be one of the world’s rarest amphibians.
One of the key factors determining population genetic diversity is effective population size, a metric that provides additional insights for management that complements census population size. Low Ne values can potentially indicate loss of genetic variability within a population (Frankham 2003; Ficetola et al. 2007; Palstra and Ruzzante 2008). Our estimates of the effective population size were low for all seven remaining populations (mainly < 100 individuals) despite reasonably observed high genetic diversity. The Ne for pond breeding amphibians is typically in tens rather than hundreds or thousands of individuals regardless of large census sizes (Beebee and Griffiths 2005). Similar estimates of the effective population size were reported for ten Natterjack toad populations in Europe (Faucher et al. 2017) and other amphibian species including the marsh frog (Rana redibunda, Zeisset and Beebee 2003), Italian agile frog (R. latastei, Ficetola et al. 2010) and common frog (Rana temporaria, Brede and Beebee 2006). This generally reflects the most common anuran breeding strategy of scramble competition where large males dominate breeding ponds and available females resulting in only a few males contributing to the gene pool of the next generation (Ficetola et al. 2010). Higher levels of polygyny and variance of male breeding success were recorded at Caherdaniel, Dooks and Glenbeigh where few, small breeding ponds are available. Variation in breeding habitat has previously been associated with differences in effective population size (Wang et al. 2011), and this is true for our study. Natterjack toad Ne was significantly associated with the breeding surface area available. The highest Ne was calculated for the Yganavan population where toads breed along the shore of a large lake, suggesting less competition among males (i.e., low polygyny). Populations with small available breeding habitat had smaller Ne, but it did not result in lower genetic diversity. Similar results were found for the California tiger salamander (A. californiense, Wang et al. 2011) highlighting the need for a more complete understanding of the parameters influencing Ne.
Despite small Ne and substantial declines in egg string numbers (breeding females) since 2004 for populations at Roscullen, Yganavan, Dooks and Glenbeigh (Reyne et al. 2019; 2021a), our analysis did not provide evidence of recent genetic bottlenecks. Dooks and Yganavan populations have declined by over 70% (Reyne et al. 2021a); however, migration between these sites may decrease the number of rare alleles, consequently masking any HE excess (Cornuet and Luikart 1996) or a lag between when population size reduction took place and an observable increase in the inbreeding coefficient suggesting that changes in the genetic diversity of the offspring are yet to be observed. The BOTTLENECK program has been reported to fail to detect known demographic bottlenecks (Whitehouse and Harley 2001) and might have limited statistical power (Peery et al. 2012). However, measures of allelic diversity have been shown to be good indicators of bottlenecks when comparable data on demographically stable populations are available (Spencer et al. 2000; Whitehouse and Harley 2001). The Magharees had the largest Natterjack toad population in Ireland accounting for 90% of the recorded egg strings deposited annually and this number has remained largely stable over time (Reyne et al. 2021a). However, allelic richness for the Magharees was among the lowest (4.79) in Ireland, though there was no significant difference in allelic richness among breeding sites. This would support the assertion of no decline in the effective population size consistent with a lack of evidence of a genetic bottleneck. Similarly, the Australian bell frog (Litoria aurea) has undergone an 80% population decline over the past 40 years with no apparent genetic bottleneck with levels of allelic richness not significantly different for 19 out of 21 populations when compared to a large demographically stable population (Burns et al. 2004).
The Natterjack toad in Ireland exhibits genetic structuring throughout its range with all sampled locations being significantly different. DAPC results imply that the Natterjack toad population in Ireland can be divided into four discrete management units: (1) Magharees, (2) Caherdaniel, (3) Roscullen, (4) Dooks, Glenbeigh, Yganavan and Inch. Amphibians exhibit strong site fidelity and limited dispersal and migration, resulting in low levels of gene flow among populations (Reading et al. 1991; Kusano et al. 1999; Pittman et al. 2008). Several studies suggested that highly structured populations are often typical of amphibians with distinction at scales less than 5 km (e.g., Shaffer and Breden 1989; Routman 1993; Driscoll 1998) questioning the notion of metapopulation dynamics (Marsh and Trenham 2001). For amphibian species with highly structured populations site-specific protection and human-mediated translocations/reintroductions may be critical management tools to preserve intraspecific genetic diversity (Shaffer et al. 2000).
Conservation recommendations should focus on maintaining high genetic diversity as well as protection of the genetic integrity of identified genetic entities. This can be achieved by maintaining adequate effective population size (Storfer et al. 2007; Wang 2009) especially in small and fragmented populations (Wang et al. 2011). The NPWS is not currently creating new ponds but should future measures to incentivise farmers to create new potential breeding ponds, and these should be clustered within and around each identified management unit. Furthermore, our results suggest that having large breeding ponds or high numbers of small ponds in close proximity can be particularly valuable for ensuring a large Ne associated with large breeding surface area and higher number of successfully mating male toads. The benefits of multiple pools in supporting Natterjack toad’s metapopulation structure have been clearly demonstrated in Britain after decades of active species conservation (Denton et al. 1997; Beebee 2014). The ongoing NPWS ‘head-start and translocation’ programme should be cognisant of our identified genetic entities when releasing toadlets back into the wild respecting their genetic provenance, not translocating individuals between genetic entities and selecting the source populations from geographically proximate sites within the same entity. The only exception is Caherdaniel, as the population is likely distinct due to founder effects, subsequent gene drift and lack of gene flow after its establishment using translocated individuals from the Magharees. Thus, should further translocations be required in the future to maintain the population these can be drawn from the original source population. The lowest observed heterozygosity and highest inbreeding coefficient were recorded for the Glenbeigh population. Inbred Natterjack toad’s tadpoles have slower growth rates and lower survival rates (Rowe and Beebee 2005). Thus, we have conservation concerns about the long-term survival of the Glenbeigh population. A genetic restoration program of a small and isolated Natterjack toad colony at Saltfleetby, Britain has led to increase in metamorph production and breeding success (Beebee 2014). Hence, consideration should be given to population supplementation at Glenbeigh with translocated individuals ideally from Yganavan as it is genetically the closest population, but alternatively from any population within the management unit. It is important to note that while genetically most relevant population is preferable as a source population, it becomes unfavourable if removal of those individuals might put the source population at risk. Hence, other factors like population size and trend, potential threats and pressures should be considered when selecting source populations.
There may be an aspiration that the Natterjack toad’s range in Ireland could be enlarged beyond its current highly restricted range, for example, by introducing animals to sites in the southwest occupied historically or introducing animals to suitable habitat in the west more generally which has never been occupied (for example, assisted migration of populations northward to track climate change). In these cases, donor populations should either be geographically proximate or, if a new location is distinct from existing genetic entities and isolated by barriers to dispersal, the Magharees population can be considered as donors due to the high genetic diversity and large population size. An experimental approach could be taken by mixing individuals from each of the genetic entity to create artificially high genetic diversity buffering any new population against local extirpation due to small initial effective population size (Beauclerc et al. 2010; IUCN/SSC 2013). Monitoring such populations would be warranted.
Our findings have important conservation implications for the management of the Natterjack toad, which is regionally Red-Listed as Endangered in Ireland. Populations appear to lack deficiency in allelic richness or heterozygosity despite low effective population sizes and there is no evidence of genetic bottlenecks despite declines in census size. We identified discrete genetic entities, which we urge species conservation programmes to consider when undertaking population supplementation, translocation or assisted migration. We also highlight the importance of long-term genetic monitoring especially of declining populations and at reintroduction sites. Apparent high levels of genetic variation gives hope for the conservation of Ireland’s rarest amphibian.
Data availability
The dataset generated during this research will be deposited on DRYAD.
Code availability
No custom codes were used; software and used codes are cited in the text.
Consent for publication
NA.
References
Akaike H (1998) Information theory and an extension of the maximum likelihood principle. In: Parzen E, Tanabe K, Kitagawa G (eds) Selected papers of Hirotugu Akaike. Springer series in statistics. Springer, New York, pp 199–213
Alford RA, Richards SJ (1999) Global amphibian declines: a problem in applied ecology. Annu Rev Ecol Evol Syst 30:133–165
Altizer S, Harrvell D, Friedle E (2003) Rapid evolutionary dynamics and disease threats to biodiversity. Trends Ecol Evol 18:589–596
Baker J, Beebee TJC, Buckley J, Gent T, Orchard D (2011) Amphibian habitat management handbook. Amphibian and Reptile Conservation, Bournemouth
Beauclerc KB, Johnson B, White BN (2010) Genetic rescue of an inbred captive population of the critically endangered Puerto Ricoan crested toad (Peltophryne lemur) by mixing lineages. Conserv Genet 11:21–32
Bécart E, Aubry A, Emmerson M (2007) Monitoring the conservation status of Natterjack Toad (Bufo calamita) in Ireland, 2004–2006 Irish Wildlife Manuals, No. 31. National Parks and Wildlife Service, Department of the Environment, Heritage and Local Government, Dublin.
Beebee TJC (1979) A review of scientific information pertaining to the Natterjack toad Bufo calamita throughout its geographical range. Biol Conserv 16:107–134
Beebee TJC (2005) Conservation genetics of amphibians. Heredity 95:423–427
Beebee TJC (2014) Amphibian conservation in Britain: a 40-year history. J Herpetol 48(1):2–12
Beebee TJC, Griffiths RA (2000) Amphibians and reptiles. A natural history of the British Herpetofauna. Harper Collins New Naturalist, London
Beebee TJC, Griffiths RA (2005) The amphibian decline crisis: a watershed for conservation biology? Biol Conserv 125:271–285
Beugin M-P, Gayet T, Pontier D, Devillard S, Jombart T (2018) A fast likelihood solution to the genetic clustering problem. Methods Ecol Evol 9:1006–1016
Blaustein AR, Wake DB, Sousa WP (1994) Amphibian declines—judging stability, persistence, and susceptibility of populations to local and global extinctions. Conserv Biol 8:60–71
Bonin A, Bellemain E, Eidesen PB, Pompanon F, Brochman C, Taberlet P (2004) How to track and assess genotyping errors in population genetics studies. Mol Ecol 13:3261–3273
Brede E, Beebee TJC (2006) Large variations in the ratio of effective breeding and census population sizes between two species of pond-breeding anurans. Biol J Linn Soc 89:365–372
Buckley J, Beebee TJC (2004) Monitoring the conservation status of an endangered amphibian: the Natterjack toad Bufo calamita in Britain. Anim Conserv 7:221–228
Burns EL, Eldridge MDB, Houlden BA (2004) Microsatellite variation and population structure in a declining Australian hylid Litoria aurea. Mol Ecol 13:1745–1757
Byrne PG, Roberts JD (2012) Evolutionary causes and consequences of sequential polyandry in anuran amphibians. Biol Rev 87:209–228
Cabido C, Garin-Barrio I, García-Azurmendi X, Rubio X, Gosá A (2010) Population differences in vulnerability to glyphosate of Natterjack Toad tadpoles. In: 2nd International symposium on the conservation of amphibians: Bufo calamita, Donostia/San Sebastian, Spain
Cabido C, Garin-Barrio I, Rubio X, Gosá A (2011) Pesticide vulnerability and genetic diversity: a cross-breeding experiment with two populations of Natterjack Toad. In: 3rd Congress of the Spanish Society of Evolutionary Biology, Madrid, Spain
Cavanaugh JE (1999) A large-sample model selection criterion based on Kullback’s symmetric divergence. Stat Probab Lett 43:33–343
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Daszak P, Cunningham AA, Hyatt AD (2003) Infectious disease and amphibian population declines. Divers Distrib 9:141–150
Denton JS, Hitchings SP, Beebee TJC, Gent A (1997) A recovery program for the Natterjack Toad (Bufo calamita) in Britain. Conserv Biol 11:1329–1338
Driscoll DA (1998) Genetic structure, metapopulation processes and evolution influence the conservation strategies for two endangered frog species. Biol Conserv 83:43–54
Driscoll DA, Hardy CM (2005) Dispersal and phylogeography of the agamid lizard Amphibolurus nobbi in fragmented and continuous habitat. Mol Ecol 14:1613–1629
Emel SL, Storfer A (2012) A decade of amphibian population genetic studies: synthesis and recommendations. Conserv Genet 13:1685–1689
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Faucher L, Godé C, Arnaud JF (2016) Development of nuclear microsatellite loci and mitochondrial single nucleotide polymorphisms for the Natterjack Toad, Bufo (Epidalea) calamita (Bufonidae), using next generation sequencing and competitive allele specific PCR (KASPar). J Hered 17(7):660–665
Faucher L, Henocq L, Vanappelghem C, Rondel S, Quevillart R, Gallina S, Gode C, Jaquiery J, Arnaud JF (2017) When new human-modified habits favour the expansion of an amphibian pioneer species: evolutionary history of the Natterjack toad (Epidalea calamita) in a coal basin. Mol Ecol 26(17):4434–4451
Ficetola GF, Garner TWJ, De Bernardi F (2007) Genetic diversity, but not hatching success, is jointly affected by post glacial colonization and isolation in the threatened frog, Rana latastei. Mol Ecol 16:1787–1797
Ficetola GF, Padoa-Schioppa E, Wang J, Garner TWJ (2010) Polygyny, census and effective population size in the threatened frog, Rana latastei. Anim Conserv 13:82–89
Frankel OH, Soule ME (1981) Conservation and evolution. Cambridge University Press, Cambridge
Frankham R (2003) Genetics and conservation biology. C R Biol 326(Supplement 1):22–29. ISSN 1631-0691
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Goudet J (2005) HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–218
Hedrick PW (2001) Conservation genetics: where are we now? Trends Evol Ecol 16:629–636
Hitchings SP, Beebee TJC (1988) Loss of genetic diversity and fitness in Common Toad (Bufo bufo) populations isolated by inimical habitat. J Evol Biol 11:269–283
IUCN (2020) The IUCN Red List of Threatened Species. Version 2020-1. https://www.iucnredlist.org. Accessed on 29 June 2020
IUCN/SSC (2013) Guidelines for reintroductions and other conservation translocations. Version 1.0. IUCN Species Survival Commission, Gland
Johnson P, Haydon DT (2007) Maximum-likelihood estimation of allelic dropout and false allele error rates from microsatellite genotypes in the absence of reference data. Genetics 175:827–842
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Res 10:551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x
Kalinowski ST (2005) Hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
King JL, Marnell F, Kingston N, Rosell R, Boylan P, Caffrey JM, Fitzpatrick Ú, Gargan PG, Kelly FL, O’Grady MF, Poole R, Roche WK, Cassidy D (2011). Ireland Red List No. 5: Amphibians, Reptiles and Freshwater Fish. National Parks and Wildlife Service, Department of Arts, Heritage and the Gaeltacht, Dublin.
Knopp T, Merilä J (2009) Multiple paternity in the moor frog, Rana arvalis. Amphib-Reptil 30:515–521
Kusano T, Maruyama K, Kanenko S (1999) Breeding site fidelity in the Japanese toad, Bufo japonicus formosus. Herpetol J 19:9–13
Mable BK (2019) Conservation of adaptive potential and functional diversity: integrating old and new approaches. Conserv Genet 20:89–100
Madsen T, Shine R, Olsson M, Wittzell H (1999) Restoration of an inbred adder population. Nature 402:34–35
Madsen T, Ujvari B, Olsson M (2004) Novel genes continue to enhance population growth in adders (Vipera berus). Biol Conserv 120:145–147
Marsh DM, Trenham PC (2001) Metapopulation dynamics and amphibian population. Conserv Biol 15:40–49
May S, Zeisset I, Beebee TJC (2011) Larval fitness and immunogenetic diversity in chytrid-infected and uninfected Natterjack toad (Bufo calamita) populations. Conserv Genet 12:805–811
McAlpine S (1993) Genetic heterozygosity and reproductive success in the green treefrog, Hyla cinerea. Heredity 70:553–558
Mills LS (2007) Conservation of wildlife populations: demography, genetics and management. Wiley-Blackwell, London
Mitton JB, Carey C, Kocher TD (1986) The relation of enzyme heterozygosity to standard and active oxygen-consumption and body size of tiger salamanders, Ambystoma tigrinum. Physiol Zool 59:574–582
Moritz C (1994) Defining ‘Evolutionary Significant Units’ for conservation. Trends Ecol Evol 9:373–375
Newman RA, Squire T (2001) Microsatellite variation and fine-scale population structure in the wood frog (Rana sylvatica). Mol Ecol 10:1087–1100
Noel S, Ouellet M, Galois P, Lapoint FJ (2007) Impact of urban fragmentation on the genetic structure of the eastern red-backed salamander. Conserv Genet 8:599–606
O’Brien SJ, Evermann JF (1988) Interactive influence of infectious disease and genetic diversity in natural populations. Trends Ecol Evol 3:254–259
Palsbøll PJ, Bérubé M, Allendorf FW (2007) Identification of management units using population genetic data. Trends Ecol Evol 22:11–16
Palstra FP, Ruzzante DE (2008) Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol 17:3428–3447
Pearman PB, Garner TWJ (2005) Susceptibility of Italian agile frog populations to an emerging strain of Ranavirus parallels population genetic diversity. Ecol Lett 8(4):401–408
Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Bëer E, Robinson S, Vasquez-Carrillo C, Pauli JN, Palsbǿll PJ (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418
Pelletier F, Reale D, Watters J, Boakes EH, Garant D (2009) Value of captive populations for quantitative genetics research. Trends Ecol Evol 24:263–270
Perl RGB, Geffen E, Malka Y, Barocas A, Renan S, Vences M, Gafny S (2018) Population genetic analysis of the recently rediscovered Hula painted frog (Latonia nigriventer) reveals high genetic diversity and low inbreeding. Sci Rep 8(1):5588
Phillips S, Geary M, Allmark M, Bennett S, Norman K, Ball RJ, Peters C, Muir AP (2020) The importance of long-term genetic monitoring of reintroduced populations: inbreeding in the Natterjack toad (Epidalea calamita). Herpetol J 30:59–167
Piry S, Luikart G, Cornuet JM (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90:502
Pittman SE, Jendrek AL, Price SJ, Dorcas ME (2008) Habitat selection and site fidelity of Cope’s gray treefrog (Hyla chrysoscelis) at the aquatic–terrestrial ecotone. J Herpetol 42(2):378–385
Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Genotyping errors: causes, consequences and solutions. Nat Rev Genet 6:847–859
R Core Team (2019) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/
Reading CJ, Loman L, Madsen T (1991) Breeding pond fidelity in the common toad, Bufo bufo. J Zool 225:201–211
Reyne M, Aubry A, Martin Y, Helyar S, Emmerson M, Reid N (2019) Natterjack toad (Epidalea calamita) monitoring and conservation status 2016–2018 Irish Wildlife Manual No. 107. National Parks and Wildlife Service, Department of Culture, Heritage and the Gaeltacht, Dublin
Reyne M, Aubry A, Emmerson M, Marnell F, Reid N (2021a) Conservation efforts fail to halt the decline of the regionally endangered Natterjack toad (Epidalea calamita) in Ireland. Biol Conserv 261:109228
Reyne M, McGowan NE, Flanagan J, Nolan P, Aubry A, Emmerson M, Marnell F, Reid N (2021b) Will predicted positive effects of climate change be enough to reverse declines of the regionally Endangered Natterjack toad in Ireland? Ecol Evol 11:5049–5064
Routman E (1993) Population structure and genetic diversity of metamorphic and paedomorphic populations of the tiger salamander, Ambystoma tigrinum. J Evol Biol 6:329–357
Rowe G, Beebee TJC (2004) Reconciling genetic and demographic estimators of effective population size in the anuran amphibian Bufo calamita. Conserv Genet 5:287–298
Rowe G, Beebee TJC (2005) Intraspecific competition disadvantages inbred Natterjack toad (Bufo calamita) genotypes over outbred ones in a shared pond environment. J Anim Ecol 74(1):71–76
Rowe G, Beebee TJC, Burke T (1997) PCR primers for polymorphic microsatellite loci in the Anuran amphibian Bufo calamita. Mol Ecol 6(4):401–402
Rowe G, Beebee TJC, Burke T (2000) A further four polymorphic microsatellite loci in the Natterjack toad Bufo calamita. Conserv Genet 1:371–372
Santymire RM, Lonsdorf EV, Lynch CM, Wildt DE, Marinari PE, Kreeger JS, Howard JG (2019) Inbreeding causes decreased seminal quality affecting pregnancy and litter size in the endangered black-footed ferret. Anim Conserv 22:331–340
Schwartz MK, Tallmon DA, Luikart G (1998) Review of DNA-based census and effective population size estimators. Anim Conserv 1:293–299
Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33
Shaffer HB (1990) Population viability analysis. Conserv Biol 4:39–40
Shaffer HB, Breden F (1989) The relationship between allozyme variation and life history: non-transforming salamanders are less variable. Copeia 4:1016–1023
Shaffer HB, Fellers GM, Magee A, Voss SR (2000) The genetics of amphibian declines: population substructure and molecular differentiation in the Yosemite toad, Bufo canorus (Anura, Bufonidae) based on single-strand conformation polymorphism analysis (SSCP) and mitochondrial DNA sequence data. Mol Ecol 9:245–257
Shaffer HB, Gidiş M, McCartney-Melstad E, Neal KM, Oyamaguchi HM, Tellez M, Toffelmier EM (2015) Conservation genetics and genomics of amphibians and reptiles. Annu Rev Anim Biosci 3(1):113–138
Smith PH, Skelcher G (2019) Effects of environmental factors and conservation measures on sand-dune population of the Natterjack toad (Epidalea calamita) in north-west England: a 31-year study. Herpetol J 29:146–154
Spencer CC, Neigel JF, Leberg PL (2000) Experimental evaluation of the usefulness of microsatellite DNA for detecting demographic bottlenecks. Mol Ecol 9:1517–1528
Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits LP (2007) Putting the ‘landscape’ in landscape genetics. Heredity 98:128–142
Sweeney P, Sweeney N, Hurley C (2013) Natterjack Toad monitoring project, 2011–2012 Irish Wildlife Manuals, No. 67. National Parks and Wildlife Service, Department of Arts, Heritage and the Gaeltacht, Dublin.
Taylor HR, Dussex N, Heezik Y (2017) Bridging the conservation genetics gap by identifying barriers to implementation for conservation practitioners. Glob Ecol Conserv 10:231–242
Tzika AC, Remy C, Gibson R, Milinkovitch MC (2009) Molecular genetic analysis of a captive-breeding program: the vulnerable endemic Jamaican yellow boa. Conserv Genet 10(1):69–77
Valbuena-Ureña E, Soler-Membrives A, Steinfartz S, Orozco-terWengel P, Carranza S (2017) No signs of inbreeding despite long-term isolation and habitat fragmentation in the critically endangered Montseny brook newt (Calotriton arnoldi). Heredity 118:424–435
Vietis DR, Nieto-Roman S, Barluenga M, Palanca A, Vences M, Meyer A (2004) Post-mating clutch piracy in an amphibian. Nature 431:305–308
Wake DB, Vredenburg VT (2008) Are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc Natl Acad Sci USA 105:11466–11473
Wang IJ (2009) Fine-scale population structure in a desert amphibian: landscape genetics of the black toad (Bufo exsul). Mol Ecol 18:3847–3856
Wang IJ, Johnson JR, Johnson BB, Shaffer HB (2011) Effective population size is strongly correlated with breeding pond size in the endangered California tiger Salamander, Ambystoma californiense. Conserv Genet 12:911–920
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Waples R, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8(4):753–756
Waples RS, Gaggiotti O (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439
Westemeier RL, Brawn JD, Simpson SA, Esker TL, Jansen RW, Walk JW, Kershner EL, Bouzat JL, Paige KN (1998) Tracking the long-term decline and recovery of an isolated population. Science 282:1695–1698
Whitehouse AM, Harley EH (2001) Post-bottleneck genetic diversity of elephant populations in South Africa, revealed using microsatellite analysis. Mol Ecol 10:2139–2149
Whiteley AR, Fitzpatrick SW, Funk WC, Tallmon DA (2015) Genetic rescue to the rescue. Trends Ecol Evol 30(1):42–49
Witzenberger KA, Hochkirch A (2011) Ex situ conservation genetics: a review of molecular studies on the genetic consequences of captive breeding programmes for endangered animal species. Biodivers Conserv 20(9):1843–1861
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Zeisset I, Beebee TJC (2003) Population genetics of a successful invader: the marsh frog Rana ridibunda in Britain. Mol Ecol 12:639–646
Kraaijeveld-Smit FJL, Beebee TJC, Griffiths RA, Moore RD, Schley L (2005) Low gene flow but high genetic diversity in the threatened Mallorcan midwife toad Alytes muletensis. Mol Ecol 14:3307–3315. https://doi.org/10.1111/j.1365-294X.2005.02614.x
Cushman SA (2006) Effects of habitat loss and fragmentation on amphibians: a review and prospectus. Biol Conserv 128:231–240. https://doi.org/10.1016/j.biocon.2005.09.031
Skerratt LF, Berger L, Speare R et al (2007) Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. EcoHealth 4:125. https://doi.org/10.1007/s10393-007-0093-5
Kalinowski ST (2004) Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet 5:539–543. https://doi.org/10.1023/B:COGE.0000041021.91777.1a
Acknowledgements
This study was commissioned and funded by the National Parks and Wildlife Service (NPWS), Department of Culture, Heritage and the Gaeltacht as a component part of a larger study won under open competitive tender by Queen’s University Belfast (QUB). Thanks to all those volunteers and work placement students who participated in Natterjack monitoring surveys. We are grateful to landowners for providing access to their land.
Funding
This study was supported by National Parks and Wildlife Service (NPWS), Department of Culture, Heritage and the Gaeltacht, Republic of Ireland.
Author information
Authors and Affiliations
Contributions
MR conducted field work, laboratory and statistical analysis, wrote the manuscript. KD provided guidelines on laboratory procedures including DNA extraction methods, selection of primers, PCR optimization and interpretation of results. CM conducted part of the laboratory analysis and contributed to the statistical analysis. AA and ME assisted with planning the fieldwork, contributed to the conception and design of the study. FM substantially contributed to revising the manuscript. NR contributed throughout from conception, methodology, fieldwork, and data analysis to manuscript editing. SH was the Principal Investigator, supervised the molecular work and statistical analysis of genetic data contributing throughout from conception, methodology, and data analysis to manuscript editing. All authors discussed the results, contributed to the draft, read the final manuscript and gave approval for publication.
Clinical trials registration
NA.
Gels and blots/image manipulation
NA.
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest or competing interests.
Ethical approval
All samples were collected under license to capture protected wildlife animals Licence No. C098/2016 issued by the National Parks and Wildlife Service (NPWS), Department of Culture, Heritage and the Gaeltacht, Republic of Ireland. Tissue samples were collected only from already dead individuals.
Informed consent
NA (no human participants in the study).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reyne, M., Dicks, K., McFarlane, C. et al. Population genetic structure of the Natterjack toad (Epidalea calamita) in Ireland: implications for conservation management. Conserv Genet 23, 325–339 (2022). https://doi.org/10.1007/s10592-021-01421-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-021-01421-7