
Abstract
Litter meadows, historically established for litter production, are species-rich and diverse ecosystems. These meadows drastically declined during the last decades along with decreasing litter use in modern livestock housing. The aim of our study was to identify the drivers of genetic variation in litter meadow species. Therefore, we tested whether genetic diversity and differentiation depend on habitat age, landscape structure, habitat quality, and/or population size. We analysed 892 individuals of Angelica sylvestris, Filipendula ulmaria, and Succisa pratensis from 20 litter meadows across the Allgäu in Baden-Württemberg (Germany) using AFLP analyses. All study species showed moderate levels of genetic diversity, while genetic differentiation among populations was low. Neither genetic diversity nor differentiation were clearly driven by habitat age. However, landscape structure, habitat quality as well as population size revealed different impacts on the genetic diversity of our study species. Past and present landscape structures shaped the genetic diversity patterns of A. sylvestris and F. ulmaria. The genetic diversity of F. ulmaria populations was, moreover, influenced by the local habitat quality. S. pratensis populations seemed to be affected only by population size. All explanatory variables represent past as well as present gene flow patterns by anthropogenic land use. Therefore, we assume that genetic diversity and differentiation were shaped by both historical creation of litter meadows via hay transfer and present mowing with agricultural machines. These land use practices caused and still cause gene flow among populations in the declining habitats.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Litter meadows constitute valuable habitats for many specialised, rare, and endangered plant and animal species (Wheeler 1988). Therefore, these semi-natural grasslands belong to the most species-rich ecosystems in Central Europe (Kull and Zobel 1991) and represent key areas for biodiversity conservation in agricultural landscapes, despite their comparably short land use history and limited spatial distribution.
According to Poschlod (2017), the construction of railway lines opened up the Alpine foreland region at the end of the nineteenth century. Agricultural goods were imported and subsistence farming efforts became redundant. Farming practices consequently changed from laborious cultivation of arable fields to more efficient grassland management for livestock farming. During this time, straw, used as bedding in stables, became scarce. Therefore, litter meadows were established, either on fodder meadows or large wetlands and peatlands. Whereas sowing and/or planting of litter plants were recommended for the establishment in drained ponds or peat-mined areas, Stebler (1898) described four management treatments for the conversion of fodder meadows into litter meadows without ploughing: (i) late cutting over several years, (ii) waiver of fertilization, (iii) irrigation, and (iv) resowing seeds or planting seedlings. Moreover, litter meadows were established by hayseed application (Müller 1752). During the 1960s, litter meadow cultivation became redundant due to massive land use changes (Poschlod 2017). Slatted floors gained more relevance in animal husbandry and thus, liquid manure replaced solid manure as preferred fertilizer. Furthermore, mineral fertilizer became comparably cheap, leading to a transformation of unproductive litter meadows into more productive fodder meadows.
Nowadays, remaining litter meadows are threatened by land use intensification, abandonment, and habitat fragmentation (Billeter et al. 2002). Habitat fragmentation limits pollen and seed exchange, restricting gene flow among populations (Schmitt 1983; Steffan-Dewenter and Tscharntke 1999; Willerding and Poschlod 2002; Honnay et al. 2006) and increasing, therefore, the likelihood of inbreeding depression, the accumulation of deleterious mutations, and the extent of genetic drift (Young et al. 1996; Picó and Van Groenendael 2007). Consequently increased genetic differentiation and reduced genetic diversity (Barrett and Kohn 1991; McKay et al. 2005), may lower individual plant fitness and thus, increase their extinction risk (Ellstrand and Elam 1993; Young et al. 1996). Hence, the knowledge about potential impact factors on genetic variation patterns becomes highly relevant to protect genetic variation, as a fundamental level of biodiversity (May 1994).
Due to an outstanding land use history, litter meadows could be found either on historically old (‘ancient’) or historically young (‘recent’) sites. In this study, ancient sites were wet grasslands that have existed since at least the 1800s, while recent sites were artificially created on drained ponds during the 1900s. High gene flow at the time of establishment and afterwards may lead to comparable levels of genetic variation among populations on sites with different habitat age (Vandepitte et al. 2010). Nevertheless, the number and origin of colonists (Wade and McCauley 1988; Whitlock and McCauley 1990) as well as the rate of gene flow and selection after colonization (Dlugosch and Parker 2008) drive genetic variation patterns of recent populations. These populations may, therefore, show both reduced genetic variation due to bottlenecks and increased divergence among populations by selection (Wade and McCauley 1988; Dlugosch and Parker 2008). Previous studies observed already comparatively decreased genetic variation levels within and among populations on recent sites (Jacquemyn et al. 2004; Dlugosch and Parker 2008; Ramakrishnan et al. 2010). Hence, we expect an impact of habitat age on the genetic variation of typical litter meadow species.
Over the past century, biodiversity decline was mainly induced by habitat loss at local, regional, and global scales (Balmford et al. 2005). Small populations, suffering from disrupted mutualistic interactions with pollinators or seed dispersers (Tscharntke and Brandl 2004), show enhanced extinction rates due to increased levels of inbreeding, loss of genetic variation through genetic erosion, fitness decline, and loss of evolutionary adaptation potential (Young et al. 1996; Adriaens et al. 2006). Nevertheless, rescue effects may lead to increased colonisation and reduced extinction rates in highly connected sites (Brown and Kodric-Brown 1977). We hypothesize, therefore, an impact of habitat size and connectivity on genetic variation. Moreover, gene flow, seed dispersal, and establishment are influenced by land use patterns (Reitalu et al. 2010; Purschke et al. 2012) representing further determinants for gene flow and genetic variation in today’s fragmented landscapes. Populations are sometimes affected more by historic than by present landscape configurations due to a time lag in species response (Adriaens et al. 2006; Reisch et al. 2017). Hence, we included past as well as present landscape structures in our analyses.
Abandonment and a lack of biomass removal led to deteriorated habitat conditions in litter meadows; moss and/or litter layers build up and act as seed traps (Ruprecht and Szabó 2012), while increased vegetation height causes ground shadowing (Jensen and Gutekunst 2003). Germination as well as establishment of seedlings are consequently restrained (Maas 1988; Špačková and Lepš 2004; Poschlod and Biewer 2005). Populations may decrease in size and a decline in genetic variation becomes more likely (Billeter et al. 2002). Therefore, we hypothesized an impact of habitat quality on the genetic variation of common litter meadow species.
In modern fragmented landscapes, remaining litter meadows are often small, fragmented, and isolated. Populations on these sites are comparatively small and more vulnerable to demographic and environmental stochasticity, despite intact vegetation structure (Hooftman et al. 2003). These populations may suffer from reduced probabilities of gene flow, increased genetic drift, and enhanced levels of inbreeding (Van Treuren et al. 2005; Aguilar et al. 2008). They may, therefore, show lower genetic variability, reduced generative (Schmidt and Jensen 2000) as well as vegetative performance (de Jong and Klinkhamer 1994), and face a higher risk of extinction (Spielman et al. 2004; Ouborg et al. 2006). Various studies observed already a positive relationship between population size and genetic variation (Leimu et al. 2006). Therefore, we would expect a positive impact of population size on genetic variation as well.
A range of studies have investigated the impact of habitat age, past and present landscape structure, habitat quality, and population size on genetic variation in dry grassland habitats (e.g. Prentice et al. 2006; Schmidt et al. 2009; Baessler et al. 2010; Rosengren et al. 2013; Reisch et al. 2017). Nevertheless, studies concerning wet grassland habitats, such as litter meadows, are still scarce.
Therefore, we analysed the genetic variation of three widespread litter meadow species using amplified fragment length polymorphism (AFLP) analyses. We chose the mainly insect-pollinated perennials Angelica sylvestris, Filipendula ulmaria, and Succisa pratensis (Kühn et al. 2004) as suitable study species. We ranked linear regression models according to AICc values to shed light on the relative importance of environmental factors on genetic variation patterns of the studied litter meadow species. Hence, the land use history and thus, the habitat age of the studied litter meadows was reconstructed using historical cadastral maps from different points in time. Moreover, past and present landscape structures including distance to the nearest settlement, area size, total area of surrounding wet grasslands, and connectivity were quantified on the basis of historic (1800s) and present (2018) cadastral maps. Local habitat quality was investigated with regards to vegetation cover data and population size. Applying these methods we aimed at answering the following questions: (i) What is the impact of habitat age on genetic diversity? Are populations of different habitat age genetically differentiated? (ii) Is genetic diversity influenced by past and/or present landscape structure? (iii) How is genetic diversity shaped by present habitat quality and/or population size?
Methods
Study design
In our study, we analysed the genetic variation of three typical litter meadow species: Angelica sylvestris (Apicaceae; 2n = 22), Succisa pratensis (Dipsacaceae; 2n = 18), and Filipendula ulmaria (Rosaceae; 2n = 14). A. sylvestris and S. pratensis flower between July and September, while F. ulmaria is flowering from June to August. All study species are perennials with a mixed mating system, showing insect (e.g. bees, syrphids, wasps, beetles) as well as self-pollination (Kühn et al. 2004).
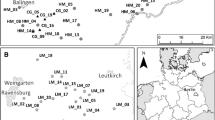
We selected 20 litter meadows distributed across the Allgäu in south-west Germany to study the effect of various environmental factors on genetic variation (Fig. 1, Table S1). The study region is characterized by a temperate climate with precipitation between 900 and 1600 mm/year and annual temperatures from 5.5 to 7.5 °C.

Geographic position and habitat age of the analysed populations of A. sylvestris, F. ulmaria, and S. pratensis
The land use history of the litter meadows was reconstructed with historical cadastral maps from three different points in time (1800s, 1910/1920s, and 1950s) to investigate the impact of habitat age on genetic variation (Table S2). We identified eleven sites as historically old (‘ancient’), which have been wet grasslands since before the 1800s, and nine sites as historically young (‘recent’), which developed from ponds during the 1900s, applying the software ArcGIS® 10.3.1 (Esri, Redlands, CA, USA).
In a next step, we digitized the oldest cadastral maps available for the area (1800s) as well as current topographical maps (2018) in a 3 km radius around each study site. Following landscape structures were chosen as potential explanatory variables for genetic diversity (Table S3): past and present distance to the nearest settlement, past and present total area of wet grasslands within each circle, and size of each study site. Moreover, we calculated past and present connectivity according to Hanski (1994) as Si = ∑j≠I exp(− αdij)Aj, where Si is the connectivity of the patch i, dij is the distance (km) between patches i and j, Aj is the area (ha) of the patch j, and α is the parameter of the exponential distribution setting the influence of distance on connectivity (Helm et al. 2006). Following Lindborg and Eriksson (2004) and Reitalu et al. (2010) α was set to one and not weighted by the dispersal abilities of the plant species in the community.
The cover of vascular plants, mosses, litter, and open soil were incorporated from vegetation surveys to examine the impact of the local habitat quality on genetic diversity (Table S4). Furthermore, we aimed to test the influence of the population size on genetic diversity. The population size of each species was, therefore, determined by counting the number of individuals in 10 to 15 1 m2 plots per study site. The average number of individuals per square meter was then multiplied with the present area size (Reisch et al. 2018). For those study sites, where no individual could be found within investigated plots although plant material was collected, the total number of individuals was set from 0 to 1 before multiplying (Table S4).
We sampled 16 individuals per population and species for molecular analyses to display more than 90% of the total genetic diversity (Leipold et al. 2018). The fresh leaf material was frozen in plastic bags in liquid nitrogen and stored at − 20 °C until DNA extraction.
Molecular analyses
The DNA extraction was carried out following the CTAB protocol from Rogers and Bendich (1994) modified by Reisch (2007). The DNA quality and concentration were determined with a spectrophotometer. Afterwards, the DNA samples were diluted to the same level of 7.8 ng DNA per µl H2O. We chose the analysis of amplified fragment length polymorphism (AFLP; Vos et al. 1995) for the analysis of the genetic variation within populations. The AFLP analyses were performed following the standardized protocol of Beckmann Coulter (Bylebyl et al. 2008). After screening 36 primer combinations per species, three species specific primer combinations were chosen for the selective amplification (Table S5). The automated sequencer GeXP (Beckmann Coulter) was used to separate the fluorescence-labelled DNA fragments by capillary gel electrophoresis. Virtual gels were analysed manually using the software Bionumerics 4.6 (Applied Maths, Kortrijk, Belgium). Only strong and clearly defined fragments were taken into account for further analyses, while samples without clear banding pattern, due to unsuccessful AFLP, were repeated or ultimately excluded.
A genotyping error rate was determined to ensure the reproducibility of the AFLP analyses (Bonin et al. 2004). Therefore, 10% of all investigated samples were analysed twice. The percentage of fragments showing differences between original and replicate lay at 3.61% (A. sylvestris), 5.36% (F. ulmaria), and 4.93% (S. pratensis).
Statistical analyses
The presence or absence of bands per particular fragment and individual was transformed into binary (0/1) matrices in Bionumerics 4.6. Based on these matrices we calculated the genetic diversity within each population in Popgene 32 (Yeh et al. 1997) as Nei’s gene diversity (GD) H = 1 − ∑(pi)2, with pi representing the allele frequency.
A Kruskal–Wallis test with a post-hoc-Dunn’s test (Dinno 2015) and following Bonferroni p-adjustment (Bland and Altman 1995) was calculated in R to compare Nei’s gene diversity on species level (R Core Team 1978). We further tested the dependence of Nei’s gene diversity on habitat age.
Hierarchical analyses of molecular variance (AMOVA) based on pairwise Euclidian distances between samples were calculated using the software GenAlEx 6.41 (Peakall and Smouse 2012). Hence, we analysed the genetic variation within and among populations as well as among populations on ancient and recent sites.
We computed Mantel tests with 999 permutations (Mantel 1967) to display correlations of geographic and genetic distances (ΦPT values calculated in the AMOVA) among populations.
Bayesian cluster analyses were performed using Structure 2.3.4 (Figure S1a–c) (Pritchard et al. 2000, 2010). Individuals from the data set are assigned randomly into groups, which are characterized by a set of allele frequencies at each locus. Hence, the number of groups was calculated using 10,000 Markov Chain Monte Carlo simulations with a burn-in period of 100,000 iterations. The analyses for the predefined value of K were run 20 times per K = 1–40, since the data set consists of an unknown number of K groups (Falush et al. 2007). The results were summarized employing the program Structure Harvester (Earl and vonHoldt 2012). The groups were assigned calculating ΔK in an ad hoc quantity procedure (Evanno et al. 2005) and the estimate of K was defined after the model, which showed the most consistent results for multiple runs and the highest probability of data.
Nei’s distance, the genetic distances among populations, was calculated with non-uniform prior distribution of allele frequencies in the program AFLPsurv, following Lynch and Milligan (1994). These Nei’s distances led to a consensus Neighbor-Net graph using the software SplitsTree 4.14.4 (Figure S2a–c) (Huson and Bryant 2006).
Wilcoxon–Mann–Whitney tests displayed possible differences between past and present landscape variables (Table S6). Correlation tests (Pearson correlation coefficients) were conducted to show potential collinearity between the explanatory variables (ii–xiii) and to avoid, therefore, false interpretation of the linear regression models (Table S7).
We formulated full linear starting models for each species in R (R Core Team 1978) describing the variation of Nei’s gene diversity in association to the scaled and centred explanatory variables: (i) habitat age (not scaled and centred), (ii) area size, (iii) past and (iv) present total area of wet meadows, (v) past and (vi) present distance to nearest settlement, and (vii) past and (viii) present connectivity, which were described above. Further data about the coverage of (ix) vascular plants, (x) mosses, (xi) litter, (xii) open soil, and (xiii) population size were included in these models. We ranked all possible linear models according to AICc values (Akaike Information Criterion corrected for small sample sizes) to detect the models with the highest information content (Burnham and Anderson 2002). Additionally, we performed a redundancy analysis (RDA) using the fragment frequency per population of all three study species and the explanatory variables as described in the linear models (Figure S3).
Results
Genetic diversity and differentiation
All studied species revealed similar levels of genetic diversity (Fig. 2). The mean genetic diversity of A. sylvestris populations was 0.216, ranging between 0.193 and 0.244. Similar values were found for F. ulmaria, whose mean genetic diversity was 0.216, with a minimum of 0.184 and a maximum of 0.248. Mean genetic diversity of S. pratensis was slightly lower with 0.210, varying from 0.167 to 0.242 (Table 1).

Boxplot showing Nei’s gene diversity of A. sylvestris (As), F. ulmaria (Fu), and S. pratensis (Sp)
Overall genetic differentiation among populations was low. The differentiation found among populations was estimated at 4% for A. sylvestris and at 5% for S. pratense. F. ulmaria showed the highest differentiation rate with 8% (Table 2). However, the AMOVAs showed no genetic differentiation among populations from ancient and recent sites.
Mantel tests revealed no significant correlation between genetic and geographic distances in either species (A. sylvestris: r = 0.0527, p = 0.052; F. ulmaria: r = 0.0003, p = 0.423; S. pratense: r = 0.0026, p = 0.334). Therefore, the studied populations are not likely to be isolated by distance.
Linear regression models
The AICc model selection generated significant models for all studied species (Table 3a–c). The model for A. sylvestris only included a negative association with the present area size, indicating a decrease of genetic diversity with increasing meadow area (Table 3a). Genetic diversity in S. pratensis was negatively affected by population size (Table 3c), explaining 21.51% of the observed variation. For F. ulmaria the model revealed more than one connection with the explanatory variables included (Table 3b). Present connectivity was the most important variable negatively influencing current genetic diversity, while past connectivity was positively associated. Present distance to the next settlement and present total area of wet meadows were positively related to genetic diversity in this species. Habitat age was also a significant predictor for genetic diversity, indicating a tendency for recent meadows to show higher genetic diversity levels. Both moss and vascular plant cover were positively associated with genetic diversity of F. ulmaria. Overall, the model accounted for 75.37% of the observed variation.
Discussion
Comprehensive biodiversity conservation should address genetic variation patterns representing the most fundamental level of biodiversity (May 1994). We aimed to identify the key drivers of genetic variation to ensure the development of applicable conservation methods and to protect the basis of biodiversity. Landscape structure, habitat quality, and population size revealed a significant impact on genetic diversity in the study species. More specifically, the surrounding landscape structure shaped genetic diversity patterns of A. sylvestris and F. ulmaria populations. Local habitat quality affected, moreover, the genetic diversity of F. ulmaria, whereas genetic diversity of S. pratensis populations was driven only by population size.
Genetic diversity and differentiation
We observed similar values of genetic variation within and among populations of all study species. The genetic diversity of these species slightly exceeded the values expected for insect pollinated species (Reisch and Bernhardt-Römermann 2014). Genetic differentiation among populations was generally low, with F. ulmaria showing the greatest differentiation. Spatial isolation did not play a major role for population differentiation.
Previous studies have shown that seeds are well transported among meadows via mowing machines (Strykstra et al. 1997). The litter meadows investigated here are typically mown by only few conservation managers once in the autumn (personal communication), enhancing gene flow by seed exchange among sites. Additionally, the occurrence of the study species is not strictly limited to litter meadows (Oberdorfer et al. 2001) and they are pollinated by a diverse group of insects (Kühn et al. 2004), providing many opportunities for gene flow by pollinators among sites, leading to an increase in genetic diversity and a decrease of genetic differentiation.
Other studies on genetic diversity and differentiation of the species analysed here are scarce. Only the effect of inbreeding and population size on the genetic variation of S. pratensis was studied previously using allozyme electrophoresis (Vergeer et al. 2003). Therefore, the genetic variation observed in these species is not directly comparable with other studies.
Effects of habitat age on genetic variation
Levels of genetic diversity in all three study species were similar among populations on ancient and recent sites. Additionally, habitat age revealed no significant impact on genetic diversity in A. sylvestris and S. pratensis in the linear regression models. This result stands in contrast to the studies of Jacquemyn et al. (2004) and Rosengren et al. (2013), who observed a comparatively lower genetic diversity on recent sites, e.g. in the moss species Homalothecium lutescens. However, historic management practices of sowing, hay and seedling transfer for the establishment and maintenance of litter meadows (Poschlod and Fischer 2016; Poschlod 2017) likely supported high levels of gene flow among ancient and recent sites. Moreover, all study species are pollinated by numerous different insects (Kühn et al. 2004), increasing the levels of gene flow among closely located sites. Thus, gene flow by pollinators and seed dispersal at the time of founding and afterwards might reduce the effects of habitat age (Vandepitte et al. 2010).
Habitat age was a significant predictor for genetic diversity patterns of F. ulmaria, revealing a tendency of more recent sites to show greater diversity values. However, the variable ‘habitat age’ was possibly included by the model selection algorithm to correct for the overestimation of past connectivity, which is significantly lower today. Therefore, we conclude that habitat age generally had little impact on genetic diversity of our study species.
Furthermore, we observed no significant differentiation among populations concerning habitat age. The practice of litter meadow establishment and traditional management practices ensured high levels of gene flow in the past. Today, seeds are still comparatively well transported via mowing machines among litter meadows (Strykstra et al. 1997). These land use practices supported and still support relatively high levels of gene flow, preventing genetic differentiation among populations on ancient and recent sites.
Effects of landscape structure on genetic diversity
Genetic diversity in A. sylvestris was negatively associated with the area of the respective litter meadow, indicating that larger meadows have lower genetic diversity. Larger habitats are expected to sustain larger populations and thus, also higher genetic diversity (Ouborg et al. 2006). In the case of A. sylvestris neither genetic diversity nor habitat size correlated with population size. A. sylvestris might colonize microsites instead of whole meadows due to variable local habitat conditions and is also not limited to litter meadows as habitat, which might falsify the impact of population size. Furthermore, habitat size was determined via topographic maps leading to a potential over- or underestimation of litter meadows’ habitat size. Therefore, we assume no or only a weak impact of habitat size on the genetic diversity of A. sylvestris populations.
Past and present landscape structures revealed the greatest impact on the genetic diversity of F. ulmaria populations. The total present area of wet meadows, past and present connectivity, and the present distance to the next settlement were associated with genetic diversity levels. All these factors have previously been shown to influence genetic diversity in grassland species (Jacquemyn et al. 2004; Reitalu et al. 2010; Münzbergová et al. 2013).
Genetic diversity in F. ulmaria increased with the present area of wet meadows around the studied populations. A large patch size and a high proportion of habitats within a geographic region is frequently found to increase genetic diversity by improving patch connectivity via pollinators or other gene flow vectors (Ouborg et al. 2006; Prentice et al. 2006). Gene flow among closely located patches decreases the effects of inbreeding and genetic drift and thus, maintains high genetic diversity (Aguilar et al. 2008).
We found a positive impact of past connectivity on the genetic diversity in F. ulmaria complying with the findings of Münzbergová et al. (2013), who observed a positive effect of historic habitat connectivity on genetic diversity of S. pratensis. In the past, traditional management of litter meadows included frequent sowing or transplanting of plant material to increase the vegetation cover of desired litter producing species (Poschlod 2017). These management practices, which may have positively affected undesired species as well, maintained high gene flow levels across the whole region. High connectivity among sites may increase colonization and reduce extinction rates, explaining the positive effect of past connectivity on the genetic diversity of F. ulmaria. However, present connectivity revealed an opposite effect on the genetic diversity in F. ulmaria. The cultivation of litter meadows became redundant during the last decades and thus, remaining species-rich litter meadows within the study region are managed by only few conservation managers today (personal communication). Moreover, seeds of all study species are fully developed during mowing season in late autumn (Poschlod et al. 2003) and are likely to be transported via mowing machines (Strykstra et al. 1997), creating ‘too much’ gene flow among populations. Exceptionally high levels of gene flow may induce an impoverishment of the local gene pool due to ‘genetic ‘swamping’ and thus, cause a negative impact of present habitat connectivity on genetic diversity in F. ulmaria.
The present distance to the nearest settlement revealed a positive impact on the genetic diversity of F. ulmaria. It is generally accepted that anthropogenic disturbance levels decrease with increasing distance to the next urban area. Since comparatively low levels of man-made disturbance led to an increase of both species and genetic diversity (Frey et al. 2016), genetic diversity levels in F. ulmaria increased with rising distance to the nearest settlement.
Effect of habitat quality and population size on genetic diversity
The genetic diversity of F. ulmaria was positively associated with moss and vascular plant cover. In a vegetation unit, the frequent abundance of moss and vascular plants is expected to decrease germination and establishment of plant species (Špačková et al. 1998; Poschlod and Biewer 2005; Drake et al. 2018). However, in wet grassland habitats mosses can act as safe sites for germination (Wang et al. 2012) by retaining seeds (Freestone 2006), producing more stable habitat conditions, and protecting seedlings from harsh climatic conditions (Donath and Eckstein 2010; Lemke et al. 2015). Similarly, grass tussocks can also retain seeds and facilitate germination, especially in wet environments (Wang et al. 2012). A high coverage of moss and vascular plants may, therefore, facilitate the germination and establishment of F. ulmaria in litter meadows and consequently increase genetic diversity levels.
Correlations between population size and genetic diversity are expected to be positive, with larger populations maintaining more genotypes (Vergeer et al. 2003; Ouborg et al. 2006). However, the genetic diversity of S. pratensis decreased with increasing population size. Grassland plant species with long life cycles, slow intrinsic dynamics, and comparatively large population size may occur as remnant populations in modern landscapes (Maurer et al. 2003). Piqueray et al. (2011) observed that historic habitat configurations may often affect the present occurrence of a species, indicating a time lag between habitat loss, fragmentation, and their consequences on genetic diversity (Helm et al. 2006). Therefore, previous studies predicted a delayed response of genetic diversity to habitat fragmentation (Honnay et al. 2007). Additionally, S. pratensis is a more specialised and less widespread species than A. sylvestris and F. ulmaria. The Pearson correlation revealed a negative impact of moss cover on the population size of S. pratensis and, moreover, a negative relationship between the cover of moss and open soil. Therefore, we hypothesise that S. pratensis depends on open soil for successful germination and establishment. Hence, genetic diversity levels were low, despite high population sizes, due to a potential bottleneck caused by conservation measures, a possible extinction debt, and/or missing niches for germination and establishment.
Conclusion
Our study revealed significant and species specific impacts of landscape structure, habitat quality, and population size on genetic diversity. While the influence of habitat size on genetic diversity in A. sylvestris remained unclear, F. ulmaria populations were significantly driven by the distance to the nearest settlement, the total area of litter meadows, and their connectivity. Moreover, the cover of mosses and vascular plants showed a significant impact on the genetic diversity of F. ulmaria populations. The genetic diversity of S. pratensis populations was affected in two ways: directly by population size and indirectly by the cover of mosses.
Abandonment of traditional land use practices changed the abundance and local habitat quality of semi-natural litter meadows during the last decades. Additionally, the practice of litter meadow establishment, traditional and also current management practices, caused and still cause man-made gene flow among litter meadows. Thus, past and present landscape structures as well as local habitat quality turned out as key variables driving genetic variation patterns of typical litter meadow species.
Recommendations for conservation
The genetic variation in the three study species was driven by different explanatory factors in different ways. Therefore, the results presented here allow no general recommendation for the conservation of genetic variation on ecosystem level. Litter meadows have a strong history of anthropogenic land use, with a high impact of periodic (re)sowing and mowing. The results of this study highlight the dynamic conditions, these comparatively young habitats experienced over time.
For the preservation of the diversity in these habitats, it is advisable to focus on the variability and dynamic processes of these ecosystems to protect and maintain the biodiversity of these habitats. Conservation measures should, therefore, focus on meadows covering the whole range of habitat conditions and spatial dimensions. They should support the variability in management practices with particular focus on traditional land use practices, such as mowing by hand or sickle bar mower. Further recommendation would include the temporal adjustment of conservation measures and the cleaning of mowing machines in between different sites to ensure moderate levels of gene flow and thus, counteract an impoverishment of the gene pool by genetic ‘swamping’. In conclusion, the future conservation of these species-rich habitats should pay reference to past as well as present processes to ensure the maintenance of litter meadows in our cultural landscape.
Data availability
Datasets generated during this study are available from the authors upon request.
Change history
13 July 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10592-021-01389-4
References
Adriaens D, Honnay O, Hermy M (2006) No evidence of a plant extinction debt in highly fragmented calcareous grasslands in Belgium. Biol Conserv 133:212–224. https://doi.org/10.1016/j.biocon.2006.06.006
Aguilar R, Quesada M, Ashworth L et al (2008) Genetic consequences of habitat fragmentation in plant populations : susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188. https://doi.org/10.1111/j.1365-294X.2008.03971.x
Baessler C, Klotz S, Durka W (2010) Temporal changes and spatial determinants of plant species diversity and genetic variation. In: Müller F, Baessler C, Schubert C, Klotz S (eds) Long-term ecological research. Springer, Dordrecht, pp 279–297
Balmford A, Bennun L, Ten Brink B et al (2005) The convention on biological diversity’s 2010 target. Science 307:212–213. https://doi.org/10.1126/science.1106281
Barrett SCH, Kohn JR (1991) Genetic and evolutionary consequences of small population size in plants: implications for conservation. In: Falk DA, Holsinger KE (eds) Genetics and conservation of rare plants. Oxford University Press, Oxford
Billeter RC, Schneller J, Diemer M (2002) Genetic diversity of Carex davalliana and Succisa pratensis in mown and abandoned fen meadows. Bull Geobot Inst ETH 68:45–54
Bland JM, Altman DG (1995) Statistics notes: multiple significance tests: the Bonferroni method. BMJ 310:170–170. https://doi.org/10.1136/bmj.310.6973.170
Bonin A, Bellemain E, Eidesen PB et al (2004) How to track and assess genotyping errors in population genetics studies. Mol Ecol 13:3261–3273. https://doi.org/10.1111/j.1365-294X.2004.02346.x
Brown JH, Kodric-Brown A (1977) Turnover rates in insular biogeography: effect of immigration on extinction. Ecology 58:445–449. https://doi.org/10.2307/1935620
Burnham KP, Anderson DR (2002) Model selection and multimodel inference : a practical information-theoretic approach, 2nd edn. Springer, New York, NY
Bylebyl K, Poschlod P, Reisch C (2008) Genetic variation of Eryngium campestre L. (Apiaceae) in Central Europe. Mol Ecol 17:3379–3388. https://doi.org/10.1111/j.1365-294X.2008.03836.x
de Jong TJ, Klinkhamer PGL (1994) Plant size and reproductive success through female and male function. J Ecol 82:399. https://doi.org/10.2307/2261307
Dinno A (2015) Nonparametric pairwise multiple comparisons in independent groups using Dunn’s Test. Stata J Promot Commun Stat Stata 15:292–300. https://doi.org/10.1177/1536867X1501500117
Dlugosch KM, Parker IM (2008) Invading populations of an ornamental shrub show rapid life history evolution despite genetic bottlenecks. Ecol Lett 11:701–709. https://doi.org/10.1111/j.1461-0248.2008.01181.x
Donath TW, Eckstein RL (2010) Effects of bryophytes and grass litter on seedling emergence vary by vertical seed position and seed size. Plant Ecol 207:257–268. https://doi.org/10.1007/s11258-009-9670-8
Drake P, Grimshaw-Surette H, Heim A, Lundholm J (2018) Mosses inhibit germination of vascular plants on an extensive green roof. Ecol Eng 117:111–114. https://doi.org/10.1016/j.ecoleng.2018.04.002
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242. https://doi.org/10.1146/annurev.es.24.110193.001245
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7:574–578. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Freestone AL (2006) Facilitation drives local abundance and regional distribution of a rare plant in a harsh environment. Ecology 87:2728–2735. https://doi.org/10.1890/0012-9658(2006)87[2728:FDLAAR]2.0.CO;2
Frey D, Arrigo N, Granereau G et al (2016) Parallel declines in species and genetic diversity driven by anthropogenic disturbance: a multispecies approach in a French Atlantic dune system. Evol Appl 9:479–488. https://doi.org/10.1111/eva.12351
Hanski I (1994) Patch-occupancy dynamics in fragmented landscapes. Trends Ecol Evol 9:131–135. https://doi.org/10.1016/0169-5347(94)90177-5
Helm A, Hanski I, Pärtel M (2006) Slow response of plant species richness to habitat loss and fragmentation. Ecol Lett 9:72–77. https://doi.org/10.1111/j.1461-0248.2005.00841.x
Honnay O, Coart E, Butaye J et al (2006) Low impact of present and historical landscape configuration on the genetics of fragmented Anthyllis vulnera populations. Biol Conserv 127:411–419. https://doi.org/10.1016/j.biocon.2005.09.006
Honnay O, Adriaens D, Coart E et al (2007) Genetic diversity within and between remnant populations of the endangered calcareous grassland plant Globularia bisnagarica L. Conserv Genet 8:293–303. https://doi.org/10.1007/s10592-006-9169-y
Hooftman DAP, Van KM, Diemer M (2003) Effects of habitat fragmentation on the fitness of two common wetland species, Carex davalliana and Succisa pratensis. Oecologia 134:350–359. https://doi.org/10.1007/s00442-002-1096-0
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. https://doi.org/10.1093/molbev/msj030
Jacquemyn H, Honnay O, Galbusera P, Roldán-Ruiz I (2004) Genetic structure of the forest herb Primula elatior in a changing landscape. Mol Ecol 13:211–219. https://doi.org/10.1046/j.1365-294X.2003.02033.x
Jensen K, Gutekunst K (2003) Effects of litter on establishment of grassland plant species: the role of seed size and successional status. Basic Appl Ecol 4:579–587. https://doi.org/10.1078/1439-1791-00179
Kühn I, Durka W, Klotz S (2004) BiolFlor—a new plant-trait database as a tool for plant invasion ecology. Divers Distrib 10:363–365. https://doi.org/10.1111/j.1366-9516.2004.00106.x
Kull K, Zobel M (1991) High species richness in an Estonian wooded meadow. J Veg Sci 2:715–718. https://doi.org/10.2307/3236182
Leimu R, Mutikainen P, Korchivera J, Fischer M (2006) How general are positive relationships between plant population size, fitness and genetic variation? J Ecol 94:942–952. https://doi.org/10.1111/j.1365-2745.2006.01150.x
Leipold M, Tausch S, Hirtreiter M et al (2018) Sampling for conservation genetics: how many loci and individuals are needed to determine the genetic diversity of plant populations using AFLP? Conserv Genet Resour. https://doi.org/10.1007/s12686-018-1069-1
Lemke T, Janßen A, Porembski S (2015) Multiple limitations to the persistence of Trollius europaeus in a fragmented agricultural landscape in the context of metapopulation theory. Plant Ecol 216:319–330. https://doi.org/10.1007/s11258-014-0439-3
Lindborg R, Eriksson O (2004) Historical landscape connectivity affects present plant species diversity. Ecology 85:1840–1845. https://doi.org/10.1890/04-0367
Lynch M, Milligan BG (1994) Analysis of population genetic structure with RAPD markers. Mol Ecol 3:91–99. https://doi.org/10.1111/j.1365-294X.1994.tb00109.x
Maas D (1988) Keimung und Etablierung von Streuwiesenpflanzen nach experimenteller Ansaat. Natur und Landschaft 63:411–415
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Maurer K, Durka W, Stöcklin J (2003) Frequency of plant species in remnants of calcareous grassland and their dispersal and persistence characteristics. Basic Appl Ecol 4:307–316. https://doi.org/10.1078/1439-1791-00162
May RM (1994) Biological diversity: differences between land and sea. Philos Trans R Soc Lond B 343:105–111. https://doi.org/10.1098/rstb.1994.0014
McKay JK, Christian CE, Harrison S, Rice KJ (2005) “How local is local?” A review of practical and conceptual issues in the genetics of restoration. Restor Ecol 13:432–440. https://doi.org/10.1111/j.1526-100X.2005.00058.x
Müller M the older called H (1752) Gründlicher Bericht, wie aus des Erdbodens Beschaffenheit vorlängstens unweit Ulm, zwischen Grimmelfingen und Gögglingen, in dem sogenannten Tauben Riedt, dass unfehlbar Turf oder Torf vorhanden seyn müssen, beurtheilet ... Ulm, Germany
Münzbergová Z, Cousins SAO, Herben T et al (2013) Historical habitat connectivity affects current genetic structure in a grassland species. Plant Biol 15:195–202. https://doi.org/10.1111/j.1438-8677.2012.00601.x
Oberdorfer E, Schwabe A, Müller T (2001) Pflanzensoziologische Exkursionsflora - Für Deutschland und angrenzende Gebiete, 8th edn. Ulmer, Stuttgart
Ouborg NJ, Vergeer P, Mix C (2006) The rough edges of the conservation genetics paradigm for plants. J Ecol 94:1233–1248. https://doi.org/10.1111/j.1365-2745.2006.01167.x
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Picó FX, Van Groenendael J (2007) Large-scale plant conservation in European semi-natural grasslands: a population genetic perspective. Divers Distrib 13:920–926. https://doi.org/10.1111/j.1472-4642.2007.00349.x
Piqueray J, Bisteau E, Cristofoli S et al (2011) Plant species extinction debt in a temperate biodiversity hotspot: community, species and functional traits approaches. Biol Conserv 144:1619–1629. https://doi.org/10.1016/j.biocon.2011.02.013
Poschlod P (2017) Geschichte der Kulturlandschaft, 2nd edn. Ulmer, Stuttgart
Poschlod P, Biewer H (2005) Diaspore and gap availability are limiting species richness in wet meadows. Folia Geobot 40:13–34. https://doi.org/10.1007/BF02803041
Poschlod P, Fischer S (2016) Das Streuwiesen-Zeitalter der Grundwassermoore und die Bewaldung der Regenmoore als Spiegel der jüngeren Landnutzungs- und Umweltgeschichte im Alpenvorland. Tuxenia Beih 9:107–117
Poschlod P, Kleyer M, Jackel A-K et al (2003) BIOPOP—a database of plant traits and internet application for nature conservation. Folia Geobot 38:263–271. https://doi.org/10.1007/BF02803198
Prentice HC, Lönn M, Rosquist G et al (2006) Gene diversity in a fragmented population of Briza media : grassland continuity in a landscape context. J Ecol 94:87–97. https://doi.org/10.1111/j.1365-2745.2005.01054.x
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:245–259. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Pritchard JK, Wen X, Falush D (2010) Documentation for structure software : Version 2 . 3
Purschke O, Sykes MT, Reitalu T et al (2012) Linking landscape history and dispersal traits in grassland plant communities. Oecologia 168:773–783. https://doi.org/10.1007/s00442-011-2142-6
R Core Team (1978) R: a language and environment for statistical computing
Ramakrishnan AP, Musial T, Cruzan MB (2010) Shifting dispersal modes at an expanding species’ range margin. Mol Ecol 19:1134–1146. https://doi.org/10.1111/j.1365-294X.2010.04543.x
Reisch C (2007) Genetic structure of Saxifraga tridactylites (Saxifragaceae) from natural and man-made habitats. Conserv Genet 8:893–902. https://doi.org/10.1007/s10592-006-9244-4
Reisch C, Bernhardt-Römermann M (2014) The impact of study design and life history traits on genetic variation of plants determined with AFLPs. Plant Ecol 215:1493–1511. https://doi.org/10.1007/s11258-014-0409-9
Reisch C, Schmidkonz S, Meier K et al (2017) Genetic diversity of calcareous grassland plant species depends on historical landscape configuration. BMC Ecol 17:19. https://doi.org/10.1186/s12898-017-0129-9
Reisch C, Schmid C, Hartig F (2018) A comparison of methods for estimating plant population size. Biodivers Conserv. https://doi.org/10.1007/s10531-018-1522-1
Reitalu T, Johansson LJ, Sykes MT et al (2010) History matters: village distances, grazing and grassland species diversity. J Appl Ecol 47:1216–1224. https://doi.org/10.1111/j.1365-2664.2010.01875.x
Rogers SO, Bendich AJ (1994) Extraction of total cellular DNA from plants, algae and fungi. In: Gelvin SB, Schilperoort RA (eds) Plant molecular biology manual. Springer, Netherlands, pp 183–190
Rosengren F, Cronberg N, Reitalu T, Prentice HC (2013) Genetic variation in the moss Homalothecium lutescens in relation to habitat age and structure. Botany 91:431–441. https://doi.org/10.1139/cjb-2012-0258
Ruprecht E, Szabó A (2012) Grass litter is a natural seed trap in long-term undisturbed grassland. J Veg Sci 23:495–504. https://doi.org/10.1111/j.1654-1103.2011.01376.x
Schmidt K, Jensen K (2000) Genetic structure and AFLP variation of remnant populations in the rare plant Pedicularis palustris (Scrophulariaceae) and its relation to population size and reproductive components. Am J Bot 87:678–689. https://doi.org/10.2307/2656854
Schmidt T, Arens P, Smulders MJM et al (2009) Effects of landscape structure on genetic diversity of Geum urbanum L. populations in agricultural landscapes. Flora Morphol Distrib Funct Ecol Plants 204:549–559. https://doi.org/10.1016/j.flora.2008.07.005
Schmitt J (1983) Flowering plant density and pollinator visitation in Senecio. Oecologia 60:97–102
Špačková I, Lepš J (2004) Variability of seedling recruitment under dominant, moss, and litter removal over four years. Folia Geobot 39:41–55. https://doi.org/10.1007/BF02803263
Špačková I, Kotorová I, Lepš J (1998) Sensitivity of seedling recruitment to moss, litter and dominant removal in an oligotrophic wet meadow. Folia Geobot 33:17–30. https://doi.org/10.1007/BF02914928
Spielman D, Brook BW, Frankham R (2004) Most species are not driven to extinction before genetic factors impact them. Proc Natl Acad Sci 101:15261–15264. https://doi.org/10.1073/pnas.0403809101
Stebler FG (1898) Die besten Streuepflanzen. Abbildungen und Beschreibungen derselben, mit einem einleitenden Teil über die Streumaterialien, einer Übersicht der wichtigsten Pflanzen der Streuewiesen und einem allgemein wirthschaftlichen Abschnitt über Produktion, Nutzung. In: IV. Teil des schweizerischen Wiesenpflanzenwerkes. K. J. Wyß, Bern, Switzerland
Steffan-Dewenter I, Tscharntke T (1999) Effects of habitat isolation on pollinator communities and seed set. Oecologia 121:432–440. https://doi.org/10.1007/s004420050949
Strykstra RJ, Verweij GL, Bakker JP (1997) Seed dispersal by mowing machinery in a Dutch brook valley system. Acta Bot Neerl 46:387–401
Tscharntke T, Brandl R (2004) Plant-insect interactions in fragmented landscapes. Annu Rev Entomol 49:405–430. https://doi.org/10.1146/annurev.ento.49.061802.123339
Van Treuren R, Bas N, Goossens PJ et al (2005) Genetic diversity in perennial ryegrass and white clover among old Dutch grasslands as compared to cultivars and nature reserves. Mol Ecol 14:39–52. https://doi.org/10.1111/j.1365-294X.2004.02391.x
Vandepitte K, Honnay O, Jacquemyn H, Roldán-Ruiz I (2010) Effects of outcrossing in fragmented populations of the primarily selfing forest herb Geum urbanum. Evol Ecol 24:1353–1364. https://doi.org/10.1007/s10682-010-9395-0
Vergeer P, Rengelink R, Copal A, Ouborg NJ (2003) The interacting effects of genetic variation, habitat quality and population size on performance of Succisa pratensis. J Ecol 91:18–26. https://doi.org/10.1046/j.1365-2745.2003.00736.x
Vos P, Hogers R, Bleeker M et al (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414. https://doi.org/10.1093/nar/23.21.4407
Wade MJ, McCauley DE (1988) Extinction and recolonization: their effects on the genetic differentiation of local populations. Evolution (N Y) 42:995. https://doi.org/10.2307/2408915
Wang Z, Nishihiro J, Washitani I (2012) Regeneration of native vascular plants facilitated by Ischaemum aristatum var. glaucum tussocks: an experimental demonstration. Ecol Res 27:239–244. https://doi.org/10.1007/s11284-011-0897-1
Wheeler BD (1988) Species richness, species rarity and conservation evaluation of rich-fen vegetation in lowland england and wales. J Appl Ecol 25:331. https://doi.org/10.2307/2403630
Whitlock MC, McCauley DE (1990) Some population genetic consequences of colony formation and extinction: genetic correlations within founding groups. Evolution (N Y) 44:1717–1724
Willerding C, Poschlod P (2002) Does seed dispersal by sheep affect the population genetic structure of the calcareous grassland species Bromus erectus? Biol Conserv 104:329–337. https://doi.org/10.1016/S0006-3207(01)00198-7
Yeh FC, Yang RC, Boyles TBJ et al (1997) POPGENE, the user-friendly shareware for population genetic analysis. Mol. Biol. Biotechnol. Centre, Univ. Alberta, Alberta
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418. https://doi.org/10.1002/cphc.201000986
Acknowledgements
The authors thank Petra Schitko for assistance in the lab and Sven Rubanschi for support in statistical approaches. We further thank Christina Manhart and Jakob Speigl, who contributed to this study during their Bachelor theses. Additionally, we would like to thank Eva Wagner, Lina Begemann, Patricia Krickl, Katrin Sagmeister, and Cornelia Straubinger for the vegetation cover data.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was financially supported by the Federal Agency for Agriculture and Food (BLE).
Author information
Authors and Affiliations
Contributions
PP and CR conceived and designed the study. EP and TAL collected the data, performed the data analyses, and wrote the manuscript. CR and PP revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to a retrospective open access order.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lehmair, T.A., Pagel, E., Poschlod, P. et al. Genetic variation of litter meadow species reflects gene flow by hay transfer and mowing with agricultural machines. Conserv Genet 21, 879–890 (2020). https://doi.org/10.1007/s10592-020-01294-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-020-01294-2