
Abstract
European wolf (Canis lupus) populations have suffered extensive decline and range contraction due to anthropogenic culling. In Bulgaria, although wolves are still recovering from a severe demographic bottleneck in the 1970s, hunting is allowed with few constraints. A recent increase in hunting pressure has raised concerns regarding long-term viability. We thus carried out a comprehensive conservation genetic analysis using microsatellite and mtDNA markers. Our results showed high heterozygosity levels (0.654, SE 0.031) and weak genetic bottleneck signals, suggesting good recovery since the 1970s decline. However, we found high levels of inbreeding (F IS = 0.113, SE 0.019) and a N e/N ratio lower than expected for an undisturbed wolf population (0.11, 95 % CI 0.08–0.29). We also found evidence for hybridisation and introgression from feral dogs (C. familiaris) in 10 out of 92 wolves (9.8 %). Our results also suggest admixture between wolves and local populations of golden jackals (C. aureus), but less extensive as compared with the admixture with dogs. We detected local population structure that may be explained by fragmentation patterns during the 1970s decline and differences in local ecological characteristics, with more extensive sampling needed to assess further population substructure. We conclude that high levels of inbreeding and hybridisation with other canid species, which likely result from unregulated hunting, may compromise long-term viability of this population despite its current high genetic diversity. The existence of population subdivision warrants an assessment of whether separate management units are needed for different subpopulations. Our study highlights conservation threats for populations with growing numbers but subject to unregulated hunting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
European wolves (Canis lupus) have experienced systematic anthropogenic culling. After experiencing a large range contraction (Randi 2011), they now undergo a process of natural re-colonisation in several regions, namely in the Alps (e.g. Fabbri et al. 2007), Iberian Peninsula (e.g. Blanco et al. 1992; Echegaray and Vilà 2010), Scandinavian Peninsula (e.g. Wabakken et al. 2001) and parts of Central-Eastern Europe (e.g. Boitani 2003). In most cases, this re-colonisation followed legal protection of the species. However, wolves are still legally hunted in many Eastern European countries, usually sustaining large numbers (Boitani 2003), meaning that most of the European wolf populations remain under hunting pressure. In spite of this, most of the conservation genetic studies to date have focused on protected populations (see Randi 2011 for review).
In Bulgaria, wolves can be found throughout most of the country, and records since the 19th century suggest widespread hunting using various methods. In the late 1950s, intensive poison use in efforts to contain rabies, resulted in a severe wolf range contraction to five areas with low human occupation by the early 1970s, with no more than an estimated 100–150 wolves remaining in the whole country (Spiridonov and Spassov 1985). Subsequent poison bans, in combination with inclusion of the wolf in the Bulgarian Red Data Book and increase in ungulate prey, allowed the population to recover to more than 1,000 individuals by the late 1990s. However, the numbers have declined since, with 700–800 individuals remaining according to most recent estimates (Spiridonov and Spassov 2011). Wolf hunting has never been banned in Bulgaria (excluding three national parks), and is presently still allowed year round with no specific quotas defined. Hunting records from the annual statistics of the Bulgarian Executive Forest Agency (http://www.nug.bg/lang/2/index) suggest an average of 250–400 individuals (25–50 % of the census population) killed annually between 2000 and 2009. An independent analysis performed by BALKANI Wildlife Society estimated the number of wolves killed annually between 2006 and 2009 at 234–249 individuals (Elena Tsingarska-Sedefcheva, personal communication). After bounty payments for killed wolves were stopped in 2010, most hunters ceased reporting on the individuals killed; therefore, the official estimates of the number of wolves killed in Bulgaria in the last few years are not as accurate as before 2010 (Tsingarska-Sedefcheva 2013). In addition to legal hunting, wolves are also killed by poachers through poisoning and trapping. However, the exact numbers are unavailable, making the assessment of the conservation status of Bulgarian wolves an important priority. In Finland, where the annual legal harvest is no more than ~15 % of the census population, a recent demographic and genetic crash has been documented, with a large, significant decline in observed heterozygosity and increase in inbreeding (Jansson et al. 2012). This crash was preceded by over 10 years of strong demographic and spatial expansion, and likely resulted from excessive hunting and poaching (Jansson et al. 2012). This raises concerns about wolf populations experiencing much higher and unregulated hunting pressure, and justifies the need for a genetic study of the Bulgarian wolf population.
Studies on mitochondrial DNA (mtDNA) and microsatellite variability in European wolves revealed haplotype and allele sharing among local populations from distant parts of the continent (Lucchini et al. 2004; Pilot et al. 2006, 2010). However, local-scale genetic structure can be detected in different regions of Europe (Pilot et al. 2006; Aspi et al. 2008; Czarnomska et al. 2013) and elsewhere in the species distribution, and appears to be determined mainly by ecological features rather than geographical barriers (e.g. Carmichael et al. 2001; Geffen et al. 2004; Pilot et al. 2006). Earlier genetic studies suggest that Bulgarian wolves contain high levels of genetic diversity compared to elsewhere in Europe (Lucchini et al. 2004; Pilot et al. 2006, 2010). This could reflect the presence of an ecologically diverse population, large effective population sizes, or historical factors, with Balkans being a Pleistocene glacial refugium. Alternatively, gene introgression from feral dogs might have artificially increased genetic diversity of Bulgarian wolves. However, this can be a valid explanation only if hybridisation occurs more frequently there than in other parts of Europe.
No study to date has looked into the fine scale wolf genetic structure in Bulgaria. Although wolf numbers have increased since the 1970s, this population still faces pressing conservation threats. The recent decreasing trend in census size together with increasing tension within local communities due to predation on domestic animals (Tsingarska-Sedefcheva 2007) warrants a more detailed assessment. This is particularly relevant within the context of IUCN reports identifying Bulgaria as a European hotspot for mammal conservation due to abundant presence of endangered mammals (Temple and Terry 2007), and the inclusion of the Bulgarian wolf in Annex II of the European Habitats Directive (Temple and Terry 2007). Detecting local scale genetic structure is of major importance for the effective conservation of this species. If genetic homogeneity over large distances is the norm, then the impact of localized mortality will be reduced. However, if strong fine-scale population division is instead widespread, then localized mortality could lead to the extirpation of unique genetic lineages that would not be easily replaced (Leonard et al. 2007).
We have thus carried out a genetic characterization of Bulgarian wolves using both mtDNA and microsatellite loci. We aimed to: (1) identify patterns of population structure within Bulgaria and consequent need for distinct conservation management units; (2) quantify genetic diversity and assess the genetic recovery of Bulgarian wolves since the 1970s demographic depletion based on contemporary data, together with the effects of increasing hunting pressure in the past 10 years; (3) assess the extent of hybridisation with feral dogs.
Methods
Study area
Bulgaria is located in the transition zone between Mediterranean and temperate continental climate. It is mainly mountainous, with lowlands of relatively low altitude in the north and southeast, and two large mountainous systems, the Stara Planina (Balkan range) running across the northern central part of the country, and the Rila-Rhodopean Massif down the central southwest (Figure S1). Forests occupy approximately 30 % of Bulgarian territory, being dominated by oak (Quercus sp.) and beech (Fagus sylvatica). A coniferous zone between 1,300–2,200 m altitude is composed of Scots pine (Pinus sylvestris), Norway spruce (Picea abies) and Silver fir (Abies alba) (Aladzhem 2000).
Four native species of ungulates exist in Bulgaria: wild boar (Sus scrofa) and roe deer (Capreolus capreolus) present throughout the country; chamois (Rupicapra rupicapra) present only in the highlands of Rila, Pirin, Central Balkan and West Rhodope Mountains, and red deer (Cervus elaphus) with a much more limited distribution. Small populations of fallow deer (Dama dama) and mouflon (Ovis musimon) have been introduced in different parts of the country, mainly hunting reserves (Peshev et al. 2004). Besides grey wolves, two other species of the genus Canis are present, the feral domestic dog Canis familiaris and the golden jackal Canis aureus, both distributed throughout the country.
Sample collection
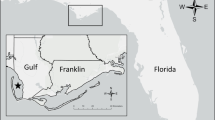
Muscle tissue samples were obtained from local hunters between May 2000 and January 2011, and samples from non-tanned skins were obtained between 2007 and 2011. All samples were placed in 95 % ethanol after the collection and eventually stored at −80 °C. Sample location against present wolf distribution is presented in Fig. 1. Exact coordinates were unavailable for some samples, and thus location often reflects the area (usually a village) around which the wolf was captured. The full dataset was comprised of 112 putative grey wolves (see results), although not all were used in all the analyses (see Table S1). In addition, 14 samples of feral dogs were obtained from roadkill carcasses, mainly in the western part of the country, and from the local breed of shepherd dogs (Karakachan dog), included in the wolf–dog hybridisation analyses (see details below).

We also included ten wolf samples from Greece genotyped in a previous study (Pilot et al. 2006), which were used to assess the level of genetic differentiation between Bulgarian wolves and neighbouring populations. We did not have access to wolf samples from other countries neighbouring with Bulgaria, and therefore only the comparison with Greek wolves was possible.
Laboratory procedures and data analysis
We analysed a 257 bp fragment of the mtDNA control region and 14 microsatellite loci: FH2001, FH2010, FH2017, FH2054, FH2079, FH2088, FH2096 (Francisco et al. 1996), C213, C250, C253, C436, C466, C642 (Ostrander et al. 1993), AHT130 (Holmes et al. 1995) and VWF (Shibuya et al. 1994). Laboratory protocols were as described by Pilot et al. (2006). Microsatellite loci were amplified in multiplex PCR reactions. In cases when two or more loci failed to be amplified, we repeated the multiplex PCR for this particular sample up to three times. If amplification of one locus from a multiplex set failed, we carried out a separate (non-multiplex) PCR for that locus. Separate PCR reactions were also carried out if there were uncertainties regarding a particular genotype at any locus amplified in the multiplex (e.g. due to stuttering or large differences in size between two allele peaks). In addition, we replicated the genotyping of 14 samples (12.5 % of the sample set) for all 14 loci. Based on these replicates, the estimated allelic dropout rate was 0.04, and estimated false allele rate (wrong allele size scored due to stuttering, PCR artefacts or human error) was 0.01. The potential presence of null alleles was assessed using MicroChecker (Van Oosterhout et al. 2004), with calculations carried out for the full wolf dataset, and for a dataset subdivided by population clusters (see results).
For mtDNA we calculated average number of nucleotide differences (k), haplotype diversity (Hd), nucleotide diversity (π), Tajima’s D, Fu and Li’s D* and F* statistics using DnaSP (Librado and Rozas, 2009). Genetic structure at mtDNA was assessed using the software Geneland (Guillot et al. 2008), which incorporates spatial information into the model. This analysis was run with four independent chains for 100,000,000 generations after 10,000,000 burn-in for K values between 1 and 5.
For microsatellite data, we estimated the expected and observed heterozygosity, inbreeding coefficient (F IS ) and Shannon information index using GenAlex (Peakall and Smouse 2006), while allelic richness was calculated using Fstat (Goudet 2001). Exact tests for Hardy–Weinberg equilibrium (HWE) were carried out in GenePop (Rousset 2008) using the MCMC method with 10,000 dememorization steps followed by 1,000 batches with 10,000 iterations per batch. Pair-wise F ST between clusters identified in Structure was calculated using Arlequin (Excoffier et al. 2005), with significance tested using 10,000 permutations.
Effective population size (N e) was calculated using two linkage disequilibrium methods, as implemented in the software packages NeEstimator (Hill 1981; Ovenden et al. 2007), LDNe (Waples and Do 2008) and OneSamp (Tallmon et al. 2008) with 50,000 iterations and initial N e range between 10 and 1,000. Samples with any missing data were excluded (Table S1). Signals for bottleneck were assessed using the software Bottleneck (Cornuet and Luikart 1996), with significance tests done with 1,000 iterations, and with 70 % proportion of the infinite allele model for the two phased model. A test for mode shift in allele frequencies was also carried out.
We used the software Structure (Pritchard et al. 2000) for the population structure analysis using microsatellite data. Structure is not well suited to data where isolation-by-distance is present, and may infer genetic structure with most individuals having mixed ancestry (Structure Manual—Pritchard et al. 2010). Therefore, we tested for the isolation by distance through a Mantel test and spatial autocorrelation as implemented in the software GenAlex (Peakall and Smouse 2006).
Structure analysis was performed with four independent runs for the predefined number of populations (K) between 1 and 15, with 100,000,000 replicates after 10,000,000 burn-in. We used no prior population information, and an admixture model with correlated allele frequencies. This analysis was repeated with the addition of the feral dog samples, in order to assess the presence of wolf–dog hybridisation, along with other analyses dedicated to this purpose (see below). StructureHarvester (Earl and vonHoldt 2011) was used to assess the results and apply the (Evanno et al. 2005) modification when applicable. Genetic structure was further assessed by using the spatial model implemented in Geneland (Guillot et al. 2008). We ran four independent chains for 10,000,000, after 1,000,000 burn-in, for K values 1–15.
Because both Structure and Geneland attempt to maximise HWE for each value of K, results can be biased in non-equilibrium populations (e.g. populations experiencing strong demographic fluctuations), we also inferred the number of clusters using the multivariate discriminant analysis of principal components method (DAPC; Jombart et al. 2010) employed in the R package Adegenet (Jombart and Ahmed 2011). We also used Adegenet to carry out a principal component analysis for a sample set including the Bulgarian wolves and 10 samples from Greece, in order to assess the level of their distinctiveness.
Possible admixture between wolves and other canids was assessed using the program NewHybrids (Anderson and Thompson 2002). Because three of the putative wolf samples were identified as golden jackals (C. aureus) (see below), the analysis was run independently for the wolf–jackal hybridisation and the wolf–dog hybridisation. The wolf–dog dataset was analysed using 30,000,000 iterations after 10,000,000 burn-in, while the wolf–jackal dataset was analysed using 40,000,000 iterations after 12,000,000 burn-in. For both datasets, the hybrid classes tested ranged from pure species to level three backcrosses for each species, including both F1 and F2 crosses (thus totalling 10 different mixing classes).
We assessed kinship for the Bulgarian wolves dataset in order to identify closely related individuals that likely formed core family groups (which could include pack members and individuals that dispersed from their natal pack). We used the Full Sibship Reconstruction method implemented in the software Kingroup (Konovalov et al. 2004) to identify clusters of individuals related at the level of Parent–offspring pairs, Full-sibs, or Half-sibs. We also carried out parentage assignment using the software Cervus (Kalinowski et al. 2007). Three different analyses were run: (1) Maternity analysis with all individuals as potential offspring, and all females as potential mothers; (2) Paternity analysis with all individuals as potential offspring, and all males as potential fathers; (3) Parent pair analysis with all individuals considered as both potential mothers or fathers and as offspring (because the age of individuals was unknown).
In the Cervus analysis, an individual was accepted as a likely parent if it was assigned to the offspring with 95 % confidence and with no more than two mismatching loci. However, in few cases, a more relaxed criteria was used (as explained below), because our goal was to identify core family groups using combined information from Cervus, Kingroup and mtDNA haplotypes, rather than establish exact genealogies within each family group. Because a wolf pack typically consists of a single couple and their offspring, most individuals (except for the father) should have the same mtDNA haplotype, in addition to all being related. Therefore, a group of individuals sharing the same mtDNA haplotype, assigned to the same mother in Cervus and to the same sibling group in Kingroup, was considered as a family group. If one of the siblings identified in Kingroup was assigned to the putative mother with only relaxed confidence level (80 %), it was still considered as belonging to the family group, because different analyses consistently identified it as a close relative of other group members, and because identifying the exact relationship was not essential for our purposes.
Results
Genetic variability of Bulgarian wolves
Three of the putative wolves had golden jackal mtDNA haplotypes; this taxonomic classification was further supported by microsatellite data (see below). These three individuals were excluded from all subsequent analysis, except where explicitly stated.
Among wolves, we found six different mtDNA haplotypes (w6, w10, w13, w16, w17, w27; frequencies in Figure S2), all previously described by Pilot et al. (2010) and representing both main haplogroups defined therein. Diversity levels were high: k = 5.03 (variance = 0.12); Hd = 0.75 (SE = 0.019); π = 0.022 (SE = 0.00029). Positive values of Tajima’s D (2.71, P < 0.01), Fu and Li’s F* (0.91, P > 0.10) and D* (1.86, P < 0.05) likely reflect the 1970s bottleneck, given that it was recent and involved fragmentation into five isolated locations.
Genetic diversity in microsatellite loci was also relatively high (I = 1.57; Allele Richness = 8.5). Observed heterozygosity (0.654, SE 0.031) was lower than expected heterozygosity (0.734, SE 0.026), resulting in high inbreeding coefficient F IS (0.113, SE 0.019) and deviations from HWE (Table S2). To assess potential biases in F IS from the inclusion of related individuals, we removed all but one individual from each family group identified in the kinship analysis. This results in a dataset of 62 individuals, and did not significantly change observed (0.633, SE 0.030) and expected heterozygosity (0.732, SE 0.025), nor F IS (0.139, SE 0.020).
Positive F IS could potentially result from the presence of null alleles. MicroChecker detected putative null alleles in 7 microsatellite loci when applied to the whole wolf dataset. However, the null allele estimation is based on the assumption that any observed deviations from HWE towards heterozygote deficit result solely from the presence of null alleles (see e.g. Chapuis and Estoup 2007). This can lead to false positives in populations that deviate from the equilibrium, e.g. due to population structure (Wahlund effect). When we applied MicroChecker to two subpopulations identified in Structure (see below), the number of loci identified as having null alleles was reduced to two in each of these subpopulations and these loci were not consistent between the subpopulations (Table S3), suggesting that null alleles are false positives resulting from violations of HWE in our study population.
Effective population size and bottleneck analysis
N e estimates varied from 162 calculated using NeEstimator to 66 calculated with LDNe using only alleles with a frequency above 0.05 (Table 1). OneSamp provided an estimate which range encompassed all the values obtained with the other methods. We thus use the mean N e obtained with OneSamp in the discussion of our results regarding N e. We found evidence for a bottleneck only for the I.A.M. mutation model implemented in Bottleneck (Table S4), and there was no change in the expected distribution of allele size class.
Isolation by distance and spatial autocorrelation
Mantel test showed weak and non-significant correlation between genetic and geographic distances (R2 = 0.0222; P = 0.992; Figure S3A). We are thus confident that the results of population structure analysis (see below) are not affected by isolation-by-distance. Spatial autocorrelation values were low for all distance classes (r ≤ 0.069), with significant positive values found for the two smallest distance classes (25 km and 50 km; Figure S3B). The positive correlations at small distances are likely due to the presence of kin groups, but this is counterbalanced by sub-adult dispersal over moderate (50–100 km) and sometimes long distances (up to about 1,000 km) (Wabakken et al. 2007).
Population structure
For the dataset without dogs, although the highest posterior probability from Structure was for K = 6 (Figure S4), no further groups with individuals assigned were added from K = 3, which was also the most likely K after ∆K correction (Evanno et al. 2005) (Figure S5). For K = 3 (Fig. 2a), most individuals clustered into two different groups, with a few individuals forming a cline of mixed ancestry. The third group consisted of three individuals identified as golden jackals based on mtDNA, further supporting their specific identification. Five wolves exhibited mixed ancestry with the jackal cluster, which may be a signature of recent hybridisation between wolves and jackals (Fig. 2).

Structure bar-plot for: a K = 3 obtained from the dataset without feral dog samples; b K = 2–4 obtained from the dataset with the added feral dog samples
Dataset including feral dog samples revealed a similar pattern of population structure. Although K = 9 had the highest posterior probability (Figure S6), no clusters with individuals assigned were added above K = 4 (Fig. 2). The division within wolf samples is consistent with the one obtained from the analysis without the dog samples (Fig. 2). In all barplots for K = 2–4, the presence of gene flow between dogs and wolves can be detected (Fig. 2), while in the barplot for K = 4 the introgression of jackal DNA into some of the wolf samples is clearly visible (Fig. 2).
No clear-cut division between the two main wolf clusters was identified in Structure, i.e. most individuals showed some level of admixture between these clusters. For the purpose of pairwise F ST calculation, individuals were assigned to one of these clusters if they had 60 % or higher ancestry in it, resulting in one cluster with N = 50 (cluster A) and one with N = 41 (cluster B). Pairwise F ST between the two clusters was moderate, but significant (F ST = 0.052, P < 0.001). F IS value was smaller compared to the overall value in cluster A (F IS = 0.063, SE 0.021), but did not significantly change in cluster B (F IS = 0.110, SE 0.042); both values were high enough to cause significant deviations from HWE (P < 0.001 in each cluster).
DAPC also identified K = 2 as the most likely number of clusters in the wolf population, although with only marginal support relative to K = 3 (Figure S7). The DAPC assignment plot (Figure S8) was mostly consistent with the one obtained in Structure (Fig. 2), although DAPC inferred a greater degree of separation between the two clusters.
Spatial analyses
Geneland results for the microsatellite dataset suggested an optimal K of 13, with little support for K = 14 and 15. However, the resulting cluster assignment map largely reflected distribution of family groups (Figure S11) rather than population structure. We thus focused on the results for K between 2 and 4, in order to compare them with the results obtained in Structure, and with the Geneland results obtained for mtDNA (see below). A map of cluster assignment for K = 2 revealed one main division between the southwestern part and the rest of the study area, with some mixed ancestry in the central geographical area (Fig. 3a). For K = 3, we obtained two large clusters corresponding to these identified for K = 2, and the third cluster that was inconsistent among runs. The clusters obtained for K = 4 (Figure S9) were broadly consistent with the mtDNA-based clusters described below.

Distribution of cluster membership probability obtained in Geneland based on a microsatellite loci for K = 2; b based on mtDNA. This represents a composite image based on interpretation of individual cluster assignment maps. See Figure S10 for the individual cluster assignment maps, and the main text for details on interpretation
Geneland results for mtDNA showed the strongest support for K = 4 (Figure S10). However, the geographical distribution of cluster four mainly encompassed an area of poor sampling coverage, and was inconsistent between runs (Figure S10).The overall pattern was consistent with the one obtained with microsatellites for K = 2 (Fig. 3a), involving the separation of southwestern samples, with a further division between the Western and the Eastern samples (Fig. 3b).
Kinship and spatial distribution of kin
We identified 14 core family groups within the study area. Seven of them consisted of individuals killed together, thus likely representing packs. Average size of identified family group was 3.5 individuals (SD = 1.7; range 2–8). This defines the average number of family members detected rather than pack size, because it is unlikely that all pack members were sampled, and because individuals that have dispersed can be still assigned to their natal pack. Most individuals were located within an area consistent with the average individual dispersal distance <100 km (Aspi et al. 2006; Fabbri et al. 2007; Figure S11). Exceptions probably represent long distance dispersal.
Hybridisation between wolves and other canids
NewHybrids results suggest that although hybridisation between wolves and dogs is apparent, introgression of dog genes into the wolf gene pool is limited (Fig. 4a). However, none of the feral dogs sampled exhibited pure dog ancestry, although this may reflect recent common ancestry between the two species (see Verginelli et al. 2005). Ten individuals with a grey wolf phenotype showed signs of admixture with dogs. They constituted 9.8 % of the wolf samples analysed (see Table S5; Figure S12 for details).

Hybridisation plot obtained using the software NewHybrids between: a wolves and dogs; b wolves and jackals
Evidence for hybridisation between wolves and jackals was much weaker, but both Structure and NewHybrids identified several wolves that were assigned a small probability of having jackal ancestry (Fig. 4b). However, individuals with jackal ancestry were not consistent between Structure and NewHybrids. Larger number of jackal samples is needed for more conclusive results.
Discussion
Genetic diversity of the Bulgarian wolf population
Diversity levels of Bulgarian wolves at mtDNA control region (Hd = 0.75, π = 0.022) were high as compared with other European wolf populations from European Russia, Poland, and Iberian Peninsula, with Hd range 0.34–0.67 and π range 0.009–0.016 (Sastre et al. 2011, Czarnomska et al. 2013) (Table S6). Observed heterozygosity at microsatellite loci (0.654) was comparable with populations from north-eastern Europe (e.g. 0.663 for European Russia, Sastre et al. 2011; 0.680 for Finland, Aspi et al. 2006), but higher than in the Apennine Peninsula, the Iberian Peninsula, and Poland (0.52–0.62; Fabbri et al. 2007; Sastre et al. 2011; Czarnomska et al. 2013) (Table S6). Although comparisons of genetic diversity between different studies are not linear, given that different mtDNA sequence lengths and different microsatellite loci were used, important information can nevertheless be obtained by such comparisons, particularly if population history and present conservation status are taken into account.
The inbreeding coefficient F IS (0.113) was comparable with those in the Finish wolf population after the recent crash (0.108; Jansson et al. 2012), and isolated populations from Italy (0.127; Verardi et al. 2006) and Iberian Peninsula (0.177; Sastre et al. 2011) (Table S6). Although F IS levels may be inflated due to Wahlund effect and the presence of closely related individuals in the dataset, this is true for all the populations mentioned above, where inbreeding has been independently confirmed using other methods (e.g. see Randi 2011 for review of studies on the Italian wolves). In our study, F IS remained high after removal of related individuals from the dataset. Moreover, similar to the Finnish wolf population (Aspi et al. 2006), we detected fine-scale genetic clusters consisting of family groups (using Geneland), which may reflect an increased genetic homogeneity of packs due to inbreeding.
Observed heterozygosity levels and other measures of genetic diversity are high in spite of the deviation from HWE towards heterozygote deficit. This suggests that high inbreeding is recent and not yet reducing genetic diversity. The observed high F IS levels together with high heterozygosity can occur if a recent increase in hunting pressure leads to incestuous mating and adoption of unrelated immigrant males that do not engage in breeding (at least not immediately after joining the pack). Both incestuous mating and the presence of unrelated adoptees have been documented in other wolf populations (e.g. Jędrzejewski et al. 2005; Liberg et al. 2005; vonHoldt et al. 2008; Rutledge et al. 2010). Incestuous mating may occur if a breeding individual is lost close to the breeding season (Liberg et al. 2005; vonHoldt et al. 2008), while adoption can occur outside the breeding season. Jędrzejewski et al. (2005) documented both incestuous mating and a presence of an unrelated, non-breeding male in one pack in an intensely hunted population. Rutledge et al. (2010) found that the percentage of packs with unrelated adoptees in the same wolf population declined from 80 to 6 % after a hunting ban. These studies provide an indirect support to the hypothesis that the high inbreeding detected in Bulgarian wolves results from a strong hunting pressure. A causative relationship could only be tested directly by comparing the inbreeding levels and pack structure in Bulgarian wolves before and after a hunting ban. This study provides data from before a hunting ban, and if a hunting ban is introduced in Bulgaria, a further study may be carried out based on a non-invasive sampling.
We should note that the inflated heterozygosity levels might be partially explained by immigration from a genetically differentiated population located outside the study area. Comparison with samples from Greece showed no differentiation from Bulgarian populations (Figure S13), although power is limited by the small sample size (Greece N = 10). Previous studies based on mtDNA showed similar haplotype composition in different countries within the Balkans (Randi et al. 2000; Pilot et al. 2010; Gomercic et al. 2010). Milenkovic et al. (2010) revealed significant morphological differentiation between wolves from the Balkans and the Carpathian Mountains, but it is unlikely that immigration from the Carpathians to Bulgaria would be widespread over such large distances. A more extensive study including the entire Balkan region is required to analyse the patterns of genetic differentiation and gene flow in this area.
Further data from Bulgarian wolves is also needed given the detected high genetic diversity and little legal restrictions to wolf hunting. Continued hunting pressure with kin structure disruption and inflated inbreeding levels can be expected to decrease heterozygosity in the future (Nilsson 2004; Liberg et al. 2005; Temple and Terry 2007). This was observed in Finland, where the expanding wolf population under moderate hunting pressure (~15 % of the census population) experienced sudden demographic decline combined with a significant loss of heterozygosity (Jansson et al. 2012). In Bulgaria, the estimated legal hunting pressure is much higher (25–50 %), and the additional mortality due to poaching, although difficult to quantify, cannot be ignored (see Lieberg et al. 2012).
Hybridisation
Hybridisation with introgression between wild wolves and feral dogs has been observed in other parts of Europe e.g. (Vilà et al. 2003; Verardi et al. 2006; Godinho et al. 2011) and is a pressing conservation concern (Boitani 2003). It is feared that introgression of dog gene variants into depleted wolf populations might lead to the displacement of gene variants exclusive to wolves, and thus compromise its long term survival in the wild (Butler 1994; Rhymer and Simberloff 1996). Although hybridisation between wolves and feral dogs can be detected in our data, it has not been extensive as most individuals were identified as pure wolves, few showed evidence for backcrossing, and no first generation hybrids were detected.
Another noteworthy result is that both Structure and NewHybrids suggest recent hybridisation between wolves and golden jackals. Wolves are known to hybridize with other canid species, particularly in areas of range overlap and in cases where population depletion reduces mate availability (Hailer and Leonard 2008). The range of grey wolves and golden jackals overlaps in southern Asia, Middle East, and the Balkans, and no hybridisation studies were performed in the areas where these two species coexist. Some north African canids originally classified as golden jackals (C. aureus) were shown to carry mtDNA haplotypes falling within the grey wolf clade (Rueness et al. 2011; Gaubert et al. 2012). This was interpreted as a taxonomic misidentification, but could also result from hybridisation and introgression of grey wolf mtDNA into golden jackal populations; analysis of nuclear DNA is needed to clarify this.
Our analysis was of limited power due to the small number of jackals, but indicates the need for further investigation on possible wolf–jackal hybridisation in Bulgaria. Annual estimates of jackal and feral dogs numbers by the Bulgarian Executive Forest Agency suggest that more than 40,000 jackals and 20,000 feral dogs exist in the country (although these numbers may be overestimated). Jackal distribution has considerably expanded during the last 20–30 years and it overlaps greatly with wolf distribution (Popov 2003). There is thus ample opportunity for hybridisation between wolves and jackals, particularly if finding suitable mating partners is made more difficult by diminishing wolf numbers. It is worth noting that the behavioural mechanism leading to hybridisation may be the same as in the case of incestuous mating, i.e. it may be triggered by the loss of a breeding individual close to the breeding season.
Demographic assessment
Results from the Bottleneck analysis are consistent with the known demographic history of Bulgarian wolves. Genetic bottleneck was not confirmed by all tests, but the method used here is known to fail detecting recent bottlenecks in populations that have recovered since the size reduction (Le Page et al. 2000; Whitehouse and Harley 2001). Although the 1970s bottleneck can be considered recent (about 13 wolf generations ago, given a mean generation time of 3 years; Mech and Seal 1987), it did not last long, as earlier estimates in 1900 suggests a larger census size of 1,000–1 600 individuals (Spiridonov and Spassov 1985). Therefore it is credible that the 1970s bottleneck is being detected in modern genetic samples, but its signal has been weakened due to recovery of genetic diversity since then.
Current estimated census size for Bulgarian wolves is between 700–800 individuals (Spiridonov and Spassov 2011), which implies an average N e/N ratio of 0.12 (95 % CI 0.08–0.29; based on N e values from OneSamp results). This is larger than that obtained for Iberian wolves (0.025-Blanco et al. 1992; 0.024-Sastre et al. 2011) but similar to the one obtained for Russian wolves (close to 0.12; Sastre et al. 2011). However, it is smaller than the N e/N calculated for Finnish wolves before the population crash (~0.28; Jansson et al. 2012) and for the reintroduced Yellowstone wolf population (0.26–0.33; vonHoldt et al. 2008) (Table S6). The average N e/N ratio expected over different taxa is 0.11 (Frankham et al. 2002), but the higher N e/N ratios observed in Finnish and Yellowstone wolves suggests this is the value expected from populations under low (Finland) or no hunting pressure (Yellowstone). Although the ratios for both Finland and Yellowstone were obtained for populations still experiencing growth, the bias introduced by this is expected to lower N e/N ratio rather than increase it. Notably, the N e/N ratio in the Finnish population dropped to 0.097 after the demographic crash (Jansson et al. 2012). Our estimates suggest that N e/N ratio in Bulgarian wolves is smaller than the values observed in more stable populations, which may indicate that Bulgarian wolves do not realise their full reproductive potential. However, we should note that there is at present considerable uncertainty in estimates of census size in Bulgaria, and confidence intervals for N e estimates were relatively high (leading to confidence intervals for N e/N ratio overlapping with estimates from Yellowstone and Finland). Also, although we compared our estimates with those obtained using the same methods in other studies if available, such direct comparison was not possible for Yellowstone wolves (vonHoldt et al. 2008). A direct comparison of N e/N ratios between Bulgarian wolves and another, non-harvested Eastern European population may help assess whether and how this ratio is affected by hunting.
Population structure
The identified population clusters correspond well to 3 of the 5 areas where wolves survived in higher numbers during the extreme range contraction of the 1970s (the remaining two areas are not sampled in this study; Spiridonov and Spassov 1985). Individuals with mixed ancestry between detected subpopulations represent either ancestral polymorphism or residual levels of gene flow as the populations diverge. The moderate but significant F ST levels between the two Structure clusters suggests the later, while the alternative scenario of extensive mixing after secondary contact would lead to heterozygote excess (the isolate breaking effect; Hartl and Clark 1997), which was not observed in our data. However, connectivity between subpopulations might have been established only recently following range expansions. It is also possible that individuals from other European regions might have migrated into Bulgaria during the 1970s bottleneck increasing the apparent differentiation, but not enough data is available in this study to assess it. We also note that the Bayesian clustering methods used showed some evidence for further substructure in Bulgarian wolves, in both microsatellite loci and mtDNA. However, four key aspects of our results led us to adopt a conservative strategy in the identification of population clusters: first, Structure results for higher K-values provided no further resolution in spite of higher likelihood scores; second, Structure results for K = 3 showed no clear genetic and spatial division between the wolf clusters, thus suggesting some level of gene flow; third, the multivariate approach implemented in DAPC (that does not rely on HWE assumptions) found no support for more than two clusters within the wolf samples; finally, Geneland results with the highest likelihood (K = 12) largely corresponded to the distribution of kin groups.
Nevertheless, there are indications that our approach might be missing further population structure. DAPC results showed much less mixing between the two clusters, and support for the existence of three clusters was only marginally inferior to two clusters. Results from mtDNA further suggest the occurrence of three (possibly four) clusters, which would be expected in case of recent differentiation, because mtDNA haplotype frequencies fixate faster in a population due to lower N e. Our sampling was opportunistic and therefore the coverage of the wolf range in Bulgaria was uneven and incomplete, and this could have compromised the detection of fine-scale population differentiation.
Population structure in wolves is known to correlate with environmental differences (e.g. Geffen et al. 2004; Pilot et al. 2006). An overlap between the three identified mtDNA clusters (Fig. 3b) and regions of distinct topography (Figure S1) can be observed, with altitude differences potentially reflecting different habitats with distinct ungulate communities, and as such, reflecting difference in prey resource availability. Therefore, the possibility of further undetected population substructure would be relevant for conservation purposes, as local habitat disturbances could affect locally adapted populations, and further reduce genetic diversity of this species. Although data from this study is insufficient to make strong statements regarding fine-scale population subdivision and its causes, it highlights the need of further studies on wolf conservation genetics in Bulgaria and the broader Balkan region.
Conclusions
Although Bulgarian wolves appear to be recovering well from a severe bottleneck, with high levels of heterozygosity, N e/N ratios are low when compared to protected wolf populations elsewhere and we thus suggest that unregulated hunting pressure is having a notable effect on the pack structure and inbreeding, and may be compromising the long term viability of the Bulgarian population. We detected the presence of population structure within the region, possibly reflecting fragmentation during the period of lower population size. Hybridisation with dogs appears to be widespread but insufficient to cause strong genetic introgression into wolf genetic pool. Unexpectedly, we also have identified signals of wolf hybridisation with golden jackals. Our results suggest that reduced mate availability due to the hunting pressure may be promoting changes in pack structure and hybridisation with closely related species. Our results emphasize the need to establish comprehensive conservation efforts for the Bulgarian wolves, and highlight important considerations when managing hunting in recovering mammalian populations in Europe.
References
Aladzhem S (2000) Miracles in the world of plants of Bulgaria. In: Aladzhem S (ed) The green gold of Bulgaria. ABV-tech Limited, Sofia, pp 59–60
Anderson EC, Thompson EA (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160:1217–1229
Aspi J, Roininen E, Ruokonen M, Kojola I, Vilà C (2006) Genetic diversity, population structure, effective population size and demographic history of the Finnish wolf population. Mol Ecol 15:1561–1576
Aspi J, Roininen E, Kiiskilä J, Ruokonen M, Kojola I, Bljudnik L, Danilov P, Heikkinen S, Pulliainen E (2008) Genetic structure of the northwestern Russian wolf populations and gene flow between Russia and Finland. Conserv Genet 10:815–826
Blanco JC, Reig S, de la Cuesta L (1992) Distribution, status and conservation problems of the wolf Canis lupus in Spain. Biol Conserv 60:73–80
Boitani L (2003) Wolf conservation and recovery. In: Mech LD, Boitani L (eds) Wolves: behaviour, ecology and conservation. University of Chicago Press, Chicago, pp 317–340
Butler D (1994) Bid to protect wolves from genetic pollution. Nature 370:497
Carmichael LE, Nagy JA, Larter NC, Strobeck C (2001) Prey specialization may influence patterns of gene flow in wolves of the Canadian Northwest. Mol Ecol 10:2787–2798
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Czarnomska SD, Jędrzejewska B, Borowik T, Niedziałkowska M, Stronen AV, Nowak S, Mysłajek RW, Okarma H, Konopiński M, Pilot M, Śmietana W, Caniglia R, Fabbri E, Randi E, Pertoldi C, Jędrzejewski W (2013) Concordant mitochondrial and microsatellite DNA structuring between Polish lowland and Carpathian Mountain wolves. Conserv Genet 14:573–588
Echegaray J, Vilà C (2010) Noninvasive monitoring of wolves at the edge of their distribution and the cost of their conservation. Anim Conserv 13:157–161
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Fabbri E, Miquel C, Lucchini V, Santini A, Caniglia R, Duchamp C, Weber J-M, Lequette B, Marucco F, Boitani L, Fumagalli L, Taberlet P, Randi E (2007) From the Apennines to the Alps: colonization genetics of the naturally expanding Italian wolf (Canis lupus) population. Mol Ecol 16:1661–1671
Francisco LV, Langsten AA, Mellersh CS, Neal CL, Ostrander EA (1996) A class of highly polymorphic tetranucleotide repeats for canine genetic mapping. Mamm Genome 7:359–362
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Gaubert P, Bloch C, Benyacoub S, Abdelhamid A, Pagani P et al (2012) Reviving the African wolf Canis lupus lupaster in north and west Africa: a mitochondrial lineage ranging more than 6,000 km wide. PLoS ONE 7:e42740
Geffen E, Anderson MJ, Wayne RK (2004) Climate and habitat barriers to dispersal in the highly mobile grey wolf. Mol Ecol 13:2481–2490
Godinho R, Llaneza L, Blanco JC, Lopes S, Álvares F, García EJ, Palacios V, Cortés Y, Talegón J, Ferrand N (2011) Genetic evidence for multiple events of hybridization between wolves and domestic dogs in the Iberian Peninsula. Mol Ecol 20:5154–5166
Gomerčić T, Sindičić M, Galov A, Arbanasić H, Kusak J, Kocijan I, Gomerčić MD, Huber D (2010) High genetic variability of the grey wolf (Canis lupus L.) population from Croatia as revealed by mitochondrial DNA control region sequences. Zool Studies 49:816–823
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ch/izea/softwares/fstat.html
Guillot G, Santos F, Estoup A (2008) Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 24:1406–1407
Hailer F, Leonard JA (2008) Hybridization among three native North American Canis species in a region of natural sympatry. PLoS ONE 3:e3333
Hartl DL, Clark AG (1997) Principles of population genetics. Sinauer Associates, Sunderland
Hill WG (1981) Estimation of effective population size from data on linkage disequilibrium. Genet Res 38:209–216
Holmes NG, Dickens HF, Parker HL, Binns MM, Mellersh CS, Sampson J (1995) Eighteen canine microsatellites. Anim Genet 26:132–133
Jansson E, Ruokonen M, Kojola I, Aspi J (2012) Rise and fall of wolf population: genetic diversity and structure during recovery, rapid expansion and drastic decline. Mol Ecol 21:5178–5193
Jędrzejewski W, Branicki W, Veit C, Medugorac I, Pilot M, Bunevich AN, Jędrzejewska B, Schmidt K, Theuerkauf J, Okarma H, Gula R, Szymura L, Förster M (2005) Genetic diversity and relatedness within packs in an intensely hunted population of wolves Canis lupus. Acta Theriol 50:3–22
Jombart T, Ahmed I (2011) Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Konovalov DA, Manning C, Henshaw MT (2004) Kingroup: a program for pedigree relationship reconstruction and kin group assignments using genetic markers. Mol Ecol Notes 4:779–782
Le Page SL, Livermore RA, Cooper DW, Taylor AC (2000) Genetic analysis of a documented population bottleneck: introduced Bennett’s wallabies (Macropus rufogriseus rufogriseus) in New Zealand. Mol Ecol 9:753–763
Leonard JA, Vilà C, Fox-Dobbs K, Koch PL, Wayne RK, Van Valkenburgh B (2007) Megafaunal extinctions and the disappearance of a specialized wolf ecomorph. Curr Biol 17:1146–1150
Liberg O, Andrén H, Pedersen H-C, Sand H, Sejberg D, Wabakken P, Åkesson M, Bensch S (2005) Severe inbreeding depression in a wild wolf (Canis lupus) population. Biol Lett 1:17–20
Liberg O, Chapron G, Wabakken P, Pedersen HC, Hobbs NT, Sand H (2012) Shoot, shovel and shut up: cryptic poaching slows restoration of a large carnivore in Europe. P Roy Soc B-Biol Sci 279:910–915
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lucchini V, Galov A, Randi E (2004) Evidence of genetic distinction and long-term population decline in wolves (Canis lupus) in the Italian Apennines. Mol Ecol 13:523–536
Mech LD, Seal US (1987) Premature reproductive activity in wild wolves. J Mammal 68:871
Milenkovic M, Sipetic VJ, Blagojevic J, Totovic S, Vujosevic M (2010) Skull variation in Dinaric–Balkan and Carpathian gray wolf populations revealed by geometric morphometric approaches. J Mammal 91:376–386
Nilsson T (2004) Integrating effects of hunting policy, catastrophic events, and inbreeding depression, in PVA simulation: the Scandinavian wolf population as an example. Biol Conserv 115:227–239
Ostrander EA, Sprague GF, Rine J (1993) Identification and characterization of dinucleotide repeat (CA)n markers for genetic mapping in dog. Genomics 16:207–213
Ovenden JR, Peel D, Street R, Courtney AJ, Hoyle SD, Peel SL, Podlich H (2007) The genetic effective and adult census size of an Australian population of tiger prawns (Penaeus esculentus). Mol Ecol 16:127–138
Peakall ROD, Smouse PE (2006) Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peshev T, Peshev D, Popov V (2004) Fauna of Bulgaria. Marin Drinov Press, Sofia
Pilot M, Jȩdrzejewski W, Branicki W, Sidorovich VE, Jȩdrzejewska B, Stachura K, Funk SM (2006) Ecological factors influence population genetic structure of European grey wolves. Mol Ecol 15:4533–4553
Pilot M, Branicki W, Jedrzejewski W, Goszczynski J, Jedrzejewska B, Dykyy I, Shkvyrya M, Tsingarska E (2010) Phylogeographic history of grey wolves in Europe. BMC Evol Biol 10:104
Popov V (2003) Mammals in Bulgaria. Vitosha Nature Park Directorate, Sofia
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Wen X, Falush D (2010) Documentation for Structure software: Version 2.3. Available from: http://pritch.bsd.uchicago.edu/structure_software/release_versions/v2.3.4/structure_doc.pdf
Randi E (2011) Genetics and conservation of wolves Canis lupus in Europe. Mammal Rev 41:99–111
Randi E, Lucchini V, Christensen MF, Mucci N, Funk SM, Dolf G, Loeschke V (2000) Mitochondrial DNA variability in Italian and East European wolves: detecting the consequences of small population size and hybridization. Conserv Biol 14:464–473
Rhymer JM, Simberloff D (1996) Extinction by hybridization and introgression. Annu Rev Ecol Syst 27:83–109
Rousset F (2008) Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Rueness EK, Asmyhr MG, Sillero-Zubiri C, Macdonald DW, Bekele A, Atickem A, Stenseth NC (2011) The cryptic African wolf: Canis aureus lupaster is not a golden jackal and is not endemic to Egypt. PLoS ONE 6:e16385
Rutledge LY, Patterson BR, Mills KJ, Loveless KM, Murray DL, White BN (2010) Protection from harvesting restores the natural social structure of eastern wolf packs. Biol Conserv 143:332–339
Sastre N, Vilà C, Salinas M, Bologov VV, Urios V, Sánchez A, Francino O, Ramírez O (2011) Signatures of demographic bottlenecks in European wolf populations. Conserv Genet 12:701–712
Shibuya H, Collins BK, Huang THM, Johnson GS (1994) A polymorphic (AGGAAT), tandem repeat in an intron of the canine von Willebrand factor gene. Anim Genet 25:122
Spiridonov G, Spassov N (1985) Wolf–Canis lupus L., 1758. In: Botev, Peshev (eds) Red Data Book of Bulgaria. Bulgarian Academy of Science, Sofia, p 132
Spiridonov G, Spassov N (2011) Canis lupus L., 1758. In: Golemanski V (ed) Red Data Book of the Republic of Bulgaria. Bulgarian Academy of Sciences & Ministry of Environment and Water, Sofia
Tallmon DA, Koyuk A, Luikart G, Beaumont MA (2008) OneSamp: a program to estimate effective population size using approximate Bayesian computation. Mol Ecol Resour 8:299–301
Temple HJ, Terry A (Compilers) (2007) The status and distribution of European mammals. Office for Official Publications of the European Communities: Luxembourg
Tsingarska-Sedefcheva E (2007) Wolf activity towards livestock in two study areas in West Bulgaria and consequential conflict with livestock breeders. In: IIIrd Congress of Ecologists of the Republic of Macedonia, Struga
Tsingarska-Sedefcheva E (2013) Wolf—Bulgaria. In: Kaczensky P, Chapron G, von Arx M, Huber D, Andren H, Linnell J (eds) Status, management and distribution of large carnivores—bear, lynx, wolf & wolverine—in Europe. European Commission, N°070307/2012/629085/SER/B3
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Verardi A, Lucchini V, Randi E (2006) Detecting introgressive hybridization between free-ranging domestic dogs and wild wolves (Canis lupus) by admixture linkage disequilibrium analysis. Mol Ecol 15:2845–2855
Verginelli F, Capelli C, Coia V, Musiani M, Falchetti M, Ottini L, Palmirotta R, Tagliacozzo A, Mazzorin ID-G, Mariani-Costantini R (2005) Mitochondrial DNA from prehistoric canids highlights relationships between dogs and South-East European wolves. Mol Biol Evol 22:2541–2551
Vilà C, Walker C, Sundqvist AK, Flagstad Ø, Andersone Z, Casulli A, Kojola I, Valdmann H, Halverson J, Ellegren H (2003) Combined use of maternal, paternal and bi-parental genetic markers for the identification of wolf–dog hybrids. Heredity 90:17–24
vonHoldt BM, Stahler DR, Smith DW, Earl DA, Pollinger JP, Wayne RK (2008) The genealogy and genetic viability of reintroduced Yellowstone grey wolves. Mol Ecol 17:252–274
Wabakken P, Sand H, Liberg O, Bjärvall A (2001) The recovery, distribution, and population dynamics of wolves on the Scandinavian peninsula, 1978–1998. Can J Zool 79:710–725
Wabakken P, Sand H, Kojola I et al (2007) Multistage, long-range natal dispersal by a global positioning system-collared Scandinavian wolf. J Wildl Manag 71:1631–1634
Waples RS, Do CHI (2008) LDne: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756
Whitehouse AM, Harley EH (2001) Post-bottleneck genetic diversity of elephant populations in South Africa, revealed using microsatellite analysis. Mol Ecol 10:2139–2149
Acknowledgments
We are very grateful to Włodzimierz Jędrzejewski for his help and support in the long-term study programme of European wolves. We thank Holly Ernest and three anonymous reviewers for their helpful comments on the manuscript. For the assistance in the collection of samples, we would like to thank Sider Sedefchev, Ivan Todev, Dimitar Dimitrov, Nina Kirova, Alexandar Dutsov, Kamen Krastanov, Todor Georgiev, Kostadin Valchev, Dimitar Arabadzhiev, Atila Sedefchev, Vergil Murarov, Georgi Stoianov, Georgi Stefanov, Panaiot Panaiotov, Yanko Yanev, Sahak Sahakian, Todor Mitev and all the other foresters and hunters who supported this activity and made this study possible. We also thank Yorgos Iliopoulos and other members of the Greek Society for the Protection and Management of Wildlife ARCTUROS for providing wolf samples from Greece. This project was funded by the former Polish State Committee for Scientific Research (Grant no. 6P04F 09421), the Bernd Thies Foundation and European Nature Heritage Fund EURONATUR (Germany). M. Pilot was supported by a fellowship from the Foundation for Polish Science.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Moura, A.E., Tsingarska, E., Dąbrowski, M.J. et al. Unregulated hunting and genetic recovery from a severe population decline: the cautionary case of Bulgarian wolves. Conserv Genet 15, 405–417 (2014). https://doi.org/10.1007/s10592-013-0547-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-013-0547-y