
Abstract
The authors’ series of studies aimed to explore the potential of terahertz time-domain spectroscopy (THz-TDS) in cellulose crystallographic studies, since THz radiation can detect most intermolecular vibrations and respond to lattice phonons. In this study, we tracked changes in four types of cellulose after ball milling. As the planetary ball milling time increases, it is observed through electron microscopy that the four types of cellulose particles are gradually destroyed into finer particles, while gel permeation chromatography can prove that the molecular weight gradually decreases after ball milling and the dispersity gradually approaches one, which indicates that the dispersion of the material was reduced. The most fascinating observation was made by THz-TDS, that is we have confirmed that after ball milling, the absorption characteristics of cellulose I and II in cellulose I treated with 10% NaOH (crystalline partial transition from cellulose I to II) exhibited an opposite trend. Specifically, the absorption of cellulose II at 2.40THz and 2.77THz increased, while the absorption of cellulose I at 2.11THz and 3.04THz decreased after ball milling, which suggests an increased conversion rate of cellulose I to cellulose II post-milling. Cellulose with different crystalline allomorphs shows different characteristic absorption in the THz region, and the peak position will not change even after the ball milling, only the absorption intensity changes. Although it can be observed through the most traditional X-ray diffraction method that the crystallinity index of all cellulose samples gradually decreases after ball milling. However, different from the THz results, the change after ball milling of cellulose I treated with 10% NaOH is only reflected in very subtle pattern changes, that is, the peak close to the 200 crystalline plane position is slightly shifted after ball milling.
Similar content being viewed by others

Introduction
The most common polymeric materials widely used in the pharmaceutical, food, and textile industries are natural cellulose (cellulose I) and regenerated cellulose (cellulose II) (Brown 2004; Kamel et al. 2008; Agarwal et al. 2013; Kafle et al. 2014; Saffarionpour 2020). An inseparable relationship is observed between the physical and chemical properties of cellulose materials and their crystalline structures and contents (Nakamura et al. 2004; Moon et al. 2011; Missoum et al. 2013; Kafle et al. 2014). Therefore, researchers have attempted to accurately measure and track changes in cellulose following physical or chemical treatments to understand and use these changes to develop new materials or application scenarios.
Numerous techniques have been used to examine the structure and properties of cellulose. For example, gel permeation chromatography (GPC) can be used to measure molecular weight and molecular distribution (Rousselle 2002). Scanning electron microscopy (SEM) allows researchers to intuitively observe the morphology of fibers at high magnifications (Zhao et al. 2007; Håkansson et al. 2014). The most widely used and almost necessary method for studying crystalline cellulose is X-ray diffraction (XRD), and several significant results have been derived from XRD measurements. For instance, the crystalline structure is determined using this technique (Wada et al. 1993; Imai and Sugiyama 1998; Imai et al. 1999), and the widely used cellulose crystallinity index is defined and provided (Segal et al. 1959; Park et al. 2010). Moreover, because of its cost effectivity, Fourier transform infrared (FTIR) spectroscopy is widely used (Kataoka and Kondo 1998; Chen et al. 2014; Hishikawa et al. 2017). Simultaneously, methods such as solid-state cross-polarization magic angle spinning carbon-13 nuclear magnetic resonance (CPMAS 13C NMR) spectroscopy and Raman spectroscopy have been used to study crystalline cellulose (Horii et al. 1987; Larsson et al. 1997; Schenzel and Fischer 2001).
In recent years, terahertz time-domain spectroscopy (THz-TDS) has attracted considerable attention. The THz radiation is characterized by frequencies ranging from 0.1 to 10 THz (wavelengths between 3 and 0.03 mm). It possesses a uniquely low photon energy (approximately 4 meV for 1 THz, whereas the photon energy range of visible light is roughly between 1.77 and 3.10 eV), leading to harmless interactions with the human body. Moreover, they have better transmission properties than infrared and visible light. With a longer wavelength than IR, the influence from scatter which is caused by the microstructure inside of the material is less in the THz radiation (Lewis 2017). THz-TDS has been used in several fields including the detection of explosives (Leahy-Hoppa et al. 2009), vibrational modes of DNA (Markelz et al. 2000; Vaks et al. 2016) and proteins (Kim et al. 2008; Robert et al. 2012; Shiraga et al. 2016). Because the THz region has a strong absorption effect on water molecules, THz can be used to map the distribution of water molecules or moisture content in materials even if the moisture content in the material is low (Federici 2012; Wang et al. 2019). However, the strong absorption of the water molecules also brings a challenge for the vivo detections, especially for the transmission measurement (Federici 2012). And the current relatively high price of the THz-TDS makes it difficult to be widely used.
THz radiation exhibited good performance in studies on cellulose due to the good response to the crystalline lattice vibrations. For example, a partial least squares (PLS) model has been used to predict the crystallinity of microcrystalline cellulose (Vieira and Pasquini 2014). We conducted a series of studies to further expand the application scenarios of THz-TDS in cellulose research, including distinguishing cellulose I allomorphs (Wang et al. 2020), determining the amount of crystalline cellulose in a sample rather than the relative crystallinity index (Wang et al. 2021), and tracing the crystalline transformation of cellulose I to cellulose II (Wang et al. 2022). There is a study that have reported the by ball milling cellulose I in a specific moisture content also will lead to a crystalline change which may be caused by the chain mobility in a cellulose–water system (Ago et al. 2004). However, there is still no report showing a crystalline change by the ball mill in a dry condition. In continuation with the previous studies, this study aims to explore the potential of employing THz-TDS in cellulose crystallography research. It is the first time we observed that after treatment with 10% NaOH and subsequent ball milling, the THz second-derivative absorption characteristics of cellulose I undergo a transition to cellulose II, which is characterized by a reduced degree of crystallinity and an increased conversion rate of cellulose II. This was also evidenced by XRD patterns showing more characteristics of cellulose II than in the initial state without ball milling. And we confirmed that the identification of cellulose II should be based on the peak at 1.32 THz, not on the more conspicuous peak at 2.77 THz that is observed in a pure cellulose II sample Distinct changes in these features were more clearly observed using the THz second-derivative absorption spectra, thus confirming this transformation without the necessity for any empirical data analysis.
Experimental
Sample preparation
To observe the crushing state and possible crushing mechanism of cellulose with different crystallines during ball milling, four different cellulose samples were prepared in this study.
-
1.
cellulose I (Avicel® PH-101, purchased from Sigma-Aldrich).
-
2.
cellulose II (prepared by treating cellulose I with 20 wt% NaOH).
-
3.
cellulose transitioning (prepared by treating cellulose I with 10 wt% NaOH).
-
4.
cellulose mixed (prepared by mixing cellulose I and cellulose II described above, both types of crystalline cellulose have a mass fraction of 50%).
For sample 2 and 3, where the microcrystalline cellulose powder treated with different concentrations of NaOH follows this protocol: NaOH treatment (different concentrations) for 30 min at room temperature 25 °C. After the reaction, the sample was washed with acetic acid and distilled water until the pH was 7 and filtered, then the washed cellulose powder was prepared by drying it in a drier containing phosphorus pentoxide. NaOH, acetic acid, and phosphorus pentoxide used in this study were purchased from Kishida Chemical Co. Ltd. The fourth sample was a mixture of cellulose I and cellulose II (both types of crystalline cellulose have a mass fraction of 50%) samples to simulate the cellulose intermediate form of cellulose I to cellulose II.
All cellulose samples were ball-milled using a benchtop ball mill (AV-2, Asahi Rika Factory. Ltd.) at 200 rpm using ceramic spheres and jars. Approximately 8 g of each sample was placed in a 450 mL ceramic jar along with 100 ceramic spheres, each with 8 mm diameter. The samples were milled for 0, 6, 12, 24, 48, 72, and 144 h. The milled powders were then collected, and 75 mg of each powder were pressed into tablets of approximately 0.35 mm thickness using a hand-pressing tableting kit (IMC-180C, Imoto Machinery Co. LTD). The process employed a tablet fixture with a diameter of 14 mm and pressure of 40 MPa. d-For reproducibility of the experiment, three tablets were prepared for each powder. The thicknesses of the tablet samples were measured using a micrometer (± 0.001 mm).
Sample measurement
GPC
The cellulose powder sample (50 mg) was vacuum dried at 40 °C for at least 2 d, 25 mg of 4-dimethylaminopyridine, and 2.5 mL of pyridine were mixed in a flask. Subsequently, 0.75 mL of propionic anhydride were added to the flask and the mixture was stirred at 110 °C for 24 h until the cellulose sample was uniformly dissolved. After the reaction, the sample was re-precipitated in ice water and shaken for 10 min. The samples were then filtered and washed with methanol in a funnel. The washed sample was dried under vacuum at 40 °C for 1 h and then dissolved in 2 mL of chloroform. The solution was then filtered through a PTFE filter and re-precipitated in methanol. The sample was filtered and washed with methanol and dried under vacuum at 40 °C (over 2 d). All dried samples were then subjected to gel permeation chromatography (GPC) measurement on a Shimadzu LC-10 system (Shimadzu) equipped with a Shimadzu RI detector (RID-10A) under the following conditions: column, Shodex K-802, K-802.5, and K-805 (Showa Denko K.K., Tokyo, Japan) arranged in series; eluent: chloroform; temperature: 40 °C; flow rate: 1.0 mL/min; standard: polystyrene standard (Showa Denko K.K.).
SEM
Morphological changes in the cellulose particles after ball-milling were observed using a desktop SEM (TM4000 PlusII, Hitachi Ltd., Japan) with backscattered electrons and an accelerating voltage of 15 kV. After different ball milling times, the four types of cellulose powders were mounted on a specimen stage with adhesive carbon tape and photographed at a 400 X magnification. To obtain the average areas of the cellulose particles in the SEM images, Fiji win64-bit version was used (Schindelin et al. 2012). First, the scale was set such that the pixel size could be converted to the actual size. Second, the Shanbhag method in the auto-threshold was utilized, which included a measurement of the distance to binary values in the entropy measures. This method can generate more accurate and meaningful thresholds, particularly for images where the foreground and background have similar intensities, such as images of particles finely ball-milled over a long period of time (Shanbhag 1994). Subsequently, Fill Holes were used to fill the gaps. Finally, the Analysis Particles function was employed to calculate the number of particles, their respective areas, and the average area of the particles in each image.
XRD
The XRD measurements of all tablet samples were performed with Cu-Kα radiation (λ = 0.1542 nm) using a diffractometer (Ultima IV, Rigaku) at a voltage of 40 kV and a current of 40 mA. Diffractograms were recorded from 5° to 30°. The scan speed was set to 5° min−1, and the sampling step was 0.05°. The background diffractograms were obtained using an empty sample holder.
THz-TDS
THz transmission spectra of all tablet samples were measured using a Tera Prospector Kit (NIPPO PRECISION Co. Ltd.), and reference signals were obtained from air measurements before and after sample measurement. The THz beam was horizontally polarized, and the bandwidth was from approximately 0.1 THz to 4.00 THz. Because cellulose II showed only weak absorption in the THz field in previous studies (Wang et al. 2022), hence, this study employs a high spectral resolution of 0.057 THz, which corresponds to the reciprocal time scanning range of 175 ps to meticulously observe spectral changes. The corresponding single-spectra measurement time was also prolonged compared to that in previous studies. The diameter of the THz-beam spot on the sample was of approximately 3 mm. Each measurement was recorded by averaging 100 scans, to improve the signal-to-noise ratio. All measurements were performed thrice for reproducibility. To avoid the influence of THz absorption of water vapor on the measurements, we placed the entire THz optical system in an almost closed acrylic box, which was constantly filled with dry air to ensure stable humidity. All samples were placed in the box for 24 h to equilibrate the ambient humidity before measurement. After placing the sample in the chamber, dry air was passed through the chamber until all THz measurements were completed.
Results and discussion
Alterations in molecular weight and particle area post ball milling
Figure 1 shows SEM images of the four types of cellulose samples at three distinct ball milling stages: initial state (0 h), intermediate state (24 h), and final state post ball milling (144 h). As the process continued, the cellulose particles underwent noticeable degradation, which is visible in the SEM images. Cellulose I, cellulose II, and mixed cellulose transformed from an initial irregular accumulation of large fibers into smaller particles. After 24 h of ball milling, some larger, non-uniform cellulose particles were still evident. After 144 h, most cellulose particles exhibited a relatively uniform spherical shape, with only a few larger particles observable.

SEM images of cellulose I (without NaOH treatment), cellulose II (20% NaOH treated), cellulose transitioning (10% NaOH treated), and cellulose mixed (50% cellulose I and 50% cellulose), from top to bottom are without ball milling, ball milling for 24 h, and ball milling for 144 h
Figures 1a and d show the swelling of cellulose induced by the NaOH treatment, where the size of cellulose II is bigger than that of cellulose I in the initial state. However, after ball milling, compared with other cellulose samples, the particle size of cellulose I was the biggest. Interestingly, the cellulose transition occurred at the stage where cellulose I began to transform into cellulose II, and its particles were rounder and larger compared with the other cellulose samples without ball milling, as shown in Fig. 1g. However, in the final stage, the particle size was similar to that of cellulose II and smaller than those of cellulose I and mixed cellulose. The particle area was used to characterize the size of the particles calculated from the SEM images, and the detail data have been summarized in Table 1. Cellulose particles break after a long time of ball milling, changing the particle size and the molecular weight. Table 1 also lists the number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (PDI, the ratio of Mw to Mn) measured by GPC.
The GPC results reveal a downward trend with increasing ball-milling time as shown in Table 1. For all the cellulose samples, the PDI was closer to one after ball milling, which indicates that the dispersion of the material was reduced, the molecular weight distribution was narrowed, and the uniformity of the material increased, which is also consistent with the results of the SEM image analysis. Because cellulose is a polymer material, the choice of Mw can better reflect the properties of macromolecules. Mw will be further analyzed and elaborated upon in a subsequent discussion.
XRD pattern and THz 2nd derivative absorption spectra changes of cellulose
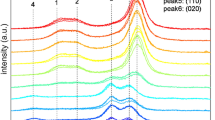
The original XRD patterns were cut in the range 10–30° and processed for background subtraction and baseline correction. The patterns are shown in Fig. 2, from top to bottom: cellulose I, cellulose II, cellulose transitioning, and cellulose mixed, where the color gradients indicate different ball milling times.

Baseline-corrected XRD patterns of a cellulose I, b cellulose II, c cellulose transitioning, and d cellulose mixed, all-in triplicate. The crystalline peaks of cellulose I and II are denoted as numbers from 1 to 6 and ball milling times are indicated by different colors
In Fig. 2a, it can be observed that the Miller indices of the three main crystalline peaks of cellulose I are (1\(\overline{1 }\)0), (110), and (200); while in Fig. 2b it can be observed that the typical cellulose II crystalline peaks correspond to the Miller indices (1\(\overline{1 }\)0), (110), and (020). For convenience of subsequent description, as shown in Fig. 2, the crystalline peaks of cellulose I are represented as peak 1, peak 2, and peak 3, and the crystalline peaks of cellulose II are represented as peak 4, peak5, and peak 6. In Fig. 2a, it can be observed that the intensity of the peaks in the XRD pattern decreased significantly after ball-milling cellulose I, and the scattering intensity corresponding to the amorphous region increased, with a maximum intensity at approximately 20° (Ling et al. 2019; French 2020; Yao et al. 2020). The crystalline peak 3 was barely discernible after 144 h of ball milling. In Fig. 2b, the intensity of the peaks of cellulose II decreases after ball milling, but not as severely as that of cellulose I, and it is not possible to observe a substantial increase in the scattering intensity corresponding to the amorphous regions approximately 16°similar to cellulose I shown in Fig. 2a. However, the belief was that the absence of crystalline diffraction at 16° meant any intensity observed there stemmed from amorphous material, which is not supported by experiment (Oh et al. 2005; French 2014). The same is true for the crystalline peaks 110 and 020, which were originally distinguishable and almost merged into a single broad and flat peak.
Cellulose transitioning occurs during the transition from cellulose I to cellulose II, and the highest intensity peak is located between peaks 3 (the 200 crystalline peak of cellulose I) and 6 (the 020 crystalline peak of cellulose II). The crystalline peak 5 showed a relatively low intensity, whereas peaks 1, 2, and 4 were only faintly visible in the XRD pattern. Meanwhile, the highest intensity peak gradually shifted to the position of peak 6, and no change was observed at the position of the peak 5. In addition, the intensity difference between peaks 5 and 6 also decreased compared to the initial state, Moreover, cellulose transitioning still exhibited clear crystalline peaks on the XRD pattern even after 144 h of ball milling.
For the cellulose mixed shown in Fig. 2d, even if the cellulose I and cellulose II contents are both 50%, the XRD pattern is still dominated by cellulose I. However, with the progress of ball milling, a peak shift similar to that in the cellulose transitioning shown in Fig. 2c is observed, and the crystalline peak originally located at peak 3 shifts to the middle position of peaks 3 and 6. Because the intensity reduction of peak 5 was not as drastic as that of peak 3, the pattern exhibited characteristics similar to the initial state of cellulose transitioning after 24 and 48 h of ball milling. However, after 72 and 144 h of ball milling, the original crystalline peak disappeared, leaving only a broad and flat scattering curve at approximately 21°.
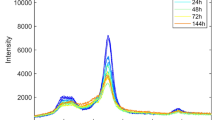
Previous studies have shown that the absorption peak of cellulose II in the THz region is smaller than that of cellulose I (Wang et al. 2022). To observe the detailed changes in the THz absorption spectra, THz 2nd order derivative absorption spectra were required. The THz 2nd derivative absorption spectra can extend the characteristics of the original absorption spectra, however its trend is opposite to that of the original spectra, that is, where the original absorption decreases, the value in the 2nd derivative absorption spectra increases. Specifically, the measured THz time-domain signal is Fourier transformed into the frequency domain, and the absorption coefficient α is calculated. Baseline fluctuations were corrected using a standard normal variation (SNV) algorithm, followed by quadratic differentiation after smoothing using a Savitzky–Golay filter with a second-order polynomial to remove noise. Because this series of studies used the same calculation method, the calculation formula was not repeated here. The specific calculation formulas used and the THz raw and SNV absorption spectra are provided in the Supplementary Information. Figure 3 is the THz absorption spectra after the 2nd order derivative of 1.15–3.25 THz, the arrangement order and the color gradient used are the same as those in Fig. 2. Similarly, the observed absorption peaks are represented by numbers 1–6, peaks 1 and 2, correspond to the absorption of cellulose I, and peaks 3 to 6 are the absorption of cellulose II, which will be discussed in detail later.

THz 2nd derivative absorption spectra of a cellulose I, b cellulose II, c cellulose transitioning, and d cellulose mixed, all-in triplicate. The THz 2nd derivative absorption peaks of cellulose I and II are denoted as numbers from 1 to 6 and ball milling times are indicated by different colors
In Fig. 3a, the same typical absorption peaks 1 (at 2.13 THz) and 2 (at 3.04 THz) exhibited by cellulose I can be observed, as in previous studies (Wang et al. 2020). As ball milling proceeded, the values of peaks 1 (at 2.13 THz) and 2 (at 3.04 THz) of its THz 2nd derivative absorption spectra gradually increased, which corresponds to the intensity decrease in its original absorption spectra. For convenience of subsequent description, the changes in the absorption spectra corresponding to the THz 2nd derivative absorption spectra will not be described in detail.
Figure 3b shows the typical absorption of cellulose II, in which the previous study is the same at peaks 3 (1.32 THz) and 6 (2.77 THz); after ball milling, we confirmed that peak 5 (2.40 THz) was also the characteristic absorption of cellulose II. In contrast to a previous study (Wang et al. 2022), peak 4 at 1.76 THz shifted to 1.71 THz, which is discussed later in the text. With an increase in the milling time, the values of peaks in the THz 2nd derivative absorption spectra increased.
Figure 3c shows the THz 2nd derivative absorption spectra of cellulose transitioning, which exhibited the most attractive spectral properties in this study. These two features are summarized below.
First, THz 2nd derivative absorption spectra show relatively uniform characteristics of both cellulose I and II, none of which are dominant. Peaks 1 and 2 correspond to the absorption of cellulose I, whereas values of peaks in the THz 2nd derivative absorption spectra increased with ball milling. In contrast, peaks 3–6, which correspond to the absorption of cellulose II, show a completely opposite trend to peaks 1 and 2, corresponding to the absorption of cellulose I, with the progress of ball milling. In other words, the corresponding values of peaks of the THz 2nd derivative absorption spectra decreased. The specific changes are shown in Figs. 4a and b To directly correspond to the actual change trend of the THz absorption peak, reversed THz 2nd derivative absorption values were used. The relative content of crystalline cellulose II during the cellulose transition may increase as the ball milling progresses. This could explain why peak 3 (200 crystalline peaks of cellulose I) in the XRD pattern of the transitioning cellulose shown in Fig. 1c gradually shifted to the position of peak 6 (020 crystalline peaks of cellulose II) as the ball milling progressed.

Change of the characteristic peaks of reversed THz 2nd derivative absorption values of cellulose transitioning of a cellulose I, marked as the regular triangle, b cellulose II, marked as the inverted triangle, with an error bar of the standard deviation. Ball milling times are indicated by different colors which are same with Figs. 2 and 3
Second, the cellulose transitioning treated with 10% NaOH used in previous studies (Wang et al. 2022) could not exhibit peaks 5 and 6 at 2.40 THz and 2.77 THz, respectively, in the THz 2nd derivative absorption spectra. Because the method of preparing cellulose transitioning is consistent with the previous study, these phenomena may be owing to the use of cellulose produced by different manufacturers with some subtle characteristic differences. This could be the reason for the shift of peak 4 from 1.76 to 1.71 THz, as shown in Fig. 3b.
Finally, Fig. 3d shows the THz 2nd derivative absorption spectra of cellulose mixed. Similar to the XRD pattern of cellulose mixed shown in Fig. 2d, the THz 2nd derivative absorption spectra are dominated by the characteristic absorption of cellulose I. All the observed values of the 2nd derivative absorption peaks are closer to zero (the corresponding absorption itself decreases) with ball milling. Notably, absorption peak 5 at 2.40 THz and even the most important absorption peak 6 at 2.77 THz are no longer visible, while the changing of the values of absorption at peak 4 at 1.71 THz only barely paraphrases the trend of increasing with the ball milling time. Without reference to the results described above; it is difficult to clearly indicate that this is the absorption peak of cellulose II based only on the results shown in Fig. 3d. Here, we can clearly observe only peak 3 at 1.32 THz which corresponds to the absorption of cellulose II. This suggests that, in the THz absorption spectra of a cellulose sample mixed with various crystalline forms, the identification of cellulose II should be based on the peak at 1.32 THz, not on the more conspicuous peak at 2.77 THz that is observed in a pure cellulose II sample.
The relationship between cellulose morphology changes and XRD pattern and THz 2nd derivative absorption spectra
To delve deeper into the interdependencies among the Mw, the sample average area obtained from SEM images, the crystallinity index (CrI) calculated from XRD, and the THz 2nd derivative absorption values, the crystallinity of each cellulose sample was first calculated, and cellulose I and II have only one CrI that represents the overall crystallinity, while for the transition and mixed cellulose samples, in addition to the overall CrI reflecting the total crystallinity, we also calculated the relative crystallinity indices CrI1 and CrI2. These indices characterize the individual crystalline extents of cellulose I and II within the samples and are calculated as follows:
where SCr and SAm denote the sum of the integrated intensities of the six crystalline peaks and the amorphous intensity curve, respectively, and the detailed process of peak deconvolution using a pseudo-Voigt profile is described in the Supplementary Information.
where SCr1 and SCr2 denote the sums of the integrated intensities of the crystalline peaks of cellulose I and II, respectively, CrI is the crystallinity index calculated using Eq. (1).
Correspondingly, in this study, we use the reversed THz 2nd derivative absorption spectra values of peak 2 at 3.04 THz and peak 3 at 1.32 THz to represent the absorption of cellulose I and II, respectively. The reasons for not using peak 6 at 2.77 THz which was used in previous studies (Wang et al. 2022) are described above. For cellulose transitioning and cellulose mixing, the sum of the reversed peaks 2 and 3 of the THz 2nd derivative absorption spectrum values was used to represent the overall spectral absorption intensity change. The interdependence between the ball milling time, Mw, sample average area obtained from SEM images, crystallinity calculated from XRD, and THz 2nd derivative absorption value are shown in Fig. 5, corresponding to different cellulose samples from left to right, from top to bottom are the relationship with the ball milling time, the relationship with the average area of samples, and the relationship with Mw, the left y-axis is CrI, marked as a diamond in the figure, and the right y-axis is the reversed THz 2nd derivative absorption spectra values as detailed above, marked as circles. Error bars represent standard deviation. Because cellulose transitioning shows a very large sample average area compared to the other samples, as can be observed in the SEM images, only the logarithmic x-axis was used in Fig. 5g to improve the visualization of the data.

Relationship between ball milled time, the average area of samples, Mw and CrI (left y-axis, marked as diamond), and reversed THz 2nd derivative absorption spectra values (right y-axis, marked as circle). Error bar shows the standard deviation, and the ball milling times are indicated by different colors, same as previous figures
It can be observed from Fig. 5 that regardless of the ball milling time, the average area of the cellulose sample, Mw, CrI, and reversed THz 2nd derivative absorption spectrum values show basically the same trend of change; only the dispersion degree of the data from the reversed THz 2nd derivative absorption spectra values is smaller. More specifically, the correlation coefficients of Mw, average area, CrI, and THz 2nd derivative absorption values were calculated and are listed in Table 2. Given the initial large average area of the cellulose transition, its correlation with the absorption value of CrI and the THz 2nd derivative absorption values is not strong. The P-values of all significant differences were > 0.05. This indicates that information from either XRD or THz 2nd derivative absorption may not precisely reflect the particle size of cellulose at a macroscopic level. In contrast, the correlation coefficient r between the THz 2nd derivative absorption value and Mw for cellulose transformation was 0.86. Except for the correlation between the average area, the CrI, and the reversed THz 2nd derivative absorption values, all other P-values are ≤ 0.05. These values are not listed in Table 2. Overall, the THz 2nd derivative absorption value and Mw, as well as the sample average size, showed a better correlation than that of CrI.
To delve deeper into why the trends of peaks 1 and 2 (located at 2.13 THz and 3.04 THz, respectively) corresponding to cellulose I, and peaks 5 and 6 (located at 2.40 THz and 2.77 THz, respectively) corresponding to cellulose II in Fig. 3c show completely opposite patterns post ball milling, we first calculate the d-spacing of cellulose transitioning. The formula used was as follows:
where λ denotes the wavelength of the X-ray radiation (0.1542 nm), θ denotes the Bragg angle, and the results are listed in Table 3, there is no big difference from the literature value, the calculation should be correct (Nakamura et al. 2004).
Simultaneously, the correlation between d-spacing and the reversed THz 2nd derivative absorption values corresponding to cellulose I is located at peaks 1 (2.11 THz) and 2 (3.04 THz), and cellulose II at peaks 5 (2.40 THz) and 6 (2.77 THz), as mentioned above. The results are also listed in Table 3 to explore the possible reasons why these peaks show an opposite trend with increasing ball milling time, that is, the THz 2nd derivative absorption values reflect the structural changes in the direction of the crystal.
It can be observed from Table 3 that THz 2nd derivative absorption values have the best correlation with the d-spacing corresponding to the 200 crystalline planes of cellulose I and the 110 crystalline planes of cellulose II, except the correlation between the THz 2nd derivative absorption values at 3.04 THz with the d-spacing of the 110 crystalline planes shows no significant difference, the p-values of the other correlations are all ≤ 0.05, and the positive and negative correlations correspond to the changing trend of reversed THz 2nd derivative absorption values.
However, the hidden danger is that after a long time of ball milling, the method of extracting the crystalline peak by deconvolution may not be able to "correctly" obtain the change of the crystalline peak in the XRD pattern, which is reflected in Table 3 is that, after ball milling for a certain period of time, d-spacing no longer changes. This was because the 2θ position reached the preset threshold during the deconvolution fitting process. In contrast, if the threshold limit is lifted, overfitting will occur; that is, it is only mathematically interpretable and not physically interpretable. In contrast, the analysis of the THz 2nd derivative spectrum does not require the introduction of deconvolution and other parts that require an artificial threshold setting; therefore, it is useful for better reproducibility of the data analysis process. As long as follow-up studies prove the specific assignment of each type of absorption, the physical explanation will also be clear.
Conclusions
This study comprehensively explored the complex relationship between the morphological changes and the XRD patterns and THz 2nd derivative absorption spectra of cellulose after ball milling. There are correlations between Mw, the average area of the sample particle, and CrI and THz 2nd derivative absorption values, which imply that these factors have a strong interdependence on the cellulose morphology. This study also confirmed for the first time that the characteristic absorption peak at 2.40 THz belongs to cellulose II, and after the cellulose treated with 10% NaOH was ball milled, it is the first time we observed that the characteristic XRD pattern and THz 2nd derivative absorption spectra were close to cellulose II. In particular, significant changes were observed in the THz 2nd derivative absorption spectra after ball milling. Peaks 1 and 2, located at 2.13 THz and 3.04 THz, respectively, correspond with cellulose I. Conversely, peaks 5 and 6, identified at 2.40 THz and 2.77 THz, respectively, are associated with cellulose II. It is important to note that these sets of peaks exhibited opposite trends after milling. That is, the intensity of cellulose I peaks decreased, whereas the intensity of cellulose II peaks increased after ball milling. The intensity changes of the four THz 2nd derivative peaks may correspond to the d-spacing changes in the 200 crystalline planes of cellulose I and the 110 crystalline planes of cellulose II. However, methods such as density functional theory (DFT) calculations may have to be introduced to further explain the variation of THz 2nd derivative absorption values at 2.11 THz, 3.04 THz, 2.40 THz, and 2.77 THz changed with the ball milling for cellulose transitioning, which treated with 10% NaOH. Overall, compared with traditional XRD, THz 2nd derivative absorption spectrum is more sensitive to possible changes in crystalline cellulose and has better reproducibility. The changes caused by the crystalline structure were intuitive. The analysis without the deconvolution process of artificially setting thresholds is not required for the personal experience of the analyst to judge whether the results make physical sense, rather than just mathematical overfitting. In particular, the THz 2nd derivative absorption spectra provide a more intuitive, effective, and feasible solution for studying the subtle structural changes in crystalline cellulose under physical treatment. A series of research results from us has shown the great potential of THz-TDS for the study of crystalline cellulose. After completing the specific assignment of confirming the absorption of cellulose in the THz region, it may become a powerful research method with the same priority as XRD.
Availability of data and materials
Not applicable.
References
Agarwal UP, Reiner RR, Ralph SA (2013) Estimation of cellulose crystallinity of lignocelluloses using near-IR FT-Raman spectroscopy and comparison of the Raman and Segal-WAXS methods. J Agric Food Chem 61:103–113. https://doi.org/10.1021/jf304465k
Ago M, Endo T, Hirotsu T (2004) Crystalline transformation of native cellulose from cellulose I to cellulose II polymorph by a ball-milling method with a specific amount of water. Cellulose 11:163–167. https://doi.org/10.1023/B:CELL.0000025423.32330.fa
Brown RM (2004) Cellulose structure and biosynthesis: what is in store for the 21st century? J Polym Sci A Polym Chem 42:487–495. https://doi.org/10.1002/pola.10877
Chen C, Luo J, Qin W, Tong Z (2014) Elemental analysis, chemical composition, cellulose crystallinity, and FT-IR spectra of Toona sinensis wood. Monatsh Chem 145:175–185. https://doi.org/10.1007/s00706-013-1077-5
Federici JF (2012) Review of moisture and liquid detection and mapping using terahertz imaging. J Infrared Milli Terahz Waves 33:97–126. https://doi.org/10.1007/s10762-011-9865-7
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896. https://doi.org/10.1007/s10570-013-0030-4
French AD (2020) Increment in evolution of cellulose crystallinity analysis. Cellulose 27:5445–5448. https://doi.org/10.1007/s10570-020-03172-z
Håkansson KMO, Fall AB, Lundell F et al (2014) Hydrodynamic alignment and assembly of nanofibrils resulting in strong cellulose filaments. Nat Commun 5:4018. https://doi.org/10.1038/ncomms5018
Hishikawa Y, Togawa E, Kondo T (2017) Characterization of individual hydrogen bonds in crystalline regenerated cellulose using resolved polarized FTIR spectra. ACS Omega 2:1469–1476. https://doi.org/10.1021/acsomega.6b00364
Horii F, Hirai A, Kitamaru R (1987) CP/MAS carbon-13 NMR spectra of the crystalline components of native celluloses. Macromolecules 20:2117–2120. https://doi.org/10.1021/ma00175a012
Imai T, Sugiyama J (1998) Nanodomains of Iα and Iβ cellulose in algal microfibrils. Macromolecules 31:6275–6279. https://doi.org/10.1021/ma980664h
Imai T, Sugiyama J, Itoh T, Horii F (1999) Almost pure iα cellulose in the cell wall of glaucocystis. J Struct Biol 127:248–257. https://doi.org/10.1006/jsbi.1999.4160
Kafle K, Greeson K, Lee C, Kim SH (2014) Cellulose polymorphs and physical properties of cotton fabrics processed with commercial textile mills for mercerization and liquid ammonia treatments. Text Res J 84:1692–1699. https://doi.org/10.1177/0040517514527379
Kamel S, Ali N, Jahangir K et al (2008) Pharmaceutical significance of cellulose: a review. Express Polym Lett 2:758–778. https://doi.org/10.3144/expresspolymlett.2008.90
Kataoka Y, Kondo T (1998) FT-IR microscopic analysis of changing cellulose crystalline structure during wood cell wall formation. Macromolecules 31:760–764. https://doi.org/10.1021/ma970768c
Kim SJ, Born B, Havenith M, Gruebele M (2008) Real-time detection of protein-water dynamics upon protein folding by terahertz absorption spectroscopy. Angew Chem Int Ed 47(34):6486–6489
Larsson PT, Wickholm K, Iversen T (1997) A CP/MAS13C NMR investigation of molecular ordering in celluloses. Carbohyd Res 302:19–25. https://doi.org/10.1016/S0008-6215(97)00130-4
Leahy-Hoppa MR, Fitch MJ, Osiander R (2009) Terahertz spectroscopy techniques for explosives detection. Anal Bioanal Chem 395:247–257. https://doi.org/10.1007/s00216-009-2803-z
Lewis RA (2017) Invited review terahertz transmission, scattering, reflection, and absorption—the interaction of THz radiation with soils. J Infrared Milli Terahz Waves 38:799–807. https://doi.org/10.1007/s10762-017-0384-z
Ling Z, Wang T, Makarem M et al (2019) Effects of ball milling on the structure of cotton cellulose. Cellulose 26:305–328. https://doi.org/10.1007/s10570-018-02230-x
Markelz AG, Roitberg A, Heilweil EJ (2000) Pulsed terahertz spectroscopy of DNA, bovine serum albumin and collagen between 0.1 and 2.0 THz. Chem Phys Lett 320:42–48. https://doi.org/10.1016/S0009-2614(00)00227-X
Missoum K, Belgacem M, Bras J (2013) Nanofibrillated cellulose surface modification: a review. Materials 6:1745–1766. https://doi.org/10.3390/ma6051745
Moon RJ, Martini A, Nairn J et al (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941. https://doi.org/10.1039/c0cs00108b
Nakamura K, Wada M, Kuga S, Okano T (2004) Poisson’s ratio of cellulose I? and cellulose II. J Polym Sci B Polym Phys 42:1206–1211. https://doi.org/10.1002/polb.10771
Oh SY, Yoo DI, Shin Y et al (2005) Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohyd Res 340:2376–2391. https://doi.org/10.1016/j.carres.2005.08.007
Park S, Baker JO, Himmel ME et al (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:10. https://doi.org/10.1186/1754-6834-3-10
Robert J, Falconer Andrea G, Markelz (2012) Terahertz spectroscopic analysis of peptides and proteins. J Infrared Millimeter Terahertz Waves 33(10):973–988. https://doi.org/10.1007/s10762-012-9915-9
Rousselle M-A (2002) Determining the molecular weight distribution of cotton cellulose: a new GPC solvent. Text Res J 72:131–134. https://doi.org/10.1177/004051750207200207
Saffarionpour S (2020) Nanocellulose for stabilization of pickering emulsions and delivery of nutraceuticals and its interfacial adsorption mechanism. Food Bioprocess Technol 13:1292–1328. https://doi.org/10.1007/s11947-020-02481-2
Schenzel K, Fischer S (2001) NIR FT Raman spectroscopy—a rapid analytical tool for detecting the transformation of cellulose polymorphs. Cellulose 8:49–57
Schindelin J, Arganda-Carreras I, Frise E et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29:786–794. https://doi.org/10.1177/004051755902901003
Shanbhag AG (1994) Utilization of information measure as a means of image thresholding. CVGIP: Gr Models Image Process 56:414–419. https://doi.org/10.1006/cgip.1994.1037
Shiraga K, Ogawa Y, Kondo N (2016) Hydrogen bond network of water around protein investigated with terahertz and infrared spectroscopy. Biophys J 111(12):2629–2641
Vaks VL, Semenova AV, Khodzitsky MK, et al (2016) Gyrotropy and absorption of DNA and amylose at THz frequencies. In: 2016 41st International conference on infrared, millimeter, and terahertz waves (IRMMW-THz). IEEE, Copenhagen, Denmark, pp 1–2
Vieira FS, Pasquini C (2014) Determination of cellulose crystallinity by terahertz-time domain spectroscopy. Anal Chem 86:3780–3786. https://doi.org/10.1021/ac4035746
Wada M, Sugiyama J, Okano T (1993) Native celluloses on the basis of two crystalline phase (Iα/Iβ) system. J Appl Polym Sci 49:1491–1496. https://doi.org/10.1002/app.1993.070490817
Wang H, Inagaki T, Hartley ID et al (2019) Determination of dielectric function of water in THz region in wood cell wall result in an accurate prediction of moisture content. J Infrared Milli Terahz Waves 40:673–687. https://doi.org/10.1007/s10762-019-00594-0
Wang H, Horikawa Y, Tsuchikawa S, Inagaki T (2020) Terahertz time-domain spectroscopy as a novel tool for crystallographic analysis in cellulose. Cellulose 27:9767–9777. https://doi.org/10.1007/s10570-020-03508-9
Wang H, Tsuchikawa S, Inagaki T (2021) Terahertz time-domain spectroscopy as a novel tool for crystallographic analysis in cellulose: the potentiality of being a new standard for evaluating crystallinity. Cellulose 28:5293–5304. https://doi.org/10.1007/s10570-021-03902-x
Wang H, Kataoka H, Tsuchikawa S, Inagaki T (2022) Terahertz time-domain spectroscopy as a novel tool for crystallographic analysis in cellulose: cellulose I to cellulose II, tracing the structural changes under chemical treatment. Cellulose 29:3143–3151. https://doi.org/10.1007/s10570-022-04493-x
Yao W, Weng Y, Catchmark JM (2020) Improved cellulose X-ray diffraction analysis using Fourier series modeling. Cellulose 27:5563–5579. https://doi.org/10.1007/s10570-020-03177-8
Zhao H, Kwak J, Conradzhang Z et al (2007) Studying cellulose fiber structure by SEM, XRD, NMR and acid hydrolysis. Carbohyd Polym 68:235–241. https://doi.org/10.1016/j.carbpol.2006.12.013
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number 21H02255 and 23K19305. The XRD measurement was supported by Radioisotope Research Center, Nagoya University, and SEM observation was supported by Dr. Takao OI.
Funding
Open Access funding provided by Nagoya University. JSPS KAKENHI Grant Number: 21H02255 and 23K19305.
Author information
Authors and Affiliations
Contributions
HW and TI conceived and designed the experiments. HW, YT, and DA performed the experiments and analyzed the data. HW wrote the paper, YT, DA, TI and ST gave final approval of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study following Compliance with Ethical Standards; this study does not involve human participants, animals and potential conflicts of interest.
Consent for publication
All authors agree to publish the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Aoki, D., Teramoto, Y. et al. Terahertz time-domain spectroscopy as a novel tool for crystallographic analysis in cellulose: tracking lattice changes following physical treatments. Cellulose (2024). https://doi.org/10.1007/s10570-024-05834-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10570-024-05834-8