
Abstract
The mechanical properties of native cellulose are critical for understanding the properties of natural biomaterials. To investigate the elastic moduli of the cellulose crystalline fraction an isotropic mechanical load using hydrostatic pressure ranging from 0.01 to 0.5 GPa was applied to flax fibers, pine wood and tension wood samples. The response of the crystalline part was monitored by using microfocused synchrotron radiation. The compressibility of the crystalline fraction of native cellulose was anisotropic and dependent on the crystal size and possibly on the composition of the sample. The compressibilities along the [001] direction, where covalent bonds hold the cellulose chains together, varied between 2.1 and 2.9 TPa−1 for the different samples and confirm similar values found for cotton fibres. However, the compressibilities along the [100]-direction range from 56.2 TPa−1 to 63.5 TPa−1, slightly exceeding the previously determined value of 50 TPa−1, which can possibly be attributed to differences between individual samples.
Similar content being viewed by others
Introduction
Native cellulose often appears in alliance with an amorphous matrix of lignin, hemicellulose and other components, building a semi-crystalline composite material with outstanding mechanical properties. Despite its low material density, it allows plants to withstand continuous high mechanical forces such as wind. Cellulose possesses a hierarchical structure from the molecular over the crystalline level to the so called microfibrils forming a helix with a certain angle to the cell axis referred to as microfibril angle (Salmén and Burgert 2009). Being synthesized in six-fold rosette complexes also referred to as terminal complexes, parallel orientated cellulose chains form crystals via hydrogen bonds and van-der-Waals force (Kölln 2004; Taylor 2008). The microfibrils have a small, specific diameter ranging from 2 to 25 nanometers and are over several micrometers in length (Nishiyama 2009). They twist around their long axis (Willhammar et al. 2021) and are considered to contain unordered regions (Fengel and Wegener 1983). The unit cell parameters for the crystal structure of the dominant allomorph cellulose Iβ provide a monoclinic unit cell with the parameters of a = 7.78 Å, b = 8.20 Å, c = 10.38 Å and γ = 96.5° (Nishiyama et al. 2002).
Determining the elastic modulus of cellulose is critical to understand the material properties of naturally occurring biomaterials. The elastic tensor of the monoclinic cellulose allomorph Iβ consists of 13 independent elements and is highly anisotropic (Nye 1985; Song et al. 2021). To understand the interplay of the different levels and the elasticity of native cellulose, in situ tensile tests have been performed on wood and flax fibers using synchrotron radiation (Reiterer et al. 1999; Kölln et al. 2005a; Clair et al. 2006). The effect on the microfibrils is visible through the shift of the Bragg reflections 200 and 004 in the diffraction pattern, roughly corresponding to the deformation in [100]- and [001] direction. In other approaches, nanoindentation and in situ bending techniques were applied to determine two diagonal elements of the elastic tensor using materials containing native cellulose (Konnerth and Gindl 2006; Orso et al. 2006). Diddens at al. performed inelastic X-ray scattering and calculated the two diagonal elastic tensor elements of native cellulose in flax via the measured sound velocity to 220 GPa in [001]-direction and 15 GPa in [100]-direction. These were also calculated theoretically with molecular dynamics simulations and vary between 98 and 202 GPa in in [001]-direction (Kroon-Batenburg and Kroon 1997; Eichhorn and Davies 2006; Dri et al. 2013; Song et al. 2021) and 14.5–19 GPa (Dri et al. 2013; Song et al. 2021) in [100]-direction.
However, it is important to take into account that often the samples used in many experiments have been chemically treated. Therefore, the results can only be transferred to a limited degree to native cellulose, where various parameters such as the interplay of the different cell wall layers, the deformation of the amorphous matrix under uniaxial load (Keckes et al. 2003; Peura et al. 2007) as well as changes in the alignment of the microfibrils towards the fiber axis (Montero et al. 2011) cannot be neglected. When interpreting the elastic modulus of native cellulose, it is important to take into account both the rotation of bond angles (Krywka et al. 2014) and hydrogen bonds (Mariano et al. 2014) at the atomic level.
In the present study native cellulose was subjected to high hydrostatic pressure ranging from 0.01 to 0.5 GPa. By exposing matrix and crystals to hydrostatic pressure, the mechanical load is considered to be homogeneous and non-directional. It is expected that the microfibrils and matrix will not be structurally affected, but rather the cellulose crystals will be compressed according to their anisotropic compressibility due to the heterogenous structure of the crystals.
The hydrostatic compression allows the direct investigation of the a- and b-axis compressibility undisturbed by effects usually present in tensile tests (e.g. the Poisson’s ratio). However, the hydrostatic method implies that the results obtained here is not directly comparable to elastic constants measured in non-hydrostatic conditions.
Materials and methods
Experimental set-up and data collection
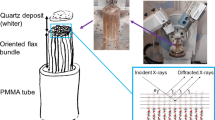
For this experiment, a bundle of industrially bleached flax fibers (Diddens et al. 2008), two untreated pieces of pine wood and two tension wood samples from poplar (Müller et al. 2006) with a thickness of 0.5 mm and a length of 20 mm each were chosen. A hydrostatic high pressure sample cell was used to perform the measurements (Krywka et al. 2008). The cell body is machined of a high tensile strength alloy, allows for pressures up to 7 kbar and contains the sample inside the pressure chamber (see Fig. 1a). Two diamond windows each with a thickness of 1 mm are stable against the pressure and allow the radiation to pass. The sample is mounted to the reusable windowless sample carrier, allowing the sample to be directly exposed to the water pressure (compare Fig. 1b).

a Assembly of the hydrostatic pressure cell with diamond windows including the sample holder with a fibre bundle; b schematic cross-section of the hydrostatic high pressure cell showing how the sample is subjected to hydrostatic pressure
The measurements with microfocused synchrotron radiation were carried out at the microfocus beamline ID13 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). The experiment was performed at an X-ray energy of 22.977 keV. After the monochromator, the beam was focussed through compound refractive lenses and conditioned by a slit system, resulting in a size of 30 × 30 µm2 with a flux of 108–109 photons/s. After passing through an ionization chamber, the beam was directed towards the sample through the diamond windows of the high pressure sample cell. The first diffraction image (compare Fig. 2) was taken at a pressure of about 10 MPa, in order to ensure that no bubbles remained in the pressure chamber. The pressure was then increased up to 0.5 GPa with a step size of 100 MPa. Afterwards, the pressure was reduced again with the same step size. The procedure with the increasing pressure was repeated without the sample to obtain the background data.

(adapted from (Diddens et al. 2008)). The linear artefacts are Kossel lines originating from the diamond windows. The fiber axis in the experiment was oriented horizontally
Diffraction image of a pine sample submerged in water at the initial pressure of 10 MPa applied. The different reflections and the referring crystal planes are shown for the 200 and the 004 reflection
Data analysis
After normalization for exposure time and ionisation chamber values the data were subjected to background subtraction. The transmission was estimated taking into account the pressure-dependent attenuation by water and further used to normalize the scattered intensity.
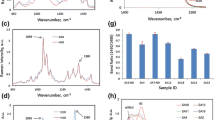
To understand the effect of pressure on the reflection position, azimuthal integration was conducted on the respective areas of the 200 reflection for the a-axis, the 110 and \(1\stackrel{-}{1}0\) reflection and the 004 reflection for the c-axis at each pressure step (compare Fig. 3). When performing a complete azimuthal integration (see Fig. 4), it can be seen that depending on the sample the 110 and \(1\stackrel{-}{1}0\) reflections are resolved differently. The variation in crystallinity, crystal sizes, and orientations in the different samples accounts for this difference. The lower limit of the material’s crystal sizes is obtained by fitting the 200 and the 004 reflection with Gauss functions using the FWHM in the Scherrer formula
not taking into account other effects broadening the peak. Table 1 shows the calculated lower limits of the mean cross section diagonal (L200) and the length of the crystals calculated from the 004 reflection (L004).
The positions of the integrated reflections in reciprocal space were determined by fitting them with Gaussian functions. The corresponding d-spacing in real space were calculated using \(d\left(p\right)=\frac{2\pi }{q\left(p\right)}\).The reflection positions were plotted as a function of pressure and fitted with a linear function. Extrapolating this function to zero pressure allows for the calculation of strain \(\epsilon = \left| {\frac{{l - l_{0} }}{{l_{0} }}} \right|\) which, since pressure is always present, can be considered as stress (\(\sigma\)) on the cross-section perpendicular to the crystal axis. With this, Young’s modulus (in specific crystallographic directions) can be calculated from Hooke’s Law: \(\varepsilon=\frac{\sigma}{E}\). Compressibilities can directly be determined for the crystallographic [100] and [001] directions of native cellulose crystals via \(\kappa=\frac{3(1-2)}{E}\) with υ = 0.377 as determined before (Nakamura et al. 2004). Comparison with data collected while decreasing the pressure showed some differences, which indicate that viscoelastic effects and potentially also humidity effects could play a role here. Therefore, only data recorded while the pressure was increased were considered. The final data point, collected after depressurization, was used for comparison with the first data point to verify that no radiation damage occurred during the data collection.

left: Tension wood raw data at 0.5 GPa; right: tension wood data at 0.5 GPa after background subtraction. The area marked in orange was integrated to determine the 200 reflection
To determine the unit cell parameter along the a-axis, the formula \(a= 2~\frac{{d}_{200}}{\sin \gamma }\) was employed., γ was assumed to be constant with a value of 96.5° according to the literature. The unit cell parameter of the c-axis was calculated by \(c=4~{d}_{004}\). Because the unit cell parameter for the b-axis cannot be determined directly from the available 110 and \(1\bar{1}0~\) reflections, which are additionally quite noisy, the d110 and \({d}_{1\bar{1}0}\) values are given directly (compare Table 2).

Radially integrated data after the subtraction of the background image showing the 110, the \(1\bar{1}0\), the 200 and the 004 reflection for flax, pine and tension wood
Results and discussion
As shown in Table 2, the values of the different samples are all slightly larger than the theoretically determined value of 7.78 Å for cellulose Iβ, as reported before for native cellulose. The expected compression in the [100] direction is clearly visible for all samples (see Fig. 5a). The compression of pine wood and tension wood samples is very similar when compared to each other. The weak van-der-Waals-bonds allow a compression between 2.83% in the case of tension wood and 3.23% in the case of pine wood when exposed to 0.5 GPa. Relating the observed compression to the crystal sizes, it becomes clear that the compression is relatively stronger for smaller crystals of pine in flax compared to the bigger crystals of tension wood.
The compressibility along the [100] direction for the bundle of flax fibers (s. Table 2) is 58.7 TPa−1. If the off-diagonal element of the elastic tensor is neglected, this corresponds to a modulus of 17.2 GPa, which is slightly higher than the value of 15 GPa obtained with inelastic X-ray scattering from a bundle of fibers (Diddens et al. 2008) and significantly lower than the 24.7 GPa obtained from a single flax fiber along the fiber direction. (Kölln et al. 2005b). Furthermore, a change of the humidity caused the surrounding water may have an influence at high pressure as Young’s modulus for flax seems to decrease with humidity (Davies and Bruce 1998).
Along the [100] direction, the compressibilities for pine wood are with 62.6 TPa−1 and 63.5 TPa−1 higher than for tension wood with 56.2 TPa−1and 58.2 TPa−1, respectively. Results from other studies (Quesada Cabrera et al. 2011; Song et al. 2021) show slightly lower values of 50 TPa−1. More systematic studies are required to investigate whether this is sample-dependent or whether there are other explanations for these values.
The length of the unit cell along the c-axis is slightly smaller, ranging from 10.33 to 10.37 Å, compared to the value of 10.38 Å previously determined by Nishiyama. The degree of compression in the [001]-direction is relatively smaller than that in the a-direction, resulting in compressions of 0.11–0.15%. The compressibilities range from 2.1 TPa−1 for pine wood sample 1 up to 2.9 TPa−1 for tension wood sample 1 and are therefore higher than the value of 1.71 TPa−1 determined by Song et al. for chemically treated tunicate, but in the range of the value of 2.8 TPa−1 determined by Quesada Cabrera et al. for cotton cellulose fibres. In the case of tension wood, the differences between samples of the same type are also significant. However, in contrast to the other samples with an error of less than 2%, the error for the compressibility of the tension wood samples amounts for 3.8% and 10.7% respectively.
Given that there are only covalent bonds in [001]-direction, one would actually expect only little compression, based on previous experiments indicating that covalent bonds in proteins are pressure insensitive up to at least 10–15 kbar (Mozhaev et al. 1996). The observed compression can most likely be explained by other processes.
High pressure studies indicate that the most likely explanation is a change of the crystal packing. The largest contributions are expected to arise from hydration changes that accompany non-covalent interactions (Mozhaev et al. 1996). Similar compressibilities found by Quesada Cabrera at al were attributed to hydrogen bonds which are likely to strengthen upon compression. However, this study was performed without a pressure-transmitting media. As water is used in the study presented here, protein-water interactions can form, promoted by pressure producing stronger and shorter bonds that decrease the overall volume (Boonyaratanakornkit et al. 2002).
The intensity of the 110 and the \(1\bar{1}0\) reflections are low compared to the background, the fits of the reflection positions come with significantly higher error bars compared to the other reflections. In Fig. 3, one can clearly see that the two reflections are best resolved for flax. The values for the \({d}_{1\bar{1}0}\) distances ranging from 5.91 Å for tension wood up to 6.06 Å for pine wood as well as the \({d}_{110}\) distances with values from 5.28 Å for pine wood and up to 5.41 Å for tension wood are in good agreement with values reported before (Quesada Cabrera et al. 2011).

Linear compression of for flax, pine and tension wood of the a [100] direction as derived from the 200 reflection for the unit cell parameter a and b of the [001] direction derived from the change of the position of the 004 reflection for the unit cell parameter c, both fitted with a straight line.
Conclusion and outlook
We have shown here that the unit cell of three different types of native cellulose shows a highly anisotropic behaviour under hydrostatic pressure, which can be attributed to the difference between covalent bonds and van-der-Waals interactions. The compressibilities along the [001] direction of the cellulose crystals are in line previous reports. For the [100] direction, the compressibilities are higher than previously reported. More systematic studies are required to investigate whether this is sample-dependent or whether this be attribute to the method applied. In order to exclude a potential influence of the pressurizing water onto the cellulose structure, similar experiments could be performed with a water-free pressurizing medium (e.g. alcohols, silicone oil).
References
Boonyaratanakornkit BB, Park CB, Clark DS (2002) Pressure effects on intra- and intermolecular interactions within proteins. Biochim et Biophys Acta (BBA) - Protein Struct Mol Enzymol 1595:235–249. https://doi.org/10.1016/S0167-4838(01)00347-8
Clair B, Alméras T, Yamamoto H et al (2006) Mechanical behavior of cellulose microfibrils in tension wood, in relation with maturation stress generation. Biophys J 91:1128–1135
Davies GC, Bruce DM (1998) Effect of environmental relative humidity and damage on the tensile properties of flax and nettle fibers. Text Res J 68:623–629. https://doi.org/10.1177/004051759806800901
Diddens I, Murphy B, Krisch M, Müller M (2008) Anisotropic elastic properties of cellulose measured using inelastic X-ray scattering. Macromolecules 41:9755–9759
Dri FL, Hector LG, Moon RJ, Zavattieri PD (2013) Anisotropy of the elastic properties of crystalline cellulose Iβ from first principles density functional theory with Van Der Waals interactions. Cellulose 20:2703–2718. https://doi.org/10.1007/s10570-013-0071-8
Eichhorn S, Davies G (2006) Modelling the crystalline deformation of native and regenerated cellulose. Cellulose 13:291–307
Fengel D, Wegener G (1983) Wood: chemistry, ultrastructure, reactions. de Gruyter, Berlin
Keckes J, Burgert I, Frühmann K et al (2003) Cell-wall recovery after irreversible deformation of wood. Nat Mater 2:810–813
Kölln K (2004) Morphologie und mechanische Eigenschaften von Zellulosefasern: Untersuchungen mit Röntgen-und Neutronenstreuung. PhD Thesis, Dissertation, Christian-Albrechts-Universität Kiel
Kölln K, Grotkopp I, Burghammer M et al (2005a) Nanobioscience mechanical properties of cellulose fibres and wood orientational aspects in situ investigated with synchrotron radiation. J Synchrotron Rad 12:739–744. https://doi.org/10.1107/S0909049505011714
Kölln K, Grotkopp I, Burghammer M et al (2005b) Mechanical properties of cellulose fibres and wood. Orientational aspects in situ investigated with synchrotron radiation. J Synchrotron Radiat 12:739–744
Konnerth J, Gindl W (2006) Mechanical characterisation of wood-adhesive interphase cell walls by nanoindentation. Holzforschung 60:429–433
Kroon-Batenburg L, Kroon J (1997) The crystal and molecular structures of cellulose I and II. Glycoconj J 14:677–690
Krywka C, Sternemann C, Paulus M et al (2008) Effect of osmolytes on pressure-induced unfolding of proteins: a high-pressure SAXS study. ChemPhysChem 9:2809–2815. https://doi.org/10.1002/cphc.200800522
Krywka C, Krasnov I, Figuli R et al (2014) Determination of silkworm silk fibroin compressibility using high hydrostatic pressure with in situ X-ray microdiffraction. Macromolecules 47:7187–7193. https://doi.org/10.1021/ma501880h
Mariano M, El Kissi N, Dufresne A (2014) Cellulose nanocrystals and related nanocomposites: review of some properties and challenges. J Polym Sci Part B: Polym Phys 52:791–806. https://doi.org/10.1002/polb.23490
Montero C, Clair B, Alméras T et al (2011) Relationship between wood elastic strain under bending and cellulose crystal strain. Compos Sci Technol
Mozhaev VV, Heremans K, Frank J et al (1996) High pressure effects on protein structure and function. Proteins 24(199601):81–91.
Müller M, Burghammer M, Sugiyama J (2006) Direct investigation of the structural properties of tension wood cellulose microfibrils using microbeam X-ray fibre diffraction. Holzforschung 60:474–479. https://doi.org/10.1515/HF.2006.078
Nakamura K, Wada M, Kuga S, Okano T (2004) Poisson’s ratio of cellulose Iβ and cellulose II Abstract. J Polym Sci Part B: Polym Phys 42(7):1206–1211. https://doi.org/10.1002/polb.10771
Nishiyama Y (2009) Structure and properties of the cellulose microfibril. J Wood Sci 55:241–249. https://doi.org/10.1007/s10086-009-1029-1
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082
Nye JF (1985) Physical properties of crystals: their representation by tensors and matrices. Oxford University Press, Oxford
Orso S, Wegst UGK, Arzt E (2006) The elastic modulus of spruce wood cell wall material measured by an in situ bending technique. J Mater Sci 41:5122–5126
Peura M, Kölln K, Grotkopp I et al (2007) The effect of axial strain on crystalline cellulose in Norway spruce. Wood Sci Technol 41:565–583
Quesada Cabrera R, Meersman F, McMillan PF, Dmitriev V (2011) Nanomechanical and structural properties of native cellulose under compressive stress. Biomacromolecules 12:2178–2183. https://doi.org/10.1021/bm200253h
Reiterer A, Lichtenegger H, Tschegg S, Fratzl P (1999) Experimental evidence for a mechanical function of the cellulose microfibril angle in wood cell walls. Philos Mag A 79:2173–2184. https://doi.org/10.1080/01418619908210415
Salmén L, Burgert I (2009) Cell wall features with regard to mechanical performance. A review COST action E35 2004–2008: Wood machining–micromechanics and fracture. Holzforschung 63:121–129
Song G, Lancelon-Pin C, Chen P et al (2021) Time-dependent elastic tensor of cellulose nanocrystal probed by hydrostatic pressure and uniaxial stretching. J Phys Chem Lett 12:3779–3785. https://doi.org/10.1021/acs.jpclett.1c00576
Taylor NG (2008) Cellulose biosynthesis and deposition in higher plants. New Phytol 178:239–252
Willhammar T, Daicho K, Johnstone DN et al (2021) Local crystallinity in twisted cellulose nanofibers. ACS Nano 15:2730–2737
Acknowledgments
We would like to acknowledge Shin-Gyu Kang for his help during this beamtime. MM would like to thank Dr Henri Chanzy for the donation of the flax fibers and for continuous support, inspiration and discussions in the field of cellulose research since 1996.
Funding
Open Access funding enabled and organized by Projekt DEAL. The authors declare that no funds, grants, or other support were received during the preparation of this manuscript. The authors declare they have no financial interests.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by CK, MB, EC and MM. The first draft of the manuscript was written by SS and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of a collection of articles in honor of Dr. Henri Chanzy on the occasion of his 90th birthday. The article was not finished in time to be bound with the rest of the papers in the Special Issue #13, September, 2023.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Storm, S.L.S., Krywka, C., Burghammer, M. et al. Investigation of native cellulose under high pressure using microfocused synchrotron radiation. Cellulose 31, 2705–2712 (2024). https://doi.org/10.1007/s10570-023-05581-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-023-05581-2