
Abstract
We have examined the role of support oxygen vacancies in the gas phase hydrogenation of furfural over Au/TiO2 and Au/CeO2 prepared by deposition–precipitation. Both catalysts exhibited a similar Au particle size distribution (1–6 nm) and mean (2.8–3.2 nm). Excess H2 consumption during TPR is indicative of partial support reduction, which was confirmed by O2 titration. Gold on CeO2 with a higher redox potential exhibited a greater oxygen vacancy density. A lower furfural turnover frequency (TOF) was recorded over Au/CeO2 than Au/TiO2 and is linked to suppressed H2 chemisorption capacity and strong –C=O interaction at oxygen vacancies that inhibited activity. Gold on non-reducible Al2O3 as benchmark exhibited greater H2 uptake and delivered the highest furfural TOF. Full selectivity to the target furfuryl alcohol was achieved over Au/TiO2 and Au/Al2O3 at 413 K and over Au/CeO2 at 473 K with hydrogenolysis to 2-methylfuran at higher reaction temperature (523 K). A surface reaction mechanism is proposed to account for the activity/selectivity response.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Oxygen vacancies in metal oxides (e.g. titanium, cerium, iron and vanadium oxides) are defects generated by the loss of lattice oxygen as a result of high temperature (≥ 673 K) annealing in ultra-high vacuum [1], chemical reduction (by H2 or CO) [2] and/or electron irradiation [3]. The formation and properties of these vacancies have been the subject of theoretical (DFT) and experimental (UPS, XPS, EELS, IR, EPR, STM) work [4,5,6]. Contributions due to oxygen vacancies have been established in catalytic water–gas shift [7], steam reforming of oxygenates [8], CO oxidation [9] and hydrodeoxygenation [10]. Moreover, the presence of these defects can modify the electronic characteristics (via electron transfer) [11], particle size (by stabilisation at vacancy sites) [12] and chemical properties (metal-support interaction) [13] of the supported metal phase (Pt [7], Ag [11], Au [13] and Pd [14]), which impact on reactant adsorption/activation. Hydrogenation is a key process in the food, petrochemical, pharmaceutical and agrochemical sectors [15]. The effect of surface oxygen vacancies in catalytic hydrogenation is still a subject of debate. Enhanced activity and (–C=O reduction) selectivity reported for Pt/CeO2 [16] and Au/Fe2O3 [17] in the hydrogenation of crotonaldehyde and benzalacetone was attributed to facilitated activation of the carbonyl function at oxygen vacancies and/or electron-rich metal nano-particles. On the other hand, a (threefold) decrease in crotonaldehyde hydrogenation activity was observed following incorporation of CeO2 (by impregnation with Ce(NO3)3) on Ru/Al2O3 and ascribed to strong –C=O interaction with oxygen deficient sites [18]. Tian et al. [19] studying the hydrogenation of cinnamaldehyde over Au/CeO2 suggested a preferential –C=C– adsorption on Auδ+ (resulting from electron transfer to support defects) to explain lower selectivity in terms of –C=O reduction. In the hydrogenation of p-chloronitrobenzene, unwanted hydrodechlorination was reported for Au/Ce0.62Zr0.38O2 and ascribed to –C–Cl scission at vacancy sites [20]. In this study, we consider the role of oxygen vacancies (on reducible TiO2 and CeO2) in determining the catalytic performance of supported Au in gas phase hydrogenation of –C=O using furfural as model reactant. Gold on non-reducible alumina serves as a benchmark catalyst.
2 Experimental
2.1 Materials and Catalyst Preparation
Commercial TiO2 (P25, Degussa) and CeO2 (Sigma-Aldrich) were used as received. The supported Au catalysts were prepared by deposition–precipitation using urea (100-fold excess, Riedel-de Haën, 99%) with HAuCl4 (1.5 × 10− 3−3.0 × 10− 3 M, 400 cm3, Sigma-Aldrich, 99%). A suspension containing the oxide carrier (10 g) was heated to 353 K (2 K min− 1) where the pH progressively increased to ca. 7 after 3–4 h as a result of urea decomposition [21]. The solid obtained was separated by filtration, washed with distilled water until chlorine free (from AgNO3 test) and dried in He (45 cm3 min− 1) at 373 K (2 K min− 1) for 5 h. The resultant sample was sieved (ATM fine test sieves) to mean particle diameter = 75 μm, activated at 2 K min− 1 to 523 K in 60 cm3 min− 1 H2, cooled to ambient temperature and passivated in 1% v/v O2/He for 1 h for ex situ characterisation. Synthesis and activation of the benchmark Au/Al2O3 catalyst is described in detail elsewhere [22].
2.2 Catalyst Characterisation
The Au content was measured by atomic absorption spectroscopy (Shimadzu AA-6650 spectrometer with an air-acetylene flame) from the diluted extract in aqua regia (25% v/v HNO3/HCl). Temperature programmed reduction (TPR), H2 and O2 chemisorption measurements were conducted on the CHEM-BET 3000 (Quantachrome Instrument) unit with data acquisition/manipulation using the TPR Win™ software. Samples were loaded into a U-shaped Pyrex glass cell (3.76 mm i.d.) and heated in 17 cm3 min− 1 (Brooks mass flow controlled) 5% v/v H2/N2 to 523 K at 2 K min− 1. The effluent gas passed through a liquid N2 trap and H2 consumption was monitored by a thermal conductivity detector (TCD). The activated samples were swept with 65 cm3 min− 1 N2 for 1.5 h, cooled to 413 K and subjected to H2 chemisorption by pulse (10 μl) titration. In blank tests, there was no measurable H2 uptake on the oxide supports alone. Oxygen chemisorption post-TPR was employed to determine the extent of support reduction [23], where the samples were reduced as described above, swept with 65 cm3 min− 1 He for 1.5 h, cooled to 413 K with pulse (50 μl) O2 titration. It has been demonstrated previously that Au contribution to total O2 adsorbed is negligible [24]. Nitrogen physisorption was performed using the commercial Micromeritics Gemini 2390p system. Samples were outgassed at 423 K for 1 h prior to analysis. Total specific surface area (SSA) was calculated using the standard single point BET method. X-ray diffractograms (XRD) were recorded on a Bruker/Siemens D500 incident X-ray diffractometer using Cu Kα radiation, scanning at 0.02° per step over the range 20° ≤ 2θ ≤ 80°. The diffractograms were identified against the JCPDS-ICDD reference standards, i.e. Au (04-0784), anatase-TiO2 (21-1272), rutile-TiO2 (21-1276), CeO2 (43-1002) and Ce2O3 (23-1048). Gold particle morphology was examined by scanning transmission electron microscopy (STEM, JEOL 2200FS field emission gun-equipped unit), employing Gatan Digital Micrograph 1.82 for data acquisition/manipulation. Samples for analysis were prepared by dispersion in acetone and deposited on a holey carbon/Cu grid (300 Mesh). The surface area weighted mean Au particle size (d) was based on a count of at least 300 particles, according to
where n i is the number of particles of diameter d i .
2.3 Catalyst Testing
Hydrogenation of furfural (Sigma-Aldrich, 99%) was carried out at atmospheric pressure and 413–523 K in situ after activation in a continuous flow fixed bed tubular reactor (15 mm i.d.). Reactions were conducted under operating conditions that ensured negligible mass/heat transport limitations. A layer of borosilicate glass beads served as preheating zone, ensuring that the furfural reactant was vaporised and reached reaction temperature before contacting the catalyst. Isothermal conditions (± 1 K) were ensured by diluting the catalyst bed with ground glass (75 µm), which was mixed thoroughly with catalyst before loading into the reactor. Reaction temperature was continuously monitored by a thermocouple inserted in a thermowell within the catalyst bed. Furfural was delivered as n-butanolic (Sigma-Aldrich, > 99%) solutions to the reactor via a glass/Teflon air-tight syringe and Teflon line using a microprocessor controlled infusion pump (Model 100 kd Scientific) at a fixed calibrated flow rate. A co-current flow of furfural and H2 was adjusted to GHSV = 1 × 104 h− 1. The molar Au to inlet reactant molar feed rate (n/F) spanned the range 4 × 10− 3–30 × 10− 3 h. Passage of furfural in a stream of H2 through the empty reactor or over support alone did not result in any detectable conversion. The reactor effluent was condensed in a liquid N2 trap for subsequent analysis using a Perkin-Elmer Auto System XL gas chromatograph equipped with a programmed split/splitless injector and a flame ionisation detector, employing a DB-1 (50 m × 0.33 mm i.d., 0.20 μm film thickness) capillary column (J&W Scientific). Data acquisition and manipulation were performed using the TurboChrom Workstation Version 6.3.2 (for Windows) chromatography data system. Furfuryl alcohol and 2-methylfuran were used as supplied (Sigma-Aldrich, 99%) for product identification/analysis. All gases (O2, H2, N2 and He) were of high purity (BOC, > 99.98%). Furfural fractional conversion (X) is defined by
and selectivity (S) to product (j) is given by
where the subscripts “in” and “out” refer to the inlet and outlet gas streams. Turnover frequency (TOF, rate per active site) was calculated using Au dispersion measurements from STEM as described elsewhere [25]. Repeated reactions with different samples from the same batch of catalyst delivered raw data reproducibility and carbon mass balances that were within ± 5%.
3 Results and Discussion
3.1 Catalyst Characterisation
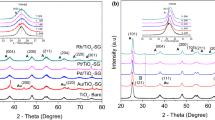
The physicochemical characteristics of Au/TiO2 and Au/CeO2 are given in Table 1; the values for Au/Al2O3 are taken from a prior publication [22]. The samples contained a similar Au loading (0.6–0.8 mol%) where the SSA match values reported for TiO2 (50 m2 g− 1) [26] and CeO2 (36–67 m2 g− 1) [11] supported group IB metal catalysts. XRD analysis (Fig. 1) of Au/TiO2 (I) revealed a mixture of tetragonal anatase [2θ = 25.3°, 37.8°, 48.1° and 62.8°, (III)] and rutile [2θ = 27.4°, 36.1°, 41.2°, 54.3°, 56.6°, 69.0° and 69.8°, (IV)] phases with an anatase : rutile ratio (5:1) consistent with Degussa P25 [27]. The XRD pattern of Au/CeO2 (II) presents principal peaks (at 2θ = 28.6°, 33.1°, 47.5°, 56.4° and 59.1°) characteristic of CeO2 (V). In both cases, there were no diffraction peaks due to Au (principal peak 2θ = 38.1°; JCPDS-ICDD card 04-0784), diagnostic of a well dispersed (< 5 nm) metal phase [24]. This was confirmed by STEM analysis (Fig. 2) where both samples exhibited quasi-spherical Au nanoparticles (IA, IB) with similar size range (1–6 nm) and mean [(II); Table 1]. The TPR profile of Au/TiO2 (Fig. 2IIIA) shows a single peak (T max = 376 K) with an associated H2 consumption that exceeded the amount required for the formation of Au0 (Table 1) but far lower than that (6200 μmol g− 1) required for Ti4+ → Ti3+. This suggests a partial reduction of the support, notably at the Au-support interface [28]. Reduction of Au/CeO2 (Fig. 2IIIB) exhibited H2 consumption at higher T max (418 K). Liu and Yang [29] reported a dependency of Au3+ reducibility on support redox properties where weaker interactions with TiO2 compared with CeO2 rendered the Au3+ component more susceptible to reduction. In the TPR of Au/CeO2, H2 consumed was greater than the requirements for Au precursor reduction but considerably less than bulk Ce4+ → Ce3+ transformation (2900 μmol g− 1). There were no signals due to Ce2O3 (main peak 2θ = 29.5°; JCPDS-ICDD card 23-1048) in the XRD pattern. Increased H2 uptake during activation of Au/CeO2 relative to Au/TiO2 suggests a greater degree of support reduction. This agrees with the higher redox potential [30] of CeO2 (E redox, Table 1). In contrast, TPR analysis of benchmark Al2O3 (with the lowest E redox) supported Au (d = 4.3 nm) generated an equivalent H2 consumption to the theoretical value for Au3+ → Au0, confirming support non-reducibility (Table 1).

XRD patterns for (I) Au/TiO2 and (II) Au/CeO2 with JCPDS-ICDD reference diffractograms for (III) anatase-TiO2 (21-1272), (IV) rutile-TiO2 (21-1276) and (V) CeO2 (43-1002)

(I) Representative STEM images with (II) associated Au particle size distribution histograms and (III) temperature programmed reduction (TPR) profiles for A Au/TiO2 (solid bars) and B Au/CeO2 (hatched bars)
The number of surface oxygen vacancies can be quantified by oxygen titration [23, 31]. Oxygen chemisorption post-TPR was employed to determine the extent of support reduction; the values are given in Table 1. Decreasing O2 uptake (Au/CeO2 > Au/TiO2 > Au/Al2O3) matched the sequence of decreasing support redox potential and H2 consumption during TPR. Oxygen vacancy formation in TiO2 has been established by in situ EPR following reduction (in H2) over 573–1073 K [32]. Boccuzzi et al. [33] using FTIR spectroscopy demonstrated H2 dissociation on Au sites supported on TiO2 (reduced at 523 K) with spillover that resulted in surface reduction. It has been established (by DFT calculation and STM) that bare ceria surfaces can be reduced (Ce4+ → Ce3+) to generate oxygen defects post-activation in H2 at 400–900 K [34, 35]. Addition of Au to ceria facilitates support reduction (273–573 K) during TPR [36]. The performance of supported Au catalysts in hydrogenation is determined by the capacity for H2 adsorption/dissociation [24]. Hydrogen chemisorption (at 413 K, Table 1) on Au/TiO2 was measurably higher than Au/CeO2. Gold particle size and support interactions impact on H2 adsorption [37,38,39]. Corner and edge sites associated with smaller Au particles (< 10 nm) have been identified as active for H2 dissociation [37]. Mean Au size is close for the three catalysts (Table 1). The order of decreasing H2 uptake (Au/Al2O3 > Au/TiO2 > Au/CeO2) matches that of increasing O2 chemisorption (Table 1). Lower uptake on Au/TiO2 and Au/CeO2 can be linked to metal encapsulation due to surface Au diffusion into the bulk (573–673 K) that is facilitated by oxygen vacancies on reducible oxides [40, 41], an effect more pronounced for Au/CeO2 with higher vacancy density.
The characterisation results demonstrate the generation of nano-scale Au particles on TiO2 and CeO2 with a greater density of surface oxygen vacancies and lower H2 uptake on Au/CeO2. Gold on non-reducible Al2O3 with similar metal size is a suitable candidate to evaluate the effect of oxygen vacancies on furfural hydrogenation over supported Au.
3.2 Catalytic Response
A search through the literature did not produce any reported application of TiO2 or CeO2 supported Au catalysts in furfural hydrogenation. We can flag the work of Ohyama et al. [42] on high pressure (38 atm) liquid phase hydrogenation of 2-hydroxylmethyl-5-furfural where reaction over Au/TiO2 resulted in furan ring opening and Au/CeO2 promoted carbonyl group reduction at a (tenfold) lower activity. No explanation was given for the observed differences in selectivity or activity. Gas phase furfural hydrogenation at 413 K over Au/TiO2 generated the target furfuryl alcohol as sole product (Fig. 3I). Reaction over Au/Al2O3 delivered an appreciably higher selective turnover frequency (TOF) and Au/CeO2 was inactive (Fig. 3II). This activity response can be linked to differences in H2 chemisorption capacity (Table 1, in the order Au/Al2O3 > Au/TiO2 > Au/CeO2) under reaction conditions. Zanella et al. [43] identified H2 dissociation as rate-determining in the chemoselective hydrogenation of aldehydes over supported Au. In this study, the TOF normalised with respect to H2 chemisorption capacity was lower for Au on reducible supports (notably Au/CeO2) relative to Au/Al2O3. This suggests a contribution due to furfural adsorption at surface oxygen vacancies. These vacancies can act as sites for strong binding of oxygenated reactants [34, 44]. The higher density of oxygen vacancies on Au/CeO2 (Table 1) can act to stabilise surface adsorbed furfural, resulting in lower reaction rates. The action of oxygen vacancies to inhibit –C=O reduction is in line with the lower activity recorded for cinnamaldehyde hydrogenation (to cinnamyl alcohol) over Au/CeO2 relative to Au/MgO-Al2O3 reported by Tian et al. [19] though this possibility was not proposed by the authors. An increase in temperature (≥ 473 K) (i) elevated TOF where Au/CeO2 consistently delivered lower rates (Fig. 3II) and (ii) resulted in a switch in selectivity from furfuryl alcohol to 2-methylfuran. Reaction over Au/TiO2 and Au/Al2O3 at 523 K generated 2-methylfuran as principal product (S > 91%). In the case of Au/CeO2, a higher reaction temperature (473 K) resulted in the selective transformation of furfural to furfuryl alcohol while a further increase (to 523 K) generated 2-methylfuran as by-product. These results suggest that elevated temperatures favour activation of –C=O for hydrogenolytic cleavage, which finds agreement in results reported for Cu/MgO [45].

Variation of I furfuryl alcohol selectivity (S Furfuryl alcohol) at an equivalent fractional furfural conversion and II turnover frequency (TOF) with temperature for reaction over Au/TiO2 (solid bars), Au/CeO2 (hatched bars) and Au/Al2O3 (grey bars). Reaction conditions: P = 1 atm; T = 413–523 K
Ceria supported Au with a greater oxygen vacancy density exhibited a distinct catalytic response compared with Au/TiO2 and Au/Al2O3. We propose a reaction mechanism that involves direct participation of surface vacancies where the carbonyl group of furfural can be ‘‘anchored’’ to a vacancy (Ce3+) site (see Fig. 4I), forming a covalent Ce–O bond with a high energy of interaction [46] that stabilises the surface reactant and lowers reactivity. The (stabilised) carbonyl group can be activated for reaction at higher temperature (523 K) where hydrogenolysis to 2-methylfuran results from hydrogen scission of –C=O. The surface Ce3+ sites are oxidised by the abstracted oxygen from the carbonyl group. Oxygen vacancies can be regenerated by H2 dissociated on Au sites that spills over to the support, resulting in a continuous creation/consumption/regeneration of these vacancies. Another possible adsorption mode is through the furan ring oxygen that interacts with the electron-rich vacancy site [47] (Fig. 4II). The energy barrier for reaction is lower relative to the covalent –C=O ‘‘anchoring’’ at vacancies. In this case, the carbonyl group is attacked by reactive hydrogen to form the target furfuryl alcohol with subsequent desorption. Oxygen defects are also present on Au/TiO2 but at a lower density with a consequent higher conversion to furfuryl alcohol at lower reaction temperature. Interaction of –C=O with Lewis acid sites (Al3+) on non-reducible Al2O3 facilitates –C=O activation [27] and results in greater reactivity and higher TOF.

Proposed surface furfural adsorption/activation and reaction for Au on reducible supports (CeO2) at oxygen vacancies via (I) the carbonyl group (grey arrows) or (II) furan ring (black arrows)
4 Conclusion
We have established structure sensitivity in the gas phase hydrogenation of furfural over (0.7–0.8 mol%) Au/TiO2 and Au/CeO2 (mean Au particle size = 2.8–3.2 nm). A surface reaction mechanism is proposed to explain the role of surface oxygen vacancies in determining hydrogenation activity and selectivity. Reaction over Au/CeO2 delivered lower furfural TOF, which can be linked to inhibited H2 chemisorption capacity. The greater oxygen vacancy density on CeO2 (with higher redox potential) post-TPR served to stabilise the –C=O function and lower reactivity. Full selectivity to the alcohol was achieved over Au/TiO2 (at 413 K) and Au/CeO2 (at 473 K) where hydrogenolysis to 2-methylfuran was promoted at 523 K. Reaction over Au on non-reducible Al2O3 delivered higher furfural TOF (at 413 K) to furfuryl alcohol with 2-methylfuran formation at T ≥ 473 K.
References
Setvín M, Aschauer U, Scheiber P, Li Y-F, Hou W, Schmid M, Selloni A, Diebold U (2013) Science 341:988
Su J, Zou X, Chen J-S (2014) RSC Adv 4:13979
Ganduglia-Pirovano MV, Hofmann A, Sauer J (2007) Surf Sci Rep 62:219
Mullins DR (2015) Surf Sci Rep 70:42
Pan X, Yang M-Q, Fu X, Zhang N, Xu Y-J (2013) Nanoscale 5:3601
Weaver JF, Zhang F, Pan L, Li T, Asthagiri A (2015) Acc Chem Res 48:1515
Vecchietti J, Bonivardi A, Xu W, Stacchiola D, Delgado JJ, Calatayud M, Collins SE (2014) ACS Catal 4:2088
Oemar U, Ang ML, Chin YC, Hidajat K, Kawi S (2015) Catal Sci Technol 5:3585
Yu Y-Y, Gong X-Q (2015) ACS Catal 5:2042
Thibodeau TJ, Canney AS, DeSisto WJ, Wheeler MC, Amar FG, Frederick BG (2010) Appl Catal A 388:86
Chang S, Li M, Hua Q, Zhang L, Ma Y, Ye B, Huang W (2012) J Catal 293:195
Ta N, Liu J, Chenna S, Crozier PA, Li Y, Chen A, Shen W (2012) J Am Chem Soc 134:20585
Maeda Y, Iizuka Y, Kohyama M (2013) J Am Chem Soc 135:906
Ge J, Zeng Z, Liao F, Zheng W, Hong X, Tsang SCE (2013) Green Chem 15:2064
Wang D, Astruc D (2015) Chem Rev 115:6621
Sepúlveda-Escribano A, Coloma F, Rodríguez-Reinoso F (1998) J Catal 178:649
Milone C, Ingoglia R, Schipilliti L, Crisafulli C, Neri G, Galvagno S (2005) J Catal 236:80
Bachiller-Baeza B, Rodríguez-Ramos I, Guerrero-Ruiz A (2001) Appl Catal A 205:227
Tian Z, Xiang X, Xie L, Li F (2013) Ind Eng Chem Res 52:288
Wang X, Perret N, Delgado JJ, Blanco G, Chen X, Olmos CM, Bernal S, Keane MA (2013) J Phys Chem C 117:994
Wang X, Perret N, Keane MA (2012) Chem Eng J 210:103
Li M, Hao Y, Cárdenas-Lizana F, Keane MA (2015) Catal Commun 69:119
Salasc S, Perrichon V, Primet M, Mouaddib-Moral N (2002) J Catal 206:82
Bond GC, Louis C, Thompson DT (2006) Catalysis by gold. Imperial College Press, London
Li M, Wang X, Perret N, Keane MA (2014) Catal Commun 46:187
Jovic V, Chen W-T, Sun-Waterhouse D, Blackford MG, Idriss H, Waterhouse GIN (2013) J Catal 305:307
Perret N, Wang X, Onfroy T, Calers C, Keane MA (2014) J Catal 309:333
Ousmane M, Liotta LF, Carlo GD, Pantaleo G, Venezia AM, Deganello G, Retailleau L, Boreave A, Giroir-Fendler A (2011) Appl Catal B 101:629
Liu SY, Yang SM (2008) Appl Catal A 334:92
Haffad D, Kameswari U, Bettahar MM, Chambellan A, Lavalley JC (1997) J Catal 172:85
Salasc S, Perrichon V, Primet M, Chevrier M, Mouaddib-Moral N (2000) J Catal 189:401
Liu H, Ma HT, Li XZ, Li WZ, Wu M, Bao XH (2003) Chemosphere 50:39
Boccuzzi F, Chiorino A, Manzoli M, Andreeva D, Tabakova T (1999) J Catal 188:176
Campbell CT, Peden CHF (2005) Science 309:713
Popa C, Ganduglia-Pirovano MV, Sauer J (2011) J Phys Chem C 115:7399
Fu Q, Saltsburg H, Flytzani-Stephanopoulos M (2003) Science 301:935
Manzoli M, Chiorino A, Vindigni F, Boccuzzi F (2012) Catal Today 181:62
Nakamura I, Mantoku H, Furukawa T, Fujitani T (2011) J Phys Chem C 115:16074
Nakamura I, Mantoku H, Furukawa T, Takahashi A, Fujitani T (2012) Surf Sci 606:1581
Akita T, Okumura M, Tanaka K, Kohyama M, Haruta M (2005) J Mater Sci 40:3101
Campo B, Volpe M, Ivanova S, Touroude R (2006) J Catal 242:162
Ohyama J, Esaki A, Yamamoto Y, Arai S, Satsuma A (2013) RSC Adv 3:1033
Zanella R, Louis C, Giorgio S, Touroude R (2004) J Catal 223:328
Paier J, Penschke C, Sauer J (2013) Chem Rev 113:3949
Nagaraja BM, Padmasri AH, Raju BD, Rao KSR (2007) J Mol Catal A 265:90
Kennedy G, Baker LR, Somorjai GA (2014) Angew Chem Int Ed 53:3405
Badawi M, Cristol S, Paul J-F, Payen E (2009) C R Chimie 12:754
Acknowledgements
We thank Dr. X. Wang for his contribution to this study. The work was supported in part by the Engineering & Physical Sciences Research Council (Grant EP/M029476/1). Financial support to Dr. M. Li through the Overseas Research Students Award Scheme (ORSAS) is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, M., Collado, L., Cárdenas-Lizana, F. et al. Role of Support Oxygen Vacancies in the Gas Phase Hydrogenation of Furfural over Gold. Catal Lett 148, 90–96 (2018). https://doi.org/10.1007/s10562-017-2228-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-017-2228-9