
Abstract
Obesity, exceptionally prevalent in the USA, promotes the incidence and progression of numerous cancer types including breast cancer. Complex, interacting metabolic and immune dysregulation marks the development of both breast cancer and obesity. Obesity promotes chronic low-grade inflammation, particularly in white adipose tissue, which drives immune dysfunction marked by increased pro-inflammatory cytokine production, alternative macrophage activation, and reduced T cell function. Breast tissue is predominantly composed of white adipose, and developing breast cancer readily and directly interacts with cells and signals from adipose remodeled by obesity. This review discusses the biological mechanisms through which obesity promotes breast cancer, the role of obesity in breast cancer health disparities, and dietary interventions to mitigate the adverse effects of obesity on breast cancer. We detail the intersection of obesity and breast cancer, with an emphasis on the shared and unique patterns of immune dysregulation in these disease processes. We have highlighted key areas of breast cancer biology exacerbated by obesity, including incidence, progression, and therapeutic response. We posit that interception of obesity-driven breast cancer will require interventions that limit protumor signaling from obese adipose tissue and that consider genetic, structural, and social determinants of the obesity–breast cancer link. Finally, we detail the evidence for various dietary interventions to offset obesity effects in clinical and preclinical studies of breast cancer. In light of the strong associations between obesity and breast cancer and the rising rates of obesity in many parts of the world, the development of effective, safe, well-tolerated, and equitable interventions to limit the burden of obesity on breast cancer are urgently needed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Epidemiology and classification of breast cancer
In 2020, breast cancer surpassed lung cancer as the leading cause of global cancer incidence in women [1]. Breast cancer is commonly stratified into molecular subtypes identified by immunohistochemistry for the presence of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2/neu) [2]. Breast tumors with detectable ER, PR, or both, with or without HER2 amplification, are defined as luminal-like tumors [3]. Tumors with HER2 overexpression, but not ER or PR, are defined as HER2 + breast cancer [4]. Triple-negative breast cancer (TNBC) is defined by a lack of expression of all three receptors [5]. About 60–90% of all breast cancers express the androgen receptor, although its potential as a therapeutic target remains controversial [6,7,8].
Luminal A tumors, defined by high ER and PR expression without HER2 amplification, is the most common molecular subtype of breast cancer with the best prognosis. Luminal B, which expresses lower levels of ER and PR and can have HER2 amplification, often presents at a higher tumor grade and has a greater risk of recurrence [9]. Both luminal subtypes are generally responsive to endocrine-based therapies, making effective treatment options more widely available. However, intrinsic and/or acquired therapeutic resistance remains pervasive [10]. HER2 + positive breast cancer, accounting for about 20% of breast cancers, is often a more aggressive tumor subtype but generally responsive to HER2-targeted therapies [11].
While TNBC accounts for only 15–20% of breast cancers, this intrinsic subtype is particularly aggressive, more likely to metastasize, and due in part to the lack of targeted therapies, results in worse clinical outcomes including greater recurrence and lower rates of overall survival [12]. Breast cancer, particularly TNBC, is a heterogeneous disease, hence transcriptomic signatures have been developed to further stratify these cancers. Basal-like breast cancer (BLBC) is a particularly aggressive subtype, defined by gene expression profiles resembling that of basal and myoepithelial breast cells [13]. While not mutually exclusive, BLBC and TNBC are frequently coincident, with TNBC making up 50–75% of BLBC tumors, and about 80% of BLBC tumors lacking ER and HER2 expression [14]. Additionally, a subset of TNBC tumors are defined as claudin-low, characterized by a stem cell-like/undifferentiated phenotype, high expression of epithelial-to-mesenchymal (EMT) markers, low levels of genomic instability, and heightened infiltration of stromal and immune cells [15]. When categorized according to intrinsic subtype, 70% of claudin-low tumors are TNBC [16]. TNBC is also characterized by deficiencies in homologous recombination with the majority of BRCA1 mutation-associated breast cancers classified as TNBC [17, 18].
2 The obesity-breast cancer link: epidemiological evidence
In 2017–2018, the age-adjusted prevalence of obesity—defined as a body mass index (BMI) of 30 kg/m2 or greater—among US adults was 42.4% [19]. Obesity promotes incidence and progression of at least 15 cancer types, including breast cancer in postmenopausal women [20]. Adipose tissue becomes the predominant site of estrogen production after menopause. Hence, women with obesity have greater postmenopausal levels of estrogen and consequently greater exposure to estrogen’s protumorigenic effects [21]. Thus, obesity-mediated exacerbation of cancer is of pressing concern. Across all breast cancer subtypes, obesity is associated with worse disease-free survival and overall survival [22]. However, the relationship between obesity and breast cancer is complicated by subtype and menopausal status across the literature. Obesity in women who are postmenopausal increases overall relative risk of developing breast cancer to 1.33, largely driven by increased rates of ER + breast cancers [23]. However, being obese is also associated with postmenopausal TNBC incidence and progression [24, 25]. The relationship between obesity and HER2 + breast cancer is still incompletely understood. Obesity is consistently associated with worse overall survival in patients with early HER2 + breast cancer, but evidence on the link between obesity and advanced HER2 + breast cancer is heterogeneous [26,27,28]. Genetic predictors of obesity, including several single nucleotide polymorphisms associated with fasting glucose and insulin, also correlate with breast cancer risk independent of family history, age, or menopausal status, pointing to the importance of the relationship between obesity, breast cancer risk, and genetics [29, 30].
2.1 Obesity and metastatic progression of breast cancer
Metastasis, the dissemination and growth of primary tumor cells in secondary sites, is the cause of 90% of tumor-related deaths in patients with breast cancer, with 5-year survival rates at 28% for affected patients [31, 32]. Typically, metastatic progression begins with local invasion of cancer cells from the primary tumor, first into the stroma surrounding the tumor and eventually to the neighboring normal tissue. Intravasation follows, as tumor cells expand their niche by entering lymphatic vessels to access the body’s systemic circulation [33]. As a distant site is reached, the cancer cells exit the bloodstream and proceed to adhere to the target organ. Metastatic outgrowth marks the final stage of metastatic progression, where quiescent tumor cells at distant sites are activated to begin proliferating [34, 35]. The mechanisms behind this activation are part of ongoing research.
For all subtypes of breast cancer, patients who are obese tend to have larger primary tumors at diagnosis and heightened risk of developing lymph node metastases [36]. Higher BMI predicts lower locoregional and distant recurrence-free survival among women with breast cancer [37], and increases association with overall mortality when compared to breast cancer patients at an ideal weight [38]. Indeed, patients with breast cancer and obesity are up to 46% more likely to have distant metastases 10 years after diagnosis [39]. Metabolic syndrome, defined in part by abdominal obesity, in patients with early breast cancer is linked to an increased risk of relapse as well as poor prognosis [40]. There are both biological and non-biological mechanisms contributing to the disparate outcomes for women with obesity and breast cancer, which have been expertly reviewed elsewhere [41].
Obesity expedites and exacerbates metastatic progression of breast cancer, supported by preclinical models [42,43,44] and clinical studies [39, 45]. Several leptin-mediated mechanisms behind this link have been established, including breast cancer invasion, migration, and immune regulation [46, 47], as well as cancer stem cell enrichment and mesenchymal stem cell dysregulation in the tumor microenvironment [31, 48]. Preclinical models of obesity demonstrate that increased myeloid-derived suppressor cell (MDSC) recruitment, collagen deposition, and changes in fibroblast phenotype in the lungs cooperate to create a favorable premetastatic niche for breast cancer [49].
3 The obesity-breast cancer link: molecular mechanisms
3.1 Obesity, adipose, and cancer interactions
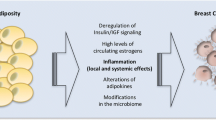
Separately, obesity and cancer are complex, integrating an incompletely understood combination of genetics, environment, and lifestyle. Hence, the relationship between obesity and cancer is immensely complicated. Despite the complexity, several mechanisms underlying the obesity-cancer link have been established. Increased white adipose tissue (WAT) mass is emerging as a nexus of tumor biology and metabolic and inflammatory dysregulation in obesity. WAT is composed of mature adipocytes, preadipocytes, endothelial cells, fibroblasts, pericytes, and immune cells [50]. Obesity also promotes hyperleptinemia, a result of dysregulated adipose tissue that can enhance inflammatory cytokine secretion [51]. In a murine model of renal cell carcinoma, hyperleptinemia has also been implicated in reduced efficacy of recombinant adenoviral/TLR agonist and anti-CTLA-4 checkpoint inhibitor immunotherapy [52]. WAT expands through adipocyte hypertrophy and hyperplasia in response to chronic nutrient excess, shifting the body’s energy balance signaling network and leading to elevated systemic insulin, estrogen, and adipokine signaling [53]. During adipose tissue expansion in the development of obesity, inflammation arises due to increases in immune infiltration, hypertrophic adipose tissue remodeling and angiogenesis, adipocyte necrosis, and dysregulated fatty acid flux due to heightened adipocyte lipolysis [54, 55]. Rapid adipocyte hypertrophy during adipose tissue expansion can create insufficient angiogenesis to achieve proper tissue vascularization, leading to hypoxic regions in WAT [56]. WAT hypoxia activates the transcription factor hypoxia-inducible transcription factor 1, which prevents preadipocyte differentiation and initiates adipose tissue fibrosis [57]. Adipose tissue macrophages engulf necrotic or damaged adipocytes to form distinct crown-like structures (CLS), a key feature of the pro-inflammatory process in adipose tissue [58]. Stressed adipose tissue, combined with hypoxia, promotes immune cell infiltration and stimulates inflammatory cytokine and chemokine release from resident macrophages in adipose tissue [59, 60] (Fig. 1). In addition to inflammatory signaling, adipocytes and their precursor mesenchymal stem cells (MSCs) support breast cancer progression by seeding the tumor microenvironment (TME) with critical supportive cell populations [61]. Adipose progenitor cells are more abundant in obese, relative to nonobese, mice [62], with greater levels recruited to the TME [63] supporting breast cancer growth and angiogenesis in vivo [64]. Cancer cells actively reprogram tumor-adjacent adipocyte matrix proteins and the inflammatory secretome, promoting the formation of cancer-associated adipocytes [65]. Cancer-associated adipocytes release free fatty acids into the TME [66], increase interstitial stiffness in breast adipose tissue [67], and enhance secretion of cytokines such as interleukin (IL)-6, interleukin (IL)-8, monocyte chemoattractant protein (MCP)-1, and tumor necrosis factor-alpha (TNFα), that promote inflammation [68]. In addition to supporting tumor cell migration, invasion [69, 70], and resistance to therapy [71, 72], cancer-associated adipocytes can transdifferentiate into adipocyte-derived fibroblasts in response to cues from the tumor. Adipocyte-derived fibroblasts, along with matrix metalloproteinases, modify extracellular matrix proteins to promote inflammation and tumor invasion [73,74,75,76]. Cancer-associated fibroblasts, which can arise from a variety of cell types including adipose-derived fibroblasts, are also key regulators of tumor development, metastatic progression, and therapy resistance [77].

The impact of obesity on the tumor microenvironment. White adipose tissue from an obese host is composed of hypertrophied adipocytes, some of which become necrotic and induce formation of crown-like structures. Adipose tissue inflammation is furthered by M1-like and metabolically activated macrophages. Tumors developing adjacent to obese adipose tissue receive numerous inflammatory and metabolic signals from adipose and are marked by immunosuppressed tumor microenvironment with ineffective tumor-infiltrating lymphocytes and immunosuppressive M2-like macrophage polarization. Adipose tissue further contributes to the tumor microenvironment via recruitment and transdifferentiation of cancer-associated fibroblasts from mesenchymal stem cells, and adipocyte-derived fibroblasts from adipocytes
In addition to modulation of adipocytes and other cells typically resident in adipose tissue, adipose-adjacent tumors actively recruit stromal cells, including MSCs, from elsewhere in the body, and reprogram their function through bidirectional communication with tumor cells to support metastatic progression [78]. Cancer cells induce lipolysis in adipocytes, releasing free fatty acids that are utilized by tumors for proliferation and migration [79] and stored within lipid droplets [66]. This transfer of fatty acids, stimulated by cytokines, sustains WAT inflammation [80] and occurs at a greater rate in breast cancer cells co-cultured with adipocytes from donors with obesity versus adipocytes from nonobese donors [79]. As breast tissue is composed of 90% WAT [81], and the human mammary epithelium is in permanent interaction with mammary adipose tissue [82], understanding the impact of excess WAT is imperative to resolving the relationship between obesity and breast cancer.
3.2 WAT and adipokines
Elevated levels of endogenous sex hormones are associated with obesity and are correlated with a risk of breast cancer in postmenopausal women [83]. After menopause, estrogen production via activity of the key enzyme aromatase becomes noncyclical and occurs mainly in adipose tissue, exacerbating estrogen production in women with obesity [84]. Obesity not only elevates estrogen production in postmenopausal women, but also increases its bioavailability through reductions in sex-hormone binding globulin [85, 86]. Increased levels of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNFα) and interleukin (IL)-6, further promote estrogen synthesis by inducing aromatase expression [87, 88].
WAT is a major endocrine organ, secreting hormones and growth factors, in addition to enzymes and metabolites. The WAT secretome is an important mediator of tumor exacerbation by obesity [81]. Adipokines, secreted by WAT, constitute a class of biologically active polypeptides with a broad range of endocrine, metabolic, and inflammatory functions [89]. Given the extensive interaction between adipocytes and tumor cells in the breast TME, adipokines play a critical role in the proliferative and invasive capacities of breast cancer [90]. While there are several adipokines (reviewed in [91]), two prominent examples are examined below.
Leptin, the polypeptide hormone primarily produced by adipocytes, is both synthesized and systemically circulates in proportion to adipose tissue mass [92]. Leptin levels are higher in patients with breast cancer compared to patients who are healthy, particularly in women who are overweight or obese [93]. Further, increased leptin associates with breast cancer risk at a standardized mean difference of 0.96 in a meta-analysis of 46 studies of over 13,500 women [94]. Leptin impacts breast cancer biology through a myriad of mechanisms that result in increased tumor volume and metastasis in preclinical and clinical models of breast cancer, including TNBC [33, 95]. Through activation of the PI3K/Akt pathway, leptin disrupts breast tissue epithelial polarity and promotes premalignant lesions [96]. High production of reactive oxygen species in TNBC is associated with lower antioxidant status to favor growth, survival, and inflammation in the presence of leptin [97]. In a patient-derived xenograft model of TNBC, leptin produced by obesity-altered adipose stem cells drove a prometastatic phenotype via upregulation of EMT-associated genes [31]. Increased leptin signaling in diet-induced obese mice results in tumoral cancer stem cell enrichment and mediates cell viability, migration, and invasion in triple-negative mammary tumor cells [48].
TNFα is a cytokine expressed in subcutaneous (and to a lower extent, visceral) adipose tissue [98] and preadipocytes [99]. In healthy breast tissue, TNFα contributes to cell proliferation and morphogenic branching [100]. As a key pro-inflammatory cytokine, TNFα is also expressed in monocytes and macrophages, and TNFα levels in adipose tissue rise 2.5-fold in individuals with obesity and have a strong positive correlation with hyperinsulinemia (r = 0.82) [101, 102]. TNFα promotes leptin secretion from adipocytes [103] and contributes to decreases in the anti-inflammatory adipokine adiponectin [104]. By inducing expression of aromatase and IL-6 in adipose tissue, TNFα also promotes estrogen synthesis [105]. TNFα promotes TNBC migration [106] and induces EMT in breast cancer stem cells while promoting a claudin-low phenotype [107] implicating this adipokine’s role in metastatic progression. Additionally, TNFα is linked to TNBC resistance to chemotherapy [108] and BLBC resistance to immune checkpoint inhibitors in vitro [109].
Thus, obesity dysregulates both endocrine and metabolic functions of WAT by promoting pro-inflammatory transformation, which is characterized by stromal remodeling, hypoxia, and altered immune profile. While significant progress has been made in understanding the relationships between these complex and intertwined processes, the relative contributions of each of these to the pathophysiology of obesity remains to be determined.
3.3 Obesity and immune function
Protumor dysregulation of the prevalence and proportion of various immune cells in the obese TME and surrounding adipose tissue promotes angiogenesis, tumor growth, metastatic spread, and immune evasion culminating in adverse outcomes for patients with obesity and breast cancer [51, 110].
Adipose tissue remodeling that occurs with weight gain promotes recruitment of adipose tissue macrophages in both subcutaneous and visceral depots [111, 112]. Classically activated (M1-like) macrophages are more abundant in obese adipose tissue and form the characteristic CLS, named after their formation of a ring-like network of macrophages surrounding necrotic, hypertrophied, and dying adipocytes in breast adipose, and other WAT [58, 92, 112]. These macrophages disrupt adipocyte signaling, increase reactive oxygen species production, and promote secretion of pro-inflammatory cytokines [58, 113]. The infiltration of macrophages and the accompanying inflammation of breast adipose tissue of patients who are obese increases the risk of mammary carcinogenesis [114, 115].
In women who are obese, the breast adipose tissue produces CCL2 (also named MCP-1) and IL-1β to recruit macrophages and secrete CXCL12, resulting in increased CLS formation [116, 117]. The presence of CLS and inflammatory mediators in breast adipose tissue of women who are obese is associated with aberrant intracellular signaling and cellular dysfunction [84]. High densities of CLS are also independently associated with an increased risk of breast cancer, in addition to their negative impact on recurrence and survival [118,119,120].
Obesity-related drivers of immunological aging are characterized in part by premature thymic involution [121]. Lipid accumulation occurring with obesity can transform thymic fibroblasts into adipocytes, leading to reduced activity of the thymus [122]. This creates a reduction in the abundance, proliferation, and diversity of T cells, the essential players of cell-mediated immune response and adaptive defense against diseases like cancer [122, 123]. Gamma delta (γδ) T cells, defined by their γ and δ T cell receptors instead of the canonical α and β T cell receptors, have an increased pro-inflammatory population in adipose tissue from obese versus nonobese mice [124]. Obesity is also associated with higher pro-inflammatory CD4 + Th1 cells relative to static levels of anti-inflammatory Th2 cells and regulatory T cells (Tregs) in adipose tissue [125]. However, due to aberrant insulin signaling, Tregs associated with obesity exacerbate adipose inflammation through alterations in cytokine production [126].
Obesity-related M1-like macrophages upregulate expression of programmed death-ligand 1 (PD-L1; an immune checkpoint protein) in TNBC, partially through enhanced secretion of IL-6 [127]. Classified by expression of the classical dendritic marker CD11c, these M1-polarized macrophages contribute to an immunosuppressive microenvironment by promoting T cell exhaustion [128]. While the breast TME comprises several types of immune cells, macrophages are the most abundant. Tumor-associated macrophages make up over 50% of TME macrophages [129] and are associated with aggressive features of TNBC tumors, including recurrence and metastases [130]. Leptin also activates IL-8 production in tumor-associated macrophages, driving tumor progression [131]. Metabolically activated macrophages are a pro-inflammatory population of macrophages unique to obesity and distinct from M1-like macrophages [132, 133]. Mammary adipose tissue macrophages from preclinical models of obesity produce inflammatory cytokines, induce a stem-like phenotype in breast cancer cells, and promote TNBC growth [132].
In adipose tissue, lower Treg abundance, coupled with an increase in CD8 + T cells, creates an obesity-specific immune profile that promotes macrophage recruitment, inflammatory cytokine production, and consequently, can contribute to tumor progression [134, 135]. The increased leptin signaling characteristic of obesity increases PD-1 expression in T cells, resulting in T cell exhaustion and contributing to heightened inflammation [136, 137]. This immune dysfunction, however, correlates with greater response to PD-1/PD-L1 treatment in patients who are obese, including improvements in CD8 + /CD4 + ratios, metastatic burden and overall survival [136, 138,139,140,141]. This relationship is not universal, as reduced anti-PD-1 therapy efficacy occurs in patients with renal cell carcinoma who are obese [142]. The impact of obesity on checkpoint blockade inhibitor response remains unclear in TNBC.
Leptin receptors are highly expressed in activated T cells, impacting their sensitivity to nutrient availability [143]. Leptin plays an important role in the increased T cell dysfunction and PD-1 expression seen with obesity [136]. Indeed, leptin-STAT3 signaling in CD8 + effector T cell metabolism promotes fatty acid ß-oxidation while inhibiting glycolysis in a model of TNBC in high fat diet (HFD)-fed mice [144]. TNBC tumors from patients who are obese have higher expression of leptin, CXCR4, and CCR9 (receptors of CXCL12 and CCL25, respectively), which negatively correlate with CD8 + T cell infiltration, as compared to tumors from patients who are not obese [44]. FasL + granulocytic MDSC are increased with obesity via leptin signaling, causing CD8 + T cell apoptosis and resistance to immunotherapy [145].
Natural killer (NK) cells, type 1 innate lymphoid cells that have a large role in tumor response, fall into two distinct categorizations of CD56 expression. CD56dim NK cells represent an activated phenotype and produce perforin and granzyme for cytotoxic functionality, while CD56bright NK cells serve a more regulatory role [146]. NK cell populations are reduced both in number and activity in obesity, with a coincident decrease in the cytotoxic CD56dim population and increase in the regulatory CD56bright population [147, 148]. Like T lymphocytes, NK cells also express the leptin receptor [149]. Chronic exposure to the elevated leptin levels associated with obesity alter post-receptor signaling in these cells, lowering JAK2 phosphorylation and decreasing production of interferon-γ [149, 150].
MDSCs are an emerging mechanistic link between obesity and cancer [151]. Inflammatory signaling pathways promote the activation and downstream immunosuppressive function of MDSCs, driving their accumulation and activity in tumors as well as adipose tissue [151, 152]. Characterized by their expression of the cell surface markers Gr1 and Cd11b, MDSC populations accumulate threefold in adipose tissue of HFD-fed mice compared to their lean counterparts after 12 weeks on diet [153]. Increased levels of circulating leptin and exogenous lipids both drive immunosuppressive MDSC accumulation in adipose tissue and the TME, all of which work together to promote tumor growth [154,155,156]. Obesity, in part through crosstalk with leptin and availability of lipids in the TME, increases the presence of MDSCs and their PD-L1 expression to enhance tumor progression [154].
Advances in our understanding of the dynamic and complex relationships of the TME, including immune cells, has led to novel therapeutic strategies [157]. The chronically activated immune response characteristic of obesity can detrimentally impact therapeutic efficacy [136, 142]. However, as TNBC, relative to other breast cancer subtypes, generally has higher levels of tumor-infiltrating lymphocytes [158] and increased expression of immune checkpoint molecules, immunotherapy has become a promising avenue for PD-1/PD-L1 blockade treatments [159, 160]. An active area of research involves combining immunotherapies with other treatment modalities, including radiation, targeted therapies such as CDK4/6 inhibitors and PARP inhibitors, and cancer vaccines, to further improve their efficacy [161,162,163,164]. Another intriguing line of work in the immunotherapy field involves the interactions between obesity, immunotherapy response and several cancers [165].
Chronic inflammatory signaling in obesity limits immune responses to numerous diseases, including cancer. Rather than a uniform depression of function, obesity promotes altered function in a host of immune cells characterized by chronic systemic inflammation and limited antitumor immunity. Thus, ongoing work to delineate how obesity mediates disruption of antitumor immunity, and to identify interventions to mitigate this is of considerable importance.
4 The obesity-breast cancer link: health disparities
4.1 Obesity biases in breast cancer care
Due to the absence of targeted therapies, chemotherapy remains the standard of care for TNBC. As many as 40% of patients who are obese receive substantially lower doses relative to patients who are not obese due to dose calculation that does not incorporate body weight [166, 167]. Compared to patients who are not obese, patients with breast cancer who are also obese are more likely to have their doses capped at an arbitrary body surface area, even in the absence of toxicity expected at full intended doses [168, 169]. This phenomenon was shown to mediate the relationship between obesity and lower breast cancer-specific survival in a large observational cohort study [170]. However, even when dosing accounts for body weight, systemic chemotherapy is less effective in patients who are obese with breast cancer [41]. Tumor-associated adipocytes can induce multidrug resistance in breast cancer by upregulating a transport-associated protein that mediates doxorubicin efflux, a mechanism amplified by obesity [171]. In a murine model of TNBC, doxorubicin treatment is less effective in HFD-fed mice compared to control-fed mice due to changes in free fatty acid availability in mammary adipose tissue [172, 173].
Despite the improvements in our understanding of the complexity of factors contributing to obesity, coupled with its increasingly high prevalence, individuals with obesity continue to experience stigma and biased treatment in the healthcare setting [174, 175]. This is compounded by the lack of clinical intervention data in patients with cancer and obesity, with obesity status reported in only 5.3% of clinical trials of obesity-related cancers, including postmenopausal breast cancer [176].
4.2 Obesity and breast cancer health disparities
Despite a relatively similar incidence of breast cancer among non-Hispanic White (NHW) and non-Hispanic Black (NHB) women, NHB women are ~ 40% more likely to die of breast cancer [177, 178] and have an overall 5-year breast cancer survival rate of 78.9% (compared to 88.6% among NHW women) [179]. While there have been substantial advances in breast cancer screening and treatment, resulting in an overall reduction in breast cancer mortality, declines are largely attributed to improvements among NHW women; the 2018-to-2019 change in the age-adjusted death rate was 19.2 vs. 18.8 per 100,000 among NHW women but unchanged among NHB women [180].
The etiology of racial disparities in breast cancer mortality are multifactorial and include contributing factors from social determinants of health and access to tumor biology and comorbidities. Obesity is an important consideration given the known association with risk, recurrence, and mortality across age categories and tumor characteristics [181]. Profound obesity-related disparities exist in the USA, where the prevalence rates of both obesity (49.6%) and severe obesity (BMI ≥ 40 kg/m2) (3.8%) among adults are most pronounced among NHB adults compared with other race and ethnic groups [19]. Notably, the obesity disparity is largely driven by NHB women; the 2017–2018 National Health and Nutrition Examination Survey showed that the prevalence of obesity among NHB women was 56.9% compared to 39.8% among NHW women [19]. Given the role of obesity in most aspects of cancer diagnosis, treatment, and progression [182], we posit that obesity could be a causal contributor to racial disparities in breast cancer outcomes, as further described below.
One factor in racial disparities in breast cancer mortality is differences in the presentation and prevalence of aggressive tumor subtypes. Specifically, adiposity increases the risk of postmenopausal ER + breast cancer and premenopausal ER-/TNBC [183]. It is well-established that NHB women compared to NHW women have a higher incidence of ER-/TNBC [184]. The Carolina Breast Cancer Study showed that BLBC occurs at a higher prevalence in premenopausal African American (AA) women (39%) compared to postmenopausal AA women (14%) and non-AA women (16%) [185]. BLBC, which progresses more quickly and has greater TP53 mutations compared to the luminal A subtype, are more prevalent in NHB women and have an unfavorable prognosis. Indeed, AA women are more likely to carry a TP53 mutation compared to White women [186].
While there is a higher prevalence of premenopausal TNBC in NHB compared to NHW women, luminal subtypes remain the most prevalent tumors among NHB women, accounting for approximately 75% of all breast cancer diagnoses [178]. After adjusting for age, NHB women with luminal A breast cancer have a 2.43 times higher rate of breast cancer mortality than their NHW counterparts [187]. Given that luminal A breast cancer — compared to TNBC — has better treatment options, insights are needed to understand drivers of such robust disparities in this relatively easier-to-treat subtype.
One potential mechanism of poor outcomes among NHB women with luminal A subtypes is obesity-related differences in DNA methylation that are associated with several clinical and histopathological features of breast cancer and clinical outcomes [188]. In a study that examined the association between obesity and DNA methylation in NHB and NHW women diagnosed with breast cancer, the authors detected interactions with ER status (PSMB1, QSOX1, and PHF1) and race (TOMM20) among the top 20 obesity-associated CpG cites. Additionally, differential methylation at the CpG sites of TOMM20, PSMB1, and QSOX1 was associated with all-cause mortality, suggesting that obesity-related dysregulation may, in part, drive mortality differences [189]. Other obesity-related mechanisms may be relevant to differential tumor progression leading to racial disparities in outcomes, including local adipose tissue inflammation [190], diversity of the gut microbiome [191], and immune parameters, each of which can affect therapeutic effectiveness [192].
While there are several biological links between obesity and racial differences in breast cancer outcomes, biology alone cannot explain the persistent disparities across age and subtypes of breast cancer. Evidence suggests that obesity-related screening biases lead to delayed diagnoses, ultimately increasing cancer mortality in patients who are obese [182]. Acute and late treatment complications are more often seen among women who are obese, and—largely due to dosing uncertainty—there remains concern of treatment efficacy in women who are obese [193]. Notably, Black women, compared to their White counterparts, are more likely to be diagnosed at a later stage and less likely to receive stage-appropriate treatment [177]. The confluence of race and obesity-related biases may profoundly affect prognostic disparities. In addition to potential obesity-related race differences at the point of care, the inclusion of systemic inequities is essential to understand obesity-related drivers of breast cancer disparities. Collin and colleagues recently reported a 1.6-fold increase in breast cancer mortality among women residing in a redlined Atlanta neighborhood defined using present-day Housing Mortgage Disclosure Act data as odds of denial of a mortgage application for a residence inside the census tract compared with those outside of the census tract. While only 20% of NHW women diagnosed with breast cancer between 2010 and 2014 lived in a redlined census tract, 80% of NHB women diagnosed during the same time frame lived in redlined areas [194]. Redlining is an important driver of the built (e.g., food deserts, green space, walkability) and lived (e.g., environmental toxicants) environments, which profoundly affect adiposity. Additional research is desperately needed to explore the role of structural mechanisms in obesity-related breast cancer disparities.
5 Dietary interventions to intercept obesity-mediated exacerbation of breast cancer
Obesity can lead to reduced efficacy of existing breast cancer therapeutics and increase treatment resistance [30, 195]. Patients with obesity experience greater risk of recurrence following several different endocrine therapies [196,197,198,199], and a patient’s BMI at diagnosis correlates with breast cancer recurrence across multiple subtypes [200]. Side effects during and after treatment, including lymphedema, also disproportionately impact women with obesity [201]. Patients with obesity also face greater risk of complications resulting from mastectomies, both with and without breast reconstruction surgery [202]. Cancer cell sensitivity to nutrient restriction, as well as their differing requirements for specific metabolites, constitute emergent hallmarks of cancer-targeted therapies [203]. Dietary interventions pose an inexpensive and effective way to manipulate availability of key nutrients for tumors and hence improve the effect of existing therapies, activate antitumor response mechanisms, and introduce tumor-specific toxicities [204]. Table 1 indicates some common dietary interventions used in clinical and preclinical studies.
5.1 Chronic calorie restriction
Chronic caloric restriction (CR), typically defined as \(\ge\) 10% reduction in caloric intake in humans and \(\ge\) 20% reduction in rodents without malnutrition or the restriction of water, is an established mechanism for extension of lifespan and healthspan in clinical and preclinical models [205, 206, 222]. CR partially reverts some of the metabolic consequences of obesity in clinical trials [223, 224]. Postmenopausal women with an increased risk of breast cancer show improved hormonal (bioavailable estradiol, testosterone, and insulin) and adipocytokine (relative adiponectin/leptin, C-reactive protein) markers of breast cancer in serum and breast tissue with weight loss of more than 10% [225]. A meta-analysis of 59 preclinical studies concluded that calorie restriction is preventive of cancer development [226]. Indeed, CR reduces the incidence of several types of tumors in rodent and rhesus monkey models [226, 227]. CR prior to tumor induction slows primary tumor growth and reduces metastatic burden in a preclinical model of TNBC, and 30% CR imposed at the time of tumor induction in this same model synergized with radiation treatment to further suppress tumor growth [228,229,230]. A 30% CR dietary regimen also mitigates chemotherapy-induced inflammation and enhances radiation response by downregulating insulin-like growth factor (IGF)-1 receptor signaling, reducing metastatic tumor burden and improving overall survival in preclinical models of TNBC [207, 231]. These results were replicated with preclinical models of estrogen-responsive breast cancer, utilizing the fasting-mimicking diet (described below) and endocrine-based therapies [232]. Some of the metabolic protective effects of CR are mediated through periods of fasting, which arise when animals rapidly consume their daily calories and then fast for the remainder of each day [221]. However, this effect has not been examined in cancer models. Although CR is not nearly as well characterized in clinical studies as it is in preclinical models [233], human trials show promise in replicating the molecular and metabolic changes established in rodents [234]. Limited adherence to CR and unsafe weight loss, however, particularly in patients with advanced stage cancer, are of major concern for the success of this intervention [235, 236]. Larger clinical trials that incorporate dietary components, patient follow-up, and standardized treatment protocols are needed to more accurately assess the impact of CR on breast and other cancers [237].
5.2 Time-restricted feeding
Time-restricted feeding (TRF), the practice of restricting time of calorie intake, rather than calories consumed, to an 8-–12-h window aligning with the circadian rhythm, is an emerging dietary pattern rapidly gaining scientific and cultural popularity [238]. As components of the circadian clock interact with nutrient-sensing pathways, inconsistent eating patterns and overeating can disrupt circadian regulation of endocrine and nutrient metabolism [239, 240]. By reducing evening energy intake and refraining from consuming food throughout the night, the TRF fasting regimen aligns meal times with the ideal postprandial hormonal response [241]. In a mouse model of postmenopausal obesity, TRF reduced body weight, improved glucose tolerance and insulin resistance, and reduced accumulation of fat in the liver [215]. These findings were corroborated by an 8-week clinical trial of time-restricted eating in participants who were overweight and obese, where a 10-h eating window resulted in clinically meaningful reductions in weight and improved fasting blood glucose [216].
There is growing evidence that circadian rhythm plays an important role in cancer pathogenesis, with chronic disruptions increasing breast cancer metastasis [242, 243]. Preclinical models of breast cancer demonstrate promising effects, with mice fed a high fat diet restricted to 12-h intervals having tumor burden reduced to that of control-fed mice [244].
5.3 Fasting
In clinical research, fasting in humans is defined as significant-to-total reprieve of caloric intake for several hours to days [208]. Due to greater feasibility and improved adherence compared to CR, periodic fasting (PF) and short-term fasting (STF), maintained for periods of 48–72 h in rodents and 2–5 days in humans, may provide an effective compromise [235]. The synergistic effect of PF in combination with cancer treatment seen in preclinical studies [245] may be due to its effect on blood glucose levels, which are reduced more profoundly in preclinical models of PF relative to CR [246]. Engaging in STF induces a differential stress response between cancer cells and healthy cells, prioritizing cellular maintenance and repair in healthy cells, and exposing a vulnerability in cancer cells due to their inability to suppress growth-promoting pathways [246, 247]. The differential stress response provides a mechanism by which fasting can promote efficacy and tolerance of chemotherapy and radiation treatments [245, 246, 248, 249]. Additionally, fasting promotes stem cell self-renewal as well as regeneration of the blood, nervous system, muscle, and liver [235, 250]. In vitro data indicates TNBC sensitization to chemotherapy treatment with 24-h fasting [209]. Two clinical pilot studies with HER2-negative breast cancer patients demonstrate STF as a feasible, tolerated intervention in both trials and a reduction in the hematological toxicity of chemotherapy treatment in one trial [248, 251].
5.4 Fasting-mimicking diet
The fasting-mimicking diet (FMD) is generally very low in proteins and carbohydrates, enriched in unsaturated fats and micronutrients, and maintained for a period of days in a cyclical fashion. It is an alternative regimen to a water-only fast with similar changes in stress resistance and blood glucose but greater tolerability and adherence [208, 250]. A combination regimen of FMD and endocrine-based therapies reduces IGF-1 receptor signaling, abates chemotherapy-induced inflammation, and enhances tumor response to radiation in preclinical models of estrogen-responsive breast cancer [232]. In vitro models of TNBC responded to a FMD, including reduced circulating glucose and IGF-1, with enhanced tumor immunogenicity and improved response to chemotherapy [210]. A clinical trial with 131 patients with HER2-negative breast cancer demonstrated that a FMD adhered to for 3 days prior to chemotherapy administration remained safe, effective, and lowered DNA damage in T cells as one metric of enhanced sensitivity to chemotherapeutic response [211]. Preclinical work has demonstrated the FMD to be effective against TNBC progression in combination with PI3K/AKT/mTOR inhibitors by potentiating kinase inhibitor response and reducing hyperglycemia, often a treatment-induced side effect [212]. Promising results from a clinical trial in which a majority of participants had breast cancer show that the FMD increases total and activated intratumor CD8 + T cells while decreasing circulating populations of immunosuppressive cells [252]. However, the impact of FMD on long-term patient survival, or its efficacy in patients with breast cancer and obesity, has not been demonstrated.
5.5 Intermittent energy restriction
Intermittent energy restriction (IER) is a broadly-encompassing term that applies to recurring patterns of fasting, either rhythmic or arhythmic, in which calorie restriction is achieved by reduction in overall eating periods rather than meal sizes [253]. The periodicity of IER, as opposed to chronic CR, has greater adherence potential while maintaining comparable reductions in body weight in patients who are overweight or obese [213, 254, 255]. Reported benefits of IER include improved glucose regulation and stress resistance as well as reduced inflammation [256]. IER protects against in vivo tumor development in a spontaneous model of mammary carcinogenesis [257].
5.6 Ketogenic diet
The ketogenic diet, composed of high fat, moderate protein, and very low carbohydrates, acquired its name in pursuit of ketogenesis: utilizing fatty acids, metabolized as the ketone bodies β-hydroxybutyrate and acetone, rather than glucose for energy [217]. The mechanistic basis for a ketogenic diet reducing breast cancer incidence and overall disease burden lies within decreased insulin signaling and overall reduced inflammation seen with prolonged nutritional ketosis [258]. Although some data exists demonstrating the feasibility of a ketogenic diet intervention during radiotherapy treatment for patients with breast cancer, such as boasting reductions in body weight and fat mass [259], there is little evidence that a ketogenic diet reduces tumor burden in patients with breast cancer. After 12 weeks of the ketogenic diet, quality of life and physical activity scores were not improved in a study of 80 patients with breast cancer undergoing chemotherapy [260]. However, in vitro and in vivo supplementation with the ketone body β-hydroxybutyrate increases tumor growth in breast cancer models, presumably by providing fuel for oxidative mitochondrial metabolism [261, 262]. However, β-hydroxybutyrate has no effect on TNBC proliferation or response to treatment via chemotherapy or radiation in vitro [263].
5.7 Mediterranean diet
Growing evidence suggests that the consumption of a Mediterranean dietary pattern has a protective effect against many chronic diseases and cancers including breast cancer [264]. The key characteristics of a Mediterranean diet include high consumption of fruits and vegetables such as green leafy vegetables, legumes, nuts, and cereals; moderate intake of fish and other meats; and low intake of sweets and eggs [219]. The majority of studies examining the protective effect of Mediterranean diet on cancer progression are largely observational studies followed by randomized clinical trials [265, 266]. Overall, high adherence to a Mediterranean dietary pattern is associated with decreased risk of cancer mortality [267] and risk of developing breast cancer [268]. While epidemiological studies suggest an inverse relationship between adherence to a Mediterranean dietary pattern and breast cancer risk, the evidence regarding breast cancer subtype is limited [265, 269]. The results of one systematic review suggest an inverse association between a Mediterranean diet pattern and breast cancer risk in postmenopausal women, and particularly postmenopausal TNBC; however, results are mixed [265]. Olive oil, the main dietary fat in Mediterranean diet, has anticancer effects in experimental studies. For example, a high extra virgin olive oil diet in a rodent model of mammary carcinogenesis increases tumor latency and decreases tumor volume, multiplicity, and incidence [270]. Additionally, olive oil consumption is associated with a lower odds of developing breast cancer [271]. Furthermore, polyunsaturated fatty acids present in a Mediterranean dietary pattern, particularly omega-3 fatty acids and specifically docosahexaenoic acid (DHA) and eicosapentaenoic (EPA), have antiproliferative effects in preclinical models of TNBC [272]. Indeed, supplementation with EPA + DHA ethyl esters reduces mammary tumor growth in obese postmenopausal (ovariectomized) mice [220].
5.8 Dietary considerations
Reducing calorie intake, through fasting or calorie restriction, prior to chemotherapy treatment reduces side effects commonly associated with chemotherapy [273] and may thereby improve quality of life for patients [251, 274, 275]. However, due to heightened risk of adverse effects associated with weight loss and alterations to inflammatory response that can occur with calorie restriction, widespread clinical intervention remains challenging [247]. Additionally, adherence to chronic CR has proven challenging for humans, with sustainability dropping off after ~ 20 weeks for participants who are not obese [276]. A long-term dietary intervention study of participants who were overweight or obese showed adherence drops of 38% for alternate-day fasting and 29% for CR over 1 year [254]. As food intake is neurologically regulated, several mechanisms are in place to drive food consumption during periods of deprivation, causing psychological stress and negatively impacting mood if restriction is too severe or extends for too long [253, 277]. Physiological changes concurrent with obesity, such as increases in orexigenic acetyl-CoA binding protein and disruption of leptin- and ghrelin-mediated appetite signaling cues, provide further barriers for adherence to restrictive diets [278, 279].
One solution to achieve the benefits of dietary restriction without encountering barriers to adherence or disturbing energy balance are calorie restriction mimetics (CRMs). The main objective of CRMs is to induce autophagy, which protects against cellular stress and damage, mobilizes energy reserves, and removes intracellular waste or debris [280]. By optimizing energy and redox metabolism, activating this endogenous mechanism may help cells avoid malignant transformation [281] or improve antitumor immunity through autophagy induction [282].
6 Conclusion
Obesity promotion of the incidence and progression of numerous cancers, including breast cancer, poses a significant public health hazard. Delineation of the mechanisms through which obesity drives cancer progression or immune evasion is critical for the development of interventions to effectively disrupt obesity-driven cancer with minimum toxicity. Dietary interventions remain of considerable interest due to their co-targeting of metabolic disruptions. Breaking the obesity-breast cancer link will require interventions that limit the protumor effects of obesity-associated adipose dysregulation and that consider other biological and genetic mediators and structural determinants of health disparities. Moreover, given high and rising rates of obesity in many parts of the world, emphasis should be placed on the development of safe and effective interventions that are acceptable and accessible to all women to reduce the burden of obesity on breast cancer.
References
Sung, H., et al. (2021). Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 71(3), 209–249.
Eliyatkin, N., et al. (2015). Molecular classification of breast carcinoma: From traditional, old-fashioned way to a new age, and a new way. Journal of Breast Health, 11(2), 59–66.
Harbeck, N., & Gnant, M. (2017). Breast cancer. Lancet, 389(10074), 1134–1150.
Loibl, S., & Gianni, L. (2017). HER2-positive breast cancer. Lancet, 389(10087), 2415–2429.
Marra, A., et al. (2020). Practical classification of triple-negative breast cancer: Intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer, 6(1), 54.
Chia, K., et al. (2015). Targeting the androgen receptor in breast cancer. Current Oncology Reports, 17(2), 4.
Kim, Y., Jae, E., & Yoon, M. (2015). Influence of androgen receptor expression on the survival outcomes in breast cancer: A meta-analysis. Journal of Breast Cancer, 18(2), 134.
Choi, J. E., et al. (2015). Androgen receptor expression predicts decreased survival in early stage triple-negative breast cancer. Annals of Surgical Oncology, 22(1), 82–89.
Li, Z.-H., et al. (2016). Luminal B breast cancer: Patterns of recurrence and clinical outcome. Oncotarget, 7(40), 65024–65033.
Higgins, M. J., & Stearns, V. (2009). Understanding resistance to tamoxifen in hormone receptor-positive breast cancer. Clinical Chemistry, 55(8), 1453–1455.
Arteaga, C. L., et al. (2012). Treatment of HER2-positive breast cancer: Current status and future perspectives. Nature Reviews Clinical Oncology, 9(1), 16–32.
Landry, I., Sumbly, V., & Vest, M. (2022). Advancements in the treatment of triple-negative breast cancer: A narrative review of the literature. Cureus, 14(2), e21970.
Rakha, E. A., Reis-Filho, J. S., & Ellis, I. O. (2008). Basal-like breast cancer: A critical review. Journal of Clinical Oncology, 26(15), 2568–2581.
Perou, C. M. (2010). Molecular stratification of triple-negative breast cancers. The Oncologist, 15(S5), 39–48.
Fougner, C., et al. (2020). Re-definition of claudin-low as a breast cancer phenotype. Nature Communications, 11(1), 1787.
Garrido-Castro, A. C., Lin, N. U., & Polyak, K. (2019). Insights into Molecular Classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discovery, 9(2), 176–198.
Foulkes, W. D., et al. (2003). Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. Journal of the National Cancer Institute, 95(19), 1482–1485.
Denkert, C., et al. (2017). Molecular alterations in triple-negative breast cancer—The road to new treatment strategies. The Lancet, 389(10087), 2430–2442.
Hales, C. M., et al. (2020). Prevalence of obesity and severe obesity among adults: United States, 2017–2018. NCHS Data Brief, 360, 1–8.
Diet, nutrition, physical activity and breast cancer. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective 2018
Bhardwaj, P., et al. (2019). Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. The Journal of Steroid Biochemistry and Molecular Biology, 189, 161–170.
Lohmann, A. E., et al. (2021). Association of obesity with breast cancer outcome in relation to cancer subtypes: A meta-analysis. jnci: Journal of the National Cancer Institute, 113(11), 1465–1475.
Chen, Y., et al. (2017). Body mass index had different effects on premenopausal and postmenopausal breast cancer risks: A dose-response meta-analysis with 3,318,796 subjects from 31 cohort studies. BMC Public Health, 17(1), 936.
White, A. J., et al. (2015). Overall and central adiposity and breast cancer risk in the Sister Study. Cancer, 121(20), 3700–3708.
Neuhouser, M. L., et al. (2015). Overweight, obesity, and postmenopausal invasive breast cancer risk. JAMA Oncology, 1(5), 611.
Modi, N.D., et al. (2021). The obesity paradox in early and advanced HER2 positive breast cancer: Pooled analysis of clinical trial data. NPJ Breast Cancer, 7(1), 30.
Widschwendter, P., et al. (2015). The influence of obesity on survival in early, high-risk breast cancer: Results from the randomized SUCCESS A trial. Breast Cancer Research, 17(1), 129.
Mazzarella, L., et al. (2013). Obesity increases the incidence of distant metastases in oestrogen receptor-negative human epidermal growth factor receptor 2-positive breast cancer patients. European Journal of Cancer, 49(17), 3588–3597.
Shu, X., et al. (2019). Associations of obesity and circulating insulin and glucose with breast cancer risk: A Mendelian randomization analysis. International Journal of Epidemiology, 48(3), 795–806.
Zhao, C., et al. (2021). Current landscape: The Mechanism and therapeutic impact of obesity for breast cancer. Frontiers in Oncology, 11, 704893.
Sabol, R.A., et al. (2019). Leptin produced by obesity-altered adipose stem cells promotes metastasis but not tumorigenesis of triple-negative breast cancer in orthotopic xenograft and patient-derived xenograft models. Breast Cancer Research, 21(1), 67.
Desantis, C. E., et al. (2019). Breast cancer statistics, 2019. CA: A Cancer Journal for Clinicians, 69(6), 438–451.
Barone, I., et al. (2020). The weight of obesity in breast cancer progression and metastasis: Clinical and molecular perspectives. Seminars in Cancer Biology, 60, 274–284.
Barkan, D., & Green, J. E. (2011). An In Vitro System to study tumor dormancy and the switch to metastatic growth. Journal of Visualized Experiments, (54), 2914.
Kaushik, S., Pickup, M. W., & Weaver, V. M. (2016). From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer and Metastasis Reviews, 35(4), 655–667.
Haakinson, D. J., et al. (2012). The Impact of Obesity on Breast Cancer: A Retrospective Review., 19(9), 3012–3018.
Bergom, C., et al. (2016). Association of locoregional control with high body mass index in women undergoing breast conservation therapy for early-stage breast cancer. International Journal of Radiation Oncology Biology Physics, 96(1), 65–71.
Petrelli, F., et al. (2021). Association of obesity with survival outcomes in patients with cancer: A systematic review and meta-analysis. JAMA Network Open, 4(3), e213520.
Ewertz, M., et al. (2011). Effect of obesity on prognosis after early-stage breast cancer. Journal of Clinical Oncology, 29(1), 25–31.
Buono, G., et al. (2020). Metabolic syndrome and early stage breast cancer outcome: Results from a prospective observational study. Breast Cancer Research and Treatment, 182(2), 401–409.
Lee, K., et al., (2019). The impact of obesity on breast cancer diagnosis and treatment. Current Oncology Reports, 21(5), 41.
Bousquenaud, M., Fico, F., Solinas, G. et al. (2018). Obesity promotes the expansion of metastasis-initiating cells in breast cancer. Breast Cancer Research, 20(1), 104.
O’Flanagan, C. H., et al. (2017). Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer, 3, 26.
Evangelista, G. C. M., S. P., Soares, S. M. A., Barros, L. R. C., Xavier, F. H. D. C., Abdo, L. M., Gualberto, A. C. M., Macedo, G. C., Clavijo-Salomon, M. A., & Gameiro, J. (2019). 4T1 Mammary Carcinoma Colonization of metastatic niches is accelerated by obesity. Frontiers Oncology, 9, 685.
Osman, M. A., & Hennessy, B. T. (2015). Obesity Correlation with metastases development and response to first-line metastatic chemotherapy in breast cancer. Clinical Medicine Insights: Oncology, 9, CMO.S32812.
Catalano, S., et al. (2015). A novel leptin antagonist peptide inhibits breast cancer growth in vitro and in vivo. Journal of Cellular and Molecular Medicine, 19(5), 1122–1132.
Naylor, C., & Petri, W. A., Jr. (2016). Leptin regulation of immune responses. Trends in Molecular Medicine, 22(2), 88–98.
Bowers, L. W., et al. (2018). Leptin signaling mediates obesity-associated csc enrichment and EMT in preclinical TNBC models. Molecular Cancer Research, 16(5), 869–879.
Hillers-Ziemer, L. E., et al. (2021). Obesity-activated lung stromal cells promote myeloid lineage cell accumulation and breast cancer metastasis. Cancers, 13(5), 1005.
Lapeire, L., et al. (2015). When fat becomes an ally of the enemy: Adipose tissue as collaborator in human breast cancer. Horm Mol Biol Clin Investig, 23(1), 21–38.
Santander, A., et al. (2015). Paracrine Interactions between adipocytes and tumor cells recruit and modify macrophages to the mammary tumor microenvironment: The role of obesity and inflammation in breast adipose tissue. Cancers, 7(1), 143–178.
Murphy, K. A., et al. (2018). Cutting edge: Elevated Leptin during diet-induced obesity reduces the efficacy of tumor immunotherapy. The Journal of Immunology, 201(7), 1837–1841.
Hopkins, B. D., Goncalves, M. D., & Cantley, L. C. (2016). Obesity and cancer mechanisms: Cancer metabolism. Journal of Clinical Oncology, 34(35), 4277–4283.
Howe, L. R., et al. (2013). Molecular pathways: Adipose inflammation as a mediator of obesity-associated cancer. Clinical Cancer Research, 19(22), 6074–6083.
Kanneganti, T.-D., & Dixit, V. D. (2012). Immunological complications of obesity. Nature Immunology, 13(8), 707–712.
Hosogai, N., et al. (2007). Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes, 56(4), 901–911.
Engin, A. (2017). Adipose tissue hypoxia in obesity and its impact on preadipocytes and macrophages: Hypoxia hypothesis. Advances in Experimental Medicine and Biology, 960, 305–326.
Faria, S. S., et al. (2020). Obesity and breast cancer: The role of crown-like structures in breast adipose tissue in tumor progression, prognosis, and therapy. Journal of Breast Cancer, 23(3), 233.
Divella, R., et al. (2016). Obesity and cancer: The role of adipose tissue and adipo-cytokines-induced chronic inflammation. Journal of Cancer, 7(15), 2346–2359.
Buechler, C., Krautbauer, S., & Eisinger, K. (2015). Adipose tissue fibrosis. World Journal of Diabetes, 6(4), 548.
D’Esposito, V., et al. (2020). Mammary adipose tissue control of breast cancer progression: Impact of obesity and diabetes. Frontiers in Oncology, 10, 1554.
Martin-Padura, I., et al. (2012). The white adipose tissue used in lipotransfer procedures is a rich reservoir of CD34<sup>+</sup> progenitors able to promote cancer progression. Cancer Research, 72(1), 325–334.
Zhang, Y., et al. (2012). Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Research, 72(20), 5198–5208.
Reggiani, F., et al. (2017). Adipose progenitor cell secretion of GM-CSF and MMP9 promotes a stromal and immunological microenvironment that supports breast cancer progression. Cancer Research, 77(18), 5169–5182.
Duong, M. N., et al. (2017). The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget, 8(34), 57622–57641.
Wang, Y. Y., et al. (2017). Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight, 2(4), e87489.
Seo, B. R., et al. (2015). Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Science Translational Medicine, 7(301), 301ra130–301ra1.
Himbert, C., et al. (2017). Signals from the adipose microenvironment and the obesity-cancer link-a systematic review. Cancer Prevention Research (Philadelphia, Pa.), 10(9), 494–506.
D’Esposito, V., et al. (2016). Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget, 7(17), 24495–24509.
Dirat, B., et al. (2011). Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Research, 71(7), 2455–2465.
Bochet, L., et al. (2011). Cancer-associated adipocytes promotes breast tumor radioresistance. Biochemical and Biophysical Research Communications, 411(1), 102–106.
Duong, M. N., et al. (2015). Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Research, 17, 57.
Bochet, L., et al. (2013). Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Research, 73(18), 5657–5668.
Eiro, N., et al. (2015). A phenotype from tumor stroma based on the expression of metalloproteases and their inhibitors, associated with prognosis in breast cancer. Oncoimmunology, 4(7), e992222.
Cid, S., et al. (2016). Expression and clinical significance of metalloproteases and their inhibitors by endothelial cells from invasive breast carcinomas. Clinical Breast Cancer, 16(4), e83–e91.
Sun, K., et al. (2013). Fibrosis and adipose tissue dysfunction. Cell Metabolism, 18(4), 470–477.
Buchsbaum, R. J., & Oh, S. Y. (2016). Breast Cancer-associated fibroblasts: Where we are and where we need to go. Cancers (Basel), 8(2), 19.
Hill, B. S., et al. (2020). Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Seminars in Cancer Biology, 60, 202–213.
Balaban, S., et al. (2017). Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer & Metabolism. 5(1), 1.
Nguyen, M. T., et al. (2007). A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. Journal of Biological Chemistry, 282(48), 35279–35292.
Pallegar, N.K. and S.L. Christian, Adipocytes in the tumour microenvironment, in Advances in Experimental Medicine and Biology. 2020, Springer International Publishing. p. 1–13
Choi, J., Cha, Y. J., & Koo, J. S. (2018). Adipocyte biology in breast cancer: From silent bystander to active facilitator. Progress in Lipid Research, 69, 11–20.
Key, T., et al. (2002). Endogenous sex hormones and breast cancer in postmenopausal women: Reanalysis of nine prospective studies. Journal of the National Cancer Institute, 94(8), 606–616.
Iyengar, N. M., Hudis, C. A., & Dannenberg, A. J. (2013). Obesity and inflammation: New insights into breast cancer development and progression. American Society of Clinical Oncology Educational Book, 33, 46–51.
Baglietto, L., et al. (2009). Circulating steroid hormone concentrations in postmenopausal women in relation to body size and composition. Breast Cancer Research and Treatment, 115(1), 171–179.
Rose, D. P., & Vona-Davis, L. (2014). Biochemical and molecular mechanisms for the association between obesity, chronic inflammation, and breast cancer. BioFactors, 40(1), 1–12.
Zhao, Y., et al. (1996). Tumor necrosis factor-alpha stimulates aromatase gene expression in human adipose stromal cells through use of an activating protein-1 binding site upstream of promoter 1.4. Mol Endocrinol, 10(11), 1350–7.
Purohit, A., et al. (1995). Aromatase activity and interleukin-6 production by normal and malignant breast tissues. Journal of Clinical Endocrinology and Metabolism, 80(10), 3052–3058.
Crudele, L., Piccinin, E., & Moschetta, A. (2021). Visceral adiposity and cancer: Role in pathogenesis and prognosis. Nutrients, 13(6), 2101.
Samuel, S. M., et al. (2018). Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treatment Reviews, 70, 98–111.
Christodoulatos, G. S., et al. (2019). The Role of adipokines in breast cancer: Current Evidence and perspectives. Current Obesity Reports, 8(4), 413–433.
Andò, S., et al. (2019). Obesity, leptin and breast cancer: Epidemiological Evidence and proposed mechanisms. Cancers, 11(1), 62.
Pan, H., et al. (2018). Association between serum leptin levels and breast cancer risk: An updated systematic review and meta-analysis. Medicine (Baltimore), 97(27), e11345.
Gui, Y., et al. (2017). The association between obesity related adipokines and risk of breast cancer: A meta-analysis. Oncotarget, 8(43), 75389–75399.
He, J. Y., et al. (2018). Adipocyte-derived IL-6 and leptin promote breast cancer metastasis via upregulation of lysyl hydroxylase-2 expression. Cell Communication and Signaling: CCS, 16(1), 100.
Tenvooren, I., et al. (2019). Elevated leptin disrupts epithelial polarity and promotes premalignant alterations in the mammary gland. Oncogene, 38(20), 3855–3870.
Mahbouli, S., et al. (2018). Activation of antioxidant defences of human mammary epithelial cells under leptin depend on neoplastic state. BMC Cancer, 18(1), 1264.
Hotamisligil, G. S., Shargill, N. S., & Spiegelman, B. M. (1993). Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science, 259(5091), 87–91.
Hube, F., & Hauner, H. (1999). The role of TNF-alpha in human adipose tissue: Prevention of weight gain at the expense of insulin resistance? Hormone and Metabolic Research, 31(12), 626–631.
Varela, L. M., & Ip, M. M. (1996). Tumor necrosis factor-alpha: A multifunctional regulator of mammary gland development. Endocrinology, 137(11), 4915–4924.
Fasshauer, M. B. (2015). Matthias, Adipokines in health and disease. Trends in Pharmacological Sciences, 36(7), 461–470.
Hotamisligil, G. S., et al. (1995). Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. Journal of Clinical Investigation, 95(5), 2409–2415.
Kirchgessner, T. G., et al. (1997). Tumor necrosis factor-alpha contributes to obesity-related hyperleptinemia by regulating leptin release from adipocytes. The Journal of Clinical Investigation, 100(11), 2777–2782.
Mauro, L., et al. (2015). Cross-talk between adiponectin and IGF-IR in breast cancer. Frontiers in Oncology, 5, 157.
Purohit, A., Newman, S. P., & Reed, M. J. (2002). The role of cytokines in regulating estrogen synthesis: Implications for the etiology of breast cancer. Breast Cancer Research, 4(2), 65–69.
Liang, M., Zhang, P., & Fu, J. (2007). Up-regulation of LOX-1 expression by TNF-alpha promotes trans-endothelial migration of MDA-MB-231 breast cancer cells. Cancer Letters, 258(1), 31–37.
Asiedu, M. K., et al. (2011). TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Research, 71(13), 4707–4719.
Zhang, Z., et al. (2018). Transmembrane TNF-alpha promotes chemoresistance in breast cancer cells. Oncogene, 37(25), 3456–3470.
Lim, S.-O., et al. (2016). Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell, 30(6), 925–939.
Donohoe, C. L., et al. (2016). Emerging concepts linking obesity with the hallmarks of cancer. Trends in Endocrinology and Metabolism: TEM, 28(1), 46-62.
Weisberg, S. P., et al. (2003). Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation, 112(12), 1796–1808.
Murano, I., et al. (2008). Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. Journal of Lipid Research, 49(7), 1562–1568.
Lumeng, C. N., Bodzin, J. L., & Saltiel, A. R. (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of Clinical Investigation, 117(1), 175–184.
Subbaramaiah, K., et al. (2012). Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discovery, 2(4), 356–365.
Morris, P. G., et al. (2011). Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prevention Research (Philadelphia, Pa.), 4(7), 1021–1029.
Engin, A. (2017) Obesity-associated breast cancer: Analysis of risk factors, in obesity and lipotoxicity. advances in experiemntal medicine and biology, C. Springer, Editor. 571–606
Mullooly, M., et al., (2017). Relationship between crown-like structures and sex-steroid hormones in breast adipose tissue and serum among postmenopausal breast cancer patients. Breast Cancer Research, 19(1), 8.
Maliniak, M. L., et al. (2020). Detection of crown-like structures in breast adipose tissue and clinical outcomes among African-American and White women with breast cancer. Breast Cancer Research, 22(1), 65.
Iyengar, N. M., et al. (2016). Systemic Correlates of white adipose tissue inflammation in early-stage breast cancer. Clinical Cancer Research, 22(9), 2283–2289.
Koru-Sengul, T., et al. (2016). Breast cancers from black women exhibit higher numbers of immunosuppressive macrophages with proliferative activity and of crown-like structures associated with lower survival compared to non-black Latinas and Caucasians. Breast Cancer Research and Treatment, 158(1), 113–126.
Yang, H., et al. (2009). Obesity accelerates thymic aging. Blood, 114(18), 3803–3812.
Dixit, V. D. (2010). Thymic fatness and approaches to enhance thymopoietic fitness in aging. Current Opinion in Immunology, 22(4), 521–528.
Coleman, M. F., et al. (2020). Cell intrinsic and systemic metabolism in tumor immunity and immunotherapy. Cancers, 12(4), 852.
Mehta, P., Nuotio-Antar, A. M., & Smith, C. W. (2015). gammadelta T cells promote inflammation and insulin resistance during high fat diet-induced obesity in mice. Journal of Leukocyte Biology, 97(1), 121–134.
Mclaughlin, T., et al. (2014). T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans. Arteriosclerosis, Thrombosis, and Vascular Biology, 34(12), 2637–2643.
Han, J. M., et al. (2014). Insulin inhibits IL-10-mediated regulatory T cell function: Implications for obesity. The Journal of Immunology, 192(2), 623–629.
Wang, Y., et al. (2020). Obesity and metabolic syndrome related macrophage promotes PD-L1 expression in TNBC through IL6/JAK/STAT pathway and can be reversed by telmisartan. Cancer Biology & Therapy, 21(12), 1179–1190.
Franklin, R. A., et al. (2014). The cellular and molecular origin of tumor-associated macrophages. Science, 344(6186), 921–925.
Lewis, C. E., & Pollard, J. W. (2006). Distinct role of macrophages in different tumor microenvironments. Cancer Research, 66(2), 605–612.
Zhang, W. J., et al. (2018). Tumor-associated macrophages correlate with phenomenon of epithelial-mesenchymal transition and contribute to poor prognosis in triple-negative breast cancer patients. Journal of Surgical Research, 222, 93–101.
Cao, H., et al. (2016). Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget, 7(40), 65441–65453.
Tiwari, P., et al. (2019). Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. Journal of Experimental Medicine, 216(6), 1345–1358.
Kratz, M., et al. (2014). Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metabolism, 20(4), 614–625.
Nishimura, S., et al. (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nature Medicine, 15(8), 914–920.
Feuerer, M., et al. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nature Medicine, 15(8), 930–939.
Wang, Z., et al. (2019). Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nature Medicine, 25(1), 141–151.
Reilly, S. M., & Saltiel, A. R. (2017). Adapting to obesity with adipose tissue inflammation. Nature Reviews Endocrinology, 13(11), 633–643.
McQuade, J. L., et al. (2018). Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. The lancet Oncology, 19(3), 310–322.
Wang, Z., Monjazeb, A. M., & Murphy, W. J. (2019). The complicated effects of obesity on cancer and immunotherapy. Immunotherapy, 11(1), 11–14.
Cortellini, A., et al. (2019). A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: When overweight becomes favorable. Journal for ImmunoTherapy of Cancer, 7(1), 57.
Dyck, L., et al. (2022). Suppressive effects of the obese tumor microenvironment on CD8 T cell infiltration and effector function. Journal of Experimental Medicine, 219(3), e20210042.
Boi, S. K., et al. (2020). Obesity diminishes response to PD-1-based immunotherapies in renal cancer. Journal for ImmunoTherapy of Cancer, 8(2), e000725.
Maciolek, J. A., Alex Pasternak, J., & Wilson, H. L. (2014). Metabolism of activated T lymphocytes. Current Opinion in Immunology, 27, 60–74.
Zhang, C., et al. (2020). STAT3 Activation-induced fatty acid oxidation in CD8+ T effector cells is critical for obesity-promoted breast tumor growth. Cell Metabolism, 31(1), 148-161.e5.
Gibson, J. T., et al. (2020). Obesity-associated myeloid-derived suppressor cells promote apoptosis of tumor-infiltrating CD8 T cells and immunotherapy resistance in breast cancer. Frontiers in Immunology, 11, 590794.
Michel, T., et al. (2016). Human CD56bright NK cells: An update. The Journal of Immunology, 196(7), 2923–2931.
Lynch, L. A., et al. (2009). Are natural killer cells protecting the metabolically healthy obese patient? Obesity (Silver Spring), 17(3), 601–605.
Bahr, I., et al. (2018). Impaired natural killer cell subset phenotypes in human obesity. Immunologic Research, 66(2), 234–244.
Laue, T., et al. (2015). Altered NK cell function in obese healthy humans. BMC Obes, 2, 1.
Wrann, C. D., et al. (2012). Short-term and long-term leptin exposure differentially affect human natural killer cell immune functions. American journal of physiology. Endocrinology and metabolism, 302(1), E108–E116.
Ostrand-Rosenberg, S. (2018). Myeloid derived-suppressor cells: Their role in cancer and obesity. Current Opinion in Immunology, 51, 68–75.
Ostrand-Rosenberg, S., & Sinha, P. (2009). Myeloid-derived suppressor cells: Linking inflammation and cancer. The Journal of Immunology, 182(8), 4499–4506.
Xia, S., et al. (2011). Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. Journal of Biological Chemistry, 286(26), 23591–23599.
Clements, V. K., et al. (2018). Frontline science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. Journal of Leukocyte Biology, 103(3), 395–407.
Turbitt, W. J., et al. (2019). Increased adiposity enhances the accumulation of MDSCs in the Tumor microenvironment and adipose tissue of pancreatic tumor-bearing mice and in immune organs of tumor-free hosts. Nutrients, 11(12), 3012.
Al-Khami, A. A., et al. (2017). Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology, 6(10), e1344804.
Iyengar, N. M., et al. (2016). Obesity and cancer mechanisms: Tumor microenvironment and inflammation. Journal of Clinical Oncology, 34(35), 4270–4276.
Hammerl, D., et al. (2020). Clonality, antigen recognition, and suppression of CD8+ T cells differentially affect prognosis of breast cancer subtypes. Clinical Cancer Research, 26(2), 505–517.
Mittendorf, E. A., et al. (2014). PD-L1 expression in triple-negative breast cancer. Cancer Immunology Research, 2(4), 361–370.
Wimberly, H., et al. (2015). PD-L1 Expression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunology Research, 3(4), 326–332.
Luke, J. J., et al. (2018). Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. Journal of Clinical Oncology, 36(16), 1611–1618.
Teo, Z. L., et al. (2017). Combined CDK4/6 and PI3Kα inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Research, 77(22), 6340–6352.
Domchek, S., et al. (2019) Abstract OT3–05–03: MEDIOLA: An open-label, phase I/II basket study of olaparib (PARP inhibitor) and durvalumab (anti-PD-L1 antibody)–Additional breast cancer cohorts. Cancer Research, 79(4_Supplement): p. OT3–05–03-OT3–05–03
Foote, J. B., et al. (2017). A STING agonist given with OX40 receptor and PD-L1 modulators primes immunity and reduces tumor growth in tolerized mice. Cancer Immunology Research, 5(6), 468–479.
Naik, A., A.M. Monjazeb, and J. Decock (2019) The Obesity paradox in cancer, tumor immunology, and immunotherapy: Potential therapeutic implications in triple negative breast cancer. Frontiers in Immunology, 10(1940)
Colleoni, M., et al. (2005). Relation between chemotherapy dose, oestrogen receptor expression, and body-mass index. Lancet, 366(9491), 1108–1110.
Griggs, J. J., Sorbero, M. E., & Lyman, G. H. (2005). Undertreatment of obese women receiving breast cancer chemotherapy. Archives of Internal Medicine, 165(11), 1267–1273.
Carroll, J. P., et al. (2014). Toxicity and tolerability of adjuvant breast cancer chemotherapy in obese women. Medical Oncology, 31(4), 881.
Morrison, V. A., et al. (2018). The impact of actual body weight-based chemotherapy dosing and body size on adverse events and outcome in older patients with breast cancer: Results from Cancer and Leukemia Group B (CALGB) trial 49907 (Alliance A151436). J Geriatr Oncol, 9(3), 228–234.
Cespedes Feliciano, E. M., et al. (2020). Body composition, adherence to anthracycline and taxane-based chemotherapy, and survival after nonmetastatic breast cancer. JAMA Oncology, 6(2), 264.
Lehuede, C., et al. (2019). Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: Role of the major vault protein (MVP). Breast Cancer Research, 21(1), 7.
Mentoor, I., et al. (2020). Decreased efficacy of doxorubicin corresponds with modifications in lipid metabolism markers and fatty acid profiles in breast tumors from obese vs. lean mice. Front Oncol, 10, 306.
Schauer, D. P., et al. (2019). Bariatric Surgery and the risk of cancer in a large multisite cohort. Annals of Surgery, 269(1), 95–101.
Phelan, S. M., et al. (2015). Impact of weight bias and stigma on quality of care and outcomes for patients with obesity. Obesity Reviews, 16(4), 319–326.
Puhl, R. M., Luedicke, J., & Grilo, C. M. (2014). Obesity bias in training: Attitudes, beliefs, and observations among advanced trainees in professional health disciplines. Obesity (Silver Spring), 22(4), 1008–1015.
Pestine, E., Stokes, A., & Trinquart, L. (2018). Representation of obese participants in obesity-related cancer randomized trials. Annals of Oncology, 29(7), 1582–1587.
Williams, D. R., Mohammed, S. A., & Shields, A. E. (2016). Understanding and effectively addressing breast cancer in African American women: Unpacking the social context. Cancer, 122(14), 2138–2149.
DeSantis, C. E., et al. (2017). Breast cancer statistics, 2017, racial disparity in mortality by state. CA: A Cancer Journal for Clinicians, 67(6), 439–448.
Prakash, O., et al. (2020). Racial disparities in triple negative breast cancer: A Review of the role of biologic and non-biologic factors. Frontiers in Public Health, 8, 576964.
Howlader, N., et al. (2018). Differences in breast cancer survival by molecular subtypes in the United States. Cancer Epidemiology, Biomarkers & Prevention, 27(6), 619–626.
Chan, D. S. M., et al. (2014). Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Annals of Oncology, 25(10), 1901–1914.
Allott, E. H., & Hursting, S. D. (2015). Obesity and cancer: Mechanistic insights from transdisciplinary studies. Endocrine-Related Cancer, 22(6), R365–R386.
Agurs-Collins, T., Ross, S. A., & Dunn, B. K. (2019). The Many faces of obesity and its influence on breast cancer risk. Frontiers in Oncology, 9, 765.
Daly, B., & Olopade, O. I. (2015). A perfect storm: How tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA: A Cancer Journal for Clinicians, 65(3), 221–238.
Carey, L. A., et al. (2006). Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA, 295(21), 2492–2502.
Keenan, T., et al. (2015). Comparison of the genomic landscape between primary breast cancer in African American versus White women and the association of racial differences with tumor recurrence. Journal of Clinical Oncology, 33(31), 3621–3627.
Collin, L. J., et al. (2019). Racial Disparities in breast cancer outcomes in the metropolitan atlanta area: New Insights and approaches for health equity. JNCI Cancer Spectr, 3(3), pkz053.
Saini, G., et al. (2019). Disadvantaged neighborhoods and racial disparity in breast cancer outcomes: The biological link. Cancer Causes and Control, 30(7), 677–686.
Do, W. L., et al. (2020). Obesity-associated methylation in breast tumors: A possible link to disparate outcomes? Breast Cancer Research and Treatment, 181(1), 135–144.
Maliniak, M. L., et al. (2021). Crown-like structures in breast adipose tissue: Early Evidence and current issues in breast cancer. Cancers (Basel), 13(9), 2222.
Hossain, F., et al., (2021). Obesity Modulates the gut microbiome in triple-negative breast cancer. Nutrients, 13(10), 3656.
Fridman, W. H., et al. (2012). The immune contexture in human tumours: Impact on clinical outcome. Nature Reviews Cancer, 12(4), 298–306.
Ross, K. H., et al. (2019). Obesity and cancer treatment efficacy: Existing challenges and opportunities. Cancer, 125(10), 1588–1592.
Collin, L. J., et al. (2021). Neighborhood-level redlining and lending bias are associated with breast cancer mortality in a large and diverse metropolitan area. Cancer Epidemiology, Biomarkers & Prevention, 30(1), 53–60.
Sun, H., et al., (2017). Triple‑negative breast cancer and its association with obesity. Molecular and Clinical Oncology, 7(6), 935–942.
Pajares, B., et al. (2013). Obesity and survival in operable breast cancer patients treated with adjuvant anthracyclines and taxanes according to pathological subtypes: A pooled analysis. Breast Cancer Research, 15(6), R105.
Cecchini, R. S., et al. (2016). Body mass index at diagnosis and breast cancer survival prognosis in clinical trial populations from NRG oncology/NSABP B-30, B-31, B-34, and B-38. Cancer Epidemiology, Biomarkers & Prevention, 25(1), 51–59.
Sestak, I., et al. (2010). Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: An exploratory analysis from the ATAC trial. Journal of Clinical Oncology, 28(21), 3411–3415.
Ewertz, M., et al. (2012). Obesity and risk of recurrence or death after adjuvant endocrine therapy with letrozole or tamoxifen in the breast international group 1–98 trial. Journal of Clinical Oncology, 30(32), 3967–3975.
Biganzoli, E., et al. (2017). Recurrence dynamics of breast cancer according to baseline body mass index. European Journal of Cancer, 87, 10–20.
Ahmed, R. L., et al. (2011). Risk factors for lymphedema in breast cancer survivors, the Iowa Women’s Health Study. Breast Cancer Research and Treatment, 130(3), 981–991.
Fischer, J. P., et al. (2013). Breast reconstruction in the morbidly obese patient: Assessment of 30-day complications using the 2005 to 2010 National Surgical Quality Improvement Program data sets. Plastic and Reconstructive Surgery, 132(4), 750–761.
Nencioni, A., et al. (2018). Fasting and cancer: Molecular mechanisms and clinical application. Nature Reviews Cancer, 18(11), 707–719.
Kanarek, N., Petrova, B., & Sabatini, D. M. (2020). Dietary modifications for enhanced cancer therapy. Nature, 579(7800), 507–517.
Rebrin, I., Forster, M. J., & Sohal, R. S. (2011). Association between life-span extension by caloric restriction and thiol redox state in two different strains of mice. Free Radical Biology & Medicine, 51(1), 225–233.
Mercken, E. M., et al. (2012). Of mice and men: The benefits of caloric restriction, exercise, and mimetics. Ageing Research Reviews, 11(3), 390–398.
Simone, B. A., et al. (2016). Caloric restriction coupled with radiation decreases metastatic burden in triple negative breast cancer. Cell Cycle, 15(17), 2265–2274.
Eriau, E., et al. (2021). Metabolic reprogramming by reduced calorie intake or pharmacological caloric restriction mimetics for improved cancer immunotherapy. Cancers (Basel), 13(6), 1260.
Lee, C., et al. (2012). Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Science Translational Medicine, 4(124), 124ra27-124ra27.
Di Biase, S., et al. (2016). Fasting-Mimicking diet reduces HO-1 to promote t cell-mediated tumor cytotoxicity. Cancer Cell, 30(1), 136–146.
de Groot, S., et al. (2020). Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nature Communications, 11(1), 3083.
Salvadori, G., et al. (2021). Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metabolism, 33(11), 2247-2259 e6.
Harvie, M. N., & Howell, T. (2016). Could intermittent energy restriction and intermittent fasting reduce rates of cancer in obese, overweight, and normal-weight subjects? A summary of evidence. Advances in Nutrition, 7(4), 690–705.
Cleary, M. P., et al. (2007). Prevention of mammary tumorigenesis by intermittent caloric restriction: Does caloric intake during refeeding modulate the response? Experimental Biology and Medicine (Maywood, N.J.), 232(1), 70–80.
Chung, H., et al. (2016). Time-restricted feeding improves insulin resistance and hepatic steatosis in a mouse model of postmenopausal obesity. Metabolism, 65(12), 1743–1754.
Peeke, P. M., et al. (2021). Effect of time restricted eating on body weight and fasting glucose in participants with obesity: Results of a randomized, controlled, virtual clinical trial. Nutrition & Diabetes, 11(1), 6.
Masood, W., P. Annamaraju, and K.R. Uppaluri, Ketogenic diet, in StatPearls. 2022: Treasure Island (FL)
Klement, R. J., et al. (2016). Anti-tumor effects of ketogenic diets in mice: A meta-analysis. PLoS ONE, 11(5), e0155050.
Davis, C., et al. (2015). Definition of the Mediterranean diet; a literature review. Nutrients, 7(11), 9139–9153.
Ford, N. A., et al. (2015). Omega-3-acid ethyl esters block the protumorigenic effects of obesity in mouse models of postmenopausal basal-like and claudin-low breast cancer. Cancer Prevention Research, 8(9), 796–806.
Pak, H. H., et al. (2021). Fasting drives the metabolic, molecular and geroprotective effects of a calorie-restricted diet in mice. Nature Metabolism, 3(10), 1327–1341.
Bales, C. W., & Kraus, W. E. (2013). Caloric restriction: Implications for human cardiometabolic health. Journal of Cardiopulmonary Rehabilitation and Prevention, 33(4), 201–208.
Campbell, K. L., et al. (2012). Reduced-calorie dietary weight loss, exercise, and sex hormones in postmenopausal women: Randomized controlled trial. Journal of Clinical Oncology, 30(19), 2314–2326.
Imayama, I., et al. (2012). Effects of a caloric restriction weight loss diet and exercise on inflammatory biomarkers in overweight/obese postmenopausal women: A randomized controlled trial. Cancer Research, 72(9), 2314–2326.
Fabian, C. J., et al. (2013). Favorable modulation of benign breast tissue and serum risk biomarkers is associated with > 10 % weight loss in postmenopausal women. Breast Cancer Research and Treatment, 142(1), 119–132.
Lv, M., et al. (2014). Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: A systematic review and meta-analysis. PLoS ONE, 9(12), e115147.
Mattison, J. A., et al. (2017). Caloric restriction improves health and survival of rhesus monkeys. Nature Communications, 8, 14063.
Saleh, A. D., et al. (2013). Caloric restriction augments radiation efficacy in breast cancer. Cell Cycle, 12(12), 1955–1963.
De Lorenzo, M. S., et al. (2011). Caloric restriction reduces growth of mammary tumors and metastases. Carcinogenesis, 32(9), 1381–1387.
Dunlap, S. M., et al. (2012). Dietary energy balance modulates epithelial-to-mesenchymal transition and tumor progression in murine claudin-low and basal-like mammary tumor models. Cancer Prevention Research (Philadelphia, Pa.), 5(7), 930–942.
Simone, B. A., et al. (2018). Caloric restriction counteracts chemotherapy-induced inflammation and increases response to therapy in a triple negative breast cancer model. Cell Cycle, 17(13), 1536–1544.
Caffa, I., et al. (2020). Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature, 583(7817), 620–624.
Champ, C. E., & Klement, R. J. (2020). Assessing successful completion of calorie restriction studies for the prevention and treatment of cancer. Nutrition, 78, 110829.
Most, J., et al. (2017). Calorie restriction in humans: An update. Ageing Research Reviews, 39, 36–45.
Brandhorst, S., & Longo, V. D. (2016). Fasting and caloric restriction in cancer prevention and treatment. Recent Results in Cancer Research, 207, 241–266.
De Groot, S., et al. (2019) Effects of short-term fasting on cancer treatment. Journal of Experimental & Clinical Cancer Research, 38(1), 209.
Alidadi, M., et al. (2021). The effect of caloric restriction and fasting on cancer. Seminars in Cancer Biology, 73, 30–44.
Hatori, M., et al. (2012). Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metabolism, 15(6), 848–860.
Eckel-Mahan, L. K., et al. (2013). Reprogramming of the Circadian Clock by Nutritional Challenge. Cell, 155(7), 1464–1478.
Zeb, F., et al. (2021). Time-restricted feeding regulates molecular mechanisms with involvement of circadian rhythm to prevent metabolic diseases. Nutrition, 89, 111244.
Patterson, R. E., & Sears, D. D. (2017). Metabolic effects of intermittent fasting. Annual Review of Nutrition, 37(1), 371–393.
Hadadi, E., et al. (2020). Chronic circadian disruption modulates breast cancer stemness and immune microenvironment to drive metastasis in mice. Nature Communications, 11(1), 3193.
Lee, Y. (2021). Roles of circadian clocks in cancer pathogenesis and treatment. Experimental & Molecular Medicine, 53(10), 1529–1538.
Sundaram, S., & Yan, L. (2018). Time-restricted feeding mitigates high-fat diet-enhanced mammary tumorigenesis in MMTV-PyMT mice. Nutrition Research, 59, 72–79.
Raffaghello, L., et al. (2008). Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A, 105(24), 8215–8220.
Longo, V. D., & Mattson, M. P. (2014). Fasting: Molecular mechanisms and clinical applications. Cell Metabolism, 19(2), 181–192.
Ibrahim, E. M., Al-Foheidi, M. H., & Al-Mansour, M. M. (2021). Energy and caloric restriction, and fasting and cancer: A narrative review. Supportive Care in Cancer, 29(5), 2299–2304.
de Groot, S., et al. (2015). The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: A randomized pilot study. BMC Cancer, 15, 652.
Icard, P., et al. (2020). Perspective: Do fasting, caloric restriction, and diets increase sensitivity to radiotherapy? A Literature Review. Advances in Nutrition, 11(5), 1089–1101.
Brandhorst, S., et al. (2015). A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metabolism, 22(1), 86–99.
Bauersfeld, S. P., et al. (2018). The effects of short-term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: A randomized cross-over pilot study. BMC Cancer, 18(1), 476.
Vernieri, C., et al. (2022). Fasting-mimicking diet is safe and reshapes metabolism and antitumor immunity in patients with cancer. Cancer Discovery, 12(1), 90–107.
Hofer, S. J., et al. (2022). The ups and downs of caloric restriction and fasting: From molecular effects to clinical application. EMBO Molecular Medicine, 14(1), e14418.
Trepanowski, J. F., et al. (2017). Effect of alternate-day fasting on weight loss, weight maintenance, and cardioprotection among metabolically healthy obese adults. JAMA Internal Medicine, 177(7), 930.
Harvie, M., & Howell, A. (2017). Potential benefits and harms of intermittent energy restriction and intermittent fasting amongst obese, overweight and normal weight subjects—A narrative review of human and animal evidence. Behavioral Sciences, 7(4), 4.
De Cabo, R., & Mattson, M. P. (2019). Effects of intermittent fasting on health, aging, and disease. New England Journal of Medicine, 381(26), 2541–2551.
Rogozina, O. P., et al. (2013). The protective effect of intermittent calorie restriction on mammary tumorigenesis is not compromised by consumption of a high fat diet during refeeding. Breast Cancer Research and Treatment, 138(2), 395–406.
Hyde, P. N. L., Maryam, B., Miller, Vincent J., Lafountain, R. A., & Volek, Jeff S. (2017). Pleiotropic effects of nutritional ketosis: Conceptual framework for keto-adaptation as a breast cancer therapy. Cancer Treatment and Research Communications, 12, 32–39.
Klement, R. J., et al. (2020). Impact of a ketogenic diet intervention during radiotherapy on body composition: III—final results of the KETOCOMP study for breast cancer patients. Breast Cancer Research, 22(1), 94.
Khodabakhshi, A., et al. (2020). Does a ketogenic diet have beneficial effects on quality of life, physical activity or biomarkers in patients with breast cancer: A randomized controlled clinical trial. Nutrition Journal, 19(1), 87.
Rodrigues, L. M., et al. (2017). The action of β-hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: evidence of a β-hydroxybutyrate paradox. Cancer & Metabolism, 5(1), 4.
Bonuccelli, G., et al. (2010). Ketones and lactate “fuel” tumor growth and metastasis. Cell Cycle, 9(17), 3506–3514.
Bartmann, C., et al. (2018). Beta-hydroxybutyrate (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer & Metabolism, 6(1), 8.
Mentella, M. C., et al. (2019). Cancer and Mediterranean diet: A review. Nutrients, 11(9), 2059.
Coughlin, S. S., Stewart, J., & Williams, L. B. (2018). A review of adherence to the Mediterranean diet and breast cancer risk according to estrogen- and progesterone-receptor status and HER2 oncogene expression. Annals of Epidemiology and Public Health, 1, 1002.
Toledo, E., et al. (2015). Mediterranean Diet and invasive breast cancer risk among women at high cardiovascular risk in the PREDIMED Trial: A randomized clinical trial. JAMA Internal Medicine, 175(11), 1752–1760.
Schwingshackl, L., et al. (2017). Adherence to Mediterranean Diet and risk of cancer: An updated systematic review and meta-analysis. Nutrients, 9(10), 1063.
Schwingshackl, L., & Hoffmann, G. (2015). Adherence to Mediterranean diet and risk of cancer: An updated systematic review and meta-analysis of observational studies. Cancer Medicine, 4(12), 1933–1947.
Morze, J., et al. (2021). An updated systematic review and meta-analysis on adherence to mediterranean diet and risk of cancer. European Journal of Nutrition, 60(3), 1561–1586.
Escrich, E., Moral, R., & Solanas, M. (2011). Olive oil, an essential component of the Mediterranean diet, and breast cancer. Public Health Nutrition, 14(12A), 2323–2332.
Psaltopoulou, T., et al. (2011). Olive oil intake is inversely related to cancer prevalence: A systematic review and a meta-analysis of 13,800 patients and 23,340 controls in 19 observational studies. Lipids in Health and Disease, 10, 127.
Donovan, M. G., et al. (2020). Do olive and fish oils of the Mediterranean Diet have a role in triple negative breast cancer prevention and therapy? An exploration of evidence in cells and animal models. Frontiers in Nutrition, 7, 571455.
Safdie, F. M., et al. (2009). Fasting and cancer treatment in humans: A case series report. Aging (Albany NY), 1(12), 988–1007.
Saxton, J. M., et al. (2014). Effects of an exercise and hypocaloric healthy eating intervention on indices of psychological health status, hypothalamic-pituitary-adrenal axis regulation and immune function after early-stage breast cancer: A randomised controlled trial. Breast Cancer Research, 16(2), R39.
Scott, E., et al. (2013). Effects of an exercise and hypocaloric healthy eating program on biomarkers associated with long-term prognosis after early-stage breast cancer: A randomized controlled trial. Cancer Causes and Control, 24(1), 181–191.
Dorling, J. L., et al. (2020). Changes in body weight, adherence, and appetite during 2 years of calorie restriction: The CALERIE 2 randomized clinical trial. European Journal of Clinical Nutrition, 74(8), 1210–1220.
Anastasiou, C. A., Karfopoulou, E., & Yannakoulia, M. (2015). Weight regaining: From statistics and behaviors to physiology and metabolism. Metabolism, 64(11), 1395–1407.
Cui, H., López, M., & Rahmouni, K. (2017). The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nature Reviews Endocrinology, 13(6), 338–351.
Bravo-San Pedro, J. M., et al. (2019). Acyl-CoA-binding protein is a lipogenic factor that triggers food intake and obesity. Cell Metabolism, 30(4), 754-767.e9.
Madeo, F., et al. (2014). Caloric restriction mimetics: Towards a molecular definition. Nature Reviews Drug Discovery, 13(10), 727–740.
Rybstein, M. D., et al. (2018). The autophagic network and cancer. Nature Cell Biology, 20(3), 243–251.
Pietrocola, F., et al. (2016). Caloric Restriction mimetics enhance anticancer immunosurveillance. Cancer Cell, 30(1), 147–160.
Funding
This work was supported by R35CA197627 and BCRF-21–073 to SDH and T32CA057726 to MSC.
Author information
Authors and Affiliations
Contributions
Conceptualization: END, MSC, LEM, MFC, SDH; writing original draft: END, MSC; writing review and editing: END, MSC, LEM, MFC, SDH; funding acquisition: MSC, SDH: supervision: SDH.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Devericks, E.N., Carson, M.S., McCullough, L.E. et al. The obesity-breast cancer link: a multidisciplinary perspective. Cancer Metastasis Rev 41, 607–625 (2022). https://doi.org/10.1007/s10555-022-10043-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-022-10043-5