
Abstract
Despite the progress made in risk stratification, sudden cardiac death and heart failure remain dreaded complications for hypertrophic cardiomyopathy (HCM) patients. Myocardial ischaemia is widely acknowledged as a contributor to cardiovascular events, but the assessment of ischaemia is not yet included in HCM clinical guidelines. This review aims to evaluate the HCM-specific pro-ischaemic mechanisms and the potential prognostic value of imaging for myocardial ischaemia in HCM. A literature review was performed using PubMed to identify studies with non-invasive imaging of ischaemia (cardiovascular magnetic resonance, echocardiography, and nuclear imaging) in HCM, prioritising studies published after the last major review in 2009. Other studies, including invasive ischaemia assessment and post-mortem histology, were also considered for mechanistic or prognostic relevance. Pro-ischaemic mechanisms in HCM reviewed included the effects of sarcomeric mutations, microvascular remodelling, hypertrophy, extravascular compressive forces and left ventricular outflow tract obstruction. The relationship between ischaemia and fibrosis was re-appraised by considering segment-wise analyses in multimodal imaging studies. The prognostic significance of myocardial ischaemia in HCM was evaluated using longitudinal studies with composite endpoints, and reports of ischaemia-arrhythmia associations were further considered. The high prevalence of ischaemia in HCM is explained by several micro- and macrostructural pathological features, alongside mutation-associated energetic impairment. Ischaemia on imaging identifies a subgroup of HCM patients at higher risk of adverse cardiovascular outcomes. Ischaemic HCM phenotypes are a high-risk subgroup associated with more advanced left ventricular remodelling, but further studies are required to evaluate the independent prognostic value of non-invasive imaging for ischaemia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease (1:200–1:500 [1]), a leading cause of sudden cardiac death (SCD) in the young, and a common cause of heart failure and atrial fibrillation in adults [2]. The clinical course of HCM is complicated by multiple factors that can interact and exacerbate the phenotype, including left ventricular outflow tract (LVOT) obstruction, mitral regurgitation, diastolic dysfunction, arrhythmias, autonomic dysfunction and myocardial ischaemia [2]. Myocardial ischaemia has been identified as an area of investigative importance [3], because of its association with adverse left ventricular (LV) remodelling and poor clinical outcomes in early studies of HCM [4] and other cardiovascular diseases.
Despite ischaemia being considered a significant contributor to the natural history of HCM [3], recommendations to assess ischaemic burden are absent from clinical guidelines, and HCM-specific strategies to mitigate ischaemia remain limited. This is in part because the treatment of ischaemia in HCM is complicated by multiple pathophysiological mechanisms, with many patients demonstrating evidence of myocardial infarction in the absence of epicardial coronary stenoses [5,6,7,8], such that multiple other pro-ischaemic mechanisms must be considered alongside therapeutic strategies other than revascularisation [6]. Despite their importance, the HCM-specific pro-ischaemic mechanisms are not yet fully understood.
Furthermore, as the major cause of SCDs in the general population, the assessment of ischaemia in HCM may address some limitations of SCD risk stratification, which has suboptimal sensitivity [9]. This may be particularly true in young HCM patients [10], who carry significant SCD burden, and in whom acute myocardial infarction is possible [11], because a structural substrate for lethal arrhythmias is frequently absent in juvenile SCDs [12]. Although ischaemia is hypothesised to contribute to SCD events, the prognostic value of imaging for ischaemia in HCM is not yet well established.
This review therefore aims to (1) reflect on the prevalence of ischaemia in HCM and its multifactorial causes, to establish potential therapeutic targets for the treatment of ischaemia without epicardial coronary stenoses; and (2) assess the prognostic impact of imaging for ischaemia in HCM in relation to other markers of disease severity (hypertrophy and fibrosis), to evaluate the role of ischaemia on imaging as a potentially novel SCD risk factor.
To address these aims, the present study first reviews the frequency of myocardial ischaemia in HCM on non-invasive assessment, with a focus on modern advancements made in cardiovascular magnetic resonance (CMR) imaging. The pathophysiological mechanisms underlying the development of ischaemia in HCM are then explored, and how these may contribute to adverse LV remodelling and symptomatic status. The role of imaging for myocardial ischaemia in HCM as a risk marker for adverse outcomes including arrhythmias and heart failure is further considered, through review of follow-up studies. Finally, the role of myocardial ischaemia is discussed as a therapeutic target, through reviewing the latest clinical trials targeting metabolic and vascular dysfunction in HCM. We expect these findings to contribute to the scientific understanding and clinical management of myocardial ischaemia in this high-risk group of patients.
Imaging of myocardial ischaemia in HCM
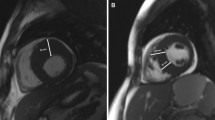
Perfusion measurements are commonly used as a surrogate of ischaemia, as integrated measures of flow through both the epicardial coronary arteries and the microcirculation. Regional perfusion defects are characteristic of the HCM phenotype and present as regions of impaired myocardial blood flow (MBF) at rest or during exercise/pharmacologically induced hyperaemia (Fig. 1). Perfusion impairment is commonly inferred from a reduced ratio of hyperaemic MBF to rest MBF, termed myocardial perfusion reserve, and is generally considered an acceptable surrogate for ischaemia [3].

Stress perfusion defects in HCM on perfusion CMR. (A, D) Basal and (B, E) adenosine-stress MBF on perfusion CMR in the (A, B) base and (D, E) mid slices of a 40-year-old woman with sarcomere mutation positive HCM, showing stress perfusion impairment (denoted with arrows) in the maximally hypertrophied anterior wall and septum. (C, F) Late gadolinium enhancement in the same patient for the base and mid slices, respectively, showing dense focal enhancement in the hypertrophied anteroseptum
Table 1 summarises the non-invasive imaging studies that have assessed the presence of perfusion defects in HCM patients, through CMR, echocardiography and nuclear imaging. However, not all imaging modalities are equally accurate. Perfusion defects on Th-201 scintigraphy, for example, correlate poorly with acidosis [13].
Since the last review of imaging techniques for myocardial ischaemia in HCM in 2009 [3], ischaemia assessment with CMR has been widely adopted. Perfusion CMR boasts MBF quantification at high resolution, without radiation exposure, and has proven high sensitivity for coronary artery disease diagnosis in the general population [45]. The capability of CMR to measure multiple modalities further enables the assessment of ischaemic and fibrotic burden in quick succession (see later Table 2). Moreover, as ischaemia results from an imbalance between oxygen supply and demand, modalities such as blood oxygen level dependent (BOLD) CMR that is sensitive to myocardial oxygenation can provide further insight to oxygen supply and demand in HCM [46].
Across the perfusion imaging studies in Table 1, ischaemia is frequently identified in HCM patients, with the largest perfusion CMR study to date identifying inducible perfusion defects in 84% of their cohort [42]. Because perfusion defects were typically identified on visual assessment, often the severity of impairment was omitted. However, quantitative assessments have identified a subset of 21–31% of patients in whom regional perfusion can fall during vasodilator stress [31, 38], a finding suggestive of severe microvascular dysfunction.
Pathophysiological mechanisms of ischaemia in HCM
With perfusion defects often remote from coronary territories [37], and in young patients likely without coronary stenoses [10], myocardial ischaemia in HCM is multifactorial in origin. Numerous post-mortem, biopsy and imaging studies have investigated the micro-, macrostructural and metabolic mechanisms underlying ischaemia in HCM.
Microstructural abnormalities in HCM
Small vessel disease
Structural abnormalities in the small blood vessels that supply the myocardium are a common finding in the histology of HCM hearts [47, 48]. As many as 56–83% of HCM patients have small vessel disease to some degree [49,50,51,52]. The abnormalities typically include marked thickening of the vessel walls, luminal narrowing of small intramural coronary arteries [53] and increased arterial stiffness [54]. The luminal area of arterioles, as a percentage of total vascular area, has been measured to be 13–30% lower in HCM patients than in controls [55,56,57], with 14% of small vessels in one study having an external diameter:lumen ratio ≥ 3 (normally < 2.5) [58]. Other studies found small vessel disease in 92% of myocardial specimens taken from 57 HCM patients [59], and that the HCM myocardium had 30 × more abnormal intramural coronary arteries per section on average than controls [51]. The degree of small vessel disease may be particularly severe in patients with heart failure [12].
Reduced density of small vessels
Numerous studies have measured the density of small vessels in hypertrophied HCM septal/LV tissue samples and have found this to be 21–44% lower than in control patients [54, 56, 57, 60,61,62,63]. With no convincing association with genotype [61], reductions in small vessel density may be linked to hypertrophy in HCM and are more severe in end-stage disease patients, such as those undergoing heart transplantation, than in patients referred for myectomy [62]. The reported association between reduced small vessel density and perfusion reserve blunting suggests that this is another cause of ischaemia [57].
Macrostructural forces in HCM
Left ventricular outflow tract obstruction
LVOT obstruction is a hallmark feature of HCM and is defined as a peak LVOT pressure gradient ≥ 30 mmHg. LVOT obstruction may be present at rest (in up to 51% of HCM patients [41, 64,65,66]) or develop during exercise (in 33–62% [65, 66]). Severe obstruction (≥ 50 mmHg) may be seen in up to 20% of patients [35]. Even in the absence of hypertrophy, LVOT obstruction may be present due to systolic motion of the anterior mitral valve leaflet towards the LVOT [67]. This typically results from lengthening of the anterior mitral valve leaflet, abnormal chordal-mitral valve attachment or bifid papillary muscle hypermobility [67].
LVOT obstruction is associated with reduced perfusion reserve [25, 35, 41, 68] particularly in the left anterior descending artery which supplies the anterior wall and septum [68, 69], reduced hyperaemic MBF [70], reduced endo-epicardial hyperaemic MBF ratio [71], reduced perfusion upslope (a measure of contrast agent wash-in time) [72] and an additional hemodynamic forward deceleration wave in systole [73]. These associations may be explained by coronary hypoperfusion and increased oxygen demand, as greater myocardial work is required to overcome the obstruction.
Despite these reported associations, other studies have found no relationship between LVOT obstruction and ischaemia, and infarction has been reported in non-obstructive HCM [11], confirming the role of other mechanisms including microvascular dysfunction [4, 10, 27, 32, 44].
Effects during diastole and systole
Diastolic dysfunction, partially related to sarcomeric mutations, is an early feature of HCM in many patients [74, 75]. With myocardial blood flow greatest during diastole, constrained diastole results in impaired perfusion [76]. In a study by Raphael et al., invasive measurements of coronary pressure and flow showed that diastolic dysfunction in HCM led to impaired decompression of the microcirculation [73], and these effects could be exacerbated by exercise [77].
During systole, hypertrophy causes excessive compression of intramyocardial blood vessels, leading to abnormal coronary haemodynamic forces [73], which can manifest as systolic flow reversal in septal perforator arteries of HCM patients [69, 78,79,80] even in the absence of LVOT obstruction [73].
Myocardial bridging
Another potential mechanism of perfusion impairment among HCM patients is myocardial bridging, which is when a segment of a coronary artery tunnels through the myocardium rather than over it. This is seen in 23–41% of HCM patients compared to 5–7% of the general population [81, 82]. Multiple case reports highlight bridging as a possible cause of ischaemia [83,84,85,86], however surgical correction of myocardial bridging remains controversial [87, 88].
Genetic and metabolic factors
In multiple studies of genotyped HCM patients, vasodilator stress BOLD CMR has detected impaired myocardial oxygenation even in pre-hypertrophic carriers of sarcomeric mutations, despite preserved perfusion [46, 89, 90]. One possible explanation for this dissociation in oxygenation and perfusion often observed in the early phase of the disease is that myocardial oxygen demand may be increased even in the absence of hypertrophy [91,92,93,94]. In line with this, experimental non-hypertrophic murine models of sarcomeric mutations have demonstrated increased oxygen expenditure arising from energetic inefficiency associated with sarcomeric mutations [95]. In HCM patients with overt hypertrophy, the degree of stress oxygenation may be as severe as that seen in severe aortic stenosis [90, 96].
Relationships between markers of disease severity and ischaemia in HCM
Myocardial hypertrophy and ischaemia
The greater the degree of hypertrophy (quantified as wall thickness or LV mass), the greater the degree of perfusion impairment (typically quantified as rest or hyperaemic MBF, perfusion reserve, flow velocity, or as the presence of visual defects). This has been reported both at a segmental level [26, 29, 31, 33, 34, 38, 41, 46, 48, 70, 72, 97,98,99,100,101] and in HCM patients on average [7, 15, 22, 27, 40, 44, 68, 70, 71, 78, 79, 102]. In addition to reductions in capillary density [61], hypertrophy was frequently associated with enhanced luminal narrowing of small vessels [50, 55, 56, 58].
A further macroscopic explanation is that the increase in muscle mass characteristic of HCM is inadequately supplied by the major coronary arteries, which have reduced luminal volume per unit myocardial mass [103,104,105]. Heterogeneous flow among major coronary arteries secondary to variable regional demand is a further consequence of hypertrophy [48, 68, 69, 105].
Although perfusion defects are overall more prevalent in cohorts with hypertrophy (Table 1), 20% of HCM mutation carriers without hypertrophy may still have perfusion defects [43], and even in hypertrophic cohorts, 30–40% of patients may have perfusion defects in segments with only mild hypertrophy [15, 106]. When compared to controls, even non-hypertrophied segments in HCM have reduced perfusion reserve on average [38].
Segmental and transmural distribution of myocardial ischaemia
Two studies reported perfusion defects [30] and exercise wall motion abnormalities (WMAs) [32] to be most frequently septal, which is consistent with the septum and anterior LV wall being the most frequently hypertrophied regions in HCM [64]. In another cohort, the septum and inferior segments were most affected by perfusion defects [101]. Perfusion defects have also been reported as primarily located in the septum in prehypertrophic HCM mutation carriers [43], which may be explained by small vessel disease reported as mostly affecting the septum in histological analyses [51, 59]. However, Villa et al. reported a more diffuse burden of hypoperfusion in a cohort without severe hypertrophy [36], consistent with the widespread distribution of small vessel disease in a different histological analysis by Varnava et al. [58].
There are also conflicting reports on the segmental distribution of perfusion defects in SPECT imaging studies [22, 23, 80, 107], which may be due to partial volume effects, given the wall thickness-dependent sensitivity of SPECT imaging. Indeed, PET studies, which have similar problems with partial volume effects due to its low resolution, sometimes report similar impairment in MBF between the septum and LV free wall [4, 70, 108], with two exceptions [28, 109].
Perfusion impairment in HCM is predominantly subendocardial [29, 31, 38, 42, 43, 70, 71, 97, 101, 106, 108], although transmural hypoperfusion has also been reported [38, 98].
Myocardial fibrosis and ischaemia
Repeated episodes of ischaemia have been implicated in fibrosis accumulation and extensive scarring in HCM [47, 53]. A longitudinal study of HCM patients with combined late gadolinium enhancement (LGE) to assess fibrosis burden and stress perfusion imaging found that patients with impaired perfusion reserve had a greater increase in LGE mass over time [39]. Indeed, multiple histopathological studies have shown that the presence of diseased small intramural coronary arteries and reductions in microvascular density are topographically correlated with the presence of fibrosis [12, 47, 51, 52, 59, 110], with one exception [58]. This was similarly reported among patients [55], with Kwon et al. reporting that the presence of small vessel disease is independently associated with 14 × increased risk of myocardial scarring [50]. The association with replacement fibrosis appears primarily in end-stage HCM [59].
However, whereas ischaemia is predominantly subendocardial in HCM [29, 31, 38, 42, 43, 70, 71, 97, 106, 108], fibrosis is predominantly mid-wall in HCM [33, 53, 99, 111,112,113,114,115,116]. This transmural dissociation has been reported directly on imaging [29, 33]. Segment-wise associations between fibrosis and ischaemia have also been reported on imaging in HCM (Table 2), however it remains to be seen whether this is due to the confounding effect of disease severity reflected by wall thickness [31], or due to difficulties in discerning regions of ischaemia and fibrosis [36].
Imaging studies that did not perform epi-endocardial segmentation, but controlled for wall thickness, found independent associations between perfusion impairment and either LGE or extracellular volume [33, 41, 99, 101]. One study with epi-endocardial segmentation and control for wall thickness reported an independent association between LGE and hyperaemic MBF [38], but two smaller similar studies found that either this association was lost after controlling for wall thickness [31], or the differences in perfusion with/without LGE were modest [106]. Patient-wise analyses typically found ischaemia-fibrosis associations [10, 28, 29, 32, 36, 42, 102, 117, 118], with reported exceptions [40, 73].
Collectively, these findings suggest colocalization of pathology (microvascular dysfunction and fibrosis) and the potential for ischaemia to promote the fibrosis phenotype. However, it is possible that imaging difficulties in discerning regions of fibrosis and hypoperfusion could overestimate the fibrosis-ischaemia association [36]. Furthermore, assessment of LGE alone can miss any association between interstitial fibrosis and small vessel disease [59] and in this regard, it is worth considering the study in which ischaema was independently associated with extracellular volume, but not LGE [41].
Extracellular matrix expansion in myocardial regions remote from microvascular dysfunction has been suggested to be triggered by pro-hypertrophic transforming growth factor beta (TGF-β) signalling secondary to the sarcomeric mutation [119]. This non-ischaemic aetiology of fibrosis would support the findings of other studies where fibrosis can be seen in hearts with normal perfusion [28, 39].
Clinical manifestations of myocardial ischaemia in HCM
The main clinical manifestations of myocardial ischaemia in HCM patients are angina and dyspnoea [17, 25, 122, 123], alongside dynamic changes on exercise/vasodilator-induced stress electrocardiogram (ECG) testing [20, 124].
Perfusion abnormalities may also be present in the absence of symptoms [14, 20, 22, 23, 40, 41, 102], so the relationship between pathology and symptoms may not always be consistent. Of interest, post-mortem studies of HCM patient hearts have noted a relative absence of historical symptoms among individuals with transmural infarction [53]. This confirms the potential for ischaemia to be silent, possibly through small fibre neuropathy reducing afferent pain signal detection. Mildly abnormal troponin levels are also common among HCM patients, reported in 74% [125], and likely represent myocyte injury or necrosis thought to be exacerbated by myocardial ischaemia.
Mechanisms of myocardial ischaemia progression in HCM
Figure 2 summarises the likely progression of factors affecting oxygen supply and demand in HCM. Early in development, ATP depletion arising from the sarcomeric mutation likely causes stress oxygenation impairment [46, 89], and could contribute to phenotype development through SERCA ATP starvation, calcium accumulation and hypertrophy via calcium signalling [126]. The cause of subsequent metabolic abnormalities in HCM is unclear [91, 127, 128]. Small vessel disease, present even in those < 1 year of age [51], could emerge during embryonic development driven by sarcomeric mutations [129], and is proposed to be one of the earliest factors in the cascade of events related to ischaemia [3]. Pre-hypertrophic diastolic dysfunction secondary to the sarcomeric mutation [74, 75] could contribute by limiting the time for myocardial relaxation, leading to increased rest MBF [76], which could promote vascular remodelling through increased shear stress. However, perfusion impairment reported in the absence of diastolic dysfunction [98] suggests that diastolic dysfunction is not the only contributor. Early perfusion defects seen on stress imaging of HCM patients may occur due to the microscopic steal phenomenon secondary to small vessel disease, or due to an abnormal vasomotor response of diseased myocardium to pharmacologically induced vasodilation [38].

Mechanisms involved in progression of ischaemia in HCM. Disease factors in HCM constrain myocardial blood supply and increase energetic demands. Likely causal (solid arrows) and possibly causal (dashed arrows) relationships between pro-ischaemic disease factors are denoted, alongside potential anti-ischaemic therapies [76]
As cells become hypertrophied, either to compensate for cells lost through ischaemia-induced fibrosis or other pathological processes, ischaemia is further promoted by local reductions in small vessel density [61], greater energetic demands, increased extravascular compression, and LVOT obstruction. Post-hypertrophy diastolic dysfunction (possibly exacerbated by ionic remodelling in hypertrophied segments [130]) may be a key contributor to ischaemia as the diastole-specific perfusion reserve is more strongly correlated with wall thickness than the time-averaged perfusion reserve [48]. Moreover, the increased oxygen demand of hypertrophy leads to resting vasodilation, giving rise to ‘maxed out’ vasodilation at rest [31, 43, 69, 71, 131] and explains why perfusion defects are more prevalent in cohorts with hypertrophy [42] than those without [43].
With disease progression, the ischaemic threshold is incrementally lowered, such that acute episodes of ischaemia are inducible despite non-stenotic epicardial coronary arteries. In some HCM patients, transient increases in energetic demands, such as during AF-induced increases in ventricular pacing [11] or during exercise [132], are sufficient to trigger an ischaemic episode, which can precipitate lethal ventricular arrhythmias.
In patients with the most severe small vessel disease and insufficient capillary density, ischaemia leads to gross macroscopic transmural scarring [3, 12, 59, 62, 111], which contributes amongst other factors (mitral regurgitation [42]) to the 2–16% subset of HCM patients that progress to end-stage disease [133, 134]. Myocyte death eventually leads to total replacement of myocardial regions with fibrosis, such that affected regions are devoid of myocytes that could hypertrophy, leading to wall thinning, LV stiffening and systolic dysfunction [39, 42, 135]. In this stage, LVOT gradients are resolved at the peril of reduced ejection fraction.
Prognostic value of myocardial ischaemia in HCM
Composite endpoints
The studies in Table 3 have analysed the association of ischaemia in HCM with adverse events using composite endpoints, where non-arrhythmic events such as heart failure or all-cause death were included. Although many studies found hypoperfusion to be associated with adverse outcomes on multivariate analysis [4, 25, 35, 121, 124, 136], they did not assess focal fibrosis burden, which is an independent predictor of mortality in HCM [137]. In the study that did account for fibrosis confounding, exercise WMAs were an independent predictor of cardiac events—not visual assessment of perfusion defects on CMR [32]. Although WMAs are not specific for myocardial ischaemia, a strong association between perfusion defects and WMAs has been reported [32]. In the largest study to date, exercise WMAs were substantiated as a possible risk factor in HCM, particularly when considered alongside non-invasive quantitative perfusion reserve measurements [35].
Overall, the studies in Table 3 demonstrate that quantitative assessment of ischaemia identifies a subgroup of HCM patients at high risk of adverse outcomes, with the hyperaemic MBF threshold on PET optimally associated with outcomes estimated as 1.1–1.35 ml/min/g [4, 134, 136]. There is mixed evidence from studies of visual perfusion assessment, suggesting that further studies may benefit from quantitative perfusion analysis.
The prognostic value of other pathophysiological mechanisms, such as myocardial bridging, is debatable. Although bridging has been shown to predict poorer prognosis in one paediatric cohort [138], this association was not consistent elsewhere [139]. Furthermore, Sorajja et al. did not observe an association between bridging and poor outcomes in adult HCM patients [140].
Arrhythmia and sudden cardiac death
Acute myocardial ischaemia (in the absence of epicardial coronary stenoses) may be an important cause of fatal arrhythmias and SCD in HCM [5,6,7, 143], as multiple case reports describe arrhythmias precipitated by acute myocardial ischaemia in young patients [11, 132, 144]. In a postmortem study of 19 young (≤ 35 years) SCD victims with HCM, 11 had physical evidence of acute-subacute myocardial ischaemia (coagulative necrosis, neutrophilic infiltrate, myocytolisis, granulation tissue healing, infarction) in the septal myocardium [47]. Multiple foci of transmural infarction have also been reported in some deceased HCM patients, despite having normal epicardial coronary arteries [53].
Numerous studies (Table 4) have analysed the association between ischaemia and arrhythmia in HCM. Many studies accounted for confounding by fibrosis and found independent associations between ischaemia measurements and arrhythmia [34, 40, 44, 109]. The specific association was variable across studies, and it is unclear the extent to which differences in imaging modality, protocol and cohort may have contributed to this variety. Importantly, however, there is some suggestion that measurements of MBF heterogeneity predict arrhythmic risk [109, 145], motivating further study of regional perfusion quantification. There is further evidence from echocardiographic and scintigraphic studies that ischaemia is associated with syncopal episodes in HCM [15, 18].
Heart failure
Some studies report an association between ischaemia and heart failure. Hamada et al. used Th-201 scintigraphy to study 48 HCM patients and found that development of heart failure was associated with perfusion impairment [107]. Similarly, Olivotto et al. used stress PET in 51 HCM patients and found that adverse LV remodelling and systolic dysfunction were predicted by quantitative assessment of perfusion [134]. SPECT imaging of 65 HCM patients also found an association between metabolic impairment and heart failure [141]. Similarly, perfusion and LGE CMR of 62 HCM patients found that reduced MBF was the only independent predictor of functional status when LGE and hypertrophy were accounted for [102]. However, in the largest longitudinal perfusion CMR study to date of 449 HCM patients, perfusion defects on visual assessment were unable to predict heart failure [42]. LGE progression may instead be a more prominent factor in the development of heart failure as an independent association has been reported [39].
Confounding by coexisting pathology
In evaluating the independent prognostic contributions of perfusion defects and presence of LGE, not only are perfusion defects confounded by presence of LGE due to the possibly causal link between ischaemia and fibrosis in HCM [39], but some perfusion defects correspond to sites of LGE [40]. It is therefore unclear whether SCDs in HCM are caused by (i) primary ventricular arrhythmia related to fibrosis [137], or (ii) secondary ventricular arrhythmia during ischaemia [34]. Simultaneous echo and perfusion CMR in 148 HCM patients showed that cardiac event rates were highest when both LGE and exercise WMAs were observed [32], hence both factors may contribute to increased risk. This is further supported by the finding that both fixed and reversible perfusion defects have prognostic value [10], as perfusion abnormalities present at rest are thought to represent severe fibrosis [34]. Ischaemia may be relevant to the cases of juvenile SCDs in which replacement fibrosis is absent [12].
As a possible cause of ischaemia, LVOT obstruction is already recognised as a risk factor for SCD in adults [149] and children [150]. Further confounding may arise from atrial fibrillation due to its association with ischaemia [147].
Exercise
Myocardial ischaemia in HCM is relevant in the context of evolving clinical guidelines on exercise restrictions. If SCD is related to exercise [151], then ischaemia is a plausible arrhythmic substrate through exercise-induced ischaemia [152], latent LVOT obstruction [66] and reduced diastolic filling time [77] during increased workload. Measurements derived from stress perfusion imaging could contribute to decisions made with patients on exercise, although at present there is limited clinical evidence that this would be useful.
Of note, two meta-analyses have shown that SCDs in young HCM patients are 75% more common in athletes than non-athletes [153, 154]. Age appears to be a factor in the association between exercise and SCD in HCM [155], with Weissler-Snir et al. reporting 80% of HCM-related SCDs in those ≤ 20 years old being related to exercise, compared to < 5% of those above 20 [151]. Interestingly, a study of 1380 HCM patients showed that, on multivariate analysis, only NSVT induced by exercise was associated with SCD—not NSVT generally [156]. In this analysis, 21% of patients with exercise-induced NSVT had preceding ST depression, and most were < 40 years of age. There are other varying reports of SCD-predictive measures derived from exercise [152, 157, 158], including the ventilation-to-CO2 (VE/ CO2) slope and anaerobic threshold during exercise, all of which could indicate prospensity for underlying myocardial ischaemia on cardiopulmonary exercise testing.
Clinical perspective: myocardial ischaemia in HCM as a therapeutic target
The potential for ischaemia as an early therapeutic target in HCM [43] is reinforced by the improvements in myocardial perfusion and patient symptoms that typically accompany invasive surgical relief of LVOT obstruction [159, 160]. By reducing the perfusion sink, relief of LVOT obstruction may cause less vasodilatory reserve to be exhausted at rest, in addition to reductions in wall stress and extravascular compression.
Multiple promising studies of pharmacologic treatments with the potential to minimise ischaemic burden in HCM are ongoing or have been completed (Fig. 2). These include angiotensin receptor blockers, vasodilators, metabolic modulators, late sodium blockers, negative inotropes as well as novel allosteric myosin inhibitors.
Angiotensin receptor blockers (‘-sartans’), which target both vascular function and the TGF-β signalling pathway (also associated with the emergence of fibrosis [119]), have shown efficacy in limiting phenotype development. Although it is unknown whether sartans affect perfusion defects in HCM [43], candesartan and valsartan may attenuate the HCM phenotype [161,162,163], with valsartan being more efficacious in those with less hypertrophic remodelling [163].
Perhexiline, a vasodilator and metabolic modulator, recently showed lack of efficacy to improve exercise capacity in HCM patients with moderate to severe heart failure (trial NCT02862600). However, the findings of this interventional study may have been significantly influenced by the advanced disease progression into heart failure of the recruited patients and the choice of primary endpoint. RESOLVE-HCM (trial NCT04426578) is another study which is assessing the impact of perhexiline on LV hypertrophy [164]. This trial includes changes in oxygen-sensitive CMR measures as a secondary endpoint, which could directly evidence anti-ischaemic pharmacologic treatment. Trientine, a modulator of copper metabolism, is also being investigated in HCM (trial ISRCTN57145331). The novel class of metabolic modulator drugs, sodium-glucose co-transporter 2 inhibitors, may also have potential in HCM (trial NCT05182658). As a further potentially novel therapeutic target due to their effects on vascular function and metabolism [165], ceramides have been implicated in the development of various cardiovascular diseases [166], which may be relevant to HCM.
Late sodium blockers, which target the pathologically increased late sodium current in HCM cardiomyocytes [130], were hypothesised to ameliorate diastolic dysfunction [167] and thus downstream ischaemic effects. However, in RESTYLE-HCM (trial 2011-004507-20), ranolazine showed no efficacy in reducing diastolic dysfunction or pro B-type natriuretic peptide in non-obstructive HCM patients, despite finding a possible antiarrhythmic effect [168]. Potential amelioration of ischaemia-induced arrhythmia by ranolazine is also described in a recent study [169]. LIBERTY-HCM (trial NCT02291237) was terminated early due to a lack of efficacy of eleclazine administration [170]. Finally, although disopyramide ameliorates symptoms and reduces LVOT obstruction in HCM, these effects are attributed to its negative inotropic action rather than late sodium block [171].
Perhaps most promising is the novel allosteric myosin inhibitor mavacamten, which (in contrast to the drugs previously introduced) specifically targets the underlying pathogenic drivers of contractile dysfunction in HCM at the sarcomeric level [172]. Both the results of EXPLORER-HCM (trial NCT03470545) [173] and additional studies [174] have proven mavacamten effective at improving cardiac function in HCM patients, including the reduction of LVOT obstruction gradients. The marked reductions in N-terminal pro B-type natriuretic peptide and cardiac troponin I during mavacamten treatment indicate that the drug may reduce the extent of ischaemic injury in HCM [175], likely through attenuation of the downstream pro-ischaemic effects of sarcomeric impairment, as shown in Fig. 2.
Future research
All the above routes towards a refined diagnosis and targeted treatment of myocardial ischaemia in HCM constitute important and promising prospects for future research into the amelioration of symptoms and risk of SCD in HCM. If successful, their integration into HCM risk stratification models and clinical guidelines is expected to yield significant advances for the management of this high-risk group of patients, as well as further insights on the overall contribution of ischaemia in cardiovascular disease. However, further research is needed to elucidate the clinical significance of ischaemia on imaging, and the relative contributions of the various pro-ischaemic mechanisms in HCM.
Although ischaemic HCM phenotypes are consistently identified as a high-risk subgroup on long term follow-up (Table 3), the role of imaging in assessing ischaemic burden for risk prediction needs rigorous testing. Reported ischaemia-arrhythmia associations are heterogeneous (Table 4) and might be explained by monitoring of ECG (Holter monitors) during conditions of rest to assess patients’ arrhythmic burden. Future work might consider whether ischaemia on non-invasive imaging is associated with arrhythmias on stress testing, particularly as some arrhythmias are preceded by ischaemic ECG changes [156]. Such multimodal approaches have already been evaluated in HCM, such as the combined use of perfusion CMR and echocardiography [32], demonstrating its potential to improve diagnosis and prognostic stratification of HCM patients.
Another consideration is that despite the strong age-dependence of SCD risk in HCM, most analyses relating ischaemia and arrhythmic risk were performed in midlife cohorts (Table 4), which may be more likely to have a fibrotic substrate due to the presence of more advanced structural LV remodelling. Future analyses of imaging for ischaemia in younger HCM cohorts may have distinct implications, given that exercise-induced ECG changes are emerging as predictive of outcomes [176].
Importantly, multi-centre studies to evaluate ischaemic burden are needed to elucidate the incremental value of perfusion imaging for ischaemic myocardial substrates over potentially irreversible substrates like LGE, particularly in subgroups where guidelines are less certain (ESC SCD risk < 6%).
Conclusion
In this review, we have presented a comprehensive discussion of the latest evidence corroborating profound links between myocardial ischaemia, disease severity and prognosis. This notably broadens former studies by covering HCM-specific ischaemic factors, the impairment of perfusion by myocardial hypertrophy, the characteristic distributions of ischaemic burden in HCM ventricles, the relationship between (hypo)perfusion and fibrosis, mechanisms of ischaemia progression in HCM, its clinical manifestations and prognostic value. Altogether, our analysis substantiates myocardial ischaemia as a strong and multifactorial contributor to adverse LV remodelling, arrhythmia, and SCD events in HCM. Despite the strong associations reported, further studies are needed to understand which non-invasive methods of ischaemia assessment have independent prognostic value in HCM, over and above co-existing myocardial fibrosis and LVOT obstruction.
References
Semsarian C, Ingles J, Maron MS, Maron BJ (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 65:1249–1254. https://doi.org/10.1016/j.jacc.2015.01.019
Ommen SR, Mital S, Burke MA et al (2020) 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy. Circulation 142:e558–e631. https://doi.org/10.1161/CIR.0000000000000937
Maron MS, Olivotto I, Maron BJ et al (2009) The case for myocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol 54:866–875. https://doi.org/10.1016/j.jacc.2009.04.072
Cecchi F, Olivotto I, Gistri R et al (2003) Coronary Microvascular Dysfunction and Prognosis in Hypertrophic Cardiomyopathy. N Engl J Med 349:1027–1035. https://doi.org/10.1056/NEJMoa025050
Liu F, Ma Y, Ge H et al (2018) Long-Term Outcomes of Acute Myocardial Infarction in Patients With Hypertrophic Cardiomyopathy. Angiology 69:900–908. https://doi.org/10.1177/0003319718778418
Yang Y-J, Fan C-M, Yuan J-Q et al (2017) Long-term survival after acute myocardial infarction in patients with hypertrophic cardiomyopathy. Clin Cardiol 40:26–31. https://doi.org/10.1002/clc.22601
Poteshkina NG, Kovalevskaya EA, Krylova NS, Fettser DV (2020) Myocardial ischemia in patients with hypertrophic cardiomyopathy. Probl Sotsialnoi Gig Zdravookhranenniiai Istor Med 27:671–676
Graziani F, Lillo R, Biagini E et al (2022) Myocardial infarction with non-obstructive coronary arteries in hypertrophic cardiomyopathy vs Fabry disease. Int J Cardiol. https://doi.org/10.1016/j.ijcard.2022.07.046
Maron MS, Rowin EJ, Wessler BS et al (2019) Enhanced American College of Cardiology/American Heart Association Strategy for Prevention of Sudden Cardiac Death in High-Risk Patients With Hypertrophic Cardiomyopathy. J Am Coll Cardiol 4:644–657. https://doi.org/10.1001/jamacardio.2019.1391
Ziolkowska L, Boruc A, Sobielarska-Lysiak D et al (2021) Prognostic Significance of Myocardial Ischemia Detected by Single-Photon Emission Computed Tomography in Children with Hypertrophic Cardiomyopathy. Pediatr Cardiol 42:960–968. https://doi.org/10.1007/s00246-021-02570-9
Limongelli G, Calabro P, Pacileo G et al (2007) Myocardial infarction in a young athlete with non-obstructive hypertrophic cardiomyopathy and normal coronary arteries. Int J Cardiol 115:e71–e73. https://doi.org/10.1016/j.ijcard.2006.07.206
Gaspari M, Basso C, Perazzolo Marra M et al (2021) Small Vessel Disease: Another Component of the Hypertrophic Cardiomyopathy Phenotype Not Necessarily Associated with Fibrosis. J Clin Med 10:575. https://doi.org/10.3390/jcm10040575
Elliott PM, Rosano GM, Gill JS et al (1996) Changes in coronary sinus pH during dipyridamole stress in patients with hypertrophic cardiomyopathy. Heart 75:179. https://doi.org/10.1136/hrt.75.2.179
O’Gara PT, Bonow RO, Maron BJ et al (1987) Myocardial perfusion abnormalities in patients with hypertrophic cardiomyopathy: assessment with thallium-201 emission computed tomography. Circulation 76:1214–1223. https://doi.org/10.1161/01.CIR.76.6.1214
Dohlen TW, Prisant LM, Frank MJ (1989) Significance of positive or negative thallium-201 scintigraphy in hypertrophic cardiomyopathy. Am J Cardiol 64:498–503. https://doi.org/10.1016/0002-9149(89)90428-1
Udelson JE, Bonow RO, O’Gara PT et al (1989) Verapamil prevents silent myocardial perfusion abnormalities during exercise in asymptomatic patients with hypertrophic cardiomyopathy. Circulation 79:1052–1060. https://doi.org/10.1161/01.CIR.79.5.1052
Takata J, Counihan PJ, Gane JN et al (1993) Regional thallium-201 washout and myocardial hypertrophy in hypertrophic cardiomyopathy and its relation to exertional chest pain. Am J Cardiol 72:211–217. https://doi.org/10.1016/0002-9149(93)90162-6
Dilsizian V, Bonow RO, Epstein SE, Fananapazir L (1993) Myocardial ischemia detected by thallium scintigraphy is frequently related to cardiac arrest and syncope in young patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 22:796–804. https://doi.org/10.1016/0735-1097(93)90193-5
Shimizu M, Yoshio H, Ino H et al (1996) Myocardial scintigraphic study with 123I 15-(p-iodophenyl)-3(R, S)-methylpentadecanoic acid in patients with hypertrophic cardiomyopathy. Int J Cardiol 54:51–59. https://doi.org/10.1016/0167-5273(95)02536-7
Elliott PM, Kaski JC, Prasad K et al (1996) Chest pain during daily life in patients with hypertrophic cardiomyopathy: an ambulatory electrocardiographic study. Eur Heart J 17:1056–1064. https://doi.org/10.1093/oxfordjournals.eurheartj.a015002
Yoshida N, Ikeda H, Wada T et al (1998) Exercise-induced abnormal blood pressure responses are related to subendocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol 32:1938–1942. https://doi.org/10.1016/S0735-1097(98)00498-7
Yamada M, Elliott PM, Kaski JC et al (1998) Dipyridamole stress thallium-201 perfusion abnormalities in patients with hypertrophic cardiomyopathy. Relationship to clinical presentation and outcome. Eur Heart J 19:500–507. https://doi.org/10.1053/euhj.1997.0769
Romero-Farina G, Candell-Riera J, Galve E et al (2004) Do myocardial perfusion SPECT and radionuclide angiography studies in adult patients with hypertrophic cardiomyopathy have prognostic implications? J Nucl Cardiol 11:578–586. https://doi.org/10.1016/j.nuclcard.2004.05.008
Melacini P, Corbetti F, Calore C et al (2008) Cardiovascular magnetic resonance signs of ischemia in hypertrophic cardiomyopathy. Int J Cardiol 128:364–373. https://doi.org/10.1016/j.ijcard.2007.06.023
Cortigiani L, Rigo F, Gherardi S et al (2008) Prognostic Implications of Coronary Flow Reserve on Left Anterior Descending Coronary Artery in Hypertrophic Cardiomyopathy. Am J Cardiol 102:1718–1723. https://doi.org/10.1016/j.amjcard.2008.08.023
Moon J, Cho IJ, Shim CY et al (2010) Abnormal Myocardial Capillary Density in Apical Hypertrophic Cardiomyopathy Can Be Assessed by Myocardial Contrast Echocardiography. Circulation 74:2166–2172. https://doi.org/10.1253/circj.CJ-10-0241
Bravo PE, Pinheiro A, Higuchi T et al (2012) PET/CT Assessment of Symptomatic Individuals with Obstructive and Nonobstructive Hypertrophic Cardiomyopathy. J Nucl Med 53:407. https://doi.org/10.2967/jnumed.111.096156
Bravo PE, Zimmerman SL, Luo H-C et al (2013) Relationship of delayed enhancement by magnetic resonance to myocardial perfusion by positron emission tomography in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging 6:210–217. https://doi.org/10.1161/CIRCIMAGING.112.000110
Tyan CC, Armstrong S, Scholl D et al (2013) Stress Hypoperfusion and Tissue Injury in Hypertrophic Cardiomyopathy. Circ Cardiovasc Imaging 6:229–238. https://doi.org/10.1161/CIRCIMAGING.112.000170
Villa A, Bettencourt N, Zarinabad N et al (2014) Stress perfusion CMR in hypertrophic cardiomyopathy: comparison with late gadolinium enhancement. J Cardiovasc Magn Reson 16:P324–P324. https://doi.org/10.1186/1532-429X-16-S1-P324
Ismail T, Hsu L-Y, Greve A et al (2014) Coronary microvascular ischemia in hypertrophic cardiomyopathy: a pixel-wise quantitative cardiovascular magnetic resonance perfusion study. J Cardiovasc Magn Reson 16:49. https://doi.org/10.1186/s12968-014-0049-1
Peteiro J, Fernandez X, Bouzas-Mosquera A et al (2015) Exercise echocardiography and cardiac magnetic resonance imaging to predict outcome in patients with hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging 16:423–432. https://doi.org/10.1093/ehjci/jeu225
Jablonowski R, Fernlund E, Aletras AH et al (2015) Regional Stress-Induced Ischemia in Non-fibrotic Hypertrophied Myocardium in Young HCM Patients. Pediatr Cardiol 36:1662–1669. https://doi.org/10.1007/s00246-015-1214-5
Chiribiri A, Leuzzi S, Conte MR et al (2015) Rest perfusion abnormalities in hypertrophic cardiomyopathy: correlation with myocardial fibrosis and risk factors for sudden cardiac death. Clin Radiol 70:495–501. https://doi.org/10.1016/j.crad.2014.12.018
Ciampi Q, Olivotto I, Gardini C et al (2016) Prognostic role of stress echocardiography in hypertrophic cardiomyopathy: the International Stress Echo Registry. Int J Cardiol 219:331–338. https://doi.org/10.1016/j.ijcard.2016.06.044
Villa ADM, Sammut E, Zarinabad N et al (2016) Microvascular ischemia in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. https://doi.org/10.1186/s12968-016-0223-8
Hernandez LE (2018) Myocardial stress perfusion magnetic resonance in children with hypertrophic cardiomyopathy. Cardiol Young 28:702–708. https://doi.org/10.1017/S1047951118000094
Camaioni C, Knott KD, Augusto JB et al (2020) Inline perfusion mapping provides insights into the disease mechanism in hypertrophic cardiomyopathy. Heart 106:824–829. https://doi.org/10.1136/heartjnl-2019-315848
Raman B, Ariga R, Spartera M et al (2019) Progression of myocardial fibrosis in hypertrophic cardiomyopathy: mechanisms and clinical implications. Eur Heart J Cardiovasc Imaging 20:157–167. https://doi.org/10.1093/ehjci/jey135
Kim EK, Lee S-C, Chang S-A et al (2020) Prevalence and clinical significance of cardiovascular magnetic resonance adenosine stress-induced myocardial perfusion defect in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson 22:30. https://doi.org/10.1186/s12968-020-00623-1
Malahfji M, Senapati A, Debs D et al (2020) Examining the impact of inducible ischemia on myocardial fibrosis and exercise capacity in hypertrophic cardiomyopathy. Sci Rep 10:15977. https://doi.org/10.1038/s41598-020-71394-z
Raphael CE, Mitchell F, Kanaganayagam GS et al (2021) Cardiovascular magnetic resonance predictors of heart failure in hypertrophic cardiomyopathy: the role of myocardial replacement fibrosis and the microcirculation. J Cardiovasc Magn Reson 23:26. https://doi.org/10.1186/s12968-021-00720-9
Hughes RK, Camaioni C, Augusto JB et al (2021) Myocardial Perfusion Defects in Hypertrophic Cardiomyopathy Mutation Carriers. J Am Heart Assoc. https://doi.org/10.1161/JAHA.120.020227
Aguiar Rosa S, Thomas B, Fiarresga A et al (2021) The Impact of Ischemia Assessed by Magnetic Resonance on Functional, Arrhythmic, and Imaging Features of Hypertrophic Cardiomyopathy. Front Cardiovasc Med. https://doi.org/10.3389/fcvm.2021.761860
Hamon M, Fau G, Née G et al (2010) Meta-analysis of the diagnostic performance of stress perfusion cardiovascular magnetic resonance for detection of coronary artery disease. J Cardiovasc Magn Reson 12:29. https://doi.org/10.1186/1532-429X-12-29
Karamitsos TD, Dass S, Suttie J et al (2013) Blunted Myocardial Oxygenation Response During Vasodilator Stress in Patients With Hypertrophic Cardiomyopathy. J Am Coll Cardiol 61:1169–1176. https://doi.org/10.1016/j.jacc.2012.12.024
Basso C, Thiene G, Corrado D et al (2000) Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol 31:988–998. https://doi.org/10.1053/hupa.2000.16659
Krams R, Cate FJ, Carlier SG et al (2004) Diastolic coronary vascular reserve: a new index to detect changes in the coronary microcirculation in hypertrophic cardiomyopathy. J Am Coll Cardiol 43:670–677. https://doi.org/10.1016/j.jacc.2003.09.046
Maron BJ, Mackey-Bojack S, Facile E et al (2020) Hypertrophic Cardiomyopathy and Sudden Death Initially Identified at Autopsy. Am J Cardiol 127:139–141. https://doi.org/10.1016/j.amjcard.2020.04.021
Kwon DH, Smedira NG, Rodriguez ER et al (2009) Cardiac Magnetic Resonance Detection of Myocardial Scarring in Hypertrophic Cardiomyopathy: Correlation With Histopathology and Prevalence of Ventricular Tachycardia. J Am Coll Cardiol 54:242–249. https://doi.org/10.1016/j.jacc.2009.04.026
Maron BJ, Wolfson JK, Epstein SE, Roberts WC (1986) Intramural (“small vessel”) coronary artery disease in hypertrophic cardiomyopathy. J Am Coll Cardiol 8:545–557. https://doi.org/10.1016/S0735-1097(86)80181-4
Moravsky G, Ofek E, Rakowski H et al (2013) Myocardial Fibrosis in Hypertrophic Cardiomyopathy: Accurate Reflection of Histopathological Findings by CMR. JACC Cardiovasc Imaging 6:587–596. https://doi.org/10.1016/j.jcmg.2012.09.018
Maron BJ, Epstein SE, Roberts WC (1979) Hypertrophic cardiomyopathy and transmural myocardial infarction without significant atherosclerosis of the extramural coronary arteries. Am J Cardiol 43:1086–1102. https://doi.org/10.1016/0002-9149(79)90139-5
Bogatyreva F, Kaplunova VRA, Kozhevnikova M et al (2021) Assessment of the structural and functional state of blood vessels in patients with hypertrophic cardiomyopathy. Kardiologiia 61:16–21
Tanaka M, Fujiwara H, Onodera T et al (1987) Quantitative analysis of narrowings of intramyocardial small arteries in normal hearts, hypertensive hearts, and hearts with hypertrophic cardiomyopathy. Circulation 75:1130–1139. https://doi.org/10.1161/01.CIR.75.6.1130
Krams R, Kofflard MJM, Duncker DJ et al (1998) Decreased Coronary Flow Reserve in Hypertrophic Cardiomyopathy Is Related to Remodeling of the Coronary Microcirculation. Circulation 97:230–233. https://doi.org/10.1161/01.CIR.97.3.230
Schwartzkopff B, Mundhenke M, Strauer BE (1998) Alterations of the Architecture of Subendocardial Arterioles in Patients With Hypertrophic Cardiomyopathy and Impaired Coronary Vasodilator Reserve: A Possible Cause for Myocardial Ischemia. J Am Coll Cardiol 31:1089–1096. https://doi.org/10.1016/S0735-1097(98)00036-9
Varnava AM, Elliott PM, Sharma S et al (2000) Hypertrophic cardiomyopathy: the interrelation of disarray, fibrosis, and small vessel disease. Heart 84:476–482. https://doi.org/10.1136/heart.84.5.476
Foà A, Agostini V, Rapezzi C et al (2019) Histopathological comparison of intramural coronary artery remodeling and myocardial fibrosis in obstructive versus end-stage hypertrophic cardiomyopathy. Int J Cardiol 291:77–82. https://doi.org/10.1016/j.ijcard.2019.03.060
Johansson B, Mörner S, Waldenström A, Stål P (2008) Myocardial capillary supply is limited in hypertrophic cardiomyopathy: A morphological analysis. Int J Cardiol 126:252–257. https://doi.org/10.1016/j.ijcard.2007.04.003
Güçlü A, Happé C, Eren S et al (2015) Left ventricular outflow tract gradient is associated with reduced capillary density in hypertrophic cardiomyopathy irrespective of genotype. Eur J Clin Invest 45:1252–1259. https://doi.org/10.1111/eci.12544
Nijenkamp LLAM, Bollen IAE, Niessen HWM et al (2020) Sex-specific cardiac remodeling in early and advanced stages of hypertrophic cardiomyopathy. PLoS ONE. https://doi.org/10.1371/journal.pone.0232427
Zhang M-K, Zhang Z, Xue H et al (2022) Microvascular Rarefaction and Myocardial Fibrosis in Hypertrophic Obstructive Cardiomyopathy: A Histopathological Comparison of Pediatric and Adult Patients. Heart Surg Forum 25:E042–E047. https://doi.org/10.1532/hsf.4277
Maron MS, Maron BJ, Harrigan C et al (2009) Hypertrophic Cardiomyopathy Phenotype Revisited After 50 Years With Cardiovascular Magnetic Resonance. J Am Coll Cardiol 54:220–228. https://doi.org/10.1016/j.jacc.2009.05.006
Maron MS, Olivotto I, Zenovich AG et al (2006) Hypertrophic Cardiomyopathy Is Predominantly a Disease of Left Ventricular Outflow Tract Obstruction. Circulation 114:2232–2239. https://doi.org/10.1161/CIRCULATIONAHA.106.644682
Shah JS, Esteban MTT, Thaman R et al (2008) Prevalence of exercise-induced left ventricular outflow tract obstruction in symptomatic patients with non-obstructive hypertrophic cardiomyopathy. Heart 94:1288. https://doi.org/10.1136/hrt.2007.126003
Patel P, Dhillon A, Popovic ZB et al (2015) Left Ventricular Outflow Tract Obstruction in Hypertrophic Cardiomyopathy Patients Without Severe Septal Hypertrophy. Circ Cardiovasc Imaging. https://doi.org/10.1161/CIRCIMAGING.115.003132
Tesic M, Djordjevic-Dikic A, Beleslin B et al (2013) Regional Difference of Microcirculation in Patients with Asymmetric Hypertrophic Cardiomyopathy: Transthoracic Doppler Coronary Flow Velocity Reserve Analysis. J Am Soc Echocardiogr 26:775–782. https://doi.org/10.1016/j.echo.2013.03.023
Kyriakidis MK, Dernellis JM, Androulakis AE et al (1997) Changes in Phasic Coronary Blood Flow Velocity Profile and Relative Coronary Flow Reserve in Patients With Hypertrophic Obstructive Cardiomyopathy. Circulation 96:834–841. https://doi.org/10.1161/01.CIR.96.3.834
Knaapen P, Germans T, Camici PG et al (2008) Determinants of coronary microvascular dysfunction in symptomatic hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 294:H986–H993. https://doi.org/10.1152/ajpheart.00233.2007
Soliman OII, Knaapen P, Geleijnse ML et al (2007) Assessment of intravascular and extravascular mechanisms of myocardial perfusion abnormalities in obstructive hypertrophic cardiomyopathy by myocardial contrast echocardiography. Heart 93:1204–1212. https://doi.org/10.1136/hrt.2006.110460
Xu H, Yang Z, Sun J et al (2014) The regional myocardial microvascular dysfunction differences in hypertrophic cardiomyopathy patients with or without left ventricular outflow tract obstruction: Assessment with first-pass perfusion imaging using 3.0-T cardiac magnetic resonance. Eur Radiol 83:665–672. https://doi.org/10.1016/j.ejrad.2014.01.008
Raphael CE, Cooper R, Parker KH et al (2016) Mechanisms of Myocardial Ischemia in Hypertrophic Cardiomyopathy: Insights From Wave Intensity Analysis and Magnetic Resonance. J Am Coll Cardiol 68:1651–1660. https://doi.org/10.1016/j.jacc.2016.07.751
Liu W, Sun D, Yang J (2017) Diastolic Dysfunction of Hypertrophic Cardiomyopathy Genotype-Positive Subjects Without Hypertrophy Is Detected by Tissue Doppler Imaging: A Systematic Review and Meta-analysis. J Ultrasound Med 36:2093–2103. https://doi.org/10.1002/jum.14250
Poutanen T, Tikanoja T, Jääskeläinen P et al (2006) Diastolic dysfunction without left ventricular hypertrophy is an early finding in children with hypertrophic cardiomyopathy–causing mutations in the β-myosin heavy chain, α-tropomyosin, and myosin-binding protein C genes. Am Heart J 151:725.e1-725.e9. https://doi.org/10.1016/j.ahj.2005.12.005
Marszalek RJ, John Solaro R, Wolska BM (2019) Coronary arterial vasculature in the pathophysiology of hypertrophic cardiomyopathy. Pflug Arch Eur J Physiol 471:769–780. https://doi.org/10.1007/s00424-018-2224-y
Plehn G, Vormbrock J, Meissner A, Trappe H-J (2009) Effects of exercise on the duration of diastole and on interventricular phase differences in patients with hypertrophic cardiomyopathy: relationship to cardiac output reserve. J Nucl Cardiol 16:233–243. https://doi.org/10.1007/s12350-008-9031-3
Tomochika Y, Tanaka N, Wasaki Y et al (1993) Assessment of flow profile of left anterior descending coronary artery in hypertrophic cardiomyopathy by transesophageal pulsed Doppler echocardiography. Am J Cardiol 72:1425–1430. https://doi.org/10.1016/0002-9149(93)90191-E
Akasaka T, Yoshikawa J, Yoshida K et al (1994) Phasic Coronary Flow Characteristics in Patients With Hypertrophic Cardiomyopathy: A Study by Coronary Doppler Catheter. J Am Soc Echocardiogr 7:9–19. https://doi.org/10.1016/S0894-7317(14)80413-6
Hirasaki S, Nakamura T, Kuribayashi T et al (1999) Abnormal course, abnormal flow, and systolic compression of the septal perforator associated with impaired myocardial perfusion in hypertrophic cardiomyopathy. Am Heart J 137:109–117. https://doi.org/10.1016/s0002-8703(99)70466-7
Basso C, Thiene G, Mackey-Bojack S et al (2009) Myocardial bridging, a frequent component of the hypertrophic cardiomyopathy phenotype, lacks systematic association with sudden cardiac death. Eur Heart J 30:1627–1634. https://doi.org/10.1093/eurheartj/ehp121
Shariat M, Thavendiranathan P, Nguyen E et al (2014) Utility of coronary CT angiography in outpatients with hypertrophic cardiomyopathy presenting with angina symptoms. J Cardiovasc Comput Tomogr 8:429–437. https://doi.org/10.1016/j.jcct.2014.09.007
Yildiz O, Altin FH, Tosun O et al (2014) Myocardial Bridging in a Child With Hypertrophic Obstructive Cardiomyopathy. World J Pediatr Congenit Heart Surg 5:611–614. https://doi.org/10.1177/2150135114536901
Olivotto I, Cecchi F, Bini R et al (2009) Tunneled left anterior descending artery in a child with hypertrophic cardiomyopathy. Nat Clin Pract Cardiovasc Med 6:134–139. https://doi.org/10.1038/ncpcardio1420
Said S (2020) Unroofing of LAD Myocardial Bridge Combined With Right Ventricular Septal Myectomy in a Child With Noonan Syndrome and HCM. World J Pediatr Congenit Heart Surg 12:659–660. https://doi.org/10.1177/2150135120943869
Popov AF, Bireta C, Schmitto JD et al (2009) Myocardial ischemia with left ventricular outflow obstruction. J Cardiothorac Surg 4:51. https://doi.org/10.1186/1749-8090-4-51
McCrindle BW, Yetman AT (2001) Myocardial bridging of the left anterior descending coronary artery in children with hypertrophic cardiomyopathy. J Am Coll Cardiol 38:921–922. https://doi.org/10.1016/S0735-1097(01)01461-9
Lameh FAMS, Joanna S (2001) Myocardial bridging of the left anterior descending coronary artery in children with hypertrophic cardiomyopathy: reply. J Am Coll Cardiol 38:922. https://doi.org/10.1016/S0735-1097(01)01462-0
Grover S, Lloyd R, Perry R et al (2019) Assessment of myocardial oxygenation, strain, and diastology in MYBPC3-related hypertrophic cardiomyopathy: a cardiovascular magnetic resonance and echocardiography study. Eur Heart J Cardiovasc Imaging 20:932–938. https://doi.org/10.1093/ehjci/jey220
Raman B, Tunnicliffe EM, Chan K et al (2021) Association Between Sarcomeric Variants in Hypertrophic Cardiomyopathy and Myocardial Oxygenation: Insights From a Novel Oxygen-Sensitive Cardiovascular Magnetic Resonance Approach. Circulation 144:1656–1658. https://doi.org/10.1161/CIRCULATIONAHA.121.054015
van der Velden J, Tocchetti CG, Varricchi G et al (2018) Metabolic changes in hypertrophic cardiomyopathies: scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc Res 114:1273–1280. https://doi.org/10.1093/cvr/cvy147
Abozguia K, Elliott P, McKenna W et al (2010) Metabolic Modulator Perhexiline Corrects Energy Deficiency and Improves Exercise Capacity in Symptomatic Hypertrophic Cardiomyopathy. Circulation 122:1562–1569. https://doi.org/10.1161/CIRCULATIONAHA.109.934059
Timmer SAJ, Germans T, Götte MJW et al (2010) Determinants of myocardial energetics and efficiency in symptomatic hypertrophic cardiomyopathy. Eur J Nucl Med Mol Imaging 37:779–788. https://doi.org/10.1007/s00259-009-1350-3
Ishiwata S, Maruno H, Senda M et al (1997) Mechanical efficiency in hypertrophic cardiomyopathy assessed by positron emission tomography with carbon 11 acetate. Am Heart J 133:497–503. https://doi.org/10.1016/s0002-8703(97)70143-1
Luedde M, Flögel U, Knorr M et al (2009) Decreased contractility due to energy deprivation in a transgenic rat model of hypertrophic cardiomyopathy. J Mol Med 87:411–422. https://doi.org/10.1007/s00109-008-0436-x
Mahmod M, Francis JM, Pal N et al (2014) Myocardial perfusion and oxygenation are impaired during stress in severe aortic stenosis and correlate with impaired energetics and subclinical left ventricular dysfunction. J Cardiovasc Magn Reson 16:29. https://doi.org/10.1186/1532-429X-16-29
Petersen SE, Jerosch-Herold M, Hudsmith LE et al (2007) Evidence for Microvascular Dysfunction in Hypertrophic Cardiomyopathy. Circulation 115:2418–2425. https://doi.org/10.1161/CIRCULATIONAHA.106.657023
Gyllenhammar T, Fernlund E, Jablonowski R et al (2014) Young patients with hypertrophic cardiomyopathy, but not subjects at risk, show decreased myocardial perfusion reserve quantified with CMR. Eur Heart J Cardiovasc Imaging 15:1350–1357. https://doi.org/10.1093/ehjci/jeu137
Huang L, Han R, Ai T et al (2013) Assessment of coronary microvascular dysfunction in hypertrophic cardiomyopathy: First-pass myocardial perfusion cardiovascular magnetic resonance imaging at 1.5 T. Clin Radiol 68:676–682. https://doi.org/10.1016/j.crad.2013.01.003
Yin L, Xu H, Zheng S et al (2017) 3.0 T magnetic resonance myocardial perfusion imaging for semi-quantitative evaluation of coronary microvascular dysfunction in hypertrophic cardiomyopathy. Int J Card Imaging 33:1949–1959. https://doi.org/10.1007/s10554-017-1189-9
Garcia Brás P, Aguiar Rosa S, Thomas B et al (2022) Associations between perfusion defects, tissue changes and myocardial deformation in hypertrophic cardiomyopathy, uncovered by a cardiac magnetic resonance segmental analysis. Rev Port Cardiol 41:559–568. https://doi.org/10.1016/j.repc.2022.03.003
Aquaro GD, Todiere G, Barison A et al (2011) Myocardial Blood Flow and Fibrosis in Hypertrophic Cardiomyopathy. J Card Fail 17:384–391. https://doi.org/10.1016/j.cardfail.2011.01.006
Kimball BP, LiPreti V, Bui S, Wigle ED (1990) Comparison of proximal left anterior descending and circumflex coronary artery dimensions in aortic valve stenosis and hypertrophic cardiomyopathy. Am J Cardiol 65:767–771. https://doi.org/10.1016/0002-9149(90)91385-J
Kaufmann P, Vassalli G, Lupi-Wagner S et al (1996) Coronary artery dimensions in primary and secondary left ventricular hypertrophy. J Am Coll Cardiol 28:745–750. https://doi.org/10.1016/0735-1097(96)00194-5
Sellers SL, Fonte TA, Grover R et al (2018) Hypertrophic Cardiomyopathy (HCM): New insights into Coronary artery remodelling and ischemia from FFRCT. J Cardiovasc Comput Tomogr 12:467–471. https://doi.org/10.1016/j.jcct.2018.08.002
Das A, Kelly C, Teh I et al (2022) Phenotyping hypertrophic cardiomyopathy using cardiac diffusion magnetic resonance imaging: the relationship between microvascular dysfunction and microstructural changes. Eur Heart J Cardiovasc Imaging 23:352–362. https://doi.org/10.1093/ehjci/jeab210
Hamada M, Shigematsu Y, Nakata S et al (2021) Predicting the clinical course in hypertrophic cardiomyopathy using thallium-201 myocardial scintigraphy. ESC Heart Fail 8:1378–1387. https://doi.org/10.1002/ehf2.13218
Lorenzoni R, Gistri R, Cecchi F et al (1998) Coronary vasodilator reserve is impaired in patients with hypertrophic cardiomyopathy and left ventricular dysfunction. Am Heart J 136:972–981. https://doi.org/10.1016/S0002-8703(98)70152-8
Lu D-Y, Yalçin H, Yalçin F et al (2018) Stress Myocardial Blood Flow Heterogeneity Is a Positron Emission Tomography Biomarker of Ventricular Arrhythmias in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol 121:1081–1089. https://doi.org/10.1016/j.amjcard.2018.01.022
Tian H, Yang C, Song Y et al (2018) Microvascular Rarefaction and Myocardial Fibrosis in Hypertrophic Obstructive Cardiomyopathy. Cardiology 141:202–211. https://doi.org/10.1159/000493005
Galati G, Leone O, Pasquale F et al (2016) Histological and histometric characterization of myocardial fibrosis in end-stage hypertrophic cardiomyopathy. Circ Heart Fail. https://doi.org/10.1161/CIRCHEARTFAILURE.116.003090
Choudhury L, Mahrholdt H, Wagner A et al (2002) Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 40:2156–2164. https://doi.org/10.1016/S0735-1097(02)02602-5
Shiozaki AA, Senra T, Arteaga E et al (2010) Myocardial fibrosis in patients with hypertrophic cardiomyopathy and high risk for sudden death. Arq Bras Cardiol 94:535–540. https://doi.org/10.1590/s0066-782x2010005000017
Deviggiano A, Carrascosa P, Zan MD et al (2016) Wall Thickness and Patterns of Fibrosis in Hypertrophic Cardiomyopathy Assessed by Cardiac Magnetic Resonance Imaging. Rev Argent Cardiol 84:208–214. https://doi.org/10.7775/RAC.84.3.7168
Bruder O, Wagner A, Jensen CJ et al (2010) Myocardial Scar Visualized by Cardiovascular Magnetic Resonance Imaging Predicts Major Adverse Events in Patients With Hypertrophic Cardiomyopathy. J Am Coll Cardiol 56:875–887. https://doi.org/10.1016/j.jacc.2010.05.007
Sotgia B, Sciagrà R, Olivotto I et al (2008) Spatial Relationship Between Coronary Microvascular Dysfunction and Delayed Contrast Enhancement in Patients with Hypertrophic Cardiomyopathy. J Nucl Med 49:1090. https://doi.org/10.2967/jnumed.107.050138
Tezuka D, Kosuge H, Terashima M et al (2018) Myocardial perfusion reserve quantified by cardiac magnetic resonance imaging is associated with late gadolinium enhancement in hypertrophic cardiomyopathy. Heart Vessels 33:513–520. https://doi.org/10.1007/s00380-017-1088-y
Esposito A, de Cobelli F, Perseghin G et al (2009) Impaired left ventricular energy metabolism in patients with hypertrophic cardiomyopathy is related to the extension of fibrosis at delayed gadolinium-enhanced magnetic resonance imaging. Heart 95:228. https://doi.org/10.1136/hrt.2008.142562
Teekakirikul P, Eminaga S, Toka O et al (2010) Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J Clin Invest 120:3520–3529. https://doi.org/10.1172/JCI42028
Ando K, Nagao M, Watanabe E et al (2020) Association between myocardial hypoxia and fibrosis in hypertrophic cardiomyopathy: analysis by T2* BOLD and T1 mapping MRI. Eur Radiol 30:4327–4336. https://doi.org/10.1007/s00330-020-06779-9
Kaimoto S, Kawasaki T, Kuribayashi T et al (2012) Myocardial perfusion abnormality in the area of ventricular septum-free wall junction and cardiovascular events in nonobstructive hypertrophic cardiomyopathy. Int J Card Imaging 28:1829–1839. https://doi.org/10.1007/s10554-011-9994-z
Camici P, Chiriatti G, Lorenzoni R et al (1991) Coronary vasodilation is impaired in both hypertrophied and nonhypertrophied myocardium of patients with hypertrophic cardiomyopathy: A study with nitrogen-13 ammonia and positron emission tomography. J Am Coll Cardiol 17:879–886. https://doi.org/10.1016/0735-1097(91)90869-B
Kim W-S, Minagoe S, Mizukami N et al (2008) No reflow-like pattern in intramyocardial coronary artery suggests myocardial ischemia in patients with hypertrophic cardiomyopathy. J Cardiol 52:7–16. https://doi.org/10.1016/j.jjcc.2008.04.004
Lazzeroni E, Picano E, Morozzi L et al (1997) Dipyridamole-Induced Ischemia as a Prognostic Marker of Future Adverse Cardiac Events in Adult Patients With Hypertrophic Cardiomyopathy. Circulation 96:4268–4272. https://doi.org/10.1161/01.CIR.96.12.4268
Gommans F, Bakker J, Cramer E et al (2013) Elevated high-sensitivity cardiac troponin is associated with hypertrophy and fibrosis assessed with CMR in patients with hypertrophic cardiomyopathy. J Cardiovasc Magn Reson 15:P144. https://doi.org/10.1186/1532-429X-15-S1-P144
Ashrafian H, Redwood C, Blair E, Watkins H (2003) Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet 19:263–268. https://doi.org/10.1016/S0168-9525(03)00081-7
Jung W-I, Sieverding L, Breuer J et al (1998) 31P NMR Spectroscopy Detects Metabolic Abnormalities in Asymptomatic Patients With Hypertrophic Cardiomyopathy. Circulation 97:2536–2542. https://doi.org/10.1161/01.CIR.97.25.2536
Sequeira V, Bertero E, Maack C (2019) Energetic drain driving hypertrophic cardiomyopathy. FEBS Lett 593:1616–1626. https://doi.org/10.1002/1873-3468.13496
Olivotto I, Cecchi F, Poggesi C, Yacoub MH (2009) Developmental origins of hypertrophic cardiomyopathy phenotypes: a unifying hypothesis. Nat Rev Cardiol 6:317–321. https://doi.org/10.1038/nrcardio.2009.9
Coppini R, Ferrantini C, Mugelli A et al (2018) Altered Ca(2+) and Na(+) Homeostasis in Human Hypertrophic Cardiomyopathy: Implications for Arrhythmogenesis. Front Physiol 9:1391. https://doi.org/10.3389/fphys.2018.01391
Youn H-J, Lee J-M, Park C-S et al (2005) The impaired flow reserve capacity of penetrating intramyocardial coronary arteries in apical hypertrophic cardiomyopathy. J Am Soc Echocardiogr 18:128–132. https://doi.org/10.1016/j.echo.2004.08.043
Gutiérrez Díez A, Tamariz-Martel Moreno A, Baño Rodrigo A, Serrano González A (2000) Muerte súbita y fibrilación ventricular de posible origen isquémico en un niño con miocardiopatía hipertrófica. Rev Esp Cardiol 53:290–293
Zhou X-L, Xiao Y, Yang K-Q et al (2015) Clinical Characteristics and Prognosis of End-stage Hypertrophic Cardiomyopathy. Chin Med J (Engl) 128:1483. https://doi.org/10.4103/0366-6999.157656
Olivotto I, Cecchi F, Gistri R et al (2006) Relevance of Coronary Microvascular Flow Impairment to Long-Term Remodeling and Systolic Dysfunction in Hypertrophic Cardiomyopathy. J Am Coll Cardiol 47:1043–1048. https://doi.org/10.1016/j.jacc.2005.10.050
Musumeci B, Tini G, Russo D et al (2021) Left Ventricular Remodeling in Hypertrophic Cardiomyopathy: An Overview of Current Knowledge. J Clin Med 10:1547. https://doi.org/10.3390/jcm10081547
Castagnoli H, Ferrantini C, Coppini R et al (2016) Role of quantitative myocardial positron emission tomography for risk stratification in patients with hypertrophic cardiomyopathy: a 2016 reappraisal. Eur J Nucl Med Mol Imaging 43:2413–2422. https://doi.org/10.1007/s00259-016-3465-7
Freitas P, Ferreira AM, Arteaga-Fernández E et al (2019) The amount of late gadolinium enhancement outperforms current guideline-recommended criteria in the identification of patients with hypertrophic cardiomyopathy at risk of sudden cardiac death. J Cardiovasc Magn Reson 21:50. https://doi.org/10.1186/s12968-019-0561-4
Yetman AT, McCrindle BW, MacDonald C et al (1998) Myocardial Bridging in Children with Hypertrophic Cardiomyopathy — A Risk Factor for Sudden Death. N Engl J Med 339:1201–1209. https://doi.org/10.1056/NEJM199810223391704
Mohiddin SA, Begley D, Shih J, Fananapazir L (2000) Myocardial bridging does not predict sudden death in children with hypertrophic cardiomyopathy but is associated with more severe cardiac disease. J Am Coll Cardiol 36:2270–2278. https://doi.org/10.1016/S0735-1097(00)00987-6
Sorajja P, Ommen SR, Nishimura RA et al (2003) Myocardial bridging in adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 42:889–894. https://doi.org/10.1016/S0735-1097(03)00854-4
Nishimura T, Nagata S, Uehara T et al (1996) Prognosis of hypertrophic cardiomyopathy: assessment by 123I-BMIPP (beta-methyl-p-(123I)iodophenyl pentadecanoic acid) myocardial single photon emission computed tomography. Ann Nucl Med 10:71–78. https://doi.org/10.1007/bf03165056
Makoto A, Toshiharu T, Naka S et al (2013) Microcirculatory dysfunction determines the long-term prognosis in asymptomatic patients with non-obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 61:E1308–E1308. https://doi.org/10.1016/S0735-1097(13)61308-X
Quarta G, Iacovoni A, Marrone C et al (2015) Microvascular ischaemia after cardiac arrest in a patient with hypertrophic cardiomyopathy. Glob Cardiol Sci Pract 2015:51. https://doi.org/10.5339/gcsp.2015.51
Rochelson E, Nappo L, Pass RH (2018) Hypertrophic cardiomyopathy: Ischemia progressing to ventricular fibrillation. Hear Case Rep 4:386–388. https://doi.org/10.1016/j.hrcr.2018.03.004
Magnusson P, Nordström J, Harms HJ et al (2020) Positron emission tomography (15O-water, 11C-acetate, 11C-HED) risk markers and nonsustained ventricular tachycardia in hypertrophic cardiomyopathy. IJC Heart Vasc. https://doi.org/10.1016/j.ijcha.2019.100452
Lorenzoni R, Gistri R, Cecchi F et al (1997) Syncope and ventricular arrhythmias in hypertrophic cardiomyopathy are not related to the derangement of coronary microvascular function. Eur Heart J 18:1946–1950. https://doi.org/10.1093/oxfordjournals.eurheartj.a015204
Sciagrà R, Sotgia B, Olivotto I et al (2009) Relationship between atrial fibrillation and blunted hyperemic myocardial blood flow in patients with hypertrophic cardiomyopathy. J Nucl Cardiol 16:92–96. https://doi.org/10.1007/s12350-008-9005-5
Komuro J, Iguchi N, Utanohara Y et al (2021) Prediction of Serious Adverse Events of Patients with Hypertrophic Cardiomyopathy by Magnetic Resonance. Int Heart J 62:135–141. https://doi.org/10.1536/ihj.20-479
Elliott PM, Gimeno JR, Tomé MT et al (2006) Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur Heart J 27:1933–1941. https://doi.org/10.1093/eurheartj/ehl041
Norrish G, Ding T, Field E et al (2020) The relationship between left ventricular outflow tract gradient and sudden cardiac death in childhood hypertrophic cardiomyopathy. Eur Heart J 41(ehaa946):0732. https://doi.org/10.1093/ehjci/ehaa946.0732
Weissler-Snir A, Allan K, Cunningham K et al (2019) Hypertrophic Cardiomyopathy-Related Sudden Cardiac Death in Young People in Ontario. Circulation 140:1706–1716. https://doi.org/10.1161/CIRCULATIONAHA.119.040271
Rodrigues T, Raposo SC, Brito D, Lopes LR (2021) Prognostic relevance of exercise testing in hypertrophic cardiomyopathy. A systematic review Int J Cardiol 339:83–92. https://doi.org/10.1016/j.ijcard.2021.06.051
Ullal AJ, Abdelfattah RS, Ashley EA, Froelicher VF (2016) Hypertrophic Cardiomyopathy as a Cause of Sudden Cardiac Death in the Young: A Meta-Analysis. Am J Med 129:486-496.e2. https://doi.org/10.1016/j.amjmed.2015.12.027
D’Ascenzi F, Valentini F, Pistoresi S et al (2021) Causes of sudden cardiac death in young athletes and non-athletes: systematic review and meta-analysis: Sudden cardiac death in the young. Trends Cardiovasc Med 32:299–308. https://doi.org/10.1016/j.tcm.2021.06.001
Drezner JA, Malhotra A, Prutkin JM et al (2021) Return to play with hypertrophic cardiomyopathy: are we moving too fast? A critical review. Br J Sports Med 55:1041. https://doi.org/10.1136/bjsports-2020-102921
Gimeno JR, Tomé-Esteban M, Lofiego C et al (2009) Exercise-induced ventricular arrhythmias and risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Eur Heart J 30:2599–2605. https://doi.org/10.1093/eurheartj/ehp327
Magrì D, Limongelli G, Re F et al (2016) Cardiopulmonary exercise test and sudden cardiac death risk in hypertrophic cardiomyopathy. Heart 102:602. https://doi.org/10.1136/heartjnl-2015-308453
Coats CJ, Rantell K, Bartnik A et al (2015) Cardiopulmonary Exercise Testing and Prognosis in Hypertrophic Cardiomyopathy. Circ Heart Fail 8:1022–1031. https://doi.org/10.1161/CIRCHEARTFAILURE.114.002248
Timmer SAJ, Knaapen P, Germans T et al (2011) Effects of alcohol septal ablation on coronary microvascular function and myocardial energetics in hypertrophic obstructive cardiomyopathy. Am J Physiol Heart Circ Physiol 301:H129–H137. https://doi.org/10.1152/ajpheart.00077.2011
Jörg-Ciopor M, Namdar M, Turina J et al (2004) Regional myocardial ischemia in hypertrophic cardiomyopathy: Impact of myectomy. J Thorac Cardiovasc Surg 128:163–169. https://doi.org/10.1016/j.jtcvs.2003.11.003
Penicka M, Gregor P, Kerekes R et al (2009) The effects of candesartan on left ventricular hypertrophy and function in nonobstructive hypertrophic cardiomyopathy: a pilot, randomized study. J Mol Diagn 11:35–41. https://doi.org/10.2353/jmoldx.2009.080082
Maqsood H, Shakeel HA, Shoukat HF et al (2020) Effects of candesartan on left ventricular hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. Eur Heart J 41(ehaa946):3360. https://doi.org/10.1093/ehjci/ehaa946.3360
Ho CY, Day SM, Axelsson A et al (2021) Valsartan in early-stage hypertrophic cardiomyopathy: a randomized phase 2 trial. Nat Med 27:1818–1824. https://doi.org/10.1038/s41591-021-01505-4
Ananthakrishna R, Lee SL, Foote J et al (2021) Randomized controlled trial of perhexiline on regression of left ventricular hypertrophy in patients with symptomatic hypertrophic cardiomyopathy (RESOLVE-HCM trial). Am Heart J 240:101–113. https://doi.org/10.1016/j.ahj.2021.06.010
Cogolludo A, Villamor E, Perez-Vizcaino F, Moreno L (2019) Ceramide and Regulation of Vascular Tone. Int J Mol Sci. https://doi.org/10.3390/ijms20020411
Kovilakath A, Cowart LA (2020) Sphingolipid Mediators of Myocardial Pathology. J Lipid Atheroscler 9:23–49
Coppini R, Ferrantini C, Pioner Josè M et al (2019) Electrophysiological and Contractile Effects of Disopyramide in Patients With Obstructive Hypertrophic Cardiomyopathy. J Am Coll Cardiol Basic Trans Science 4:795–813. https://doi.org/10.1016/j.jacbts.2019.06.004
Olivotto I, Camici PG, Merlini PA et al (2018) Efficacy of Ranolazine in Patients With Symptomatic Hypertrophic Cardiomyopathy. Circ Heart Fail. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004124
Argirò A, Zampieri M, Dei L-L et al (2022) Safety and efficacy of ranolazine in hypertrophic cardiomyopathy: real-world experience in a National Referral Center. Int J Cardiol. https://doi.org/10.1016/j.ijcard.2022.10.014
Olivotto I, Hellawell JL, Farzaneh-Far R et al (2016) Novel Approach Targeting the Complex Pathophysiology of Hypertrophic Cardiomyopathy. Circ Heart Fail. https://doi.org/10.1161/CIRCHEARTFAILURE.115.002764
Sherrid MV, Barac I, McKenna WJ et al (2005) Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 45:1251–1258. https://doi.org/10.1016/j.jacc.2005.01.012
Palandri C, Santini L, Argirò A et al (2022) Pharmacological Management of Hypertrophic Cardiomyopathy: from Bench to Bedside. Drugs 82:889–912. https://doi.org/10.1007/s40265-022-01728-w
Olivotto I, Oreziak A, Barriales-Villa R et al (2020) Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 396:759–769. https://doi.org/10.1016/S0140-6736(20)31792-X
Pysz P, Rajtar-Salwa R, Smolka G et al (2021) Mavacamten - a new disease-specific option for pharmacological treatment of symptomatic patients with hypertrophic cardiomyopathy. Kardiol Pol 79:949–954
Ho CY, Mealiffe ME, Bach RG et al (2020) Evaluation of Mavacamten in Symptomatic Patients With Nonobstructive Hypertrophic Cardiomyopathy. J Am Coll Cardiol 75:2649–2660. https://doi.org/10.1016/j.jacc.2020.03.064
Conway J, Min S, Villa C et al (2022) The Prevalence and Association of Exercise Test Abnormalities with Sudden Cardiac Death and Transplant-Free Survival in Childhood Hypertrophic Cardiomyopathy. Circulation 147:718–727. https://doi.org/10.1161/CIRCULATIONAHA.122.062699
Funding
JAC is supported by a scholarship from the UK Engineering and Physical Sciences Research Council (2421745). ZA is a recipient of a Clinical Research Training Fellowship from the British Heart Foundation (BHF) (FS/CRTF/21/24144). BR is funded by the Oxford BHF Centre of Research Excellence (RE/18/3/34214). ABO acknowledges support from a BHF Intermediate Basic Science Fellowship (FS/17/22/32644). The authors acknowledge additional support from the Oxford BHF Centre of Research Excellence (RE/13/1/30181). For the purpose of Open Access, the authors have applied a CC BY public copyright license to any Author Accepted Manuscript (AAM) version arising from this submission.
Author information
Authors and Affiliations
Contributions
JAC, BR and ABO outlined, drafted, and contributed to the writing of the manuscript. ZA supplied perfusion imaging figure, gave critical feedback, and contributed to revisions of the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Coleman, J.A., Ashkir, Z., Raman, B. et al. Mechanisms and prognostic impact of myocardial ischaemia in hypertrophic cardiomyopathy. Int J Cardiovasc Imaging 39, 1979–1996 (2023). https://doi.org/10.1007/s10554-023-02894-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10554-023-02894-y