
Abstract
Background
Remethylation defects are rare inherited disorders in which impaired remethylation of homocysteine to methionine leads to accumulation of homocysteine and perturbation of numerous methylation reactions.
Objective
To summarise clinical and biochemical characteristics of these severe disorders and to provide guidelines on diagnosis and management.
Data sources
Review, evaluation and discussion of the medical literature (Medline, Cochrane databases) by a panel of experts on these rare diseases following the GRADE approach.
Key recommendations
We strongly recommend measuring plasma total homocysteine in any patient presenting with the combination of neurological and/or visual and/or haematological symptoms, subacute spinal cord degeneration, atypical haemolytic uraemic syndrome or unexplained vascular thrombosis. We strongly recommend to initiate treatment with parenteral hydroxocobalamin without delay in any suspected remethylation disorder; it significantly improves survival and incidence of severe complications. We strongly recommend betaine treatment in individuals with MTHFR deficiency; it improves the outcome and prevents disease when given early.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
This guideline development process was initiated within the frame of the “European network and registry for homocystinurias and methylation defects” (E-HOD) project, which was started in February 2013. The main aims of the project are the formation of a sustainable international collaboration of experts and clinically active centres, development of guidelines and establishment of a disease registry for homocystinurias and methylation defects. Given the often significant delays in diagnosis and the absence of standardised treatment protocols, the evaluation of the published knowledge and delineation of guidelines for diagnosis and treatment of these rare diseases are urgently needed. Most of the existing studies and reports are non-systematic, observational studies, case series or case reports, which are generally considered to be low quality evidence. However, following collation of the available evidence some very consistent patterns evolved. Confirmation and validation of our observations by insights gained from other fields of research (e.g. vitamin B12 deficiency in the elderly) additionally informed our interpretation of the evidence.
Homocysteine (Hcy) is an amino acid formed from methionine (Met). Under normal circumstances Hcy is converted into cysteine (transulfuration pathway) or remethylated (remethylation pathway) forming Met.
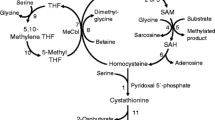
All genetic remethylation defects share deficient activity of methionine synthase due to various reasons: decreased function of the methionine synthase enzyme protein itself or of the associated enzyme methionine synthase reductase; deficient production of the cofactor, methyl-cobalamin; or disturbed supply of the substrate methyl-tetrahydrofolate (MTHF). Some genetic disorders of intracellular cobalamin (cbl) transport and processing (cblC, cblD-MMA/Hcy, cblF and cblJ) cause deficient synthesis not only of methylcobalamin but also of adenosylcobalamin, the cofactor for methylmalonyl-CoA mutase. Those combined remethylation disorders are associated with increased Hcy and methylmalonic acid (MMA). 5,10-methylenetetrahydrofolate reductase (MTHFR) deficiency leads to impaired provision of 5-MTHF resulting in decreased function of methionine synthase (Fowler 2005) (Fig. 1)

Remethylation disorders: metabolic pathways. MTHFR methylenetetrahydrofolate reductase, THF tetrahydrofolate, Gly glycine, Ser serine, DHF dihydrohydrofolate, Met methionine, Hcy homocysteine, AdoMet adenosylmethionine, AdoHcy adenosylhomocysteine, MS methionine synthase, MSR methionine synthase reductase, BHMT betaine homocysteine methyltransferase, CBS cystathionine-beta-synthase, DMG dimethylglycine, MeCbl methylcobalamin, AdoCbl adenosyl-Cbl
.
Our understanding of the pathophysiology of the remethylation disorders and MTHFR deficiency is incomplete. At present, there are five main hypotheses: (A) direct toxicity of metabolites, (B) missing products, (C) impaired methylation capacity, (D) oxidative stress, (E) impaired non-enzymatic protein functions.
-
(A)
Hyperhomocysteinaemias are predominantly associated with neurocognitive and vascular pathology. Homocysteine and its metabolic product, homocysteic acid, have been implicated in provocation of seizures in rats (Kubová et al 1995; Mares et al 1997). Besides its impact on renal function, accumulation of MMA along with dicarboxylic acids like 2-methylcitrate and propionyl-CoA have been found to induce synergistic mitochondrial dysfunction (Morath et al 2008; Zsengellér et al 2014).
-
(B)
Impaired methionine synthase activity with accumulation of intracellular 5-MTHF (folate trap) and subsequent disruption of nucleotide synthesis compromise rapidly proliferating tissues such as bone marrow or epithelia. In MTHFR deficiency, this trapping of folates is not found but 5-methylTHF synthesis is impaired, resulting in low levels of methyl-tetrahydrofolate (CH3-THF) in the central nervous system of unknown significance (Schiff and Blom 2012).
-
(C)
Remethylation of homocysteine (Hcy) is an important mechanism that maintains the methylation capacity of the organism and removes excess Hcy. Methylation reactions such as DNA methylation, creatine and myelin synthesis utilise S-adenosylmethionine (SAM), a metabolite formed from Met as the methyl group donor. Accordingly, any disorder or disruption of the Met-Hcy-SAM pathway affects methylation capacity by impairing multiple metabolic systems and processes (King et al 2012; Surtees et al 1991).
-
(D)
Enhanced oxidative stress due to a disturbance of glutathione metabolism has been shown in patients with the cblC defect and may contribute to the underlying pathophysiology (Pastore et al 2014).
-
(E)
Recent data suggest that not only the accumulation of metabolites but also the perturbed non-enzymatic functions, e.g. of the MMACHC protein may add to the pathophysiology behind the disease (Moreno-Garcia et al 2014; Brooks et al 2016).
Methods
Panelists involved in the guideline process were invited by the EHOD project leaders according to their expertise regarding the diseases in terms of biochemistry; genetics; clinical, psychosocial and laboratory diagnosis and patient management and/or research activities.
The PubMed and Cochrane databases were searched using the following terms: remethylation defects OR severe mthfr OR severe methylenetetrahydrofolate reductase OR cblc OR cbld OR cblf OR cblj OR cble OR cblg OR methylmalonic aciduria homocystinuria OR methylmalonic aciduria homocystinuria OR methylmalonic acidemia homocysteinemia OR methylmalonic aciduria homocysteinemia OR “Cobalamin C OR Cobalamin D OR Cobalamin E OR Cobalamin F OR Cobalamin G OR Cobalamin J” (last update February 2016: 834 publications).
The resulting list of publications was disposed of all articles predominantly or only addressing other diseases (e.g. classical homocystinuria) or MTHFR polymorphisms (e.g. C677T) or without information relevant to the outcomes of interest (e.g. mutation reports only) by screening of abstracts. In total, 174 publications were included in the qualitative data analysis. The clinical characterisation of the diseases (Table 1) was based on 117 publications.
The following main questions and outcomes of interest in males and females with any type of onset (early or late) of one of the diseases of interest were addressed (outcomes in parentheses).
-
Which clinical signs are characteristic and allow timely diagnosis (Outcome: timely clinical diagnosis; this outcome is considered important)
-
Which biochemical parameters allow timely and valid diagnosis? (Outcome: valid, timely laboratory diagnosis; this outcome is considered important)
-
How can we prevent death and avoid/treat severe organ damage (Outcome: survival, severe organ complications; this outcome is considered critical)
-
How can we prevent or treat eye disease and neurocognitive impairment? (Outcome: visual and neurocognitive function; this outcome is considered critical)
In a first step, all reports made available to the panelists were graded according to the Scottish Intercollegiate Guidelines Network (SIGN) criteria by the authors who had assigned themselves according to their expertise to five major working groups focusing on the topics “clinical signs and symptoms”; “differential diagnosis”; “laboratory/biochemical parameters”; “disease course and outcome”; “treatment”. Due to the rarity of the diseases, studies and reports of all types of designs (from case reports and case series to metaanalyses) were included; randomised controlled trials were not available.
The working groups prepared drafts on their topics, which were carefully evaluated in moderated discussion groups. Elaboration and grading of recommendations was accomplished by moderated, consensus-oriented discussions according to the “Grading of Recommendations Assessment, Development and Evaluation (GRADE) working group” approach (http://www.gradeworkinggroup.org/; Guyatt et al 2011a, b, c, 2012); a patient representative was present at these meetings. Finally, for each outcome the quality of the evidence was rated in one of three categories (low, moderate, high) as defined by the GRADE working group. The strength of a recommendation (strong recommendation, recommendation and suggestion; for more information see Supplementary material) was summarised from written assessments from the panelist using the following set of questions:
-
Are you confident that the benefits outweigh the harms/burden or vice versa?
-
Is there high, moderate or low quality evidence? Please consider:
-
Risk of bias
-
Study design
-
Directness and consistency of results
-
Magnitude of effect
-
Dose–response gradient
-
Publication bias
-
-
Are you confident that the recommendation meets typical values and preferences of the target population (e.g. patients, parents)?
-
Are the resources worth the expected net benefit following the recommendation?
-
Overall strength of recommendation: weak or strong?
Data derived from case reports, case series or any other type of observational studies were by principle rated as low quality evidence. However, evidence for some outcomes was considered moderate on the basis of high consistency of results. High quality of evidence was only available for betaine treatment in MTHFR deficiency. This evidence was based on a metaanalysis, which revealed direct and consistent results as well as a relationship between time of onset of treatment and outcome.
Which clinical signs are characteristic for remethylation defects and allow for timely diagnosis?
General clinical patterns of remethylation disorders
The published clinical presentations of patients suffering from remethylation disorders (summarised in Table 1) reveal no specific, distinct signs and symptoms but rather highlight that these conditions affect multiple systems and thus often present in a rather multifaceted manner. Nevertheless, a few patterns of clinical presentations can be identified.
Most prominently, the central and peripheral nervous system and the bone marrow are affected. Developmental and neurocognitive impairment, feeding problems, neurological symptoms including seizures, movement disorders, abnormal muscle tone, visual impairment, neuropathy and haematological abnormalities are present in the majority of patients. In many patients, renal manifestations, e.g. atypical haemolytic uraemic syndrome (HUS) or glomerulopathy, mostly related to microangiopathy, have been identified.
Clinical presentations may vary considerably. Severe acute encephalopathy, macrocytic anaemia, atypical HUS, cardiopulmonary signs, subacute combined degeneration of the cord or psychiatric symptoms, each isolated or in combination with other symptoms may be present. Therefore, in particular, a combination of neurological and haematological symptoms, often in the presence of failure to thrive or feeding difficulties should raise suspicion of a remethylation disorder (see references to Table 1).
Age specific patterns of remethylation disorders
Findings from published case reports indicate that the patterns of clinical manifestation of remethylation disorders vary with age (Table 2).
Clinical patterns specific for combined remethylation disorders (cblC, cblD-MMA/HC, cblF, cblJ)
Severe intrauterine growth retardation is rare, whereas postnatal growth failure and feeding difficulties are very frequent. Three quarters of children with combined disorders manifest during the neonatal period or in early infancy. Neonates typically show lethargy, seizures and muscular hypotonia, often in combination with megaloblastic anaemia or neutropenia/pancytopenia. Up to 50 % of infants exhibit signs of visual impairment such as nystagmus on the background of retinopathy or optic atrophy (Bonafede et al 2015; Weisfeld-Adams et al 2015; Brooks et al 2016). Microangiopathic renal or pulmonary disease mostly manifests early but may also be present in older children or adults (Grangé et al 2015; Kömhoff et al 2013; Sharma et al 2007).
Older infants and young children often show signs of acute encephalopathy; visual and cognitive impairment are prevalent. Older children, adolescents and adults may present with acute or chronic behavioural or psychiatric abnormalities, cognitive impairment, signs of peripheral neuropathy and ataxia that reflect subacute degeneration of the spinal cord or rarely venous thromboembolism (Rahmandar et al 2014; Huemer et al 2014; Grangé et al 2015). Other than optic pallor, ocular manifestations are rare in late-onset cblC (Gizicki et al 2014; Weisfeld-Adams et al 2015).
Clinical patterns specific for isolated remethylation disorders (cblD-HC, cblE, cblG)
The less prevalent isolated disorders of methionine synthase generally present similar to combined disorders. The typical clinical patterns of the cblE and the cblG defect are indistinguishable. Affected individuals usually manifest in infancy, mostly with haematological abnormalities, muscular hypotonia and neurocognitive impairment. One third of all patients present with impaired consciousness, seizures, and one quarter with signs of visual impairment. Older children and adults can manifest with signs reflecting spinal cord involvement (subacute degeneration of the spinal cord) or psychiatric symptoms (Huemer et al 2015a). Single cases with isolated macrocytic anaemia without neurocognitive impairment have been reported (Vilaseca et al 2003; Ruiz-Mercado et al 2015).
Clinical patterns specific for MTHFR deficiency
MTHFR deficiency mainly presents in early childhood but can potentially present at any age with a high prevalence of cognitive impairment. Neonates and young infants often manifest with feeding difficulties, decreased consciousness, hydrocephalus and muscular hypotonia. Apnoea is a frequent complication (Broomfield et al 2014; Diekman et al 2014; Huemer et al 2016; Schiff et al 2011; Strauss et al 2007).
Older infants and children frequently manifest with seizures and cognitive impairment, often displaying acquired microcephaly. With increasing age, peripheral neuropathy, gait abnormalities and spasticity may become evident and some older patients manifest with additional or even isolated behavioural or psychiatric disorders (Birnbaum et al 2008; Michot et al 2008).
Individuals with MTHFR deficiency do not usually present with haematological abnormalities. There are just a few reports of older children and adults with megaloblastic anaemia in association with long-standing neurological manifestations (Broomfield et al 2014; Schiff et al 2011).
Outcome: timely clinical diagnosis
-
Recommendation 1:
We strongly recommend consideration of an acquired or genetic disorder of remethylation in the case of neurological and/or visual and/or haematological symptoms. (Quality of the evidence: moderate)
-
Recommendation 2:
We strongly recommend considering an acquired or genetic disorder of remethylation in the case of unexplained thrombosis and/or spinal cord degeneration and/or atypical HUS. (Quality of the evidence: moderate)
Clinical differential diagnosis
All clinical presentations of genetic disorders of remethylation can be mimicked by common conditions, which reduce the availability of folate or cobalamin such as nutritional deficiency, acquired malabsorption or drug-associated effects (Table 3). Maternal vitamin B12 deficiency due to a strict vegan diet or to undiagnosed pernicious anaemia needs to be excluded in neonates and young infants presenting with signs of a remethylation disorder or identified by newborn screening (NBS) and subnormal vitamin B12 concentrations (Scolamiero et al 2014).
A number of other rare genetic diseases can mimic defects in intracellular cobalamin metabolism and should be considered in children with clinical signs and biochemical hallmarks of a remethylation disorder but with normal serum concentrations of vitamin B12 and folates: transcobalamin (TC) deficiency is biochemically and clinically indistinguishable from combined cobalamin disorders (Trakadis et al 2014). The recently discovered MTHFD1 deficiency (Watkins and Rosenblatt 2012) affects folate metabolism and can cause mild hyperhomocysteinaemia. The five patients described so far displayed severe megaloblastic anaemia and pancytopenia, immunological problems and renal microangiopathy (Burda et al 2015).
The HCFC1 X-linked defect (cbl X, Yu et al 2013) is sometimes associated with disturbed function of the CblC protein but its diagnosis can be challenging because not all affected boys have elevated homocysteine concentrations. Clinical hallmarks are neonatal or early infantile onset of severe developmental disorder, prominent epileptic seizures, cognitive impairment and a dystonic-choreatic movement disorder; moderately elevated MMA was observed in all 14 patients reported by Yu et al 2013 (Table 4).
Which parameters allow valid and timely laboratory diagnosis?
Patients with a suspected remethylation disorder require immediate and urgent biochemical investigation (Fig. 2). The diagnosis of remethylation disorders can be confirmed by investigations at the levels of metabolites, enzymatic studies and/or molecular genetic analysis.

Diagnostic pathway and management of the patient with a suspected remethylation disorder. All biochemical results refer to pre-treatment samples. N = normal
Biochemical metabolites
Plasma total homocysteine (tHcy) is the first biochemical parameter to assess when a remethylation disorder is suspected. THcy levels usually are >50 but mostly >100 μmol/L in untreated patients with remethylation disorders (Carrillo-Carrasco et al 2009; Van Hove et al 2002; D’Aco et al 2014; Miousse et al 2009).
Routinely, tHcy is measured by chromatographic methods or immunoassays. Chromatographic methods are more specific than immunoassays and are usually coupled with tandem mass spectrometry (MS/MS) detection allowing simultaneous determination of other compounds of interest for diagnosis and follow up of these diseases, e.g. Met, cysteine (Refsum et al 2004) or possibly cystathionine (Stabler et al 1993; Stabler et al 2013).
There is no influence of the type of collection tube used (EDTA, heparinised, citrate, serum, gel separator tubes). However, blood samples should be centrifuged within 1 h or kept cold until centrifugation (<6 h). After removal of blood cells, tHcy is stable for several weeks at 4 °C and for several years after freezing (Refsum et al 2004).
Free homocystine in plasma or urine measured by conventional ion-exchange chromatography is not the method of choice since it may remain undetectable even in the presence of significantly elevated tHcy in plasma (Fowler and Jakobs 1998; Moat et al 1999).
To further discriminate remethylation disorders from other hyperhomocysteinaemias, determination of urinary (or plasma) MMA (Fowler and Jakobs 1998), plasma Met, blood acylcarnitine profile, serum vitamin B12 and folate are required and should promptly be determined (see Figs. 1 and 2). In case shipment of urine or blood samples is difficult, an experienced laboratory should give advice on the investigation of the parameters from DBS. Treatment should not be delayed by waiting for confirmation of the exact defect.
Outcome: valid, timely laboratory diagnosis
-
Recommendation 3
-
Elevated plasma tHcy is the hallmark of remethylation disorders. We strongly recommend that investigations in patients with a suspected remethylation disorder should start with the measurement of total homocysteine in blood. We recommend the blood sample for tHcy to be centrifuged within an hour and kept at +4° or frozen until analysis. Immunoassays or chromatographic methods are suitable for tHcy measurement. (Quality of the evidence: moderate)
-
-
Recommendation 4
-
We strongly recommend against measuring free homocysteine instead of total homocysteine. (Quality of the evidence: moderate)
-
-
Recommendation 5
-
We strongly recommend that in the case of high total homocysteine, plasma and urine samples for determination of MMA, methionine, folate and vitamin B12 are to be obtained before treatment is started. (Quality of the evidence: moderate)
-
Differential diagnosis of biochemical parameters
Hyperhomocysteinaemia is the hallmark of the remethylation and transulfuration disorders. Mild to moderate hyperhomocysteinaemia is common in vitamin B12 or folate deficiencies but also in very severe vitamin B6 deficiency and in patients with renal failure or hypothyroidism. While megaloblastic anaemia with increased MCV and hypersegmented neutrophils on blood film are found in both vitamin B12 and folate deficiencies, elevated MMA is only observed in functional vitamin B12 deficiency (Whitehead 2006).
In the case of normal serum vitamin B12 and folate levels, TC deficiency, primary remethylation and transulfuration disorders should be considered. Holotranscobalamin measurement, which unfortunately is performed only in a few laboratories shows low levels in TC deficiency; this disorder can be confirmed by molecular genetics investigation (Trakadis et al 2014).
The plasma Met concentration differentiates between remethylation disorders and deficiency of CBS, a key enzyme of the transulfuration pathway. Methionine is low or normal in remethylation disorders while it is usually elevated or at least borderline normal in CBS deficiency (Mudd 2011).
Megaloblastic anaemia with increased MCV is often observed in the neonatal forms of intracellular cobalamin defects (cblF, cblJ, cblC, cblD-MMA/HC, cblD-HC, cblE and cblG) but less frequently in the late onset forms of these disorders and not normally in MTHFR deficiency (Rosenblatt et al 1997; Carrillo-Carrasco et al. 2013).
The recently described HCFC1 defect (cbl X) has a different pathophysiological background and is beyond the scope of these guidelines. All 14 patients presented with moderately increased MMA and five of these patients had hyperhomocysteinaemia (Yu et al 2013). In two of the five published patients with methylenetetrahydrofolate dehydrogenase 1 deficiency, tHcy concentrations were moderately elevated (Burda et al 2015) (Table 4).
Enzymatic studies
Direct enzyme assays are only available for MTHFR, methionine synthase and methionine synthase reductase (Suormala et al 2002; Gulati et al 1997; Olteanu and Banerjee 2001). These direct enzyme assays can be problematic. MTHFR should be assayed in cultured skin fibroblasts preferably in the physiological direction of the reaction (Suormala et al 2002). Apo-methionine synthase and holo-methionine synthase (methionine synthase plus methionine synthase reductase) activities can be measured by the same reaction but with different conditions of reduction (Yamada et al 2006).
Indirect assays measuring the integrity of several enzymes in the same pathway are useful for remethylation disorders investigations. Incorporation of 1-[14C]-propionate into amino acids and then cell proteins via the propionate pathway reflects the level of intracellular synthesis of adenosylcobalamin. Incorporation of [14C]-methyltetrahydrofolate into methionine explores the methionine synthase and methionine synthase reductase activities and the capacity of the cells to produce methylcobalamin. Incorporation of [14C]-formate into methionine or serine reflects the activities of the methylene-tetrahydrofolate dehydrogenase, methylene-tetrahydrofolate reductase and the methionine synthase/methionine synthase reductase activities (Table 5). These tests also allow the assessment of possible in vitro vitamin-responsiveness of the relevant disorders (Suormala et al 2002; Gulati et al 1997; Olteanu and Banerjee 2001). Enzyme assays are needed to characterise functional consequences of new variants identified by molecular genetic investigations including next generation sequencing.
Molecular genetic analysis
The cblC gene (MMACHC OMIM 609831) is located in chromosome region 1p34.1 and has five exons (Lerner-Ellis et al 2006). The three most common mutations c.271dupA, c.394C>T and c.331C>T have been identified in the MMACHC gene. The c.271dupA that represents about 30 % of the identified alleles and c.331C>T mutations are associated with early onset disease. The c.394C>T mutation is associated mainly with late onset disease. The c.609G>A mutation accounts for 85 % of the identified alleles in Chinese patients, and even though it leads to a premature termination codon it remains predominantly associated with late onset disease in this population (Wang et al 2010) but has been observed in some early-onset cases in other populations (Weisfeld-Adams et al 2013). Other mutations have been shown to cluster according to ethnicity (Morel et al 2006; Nogueira et al 2008).
The precise function of MMACHC is not completely understood. Recombinant MMACHC binds cobalamin, can function in vitro as a cyanocobalamin (CNCbl) decyanase (Kim et al 2008) and can dealkylate other cobalamin forms (Hannibal et al 2009). MMACHC may act as an intracellular cobalamin trafficking chaperone that carries out targeted delivery of cobalamin to and from other cobalamin-related proteins (Lerner-Ellis et al 2009).
In contrast to the cblC defect which presents exclusively as combined hyperhomocysteinaemia with MMA-uria, cblD patients can have three distinct biochemical phenotypes: isolated hyperhomocysteinemia (cblD-HC), isolated MMA-uria (cblD-MMA, beyond the scope of these guidelines), or combined hyperhomocysteinemia and MMA-uria (cblD-MMA/HC). MMADHC (OMIM 611935) is the gene responsible for cblD defect (Suormala et al 2004; Coelho et al 2008). Genotype–phenotype correlation analysis of the three cblD variants suggests that the N- and C-terminal regions of MMADHC have specific effects on functions in the mitochondrion and cytoplasm, respectively (Stucki et al 2012; Miousse et al 2009). cblD-HC retains mitochondrial function with a full-length protein which harbours only C-terminal missense mutations in conserved residues; cblD-MMA retains cytoplasmic function due to translation of an error-free C-terminus facilitated by downstream re-initiation; and in cblD-MMA/HC complete loss of functionality occurs with deleterious mutations that are localised downstream of Met116 (Jusufi et al 2014).
To date, there have been no obvious genotype-phenotype correlations in cblF (LMBRD1, OMIM 612625) (Armour et al 2013), cblJ (ABCD4, OMIM 603214) (Coelho et al 2012), cblE (MTRR, OMIM 602568) and cblG (MTR, OMIM 156570) (Watkins and Rosenblatt 1989; Huemer et al 2015a).
If clinical and biochemical parameters are characteristic for a combined remethylation disorder it may be—dependent on the population—a pragmatic approach to look first for cblC, the most frequent remethylation disorder by searching for the common mutation (c.271dupA) or MMACHC sequencing. If these analyses however prove negative, functional and/or complementation studies in fibroblasts or extended molecular genetic testing for other defects are needed.
For MTHFR deficiency (OMIM 6070993) the majority of mutations are private and neither type nor location of mutation correlates with clinical phenotype (Froese et al 2016). Genetic testing for MTFHR deficiency should be interpreted with caution since there are numerous polymorphisms in this gene, which have been related to various common disorders and conditions. Polymorphisms in general including the most investigated thermolabile variant c.677C>T in MTHFR are not responsible for severe remethylation disorders (Tsang et al 2015).
Outcome: valid, timely laboratory diagnosis
-
Recommendation 6
-
We strongly recommend diagnostic confirmation by molecular genetic analysis and/or direct or indirect enzyme assays in cultured skin fibroblasts (or lymphocytes) in experienced laboratories. (Quality of the evidence: moderate)
-
Prenatal diagnosis
Metabolites concentration (tHcy and MMA) in cell-free amniotic fluid, enzyme activity of MTHFR and methionine synthase or methionine synthase reductase in cultured amniotic cells and incorporation of propionate and methyltetrahydrofolate have been used for many years (Fowler and Jakobs 1998; Merinero et al 1998) and their combined use is reliable (Morel et al 2005). Indirect enzyme assays in chorion biopsy should be avoided (Morel et al 2005; Merinero et al 1998).
However nowadays, the molecular genetic diagnosis is the most advisable method provided the causal mutations in the index case and carrier status in the parents have been identified. Molecular genetic testing can be performed from chorionic villi or amniotic fluid samples (Morel et al 2005).
Outcome: valid, timely laboratory diagnosis
-
Recommendation 7
-
If prenatal diagnosis is considered in individual cases we recommend to perform molecular genetic analysis from chorionic villi or amniotic fluid samples given that mutations in the index case and carrier status in the parents have been identified (Quality of the evidence: low)
-
Feasibility and impact on outcome of newborn screening (NBS) for combined remethylation disorders
NBS for the cblC defect should be considered since survival and prevention of severe complications such as HUS, hydrocephalus and haematological abnormalities in early-onset patients can be improved by early treatment (Huemer et al 2015b). The impact of early treatment on neurocognitive development is unclear and early treatment has little influence on eye disease (Weisfeld-Adams et al 2013). In most late-onset cblC patients, dementia, renal function, myelopathy and axonal neuropathy improve on treatment and long-lasting disease existing before treatment initiation correlates with residual pathologies (Huemer et al 2014). There is insufficient data on NBS for other combined remethylation disorders.
Outcome: survival, severe organ complications; visual and neurocognitive function
-
Recommendation 8
-
We strongly recommend early treatment in patients with the cblC defect as it improves survival, corrects haematological abnormalities and may prevent HUS and hydrocephalus. However, early treatment has little influence on eye disease and unclear impact on neurocognitive outcome (Quality of the evidence: moderate)
-
Present knowledge allows no conclusion concerning the clinical benefit of early treatment for the cblD-MMA/HC, cblF and cblJ defects.
The Region 4 Stork (R4S) data indicate that the sensitivity of C3/C2 as primary markers is superior to Met and Met/Phe (McHugh et al 2011). However, specificity of C3 and/or C3/C2 for detecting these diseases is generally low (la Marca et al 2007; Tortorelli et al 2010). The positive predictive value substantially increased from 4 to 100 % by measurement of MMA and 11 to 36 % by measuring tHcy as second tier analytes in DBS (la Marca et al 2007; Tortorelli et al 2010). The sensitivity of C3 and C3/C2 for mild/late-onset forms is unknown. It is of note that neonatal metabolic disturbances due to maternal vitamin B12 deficiency may also be detected. It has recently been shown that heptadecanoyl-carnitine (C17) may have an even higher predictive value for perturbations of MMA metabolism including combined remethylation defects and may thus be a promising first tier analyte (Malvagia et al 2015).
Outcome: timely and valid laboratory diagnosis
-
Recommendation 9
-
We strongly recommend to obtain plasma for determination of serum vitamin B12 before treatment is started in cases identified by NBS as part of studies to exclude maternal vitamin B12 deficiency. (Quality of the evidence: moderate)
-
-
Recommendation 10
-
We recommend use of C3 acylcarnitine and the C3/C2 ratio as primary markers to screen for early onset cblC defect. (Quality of the evidence: moderate)
-
-
Recommendation 11
-
We suggest consideration of C17 acylcarnitine as a promising primary marker to screen for early onset cblC defect. (Quality of the evidence: low)
-
-
Recommendation 12
-
We strongly recommend performing second tier testing using tHcy and MMA to improve specificity and to differentiate the defects from other disorders. (Quality of the evidence: moderate)
-
Feasibility and impact on outcome of NBS for isolated remethylation disorders and MTHFR deficiency
The use of NBS for detection of the isolated remethylation disorders and the MTHFR defect appears worthy of consideration. In the cblE and cblG defect, macrocytic anaemia (Vilaseca et al 2003; Kvittingen et al 1997) and neurocognitive performance (Harding et al 1997; Rosenblatt et al 1985; Schiff et al 2011; Kvittingen et al 1997; Müller et al 2007) often respond to treatment (Müller et al 2007; Schiff et al 2011). Eye disease seems not to be responsive (Huemer et al 2014). Early detection by NBS and timely treatment improved short-term outcomes of two asymptomatic patients with the cblE and cblG defect and three symptomatic patients with MTHFR deficiency (Wong et al 2016). Moreover, early betaine treatment has a clear positive impact on outcome in MTHFR deficiency (Diekman et al 2014).
Neonatal screening for cblD-HC, cblE and cblG and for MTHFR deficiency appears to be feasible by detecting decreased methionine and methionine-to-phenylalanine ratio (Bowron et al 2005) in DBS. The second-tier marker tHcy clearly differentiates patients from controls (McHugh et al 2011; Tortorelli et al 2010). However, sufficient data on efficacy of NBS programs for these disorders is lacking.
Outcome: survival, severe organ damage; neurocognitive impairment
-
Recommendation 13
-
We strongly recommend early identification and treatment with betaine for MTHFR deficiency. Presymptomatic betaine treatment prevents severe neurological impairment (Quality of the evidence: high)
-
Disease course and outcome of combined remethylation disorders
Most information on complications and outcome is based on data from patients with cblC disease. There is limited data about the natural history of the other combined disorders (Table 6).
Patients with the cblC defect usually present with severe complications and historically have a poor long-term outcome (Rosenblatt et al 1997; Andersson et al 1999; Fischer et al 2014). It is well recognised that despite treatment and improved metabolic parameters, severe complications such as developmental delay and progressive visual loss may still develop (Andersson et al 1999; Enns et al 1999; Patton et al 2000; Fischer et al 2014; Weisfeld-Adams et al 2015). Even if the neurologic status stabilises or improves with therapy, the sequelae remain in a large proportion of patients (Whitehead 2006; Watkins and Rosenblatt 1989), especially if the initiation of treatment was delayed or insufficient (Huemer et al 2014). Haematological symptoms and failure to thrive generally resolve with treatment (Martinelli et al 2011; Fischer et al 2014).
The late-onset phenotype has a more favourable outcome than early-onset cblC disease but is still associated with residual sequelae such as learning difficulties, neurobehavioural symptoms, neurogenic bladder and gait abnormalities (Rosenblatt et al 1997) and at least one patient, in whom treatment was ceased, deteriorated and died (Thauvin-Robinet et al 2008).
Mortality
Early detection and treatment with parenteral OHCbl appear to have decreased the mortality in newborns with cblC. A retrospective analysis of 50 patients (44 presented in the first year of life) with cblC disease reported an overall mortality rate of 30 % (Rosenblatt et al 1997). Of the 13 patients that died, four were not treated, two received only cyanocobalamin (CNCbl), and three were initially treated with CNCbl and then switched to OHCbl (Rosenblatt et al 1997; Carrillo-Carrasco et al 2012).
Later, in the Fischer study about outcome in a series of 88 patients with the cblC defect, the mortality was 11.4 % (90 % of them with infantile onset). Treatment following various regimes of parenteral OHCbl complemented in some cases by betaine, folate/folinic acid and carnitine resulted in improvement of biochemical abnormalities, non-neurological signs and mortality (Fischer et al 2014). It has been suggested that daily treatment with parenteral OH-Cbl combined with betaine would be associated with a favourable outcome (Carrillo-Carrasco et al 2012).
Renal disease and microangiopathy
Thrombotic microangiopathy (TMA) leading to atypical HUS is the most common renal manifestation in cblC; it has also been observed in cblD-MMA/HC patients. HUS may cause hypertension, intravascular haemolysis, microscopic haematuria, proteinuria and renal function deterioration, occasionally proceeding to renal failure (Van Hove et al 2002; Guigonis et al 2005; Sharma et al 2007; Geraghty et al 1992; Morath et al 2013). A case of focal segmental glomerulosclerosis and atypical glomerulopathy (Brunelli et al 2002) has been reported. Histological findings of the kidneys include widening of the mesangium, swelling of endothelial cells with detachment from the basement membrane, and granular deposits in the subendothelial space (Van Hove et al 2002; Russo et al 1992; Brunelli et al 2002; McCully 1969). Isolated pulmonary hypertension has been observed (Iodice et al 2013; Gündüz et al 2014) in cblC patients. Early onset combined pulmonary hypertension and renal thrombotic microangiopathy with a fatal course in several untreated cases has been reported (Kömhoff et al 2013), this picture is also seen in rare cases of late onset cblC patients (Grangé et al 2015). Timely treatment can improve outcome significantly (Grangé et al 2015; Kömhoff et al 2013; Gündüz et al 2014).
Outcome: survival, severe organ complications
-
Recommendation 14
-
We recommend monitoring for all aspects of renal disease including arterial blood pressure in patients with cobalamin related remethylation disorders. (Quality of the evidence: low)
-
Vascular problems
Thromboembolic complications are an important cause for morbidity and mortality in patients with cblC disease (Martinelli et al 2011; Thauvin-Robinet et al 2008). These include recurrent venous thrombosis (Thauvin-Robinet et al 2008; Roze et al 2003; Augoustides-Savvopoulou et al 1999; Bodamer et al 2001; Powers et al 2001; Guigonis et al 2005), pulmonary thrombosis (Powers et al 2001; Thauvin-Robinet et al 2008; McCully 1969; Baumgartner et al 1979a, b; Van Hove et al 2002), cor pulmonale (Profitlich et al 2009a, b) and cerebrovascular complications (Brunelli et al 2002; Geraghty et al 1992).
tHcy levels above 45 μmol/L have been reported to be associated with the development of vascular complications in several patients (Carrillo-Carrasco et al 2009). Remarkably, the original case of cblC disease presenting diffuse vascular lesions with proliferative fibrous intimal plaques and focal necrosis of the artery wall contributed to the development of the homocysteine theory of arteriosclerosis (McCully 1992).
With appropriate treatment, the incidence of thromboembolic complications can be reduced and may even be prevented in late onset patients (Huemer et al 2014).
Outcome: survival, severe organ complications
-
Recommendation 15
-
The incidence of vascular complications is significantly reduced with appropriate treatment in late onset patients and may be prevented in early onset patients with remethylation disorders. (Quality of the evidence: moderate)
-
Neurocognitive and psychiatric problems
Neurodevelopmental impairment of varying degrees is common in combined remethylation defects. Patients with early-onset cblC disease present with a wide range of neurological manifestations that include microcephaly, hydrocephalus, hypotonia, cognitive defects and seizures. Cognitive defects can be variable, are related to disease severity and treatment onset but may also continue to worsen despite treatment. Two treated patients with early-onset cblC disease were longitudinally followed using neuropsychological testing. These tests showed a decline in attention and executive functions, while other skills were relatively spared (Beauchamp et al 2009). Developmental delay with impairment of verbal and non-verbal cognitive skills was also documented at follow-up of cblF patients despite treatment (Alfadhel et al 2011; Gailus et al 2010), but in some early treated patients, neurocognitive outcome was satisfactory (Miousse et al 2011; Armour et al 2013). Information on outcome in the cblJ defect (Coelho et al 2012) is extremely limited; two reported patients responded well to methylcobalamin (Kim et al 2012). In the cblD-MMA/HC defect, some survivors showed improvement or even normalisation of neurocognitive function (Suormala et al 2004; Miousse et al 2009).
Patients with late-onset disease can show progressive encephalopathy with regression, deterioration in school or work performance, behavioural and personality changes possibly resulting in dementia, psychosis, episodes of acute mental confusion, lethargy and seizures that improve under specific treatment (Huemer et al 2014).
Epilepsy is a common but nonspecific occurrence in patients with remethylation disorders. The EEG and seizure patterns are nonspecific. Severely affected patients may be difficult to treat and may have recurrent status epilepticus (Biancheri et al 2002).
Brain MRI abnormalities are common in remethylation defects and include diffuse cerebral atrophy, white matter changes, basal ganglia lesions and hydrocephalus.
The most common imaging findings in the early-onset cblC cases are variable degrees of white matter abnormality with T2 hyperintensity in periatrial and periventricular white matter, with thinning of the corpus callosum. Basal ganglia lesions and hydrocephalus have also been described, and may be attributable to microvascular abnormalities (Greitz 2007). In one cohort of young early-onset cblC patients, there was a high incidence of craniocaudal shortening of the pons on sagittal MR images (Weisfeld-Adams et al 2013).
Cerebral atrophy and deep white matter bulk loss, which is more pronounced posteriorly, were significant findings in late-onset cblC disease patients (Ogier de Baulny et al 1998; Longo et al 2005; Rossi et al 2001; Wang et al 2012). Bilateral abnormalities of the cerebellar cortex were found in one patient (Wang et al 2012).
The pathophysiological mechanisms underlying the white matter abnormalities detected by MR imaging have been associated with oedema and abnormal myelination. Myelination is closely related to methylation capacity whereby deficiency of SAM in the CSF was shown to be associated with impaired myelinisation (Rossi et al 2001). Moreover, a decrease in N-acetyl aspartate (NAA) and increased lactate in the basal ganglia or in the periventricular white matter has been observed on brain MR spectroscopy (Longo et al 2005; Wang et al 2012).
Spongiform white matter degeneration and demyelination of the dorsal and lateral columns of the cord or subacute combined degeneration of the spinal cord (SACD) are known complications in remethylation disorders (Huemer et al 2014; Liu et al 2015). Changes are similar to classical adult spinal cord degeneration due to, e.g. severe nutritional vitamin B12 deficiency (Scalabrino et al 2007; Maamar et al 2008). Subacute combined degeneration of the spinal cord may be the presenting symptom in late onset patients or the consequence of insufficient treatment. Symptoms are progressive and include numbness of lower extremities, gait disturbances, incontinence, progressive leg weakness with spastic paraparesis and tetraplegia (Mitchell et al 1986; Ben-Omran et al 2007; Thauvin-Robinet et al 2008; Augoustides-Savvopoulou et al 1999; Powers et al 2001; Roze et al 2003; Tsai et al 2007; Bodamer et al 2001; Shinnar and Singer 1984).
Behavioural problems are frequent in patients with early-onset remethylation disorders. Psychiatric manifestations may be the presenting sign in patients with late-onset remethylation disorders. Dementia, behavioural problems and psychiatric symptoms have frequently been observed in the combined (Roze et al 2003; Liu et al 2015; Huemer et al 2014) as well in isolated remethylation disorders (Huemer et al 2015a) and MTHFR deficiency (Birnbaum et al 2008). In early onset cases, behavioural problems and psychiatric symptoms can evolve over time despite treatment (Fischer et al 2014). Late-onset cases may present with rather non-specific psychiatric and behavioural symptoms, which respond well to timely treatment (Ben-Omran et al 2007).
Ophthalmological problems
Visual dysfunction is very frequent in patients with early onset remethylation disorders and can be rapidly progressive while late-onset patients rarely exhibit eye disease. Current treatment does not seem to modify the progression of visual disease significantly.
Maculopathy, progressive retinal dysfunction, visual impairment, nystagmus, strabismus (Ricci et al 2005; Schimel and Mets 2006; Gerth et al 2008; Weisfeld-Adams et al 2015) and less frequently optic atrophy (Patton et al 2000; Weisfeld-Adams et al 2015) are seen in most patients with early onset cblC disease. Although rod cell sensitivity was reported to improve in a patient with cblC disease following normalisation of plasma Met levels (Tsina et al 2005) generally retinal dysfunction can be progressive and even lead to blindness despite treatment and stabilised systemic function (Fischer et al 2014).
The first signs of eye disease are frequently noted several weeks after birth and include “wandering eye movements,” inability to fixate and nystagmus. The fundoscopic exam can reveal the salt-and-pepper pigmentary macular changes that progresses to the characteristic “bull’s eye” maculopathy characterised by a hypopigmented perimacular zone surrounded by a hyperpigmented ring or coloboma like lesions (Robb et al 1984; Grant et al 2009; Weisfeld-Adams et al 2015). Retinopathy can be revealed by the electroretinogram well before clinical signs are apparent (Ogier de Baulny et al 1998; Weisfeld-Adams et al 2015) showing a progressive decline in scotopic (cones) and/or photopic (rods) responses (Schimel and Mets 2006; Robb et al 1984; Gerth et al 2008; Gaillard et al 2008; Weisfeld-Adams et al 2015).
Ocular coherence tomography, electroretinogram and visual evoked potentials, are extremely useful diagnostic and monitoring tools (Weisfeld-Adams et al 2015). Knowledge and awareness of visual dysfunction, particularly in children with early-onset disease, allows initiation of appropriate early vision intervention programs and support (Gerth et al 2008).
Late onset patients rarely have severe visual disease (Fuchs et al 2012; Fischer et al 2014; Gerth et al 2008). Minimal pigmentary retinal abnormalities have been observed in some cases (Gerth et al 2008; Weisfeld-Adams et al 2015) and the classical “bull’s eye” picture has only been described in one late-onset patient (Collison et al 2015).
Outcome: visual and neurocognitive function
-
Recommendation 16
-
As knowledge and awareness of visual dysfunction progression allows timely initiation of appropriate vision intervention programs and support, we recommend that every patient newly diagnosed with a remethylation disorder should receive an ophthalmological consultation independent of the age at diagnosis and severity of disease. (Quality of the evidence: low)
-
Disease course and outcome in isolated remethylation disorders (cblD-HC, cblE, cblG)
Disease course and outcome in isolated remethylation disorders generally share many features with the combined disorders.
Mortality
Deceased patients have been reported in the literature but the database is too small to make a more general statement on mortality.
Renal disease, microangiopathy and anaemia
Atypical HUS and glomerulopathy have been reported in single patients (Paul et al 2013; Huemer et al 2015a). Macrocytic anaemia in isolated remethylation defects often responds to treatment (Harding et al 1997; Vilaseca et al 2003).
Vascular problems
Thromboembolism may lead to severe clinical pathology but has only rarely been reported (Huemer et al 2015a).
Neurocognitive and psychiatric problems
Muscular hypotonia, seizures, cognitive impairment, behavioural problems, psychiatric symptoms, feeding problems (Watkins and Rosenblatt 1989; Huemer et al 2015a) as well as microcephaly, brain atrophy and white matter changes (Zavadáková et al 2005) are characteristic findings during the disease course (Watkins and Rosenblatt 1989). Hydrocephalus has rarely been reported (Huemer et al 2015a).
Ophthalmological problems
Eye disease, mainly comprising retinopathy, nystagmus and strabismus is frequently observed and seems not to respond well to treatment (Watkins and Rosenblatt 1989; Huemer et al 2015a).
Disease course and outcome in MTHFR deficiency
Mortality
Recent experience suggests that betaine treatment if initiated early reduces mortality in patients with MTHFR deficiency (Diekman et al 2014)
Vascular problems
Arterial and venous thrombosis are generally rare and predominantly encountered in adolescent or adult patients (Visy et al 1991; Tonetti et al 2002).
Neurocognitive problems
Severe neurological signs with some age-related variations characterise MTHFR deficiency. Neurological symptoms in MTHFR range from seizures, lethargy, apnoea and coma in early infancy to gait disturbances with lower limb spasticity, weakness, psychiatric behaviour such as schizophrenia-like psychosis (Mudd and Freeman 1974; Regland et al 1994; Pasquier et al 1994), and visual problems in adolescence/adulthood (Haworth et al 1993; Birnbaum et al 2008). Neonates and young infants exhibit acute neurological deterioration, which may be fatal or leave severe neurological deficits. Older individuals typically exhibit progressive triphasic deterioration: an initial period of normal development, then a second phase with acquired microcephaly and psychomotor delay, followed by abrupt deterioration associated with respiratory failure that may be fatal.
In early onset severe forms clinical sequelae associated with MTHFR deficiency include microcephaly, hydrocephalus, seizures, hypotonia and global development delay, whereas in the adolescence onset form mental retardation and progressive encephalopathy are present (Arn et al 1998; Bishop et al 2008; Schiff et al 2011).
Some patients develop leukoencephalopathy and lower limb-dominant demyelinating polyneuropathy (Walk et al 1994) and may have subacute degeneration of the spinal cord (Hyland et al 1988). The peripheral neuropathy has been related to the low level of serum folic acid (Nishimura et al 1985). Seizures are variable in type and include myoclonic, clonic and/or tonic episodes. Infantile spasms have been described (Prasad et al 2011). Cerebral atrophy and white matter changes are common (Arn et al 1998; Birnbaum et al 2008).
Early treated patients exhibit a more favourable outcome (Schiff et al 2011; Diekman et al 2014).
In MTHFR deficiency, early diagnosis and treatment are usually associated with better neurological outcome.
The effects of treatment on clinical outcome
Impact of prenatal treatment on outcome
Prenatal maternal treatment with parenteral OHCbl has anecdotally been used. For cblC disease, Trefz et al (2016) report a favourable outcome of a pregnancy with an affected fetus (sibling to a severely affected child) after treatment of the mother with high-dose OHCbl (30 mg/week) and 5 mg/day folic acid from week 15 of pregnancy. Prenatal treatment with lower OHCbl doses resulted in biochemical improvement. Organ complications such as microangiopathy as present in the patient’s sibling were not observed but the patient developed eye disease and neurocognitive impairment (Huemer et al 2005). There is no evidence regarding prenatal treatment for the other diseases.
Outcome: survival, severe organ damage
-
Recommendation 17
-
We suggest that prenatal maternal treatment may be considered in a pregnancy with a fetus with proven cblC disease. (Quality of the evidence: low)
-
Improving the outcome in the acutely ill patient with a suspected or proven remethylation disorder
Remethylation disorders generally present as chronic, slowly progressive neurological disease but may also present with acute deterioration on the background of chronic disease or with new-onset life-threatening complications. Individuals of all ages may manifest with a sudden thromboembolic event, microangiopathic kidney or pulmonary disease, cardiomyopathy, loss of ambulation or more commonly with acute encephalopathy, displaying epileptic seizures impaired consciousness or acute behavioural deterioration. Restoration of methylation capacity with appropriate medication may reverse many of the acute clinical signs (Fischer et al 2014; Huemer et al 2014) (Fig. 2).
Outcome: survival, severe organ damage; neurocognitive impairment
-
Recommendation 18
-
We strongly recommend immediate treatment with parenteral cobalamin in suspected cases. (Quality of the evidence: moderate)
-
-
Recommendation 19
-
We recommend consideration of betaine treatment as soon as hyperhomocysteinaemia is proven and normal/low methionine confirmed. (Quality of the evidence: moderate)
-
-
Recommendation 20
-
We suggest consideration of additional enteral supplementation with folinic acid or L-methionine in individual cases. (Quality of the evidence: low)
-
Improving the outcome: long-term management of cobalamin-related combined and isolated remethylation disorders
Most of the knowledge about the management of remethylation disorders is derived from the experience with individuals with the cblC defect, the most frequent of the disorders. In clinical practice the rarer combined disorders of the cblD-MMA/HC, cblF and cblJ types as well as the isolated remethylation disorders are generally managed identically but experience concerning treatment efficacy is considerably less and weaker.
Treatment of individuals with the remethylation disorders aims to improve clinical features and metabolic abnormalities by reducing tHcy and normalising Met and—in combined disorders—MMA (Table 7).
Cobalamin
Cobalamin is the cofactor of methionine synthase. In the cblC defect, hydroxocobalamin (OHCbl) appears to be more effective than cyanocobalamin (CNCbl) (Andersson and Shapira 1998; Bodamer et al 2001). Froese et al (2010) describe that, e.g. MMAMHC with the c.482G>A (R161Q) mutation binds OHCbl with more affinity than CNCbL. The parental route of administration (IV, SQ or IM) has been proved to be beneficial in patients with the cblC defect (Van Hove et al 2002), whereas oral OHCbl alone seems to be ineffective (Brunelli et al 2002; Bartholomew et al 1988; Gold et al 1996; Frattini et al 2010). Long-term IV application is impracticable and the subcutaneous route seems to be less effective than IM injections (Thauvin-Robinet et al 2008). Many centres use 1 mg of parenteral OHCbl daily in neonates (assuming a body weight of 3 kg, the dosage is 0.33 mg/kg/day). In the long-term treatment, as patients stabilise, the frequency of administration is decreased to minimise the frequency of injections. However, determination of optimal doses and intervals between injections is hampered by very great variation in patient features (Dionisi-Vici et al 2013; Ogier de Baulny et al 1998). Some recent reports suggest that higher OHCbl doses (plasma levels near 1,000,000 pg/ml) can provide a better metabolic control and dose escalation seems to be effective in several reports, including a case of pregnancy in a cblC affected female (Brunel-Guitton et al 2010). In 2002 Van Hove et al (2002) described a good response to OHCbl dose escalation regarding metabolic and clinical condition in two cblC patients presenting with thrombotic microangiopathies (TMAs). During 13 years of follow up in a cblC patient Carrillo-Carrasco et al (2009) observed a considerable dose-dependent reduction of MMA and tHcy plasma levels and an elevation of methionine levels as the dose of IM OHCbl was increased. Matos et al 2013 performed an open-label study in five cblC patients to determine the effect of OHCbl dose escalation on the clinical and biochemical condition. They observed a good metabolic response in 2/5 patients, whereas clinical response was quite variable and not clearly related to metabolic improvement. In early onset cblC disease, tHcy mostly cannot be normalised; levels between 40 and 60 μmol/L are typically reached. In some late onset patients, tHcy has been normalised.
There is limited experience with OHCbl (1000 μg/day) treatment in cblD-MMA/HC disease but there seems to be a response to treatment (Miousse et al 2009; Suormala et al 2004). Most patients with cblE and cblG disease were treated with OH-Cbl and seemed to benefit (Huemer et al 2015a, b). As OH-Cbl is generally used to treat other cobalamin-related remethylation disorders than cblC disease, no sufficient data is available on the clinical efficacy of other cobalamin preparations .
In conclusion, OHCbl treatment is usually started at a dosage of 1 mg IM daily and then it should be titrated individually based on metabolic response.
Outcome: survival and severe organ damage
-
Recommendation 21
-
We strongly recommend using parenteral OHCbl in treating patients with the cblC defect and other cobalamin-related remethylation disorders. (Quality of the evidence: high)
-
-
Recommendation 22
-
We recommend applying a starting dose of 1000 μg (1 mg) OHCbl daily given parenterally in patients with the cblC defect. This regime has also been applied in other cobalamin-related remethylation defects. (Quality of the evidence: low)
-
-
Recommendation 23
-
We suggest that the minimum effective OHCbl dose and frequency of administration should be individually titrated. Escalating doses of OHCbl may result in biochemical improvement; however, significant clinical benefit remains to be proven. Frequency of administration of OHCbl ranges between daily and weekly without evidence of advantage of one over the other. (Quality of the evidence: low)
-
Betaine
Betaine derives from choline and is a potent methyl group donor involved in the process of remethylation of Hcy to Met via betaine-homocysteine methyltransferase in the liver, thus bypassing the MeCbl-dependent pathway. Betaine needs to be supplemented orally and decreases Hcy levels. The knowledge about betaine pharmacokinetics is limited (Schwahn et al 2003). It has been suggested that frequent administration of a moderate dose may provide clinical and biochemical benefit (Ucar et al 2010). Betaine or OHCbl alone may not result in sufficient metabolic control, but they seem to have synergistic effects (Bartholomew et al 1988). Betaine is usually administered at a dose of up to 250 mg/kg/day in children (Beauchamp et al 2009; Schiff et al 2011), but has been used at a dose of 850 mg/kg/day to treat cor pulmonale in a patient with the cblC defect (Profitlich et al 2009a, b). Betaine has also been used during pregnancy with no neonatal adverse effect (Pierre et al 2006). Anhydrous betaine is the preferred formulation.
Outcome: survival and severe organ damage
-
Recommendation 24
-
We recommend oral betaine treatment in cblC disease and other cobalamin-related remethylation disorders (Quality of the evidence: low)
-
-
Recommendation 25
-
We suggest that the minimum effective betaine dose should be individually titrated to improve the levels of tHcy and methionine. (Quality of the evidence: low)
-
Side effects of treatment with cobalamin and betaine
Few side effects related to cblC disease treatment have been reported. These are usually scarce and reversible as soon as the treatment is withdrawn.
OHCbl side effects such as red discoloration of the urine and skin, hypersensitivity and photosensitivity reactions, nausea, infusion site reactions and headache have rarely been reported in patients receiving high doses. Mostly, parenteral OHCbl even in doses as high as 20 mg per day have been tolerated without side effects (Carrillo-Carrasco et al 2009).
In a single case, teeth discoloration occurred after 1 month of OHCbl (1 mg/day IM) and folinic acid (10 mg/day) treatment and improved after folinic acid discontinuation (Augoustides-Savvopoulou et al 1999).
Long-tem follow up case series and case reports indicate that betaine is generally well tolerated and safe even at high doses (3–6 g/day in infants; 9 g/day in adults) (Ogier De Baulny et al 1998; Thauvin-Robinet et al 2008). However, in a 15-month-old, epileptic crises have been observed soon after initiation of betaine treatment (250 mg/kg/day) (Enns et al 1999).
Betaine decreases Hcy levels at the expense of increasing Met. The clinical relevance of high Met is under discussion but very high levels have been related to cerebral oedema. In a 10-year-old female with classical homocystinuria plasma Met increased to >3000 μmoL/L under betaine treatment (200 mg/kg/day). The girl developed clinical signs and radiological evidence of cerebral oedema which resolved when betaine was discontinued and Met levels returned to normal (Yaghmai et al 2002). Devlin et al (2004) reported on a similar clinical course in a boy at a plasma Met level of 1190 μmol/L. Therefore, caution is required and betaine should immediately be withdrawn in any case of any symptoms suggesting increased intracranial pressure or very high Met levels (Lawson-Yuen and Levy 2006).
Folates
Folate has been frequently used as adjunctive therapy in patients with cblC disease. Folinic acid (5-formyl-THF) is the most stable form of the reduced vitamin and crosses the blood brain barrier more efficiently than folic acid (Spector 1979). Daily doses from 5 to 30 mg, divided in 2–3 administrations/week of folate or folinic acid (Carrillo-Carrasco et al 2012; Schiff et al 2011) have been used. In the majority of reported patients, folic acid rather than folinic acid has been used in the long-term treatment. In some cases no beneficial effect of the adjunctive therapy with folic or folinic acid has been reported (Enns et al 1999; Bartholomew et al 1988; Fischer et al 2014) but no long-term studies have been performed.
-
Recommendation 26
-
Results failed to demonstrate or exclude a beneficial or detrimental effect of folic and/or folinic acid as adjunctive therapy in patients with cblC disease and other cobalamin-related remethylation disorders. (Quality of the evidence: low)
-
Levocarnitine
Levocarnitine facilitates the excretion of propionyl groups and prevents carnitine deficiency, since the de novo synthesis of carnitine depends on methionine and may be decreased in cblC patients. Some reports in literature show no beneficial effect of adding carnitine to the long-term treatment of cblC disease (Enns et al 1999; Bartholomew et al 1988). There is no clear consensus about the recommended dose and this can range between 50 and 200 mg/kg/day.
-
Recommendation 27
-
Results failed to demonstrate or exclude a beneficial or detrimental effect of oral carnitine as adjunctive therapy in patients with cblC disease and other cobalamin-related remethylation disorders. (Quality of the evidence: low)
-
Dietary restrictions
Few reports describe the use of dietary restriction in cblC patients, mainly with no beneficial effect on metabolic control (Carrillo-Carrasco et al 2009; Enns et al 1999). Huemer et al (2005) report significantly lower MMA excretion upon protein restriction (methionine-free, threonine-free, valine-free, isoleucine-low formula). However, there is evidence suggesting a possible role of low methionine as a contributing factor for the pathogenesis of cblC disease (Martinelli et al 2011) and other remethylation disorders. Met is essential in patients with remethylation defects and its plasma levels should be maintained in the normal range. Patients on protein-restricted diets had lower height-for-age z-score. Patients consuming medical foods with presumably low Met supply had lower head circumference and lower plasma Met concentrations (Manoli et al 2015). For these reasons, methionine-free formulas and protein restriction leading to low methionine plasma levels is contraindicated in cblC patients (Ogier de Baulny et al 1998; Manoli et al 2015).
Outcome: survival, severe organ damage; neurocognitive impairment
-
Recommendation 28
-
We strongly recommend not to restrict protein in cblC disease and other remethylation disorders. (Quality of the evidence: moderate)
-
Amino acid supplementation
In patients with cblC defects methionine and cysteine supplements are mostly not required to maintain normal plasma levels of these two amino acids and in a recent retrospective multicentre study evaluating clinical, biochemical and therapeutic measures in 88 cblC patients (Fischer et al 2014) supplemental Met was used only in one patient; cysteine supplementation was never applied. Some patients were treated with betaine and Met in combination when plasma Met was low (Smith et al 2006).
Outcome: survival, severe organ damage; neurocognitive impairment
-
Recommendation 29
-
Met is essential in patients with remethylation defects and we recommend maintaining its plasma levels in the normal range; if necessary this may be achieved by oral methionine supplementation. (Quality of the evidence: low)
-
Improving the outcome: long-term management of MTHFR deficiency
Betaine
In MTHFR deficiency betaine in a variable dose of 100–250 mg/kg/day in children and 5–20 g/day in adults is used to increase systemic Met levels. A meta-analysis of case reports (Diekman et al 2014) revealed that betaine treatment at a dosage of 100 mg/kg/day or more was clearly associated with increased survival. Only two of 26 patients died after initiation of treatment. Furthermore, early treatment was associated with normal psychomotor development in all five patients. In contrast, none of the 19 patients in the delayed-treatment group (untreated patients excluded) had normal psychomotor development. Nevertheless, psychomotor development stabilised or improved in all 17 surviving patients in whom betaine treatment was initiated late. Anhydrous betaine is the preferred formulation.
Outcome: survival, severe organ damage; neurocognitive impairment
-
Recommendation 30
-
We strongly recommend early treatment with betaine as it improves clinical outcome and prevents neurological deterioration in MTHFR deficiency. (Quality of the evidence: high)
-
See also recommendation 13
Folates
In MTHFR deficiency 5-methylTHF synthesis is impaired, resulting in low levels of methyl-tetrahydrofolate (CH3-THF) in the central nervous system. There have been some reports describing the use of folic acid (5–80 mg/day) with good (Al Tawari et al 2002; Munoz et al 2015) or poor (Holme and Ronge 1989; Ucar et al 2010) response. A dose–response relationship was not observed (Holme and Ronge 1989). A combination of folic and folinic acid was associated with favourable outcome (Tallur et al 2005; Lossos et al 2014). It has recently been shown that folic acid may exacerbate cerebral CH3-THF deficiency (Hyland et al 2010) and it has therefore been suggested to avoid folic acid and preferably use folinic acid or 5-CH3-THF. However, therapeutic 5-CH3-THF in a daily dose as high as 45 mg failed to correct the low CSF levels in a patient with MTHFR deficiency (Knowles et al 2016; Schiff and Blom 2012). There is at present no consensus concerning the most effective dosage of folinic acid or 5-CH3-THF and the problem of the assumed instability of 5-CH3-THF has not yet been solved.
Outcome: neurocognitive impairment
-
Recommendation 31
-
Results failed to demonstrate or exclude a beneficial or detrimental effect of folic or folinic acid or 5-CH3THF as adjunctive therapy to restore cellular and cerebral folate deficiency in MTHFR deficiency on clinical outcome. (Quality of the evidence: low).
-
Other substances
The data basis addressing the efficacy of vitamin B6, cobalamin, riboflavin and carnitine is not reliable; various dosages and combinations of these substances have been used and their clinical efficacy cannot be assessed (Huemer et al 2016).
Methionine supplementation
In a very small number of patients, the clinical condition improved with supplementation of methionine (Schiff et al 2011; Tortorelli et al 2010), however, the data basis is not reliable enough to allow delineation of a recommendation.
Improving clinical outcome: management of general anaesthesia in remethylation disorders
A fatal adverse event was reported in a patient with MTHFR deficiency anaesthetised with nitrous oxide (N2O) which is a known inhibitor of methionine synthase (Selzer et al 2003; Erbe and Salis 2003). In seven patients with the cblC and one with the cblG defect, propofol was safe and effective as an induction and maintenance agent for elective short procedures in metabolically and hemodynamically stable patients (Ktena et al 2015).
-
Recommendation 32
-
We strongly recommend against the use of nitrous oxide in patients with remethylation disorders. (Quality of the evidence: high)
-
Improving clinical outcome: management of pregnancy in remethylation disorders
There are only three reported cases of pregnancies in women with the cblC defect and thus no recommendations can be delineated. An asymptomatic woman was identified when low carnitine levels were found in her healthy child’s newborn screening sample (Lin et al 2009). Brunel-Guitton et al (2010) describe successful outcome of a pregnancy in a treated woman with the cblC defect. Acetylsalicylic acid at the antiplatelet oral dose of 80 mg/day was applied during pregnancy and delivery and specific treatment with OHCbl, folic acid and carnitine was started at 15 weeks of gestation. The OHCbl and carnitine administration was adjusted to maintain a decrease of tHcy and an increase of total carnitine plasma levels. Recently a woman with late-onset cblC, also treated from the 15th week of gestation and giving birth to a healthy child has been reported (Liu et al 2015). So far, pregnancies in women with other remethylation defects have not been reported.
Closing remarks
These guidelines are the result of the evaluation of available information from the literature based on the GRADE methodology and are aimed at delivering recommendations based on the best available data. The rarity of cobalamin-related remethylation disorders and MTHFR deficiency, the absence of larger data samples from international registries until recently and data mainly derived from case reports, case series or expert opinion results in a low quality of the evidence for several of the recommendations made. The working group responsible for this guideline commits itself to revise this work in the future.
References
Al Essa MA, Al Amir A, Rashed M et al (1999) Clinical, fluorine-18 labeled 2-fluoro-2-deoxyglucose positron emission tomography of the brain, MR spectroscopy, and therapeutic attempts in methylenetetrahydrofolate reductase deficiency. Brain Dev 21:345–349
Al Tawari AA, Ramadan DG, Neubauer D, Heberle LC, Al Awadi F (2002) An early onset form methylenetetrahydrofolate reductase deficiency: a report of a family from Kuwait. Brain Dev 24:304–309
Alfadhel M, Lillquist YP, Davis C, Junker AK, Stockler-Ipsiroglu S (2011) Eighteen-year follow-up of a patient with cobalamin F disease (cblF): report and review. Am J Med Genet 155A:2571–2577
Andersson HC, Shapira E (1998) Biochemical and clinical response to hydroxocobalamin versus cyanocobalamin treatment in patients with methylmalonic acidemia and homocystinuria (cblC). J Pediatr 132:121–124
Andersson HC, Marble M, Shapira E (1999) Long-term outcome in treated combined methylmalonic acidemia and homocystinemia. Genet Med 1:146–150
Arai M, Osaka H (2011) Acute leukoencephalopathy possibly induced by phenytoin intoxication in an adult patient with methylenetetrahydrofolate reductase deficiency. Epilepsia 52:e58–61
Armour CM, Brebner A, Watkins D, Geraghty MT, Chan A, Rosenblatt DS (2013) A patient with an inborn error of vitamin B12 metabolism (cblF) detected by newborn screening. Pediatrics 132:e257–261
Arn PH, Williams CA, Zori RT, Driscoll DJ, Rosenblatt DS (1998) Methylenetetrahydrofolate reductase deficiency in a patient with phenotypic findings of Angelman syndrome. Am J Med Genet 77:198–200
Atkinson C, Miousse IR, Watkins D, Rosenblatt DS, Raiman JA (2014) Clinical, biochemical end molecular presentation in a patient with the CblD Homocystinuria error of cobalamin metabolism. JIMD Rep 17:77–81
Augoustides-Savvopoulou P, Mylonas I, Sewell AC, Rosenblatt DS (1999) Reversible dementia in an adolescent with cblC disease: clinical heterogeneity within the same family. J Inherit Metab Dis 22:756–758
Backe PH, Ytre-Arne M, Røhr AK et al (2013) Novel deletion mutation identified in a patient with late-onset combined methylmalonic acidemia and homocystinuria, cblC Type. JIMD Rep 11:79–85
Bartholomew DW, Batshaw ML, Allen RH et al (1988) Therapeutic approaches to cobalamin-C methylmalonic acidemia and homocystinuria. J Pediatr 112:32–9
Baumgartner ER, Wick H, Linnell JC, Gaull GE, Bachmann C, Steinmann B (1979a) Congenital defect in intracellular cobalamin metabolism resulting in homocystinuria and methylmalonic aciduria. II. Biochemical investigations. Helv Paediatr Acta 34:483–496
Baumgartner ER, Wick H, Maurer R, Egli N, Steinmann B (1979b) Congenital defect in intracellular cobalamin metabolism resulting in homocysteinuria and methylmalonic aciduria. I. Case report and histopathology. Helv Paediatr Acta 34:465–482
Beauchamp MH, Anderson V, Boneh A (2009) Cognitive and social profiles in two patients with cobalamin C disease. J Inherit Metab Dis 32:327–334
Bellini C, Cerone R, Bonacci W et al (1992) Biochemical diagnosis and outcome of 2 years treatment in a patient with combined methylmalonic aciduria and homocystinuria. Eur J Pediatr 151:818–820
Ben-Omran TI, Wong H, Blaser S, Feigenbaum A (2007) Late-onset cobalamin-C disorder: a challenging diagnosis. Am J Med Genet 143A:979–984
Ben-Shachar S, Zvi T, Rolfs A et al (2012) A founder mutation causing a severe methylenetetrahydrofolate reductase (MTHFR) deficiency in Bukharian Jews. Mol Genet Metab 107:608–610
Biancheri R, Cerone R, Schiaffino MC et al (2001) Cobalamin (Cbl) C/D deficiency: clinical, neurophysiological and neuroradiologic findings in 14 cases. Neuropediatrics 32:14–22
Biancheri R, Cerone R, Rossi A et al (2002) Early-onset cobalamin C/D deficiency: epilepsy and electroencephalographic features. Epilepsia 43:616–622
Biotti D, Esteban-Mader M, Diot E et al (2014) Clinical reasoning: a young woman with rapid mental deterioration and leukoencephalopathy: a treatable cause. Neurology 83:e182–186
Birnbaum T, Blom HJ, Prokisch H, Hartig M, Klopstock T (2008) Methylenetetrahydrofolate reductase deficiency (homocystinuria type II) as a rare cause of rapidly progressive tetraspasticity and psychosis in a previously healthy adult. J Neurol 255:1845–1846
Bishop L, Kanoff R, Charnas L, Krenzel C, Berry SA, Schimmenti LA (2008) Severe asymptomatic methylenetetrahydrofolate reductase (MTHFR) deficiency: a case report of nonclassical homocystinuria. J Child Neurol 23:823–828
Bodamer OA, Rosenblatt DS, Appel SH, Beaudet AL (2001) Adult-onset combined methylmalonic aciduria and homocystinuria (cblC). Neurology 56:1113
Bonafede L, Ficicioglu CH, Serrano L, Han G, Morgan J, Mills MD (2015) Cobalamin C deficiency shows a rapidly progressing maculopathy with severe photoreceptor and ganglion cell loss. IOVS 56:7876
Bowron A, Barton A, Scott J, Stansbie D (2005) Blood spot homocysteine: a feasibility and stability study. Clin Chem 51:257–258
Brandstetter Y, Weinhouse E, Splaingard ML, Tang TT (1990) Cor pulmonale as a complication of methylmalonic acidemia and homocystinuria (Cbl-C type). Am J Med Genet 36:167–171
Brooks BP, Thompson AH, Sloan JL et al (2016) Ophthalmic manifestations and long-term visual outcomes in patients with cobalamin C deficiency. Ophthalmology 123:571–82
Broomfield A, Abulhoul L, Pitt W, Jameson E, Cleary M (2014) Reversal of respiratory failure in both neonatal and late onset isolated remethylation disorders. JIMD Rep 16:51–56
Brunel-Guitton C, Costa T, Mitchell GA, Lambert M (2010) Treatment of cobalamin C (cblC) deficiency during pregnancy. J Inherit Metab Dis 33:409–412
Brunelli SM, Meyers KE, Guttenberg M, Kaplan P, Kaplan BS (2002) Cobalamin C deficiency complicated by an atypical glomerulopathy. Pediatr Nephrol 17:800–803
Burda P, Kuster A, Hjalmarson O, Suormala T, Bürer C, Lutz S (2015) Characterization and review of MTHFD1 deficiency: four new patients, cellular delineation and response to folic and folinic acid treatment. J Inherit Metab Dis 38:863–872
Cappuccio G, Cozzolino C, Frisso G (2014) Pearls & oysters: familial epileptic encephalopathy due to methylenetetrahydrofolate reductase deficiency. Neurology 83:e41–44
Carmel R, Bedros AA, Mace JW, Goodman SI (1980) Congenital methylmalonic aciduria--homocystinuria with megaloblastic anemia: observations on response to hydroxocobalamin and on the effect of homocysteine and methionine on the deoxyuridine suppression test. Blood 55:570–579
Carmel R, Watkins D, Goodman SI, Rosenblatt DS (1988) Hereditary defect of cobalamin metabolism (cblG mutation) presenting as a neurologic disorder in adulthood. N Engl J Med 318:1738–1741
Carrillo-Carrasco N, Venditti CP (2012a) Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis 35:103–114
Carrillo-Carrasco N, Chandler RJ, Venditti CP (2012b) Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J Inherit Metab Dis 35:91–102
Carrillo-Carrasco N, Adams D, Venditti CP (2013) Disorders of Intracellular Cobalamin Metabolism. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. 2008 Feb 25 [updated 2013 Nov 21]
Carrillo-Carrasco N, Sloan J, Valle D, Hamosh A, Venditti CP (2009) Hydroxocobalamin dose escalation improves metabolic control in cblC. J Inherit Metab Dis 32:728–731
Cerone R, Schiaffino MC, Caruso U, Gatti R (2000) Facial anomalies in combined methylmalonic aciduria and homocystinuria. Am J Med Genet 6:87
Chang JT, Chen YY, Liu TT, Liu MY, Chiu PC (2011) Combined methylmalonic aciduria and homocystinuria cblC type of a Taiwanese infant with c.609G>A and C.567dupT mutations in the MMACHC gene. Pediatr Neonatol 52:223–226
Clayton PT, Smith I, Harding B, Hyland K, Leonard JV, Leeming RJ (1986) Subacute combined degeneration of the cord, dementia and parkinsonism due to an inborn error of folate metabolism. J Neurol Neurosurg Psychiatry 49:920–927
Coelho D, Suormala T, Stucki M et al (2008) Gene identification for the cblD defect of vitamin B12 metabolism. N Engl J Med 358:1454–1464
Coelho D, Kim JC, Miousse IR et al (2012) Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat Genet 44:1152–1155
Cogan DG, Schulman J, Porter RJ, Mudd SH (1980) Epileptiform ocular movements with methylmalonic aciduria and homocystinuria. Am J Ophthalmol 90:251–253
Collison FT, Xie YA, Gambin T et al (2015) Whole exome sequencing identifies an adult-onset case of methylmalonic aciduria and homocystinuria type C (cblC) with non-syndromic bull’s eye maculopathy. Ophthalmic Genet 36:270–275
Carrillo-Carrasco N, Chandler RJ, Venditti CP (2012b) Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J Inherit Metab Dis 35:91–102
D’Aco KE, Bearden D, Watkins D, Hyland K, Rosenblatt DS, Ficicioglu C (2014) Severe 5,10- Methyltetrahydrofolate reductase deficiency and two MTHFR variants in an adolescent with progressive myoclonic epilepsy. Pediatr Neurol 51:266–270
D’Alessandro G, Tagariello T, Piana G (2010) Oral and craniofacial findings in a patient with methylmalonic aciduria and homocystinuria: review and a case report. Minerva Stomatol 59:129–137
De Bie I, Nizard SD, Mitchell GA (2009) Fetal dilated cardiomyopathy: an unsuspected presentation of methylmalonic aciduria and hyperhomocystinuria, cblC type. Prenat Diagn 29:266–270
Devlin AM, Hajipour L, Gholkar A, Fernandes H, Ramesh V, Morris AA (2004) Cerebral edema associated with betaine treatment in classical homocystinuria. J Pediatr 144:545–548
Diekman EF, de Koning TJ, Verhoeven-Duif NM, Rovers MM, van Hasselt PM (2014) Survival and psychomotor development with early betaine treatment in patients with severe methylenetetrahydrofolate reductase deficiency. JAMA Neurol 71:188–194
Dionisi-Vici C, Martinelli D, Ceravolo F, Boenzi S, Pastore A (2013) Optimizing the dose of hydroxocobalamin in cobalamin C (cblC) defect. Mol Genet Metab 109:329–330
Ellaway C, Christodoulou J, Kamath R, Carpenter K, Wilcken B (1998) The association of protein-losing enteropathy with cobalamin C defect. J Inherit Metab Dis 21:17–22
Engelbrecht V, Rassek M, Huismann J, Wendel U (1997) MR and proton MR spectroscopy of the brain in hyperhomocysteinemia caused by methylenetetrahydrofolate reductase deficiency. AJNR Am J Neuroradiol 18:536–539
Enns GM, Barkovich AJ, Rosenblatt DS et al (1999) Progressive neurological deterioration and MRI changes in cblC methylmalonic acidaemia treated with hydroxocobalamin. J Inherit Metab Dis 22:599–607
Erbe RW, Salis RJ (2003) Severe methylenetetrahydrofolate reductase deficiency, methionine synthase, and nitrous oxide--a cautionary tale. N Engl J Med 349:5–6
Fischer S, Huemer M, Baumgartner M et al (2014) Clinical presentation and outcome in a series of 88 patients with the cblC defect. J Inherit Metab Dis 37:831–840
Fowler B (2005) Homocysteine: overview of biochemistry, molecular biology, and role in disease processes. Semin Vasc Med 5:77–86
Fowler B, Jakobs C (1998) Post- and prenatal diagnostic methods for the homocystinurias. Eur J Pediatr 157:S88–93
Frattini D, Fusco C, Ucchino V, Tavazzi B, Della Giustina E (2010) Early onset methylmalonic aciduria and homocystinuria cblC type with demyelinating neuropathy. Pediatr Neurol 43:135–138
Froese DS, Healy S, McDonald M et al (2010) Thermolability of mutant MMACHC protein in the vitamin B12-responsive cblC disorder. Mol Genet Metab 100:29–36
Froese DS, Huemer M, Suormala T et al (2016) Mutation update and review of severe MTHFR deficiency. Hum Mutat. doi:10.1002/humu.22970
Fuchs LR, Robert M, Ingster-Moati I et al (2012) Ocular manifestations of cobalamin C type methylmalonic aciduria with homocystinuria. J AAPOS 16:370–375
Gaillard MC, Matthieu JM, Borruat FX (2008) Retinal dysfunction in combined methylmalonic aciduria and homocystinuria (Cblc) disease: a spectrum of disorders. Klin Monbl Augenheilkd 225:491
Gailus S, Suormala T, Malerczyk-Aktas AG, Toliat MR, Wittkampf T, Stucki M (2010) A novel mutation in LMBRD1 causes the cblF defect of vitamin B(12) metabolism in a Turkish patient. J Inherit Metab Dis 33:17–24
Geraghty MT, Perlman EJ, Martin LS et al (1992) Cobalamin C defect associated with hemolytic-uremic syndrome. J Pediatr 120:934–937
Gerth C, Morel CF, Feigenbaum A, Levin AV (2008) Ocular phenotype in patients with methylmalonic aciduria and homocystinuria, cobalamin C type. J AAPOS 12:591–596
Gizicki R, Robert MC, Gómez-López L et al (2014) Long-term visual outcome of methylmalonic aciduria and homocystinuria, cobalamin C type. Ophthalmology 121:381–386
Gold R, Bogdahn U, Kappos L et al (1996) Hereditary defect of cobalamin metabolism (homocystinuria and methylmalonic aciduria) of juvenile onset. J Neurol Neurosurg Psychiatry 60:107–108
Goodman SI, Moe PG, Hammond KB, Mudd SH, Uhlendorf BW (1970) Homocystinuria with methylmalonic aciduria: two cases in a sibship. Biochem Med 4:500–515
Goyette P, Sumner JS, Milos R et al (1994) Human methylenetetrahydrofolate reductase: isolation of cDNA, mapping and mutation identification. Nat Genet 7:195–200
Goyette P, Frosst P, Rosenblatt DS, Rozen R (1995) Seven novel mutations in the methylenetetrahydrofolate reductase gene and genotype/phenotype correlations in severe methylenetetrahydrofolate reductase deficiency. Am J Hum Genet 56:1052–1059
Goyette P, Christensen B, Rosenblatt DS, Rozen R (1996) Severe and mild mutations in cis for the methylenetetrahydrofolate reductase (MTHFR) gene, and description of five novel mutations in MTHFR. Am J Hum Genet 59:1268–1275
Grangé S, Bekri S, Artaud-Macari E et al (2015) Adult-onset renal thrombotic microangiopathy and pulmonary arterial hypertension in cobalamin C deficiency. Lancet 386:1011–1012
Grant LW, McCandless SE, Traboulsi EI (2009) Maculopathy due to cobalamin C (cblC) disease in an Amish child. J Pediatr Ophthalmol Strabismus. doi:10.3928/01913913-20090918-10
Greitz D (2007) Paradigm shift in hydrocephalus research in legacy of Dandy’s pioneering work: rationale for third ventriculostomy in communicating hydrocephalus. Childs Nerv Syst 23:487–489
Grünert SC, Fowler B, Superti-Furga A, Sass JO, Schwab KO (2011) Hyperpyrexia resulting in encephalopathy in a 14-month-old patient with cblC disease. Brain Dev 33:432–436
Guigonis V, Frémeaux-Bacchi V, Giraudier S et al (2005) Late-onset thrombocytic microangiopathy caused by cblC disease: association with a factor H mutation. Am J Kidney Dis 45:588–595
Gulati S, Chen Z, Brody LC, Rosenblatt DS, Banerjee R (1997) Defects in auxiliary redox proteins lead to functional methionine synthase deficiency. J Biol Chem 272:19171–19175
Gündüz M, Ekici F, Özaydın E, Ceylaner S, Perez B (2014) Reversible pulmonary arterial hypertension in cobalamin-dependent cobalamin C disease due to a novel mutation in the MMACHC gene. Eur J Pediatr 173:1707–1710
Guyatt G, Oxman AD, Akl EA et al (2011a) GRADE guidelines: 1. Introduction-GRADE evidence profiles and summary of findings tables. J Clin Epidemiol 64:383–94
Guyatt GH, Oxman AD, Kunz R et al (2011b) GRADE guidelines: 2. Framing the question and deciding on important outcomes. J Clin Epidemiol 64:395–400
Guyatt GH, Oxman AD, Schünemann HJ, Tugwell P, Knottnerus A (2011c) GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol 64:380–382
Guyatt GH, Norris SL, Schulman S et al (2012) Methodology for the development of antithrombotic therapy and prevention of thrombosis guidelines: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest 141:53S–70S
Haan EA, Rogers JG, Lewis GP, Rowe PB (1985) 5,10-Methylenetetrahydrofolate reductase deficiency. Clinical and biochemical features of a further case. J Inherit Metab Dis 8:53–57
Hannibal L, Kim J, Brasch NE et al (2009) Processing of alkylcobalamins in mammalian cells: a role for the MMACHC (cblC) gene product. Mol Genet Metab 97:260–266
Harding CO, Arnold G, Barness LA, Wolff JA, Rosenblatt DS (1997) Functional methionine synthase deficiency due to cblG disorder: a report of two patients and a review. Am J Med Genet 71:384–390
Harpey JP, Rosenblatt DS, Cooper BA, Le Moël G, Roy C, Lafourcade J (1981) Homocystinuria caused by 5,10-methylenetetrahydrofolate reductase deficiency: a case in an infant responding to methionine, folinic acid, pyridoxine, and vitamin B12 therapy. J Pediatr 98:275–278
Haworth JC, Dilling LA, Surtees RA et al (1993) Symptomatic and asymptomatic methylenetetrahydrofolate reductase deficiency in two adult brothers. Am J Med Genet 45:572–575
Holme E, Ronge E (1989) Betaine for treatment of homocystinuria caused by methylenetetrahydrofolate reductase deficiency. Arch Dis Child 64:1061–1064
Howard R, Frieden IJ, Crawford D et al (1997) Methylmalonic acidemia, cobalamin C type, presenting with cutaneous manifestations. Arch Dermatol 133:1563–1566
Huemer M, Simma B, Fowler B, Suormala T, Bodamer OA, Sass JO (2005) Prenatal and postnatal treatment in cobalamin C defect. J Pediatr 147:469–472
Huemer M, Scholl-Bürgi S, Hadaya K et al (2014) Three new cases of late-onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy. Orphanet J Rare Dis 15:161
Huemer M, Bürer C, Ješina P et al (2015a) Clinical onset and course, response to treatment and outcome in 24 patients with the cblE or cblG remethylation defect complemented by genetic and in vitro enzyme study data. J Inherit Metab Dis 38:957–967
Huemer M, Kožich V, Rinaldo P et al (2015b) Newborn screening for homocystinurias and methylation disorders: systematic review and proposed guidelines. J Inherit Metab Dis 38:1007–1019
Huemer M, Mulder-Bleile R, Burda P et al (2016) Clinical pattern, mutations and in vitro residual activity in 33 patients with severe 5, 10 methylenetetrahydrofolate reductase (MTHFR) deficiency. J Inherit Metab Dis 39:115–124
Hyland K, Smith I, Bottiglieri T et al (1988) Demyelination and decreased S-adenosylmethionine in 5,10-methylenetetrahydrofolate reductase deficiency. Neurology 38:459–462
Hyland K, Shoffner J, Heales SJ (2010) Cerebral folate deficiency. J Inherit Metab Dis 33:563–570
Iodice FG, Di Chiara L, Boenzi S et al (2013) Cobalamin C defect presenting with isolated pulmonary hypertension. Pediatrics 132:e248–251
Jusufi J, Suormala T, Burda P, Fowler B, Froese DS, Baumgartner MR (2014) Characterization of functional domains of the cblD (MMADHC) gene product. J Inherit Metab Dis 37:841–849
Kanwar YS, Manaligod JR, Wong PW (1976) Morphologic studies in a patient with homocystinuria due to 5, 10-methylenetetrahydrofolate reductase deficiency. Pediatr Res 10:598–609
Kim J, Gherasim C, Banerjee R (2008) Decyanation of vitamin B12 by a trafficking chaperone. Proc Natl Acad Sci U S A 23:14551–14554
Kim JC, Lee NC, Hwu PW et al (2012) Late onset of symptoms in an atypical patient with the cblJ inborn error of vitamin B12 metabolism: diagnosis and novel mutation revealed by exome sequencing. Mol Genet Metab 107:664–668
Kind T, Levy J, Lee M, Kaicker S, Nicholson JF, Kane SA (2002) Cobalamin C disease presenting as hemolytic-uremic syndrome in the neonatal period. J Pediatr Hematol Oncol 24:327–329
King GD, Rosene DL, Abraham CR (2012) Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age 34:1405–1419
Knowles L, Morris AA, Walter JH (2016) Treatment with mefolinate (5-Methyltetrahydrofolate), but not folic acid or folinic acid, leads to measurable 5-methyltetrahydrofolate in cerebrospinal fluid in methylenetetrahydrofolate reductase deficiency. JIMD Rep 29:103–107
Kömhoff M, Roofthooft MT, Westra D et al (2013) Combined pulmonary hypertension and renal thrombotic microangiopathy in cobalamin C deficiency. Pediatrics 132:e540–544
Ktena YP, Ramstad T, Baker EH et al (2015) Propofol administration in patients with methylmalonic acidemia and intracellular cobalamin metabolism disorders: a review of theoretical concerns and clinical experiences in 28 patients. J Inherit Metab Dis 38:847–853
Kubová H, Folbergrová J, Mares P (1995) Seizures induced by homocysteine in rats during ontogenesis. Epilepsia 36:750–756
Kvittingen EA, Spangen S, Lindemans J, Fowler B (1997) Methionine synthase deficiency without megaloblastic anaemia. Eur J Pediatr 156:925–930
la Marca G, Malvagia S, Pasquini E, Innocenti M, Donati MA, Zammarchi E (2007) Rapid 2nd-tier test for measurement of 3-OH-propionic and methylmalonic acids on dried blood spots: reducing the false-positive rate for propionylcarnitine during expanded newborn screening by liquid chromatography-tandem mass spectrometry. Clin Chem 53:1364–1369
Lawson-Yuen A, Levy HL (2006) The use of betaine in the treatment of elevated homocysteine. Mol Genet Metab 88:201–207
Leclerc D, Wilson A, Dumas R et al (1998) Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc Natl Acad Sci 95:3059–3064
Lerner-Ellis JP, Tirone JC, Pawelek PD et al (2006) Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet 38:93–100
Lerner-Ellis JP, Anastasio N, Liu J et al (2009) Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum Mutat 30:1072–1081
Lesesve JF, Latger-Cannard V (2013) Crucial microscopic features leading to diagnosis of cobalamin C deficiency in a new-born. Br J Haematol 161:608
Levy HL, Mudd SH, Schulman JD, Dreyfus PM, Abeles RH (1970) A derangement in B12 metabolism associated with homocystinemia, cystathioninemia, hypomethioninemia and methylmalonic aciduria. Am J Med 48:390–397
Li YN, Gulati S, Baker PJ, Brody LC, Banerjee R, Kruger WD (1996) Cloning, mapping and RNA analysis of the human methionine synthase gene. Hum Mol Genet 5:1851–1858
Lin HJ, Neidich JA, Salazar D et al (2009) Asymptomatic maternal combined homocystinuria and methylmalonic aciduria (cblC) detected through low carnitine levels on newborn screening. J Pediatr 155:924–927
Liu Y, Wang Q, Li X, Ding Y, Song J, Yang Y (2015) First Chinese case of successful pregnancy with combined methylmalonic aciduria and homocystinuria, cblC type. Brain Dev 37:286–291
Longo D, Fariello G, Dionisi-Vici C et al (2005) MRI and 1H-MRS findings in early-onset cobalamin C/D defect. Neuropediatrics 36:366–372
Lossos A, Omri T, Tsipi M, Vardiella M, Rima R, Daniel L (2014) Severe methylenetetrahydrofolate reductase deficiency clinical clues to a potentially treatable cause of adult onset hereditary spastic paraplegia. JAMA Neurol 71:901–904
Maamar M, Mezalek ZT, Harmouche H, Adnaoui M, Aouni M, Maaouni A (2008) Contribution of spinal MRI for unsuspected cobalamin deficiency in isolated sub-acute combined degeneration. Eur J Intern Med 19:143–145
Mah W, Deme JC, Watkins D et al (2013) Subcellular location of MMACHC and MMADHC, two human proteins central to intracellular vitamin B(12) metabolism. Mol Gen Metab 108:112–118
Malvagia S, Haynes CA, Grisotto L et al (2015) Heptadecanoylcarnitine (C17) a novel candidate biomarker for newborn screening of propionic and methylmalonic acidemias. Clin Chim Acta 23:342–348
Manoli I, Myles JG, Sloan JL et al (2015) A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 2: cobalamin C deficiency. Genet Med. doi:10.1038/gim.2015.107
Mares P, Folbergrová J, Langmeier M, Haugvicová R, Kubová H (1997) Convulsant action of D, L-homocysteic acid and its stereoisomers in immature rats. Epilepsia 38:767–776
Martinelli D, Deodato F, Dionisi-Vici C (2011) Cobalamin C defect: natural history, pathophysiology, and treatment. J Inherit Metab Dis 34:127–135
Matos IV, Castejón E, Meavilla S et al (2013) Clinical and biochemical outcome after hydroxocobalamin dose escalation in a series of patients with cobalamin C deficiency. Mol Genet Metab 109:360–365
McCully KS (1969) Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 56:111–128
McCully KS (1992) Homocystinuria, arteriosclerosis, methylmalonic aciduria, and methyltransferase deficiency: a key case revisited. Nutr Rev 50:7–12
McHugh D, Cameron CA, Abdenur JE et al (2011) Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med 13:230–254
Menni F, Testa S, Guez S, Chiarelli G, Alberti L, Esposito S (2012) Neonatal atypical hemolytic uremic syndrome due to methylmalonic aciduria and homocystinuria. Pediatr Nephrol 27:1401–1405
Merinero B, Pérez-Cerdá C, Garcia MJ et al (1998) Reliability of biochemical parameters used in prenatal diagnosis of combined methylmalonic aciduria and homocystinuria. Prenat Diagn 18:947–952
Michot JM, Sedel F, Giraudier S, Smiejan JM, Papo T (2008) Psychosis, paraplegia and coma revealing methylenetetrahydrofolate reductase deficiency in a 56-year-old woman. J Neurol Neurosurg Psychiatry 79:963–964
Miousse IR, Watkins D, Coelho D et al (2009) Clinical and molecular heterogeneity in patients with the cblD inborn error of cobalamin metabolism. J Pediatr 154:551–556
Miousse IR, Watkins D, Rosenblatt DS (2011) Novel splice site mutations and a large deletion in three patients with the cblF inborn error of vitamin B12 metabolism. Mol Genet Metab 102:505–507
Mitchell GA, Watkins D, Melançon SB et al (1986) Clinical heterogeneity in cobalamin C variant of combined homocystinuria and methylmalonic aciduria. J Pediatr 108:410–415
Moat SJ, Bonham JR, Tanner MS, Allen JC, Powers HJ (1999) Recommended approaches for the laboratory measurement of homocysteine in the diagnosis and monitoring of patients with hyperhomocysteinaemia. Ann Clin Biochem 36:372–379
Morath MA, Okun JG, Müller IB, Sauer SW, Hörster F, Hoffmann GF, Kölker S (2008) Neurodegeneration and chronic renal failure in methylmalonic aciduria—a pathophysiological approach. J Inherit Metab Dis 31:35–43
Morath MA, Hörster F, Sauer SW (2013) Renal dysfunction in methylmalonic acidurias: review for the pediatric nephrologist. Pediatr Nephrol 28:227–235
Morel C, Watkins D, Scott P, Rinaldo P, Rosenblatt DS (2005) Prenatal diagnosis for methylmalonic acidemia and inborn errors of vitamin B12 metabolism and transport. Mol Genet Metab 86:160–171
Morel CF, Lerner-Ellis JP, Rosenblatt DS (2006) Combined methylmalonic aciduria and homocystinuria (cblC): phenotype-genotype correlations and ethnic-specific observations. Mol Genet Metab 88:315–321
Moreno-Garcia MA, Pupavac M, Rosenblatt DS, Tremblay ML, Jerome-Majewska LA (2014) The Mmachc gene is required for pre-implantation embryogenesis in the mouse. Mol Genet Metab 112:198–204
Mudd SH (2011) Hypermethioninemias of genetic and non-genetic origin: a review. Am J Med Genet C Semin Med Genet 15:3–32
Mudd SH, Freeman JM (1974) N-5,10-methylenetetrahydrofolate reductase deficiency and schizophrenia: a working hypothesis. J Psychiatr Res 11:259–262
Müller P, Horneff G, Hennermann JB (2007) A rare inborn error of intracellular processing of cobalamine presenting with microcephalus and megaloblastic anemia: a report of 3 children. Klin Padiatr 219:361–367
Munoz T, Patel J, Badilla-Porras R, Kronick J, Mercimek-Mahmutoglu S (2015) Severe scoliosis in a patient with severe methylenetetrahydrofolate reductase deficiency. Brain Dev 37:168–170
Nishimura M, Yoshino K, Tomita Y et al (1985) Central and peripheral nervous system pathology of homocystinuria due to 5,10-methylenetetrahydrofolate reductase deficiency. Pediatr Neurol 1:375–378
Nogueira C, Aiello C, Cerone R et al (2008) Spectrum of MMACHC mutations in Italian and Portuguese patients with combined methylmalonic aciduria and homocystinuria, cblC type. Mol Genet Metab 93:475–480
Ogier de Baulny H, Gérard M, Saudubray JM, Zittoun J (1998) Remethylation defects: guidelines for clinical diagnosis and treatment. Eur J Pediatr 157:S77–83
Olteanu H, Banerjee R (2001) Human methionine synthase reductase, a soluble P-450 reductase-like dual flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J Biol Chem 276:35558–35563
Pasquier F, Lebert F, Petit H, Zittoun J, Marquet J (1994) Methylenetetrahydrofolate reductase deficiency revealed by a neuropathy in a psychotic adult. J Neurol Neurosurg Psychiatry 57:765–756
Pastore A, Martinelli D, Piemonte F et al (2014) Glutathione metabolism in cobalamin deficiency type C (cblC). J Inherit Metab Dis 37:125–129
Patton N, Beatty S, Lloyd IC, Wraith JE (2000) Optic atrophy in association with cobalamin C (cblC) disease. Ophthalmic Genet 21:151–154
Paul EA, Guttenberg M, Kaplan P et al (2013) Atypical glomerulopathy associated with the cblE inborn error of vitamin B12 metabolism. Pediatr Nephrol 28:1135–1139
Pierre G, Gissen P, Chakrapani A, McDonald A, Preece M, Wright J (2006) Successful treatment of pyridoxine-unresponsive homocystinuria with betaine in pregnancy. J Inherit Metab Dis 29:688–689
Powers JM, Rosenblatt DS, Schmidt RE et al (2001) Neurological and neuropathologic heterogeneity in two brothers with cobalamin C deficiency. Ann Neurol 49:396–400
Prasad AN, Rupar CA, Prasad C (2011) Methyltetrahydrofolate reductase (MTHFR)deficiency and infantile epilepsy. Brain Dev 33:758–769
Profitlich L, Kirmse B, Wasserstein MP, Diaz G, Srivastava S (2009a) Resolution of cor pulmonale after medical management in a patient with cblC-type methylmalonic aciduria and homocystinuria: a case report. Cases J 30:8603
Profitlich LE, Kirmse B, Wasserstein MP, Diaz GA, Srivastava S (2009b) High prevalence of structural heart disease in children with cblC-type methylmalonic aciduria and homocystinuria. Mol Genet Metab 98:344–348
Rahmandar MH, Bawcom A, Romano ME, Hamid R (2014) Cobalamin C deficiency in an adolescent with altered mental status and anorexia. Pediatrics 134:e1709–1714
Refsum H, Grindflek AW, Ueland PM et al (2004) Screening for serum total homocysteine in newborn children. Clin Chem 50:1769–1784
Regland B, Johansson BV, Gottfries CG (1994) Homocysteinemia and schizophrenia as a case of methylation deficiency. J Neural Transm Gen Sect 98:143–152
Ribes A, Briones P, Vilaseca MA et al (1990) Methylmalonic aciduria with homocystinuria: biochemical studies, treatment, and clinical course of a Cbl-C patient. Eur J Pediatr 149:412–415
Ricci D, Pane M, Deodato F et al (2005) Assessment of visual function in children with methylmalonic aciduria and homocystinuria. Neuropediatrics 36:181–185
Robb RM, Dowton SB, Fulton AB, Levy HL (1984) Retinal degeneration in vitamin B12 disorder associated with methylmalonic aciduria and sulfur amino acid abnormalities. Am J Ophthalmol 97:691–696
Ronge E, Kiellman (1996) Long term treatment with betaine in methylenetetrahydrofolate reductase deficiency. Arch Dis Child 74:239–241
Rosenblatt DS, Cooper BA, Schmutz SM, Zaleski WA, Casey RE (1985) Prenatal vitamin B12 therapy of a fetus with methylcobalamin deficiency (cobalamin E disease). Lancet 18:1127–1129
Rosenblatt DS, Laframboise R, Pichette J, Langevin P, Cooper BA, Costa (1986) New disorder of vitamin B12 metabolism (cobalamin F) presenting as methylmalonic aciduria. Pediatrics 78:51–54
Rosenblatt DS, Aspler AL, Shevell MI, Pletcher BA, Fenton WA, Seashore MR (1997) Clinical heterogeneity and prognosis in combined methylmalonic aciduria and homocystinuria (cblC). J Inherit Metab Dis 20:528–538
Rossi A, Cerone R, Biancheri R et al (2001) Early-onset combined methylmalonic aciduria and homocystinuria: neuroradiologic findings. AJNR Am J Neuroradiol 22:554–563
Roze E, Gervais D, Demeret S et al (2003) Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol 60:1457–1462
Ruiz-Mercado M, Vargas MT, Pérez de Soto I et al (2015) Methionine synthase reductase deficiency (CblE): a report of two patients and a novel mutation. Hematology doi: 10.1179/1607845415Y.0000000017
Russo P, Doyon J, Sonsino E, Ogier H, Saudubray JM (1992) A congenital anomaly of vitamin B12 metabolism: a study of three cases. Hum Pathol 23:504–512
Rutsch F, Gailus S, Miousse IR et al (2009) Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat Genet 41:234–239
Scalabrino G, Veber D, Mutti E (2007) New pathogenesis of the cobalamin-deficient neuropathy. Med Secoli 19:9–18
Schiff M, Blom HJ (2012) Treatment of inherited homocystinurias. Neuropediatrics 43:295–304
Schiff M, Benoist JF, Tilea B, Royer N, Giraudier S, Ogier de Baulny H (2011) Isolated remethylation disorders: do our treatments benefit patients? J Inherit Metab Dis 34:137–145
Schimel AM, Mets MB (2006) The natural history of retinal degeneration in association with cobalamin C (cbl C) disease. Ophthalmic Genet 27:9–14
Scolamiero E, Villani GR, Ingenito L et al (2014) Maternal vitamin B12 deficiency detected in expanded newborn screening. Clin Biochem 47:312–317
Schwahn BC, Hafner D, Hohlfeld T, Balkenhol N, Laryea MD, Wendel U (2003) Pharmacokinetics of oral betaine in healthy subjects and patients with homocystinuria. Br J Clin Pharmacol 55(1):6–13
Selzer RR, Rosenblatt DS, Laxova R, Hogan K (2003) Adverse effect of nitrous oxide in a child with 5,10-methylenetetrahydrofolate reductase deficiency. N Engl J Med 349:45–50
Sharma AP, Greenberg CR, Prasad AN, Prasad C (2007) Hemolytic uremic syndrome (HUS) secondary to cobalamin C (cblC) disorder. Pediatr Nephrol 22:2097–2103
Shih VE, Axel SM, Tewksbury JC, Watkins D, Cooper BA, Rosenblatt DS (1989) Defective lysosomal release of vitamin B12 (cb1F): a hereditary cobalamin metabolic disorder associated with sudden death. Am J Med Genet 33:555–563
Shinnar S, Singer HS (1984) Cobalamin C mutation (methylmalonic aciduria and homocystinuria) in adolescence. A treatable cause of dementia and myelopathy. N Engl J Med 311:451–454
Sibani S, Christensen B, O’Ferrall E et al (2000) Characterization of six novel mutations in the methylenetetrahydrofolate reductase (MTHFR) gene in patients with homocystinuria. Hum Mutat 15:280–287
Smith DL, Bodamer OA (2002) Practical management of combined methylmalonicaciduria and homocystinuria. J Child Neurol 17:353–356
Smith SE, Kinney HC, Swoboda KJ, Levy HL (2006) Subacute combined degeneration of the spinal cord in cblC disorder despite treatment with B12. Mol Genet Metab 88:138–145
Spector R (1979) Affinity of folic acid for the folate-binding protein of choroid plexus. Arch Biochem Biophys 194:632–634
Stabler SP, Lindenbaum J, Savage DG, Allen RH (1993) Elevation of serum cystathionine levels in patients with cobalamin and folate deficiency. Blood 81:3404–3413
Stabler SP, Korson M, Jethva R et al (2013) Metabolic profiling of total homocysteine and related compounds in hyperhomocysteinemia: utility and limitations in diagnosing the cause of puzzling thrombophilia in a family. JIMD Rep 11:149–63
Steen C, Rosenblatt DS, Scheying H, Braeuer HC, Kohlschütter A (1997) Cobalamin E (cblE) disease: a severe neurological disorder with megaloblastic anaemia, homocystinuria and low serum methionine. J Inherit Metab Dis 20:705–706
Strauss KA, Morton DH, Puffenberger EG et al (2007) Prevention of brain disease from severe 5,10 methyltetrahydrofolate reductase deficiency. Mol Genet Metab 91:165–175
Stucki M, Coelho D, Suormala T, Burda P, Fowler B, Baumgartner MR (2012) Molecular mechanisms leading to three different phenotypes in the cblD defect of intracellular cobalamin metabolism. Hum Mol Genet 21:1410–1418
Suormala T, Gamse G, Fowler B (2002) 5,10-methylenetetrahydrofolate reductase (MTHFR) assay in the forward direction: residual activity in MTHFR deficiency. Clin Chem 48:835–843
Suormala T, Baumgartner MR, Coelho D et al (2004) The CbD defect causes either isolated or combinated deficiency of Methylcobalamin and adenosylcobalamin synthesis. J Biol Chem 279:42742–42749
Surtees R, Leonard J, Austin S (1991) Association of demyelination with deficiency of cerebrospinal fluid S-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet 338:1550–1554
Tallur KK, Johnson DA, Kirk JM, Sandercock PA, Minns RA (2005) Folate-induced reversal of leukoencephalopathy and intellectual decline in methylene-tetrahydrofolate reductase deficiency: variable response in siblings. Dev Med Child Neurol 47:53–56
Tanpaiboon P, Sloan JL, Callahan PF et al (2013) Noncompaction of the ventricular myocardium and hydrops fetalis in cobalamin C disease. JIMD Rep 10:33–38
Thauvin-Robinet C, Roze E, Couvreur G et al (2008) The adolescent and adult form of cobalamin C disease: clinical and molecular spectrum. J Neurol Neurosurg Psychiatry 79:725–728
Tomaske M, Bosk A, Heinemann MK et al (2001) CblC/D defect combined with haemodynamically highly relevant VSD. J Inherit Metab Dis 24:511–512
Tonetti C, Ruivard M, Rieu V, Zittoun J, Giraudier S (2002) Severe methylenetetrahydrofolate reductase deficiency revealed by a pulmonary embolism in a young adult. Br J Haematol 119:397–379
Tonetti C, Saudubray JM, Echenne B, Landrieu P, Giraudier S, Zittoun J (2003) Relations between molecular and biological abnormalities in 11 families from siblings affected with methylenetetrahydrofolate reductase deficiency. Eur J Pediatr 162:466–475
Tortorelli S, Turgeon CT, Lim JS et al (2010) Two-tier approach to the newborn screening of methylenetetrahydrofolate reductase deficiency and other remethylation disorders with tandem mass spectrometry. J Pediatr 157:271–275
Traboulsi EI, Silva JC, Geraghty MT, Maumenee IH, Valle D, Green WR (1992) Ocular histopathologic characteristics of cobalamin C type vitamin B12 defect with methylmalonic aciduria and homocystinuria. Am J Ophthalmol 113:269–280
Trakadis YJ, Alfares A, Bodamer OA, Buyukavci M, Christodoulou J, Connor P (2014) Update on transcobalamin deficiency: clinical presentation, treatment and outcome. J Inherit Metab Dis 37:461–473
Trefz FK, Scheible D, Frauendienst-Egger G et al (2016) Successful intrauterine treatment of a patient with cobalamin C defect. Mol Genet Metab Rep 6:55–59. doi:10.1016/j.ymgmr.2016.01.005.eCollection
Tsai AC, Morel CF, Scharer G et al (2007) Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am J Med Genet A 143A:2430–2434
Tsang BL, Devine OJ, Cordero AM et al (2015) Assessing the association between the methylenetetrahydrofolate reductase (MTHFR) 677C>T polymorphism and blood folate concentrations: a systematic review and meta-analysis of trials and observational studies. Am J Clin Nutr 101:1286–1294
Tsina EK, Marsden DL, Hansen RM, Fulton AB (2005) Maculopathy and retinal degeneration in cobalamin C methylmalonic aciduria and homocystinuria. Arch Ophthalmol 123:1143–1146
Ucar SK, Koroğlu OA, Berk O et al (2010) Titration of betaine therapy to optimize therapy in an infant with 5,10-methylenetetrahydrofolate reductase deficiency. Eur J Pediatr 169:241–243
Urbón Artero A, Aldana Gómez J, Reig Del Moral C, Nieto Conde C, Merinero Cortés B (2002) Neonatal onset methylmalonic aciduria and homocystinuria: biochemical and clinical improvement with betaine therapy. An Esp Pediatr 56:337–341
Urreizti R, Moya-García AA, Pino-Ángeles A et al (2010) Molecular characterization of five patients with homocystinuria due to severe methylenetetrahydrofolate reductase deficiency. Clin Genet 78:441–448
Van Hove JL, Van Damme-Lombaerts R, Grünewald S et al (2002) Cobalamin disorder Cbl-C presenting with late-onset thrombotic microangiopathy. Am J Med Genet 111:195–201
Vilaseca MA, Vilarinho L, Zavadakova P et al (2003) CblE type of homocystinuria: mild clinical phenotype in two patients homozygous for a novel mutation in the MTRR gene. J Inherit Metab Dis 26:361–369
Visy JM, Le Coz P, Chadefaux B et al (1991) Homocystinuria due to 5,10-methylenetetrahydrofolate reductase deficiency revealed by stroke in adult siblings. Neurology 41:1313–1315
Waggoner DJ, Ueda K, Mantia C, Dowton SB (1998) Methylmalonic aciduria (cblF): case report and response to therapy. Am J Med Genet 79:373–375
Walk D, Kang SS, Horwitz A (1994) Intermittent encephalopathy, reversible nerve conduction slowing, and MRI evidence of cerebral white matter disease in methylenetetrahydrofolate reductase deficiency. Neurology 44:344–347
Wang F, Han L, Yang Y et al (2010) Clinical, biochemical, and molecular analysis of combined methylmalonic acidemia and hyperhomocysteinemia (cblC type) in China. J Inherit Metab Dis 33:S435–S442
Wang X, Sun W, Yang Y, Jia J, Li C (2012) A clinical and gene analysis of late-onset combined methylmalonic aciduria and homocystinuria, cblC type, in China. J Neurol Sci 15:155–159
Wang Q, Liu J, Liu YP et al (2014) Methylenetetrahydrofolate reductase deficiency-induced schizophrenia in a school-age boy. Zhongguo Dang Dai Er Ke Za Zhi 16:62–66
Watkins D, Rosenblatt DS (1989) Functional methionine synthase deficiency (cblE and cblG): clinical and biochemical heterogeneity. Am J Med Genet 34:427–434
Watkins D, Rosenblatt DS (2012) Update and new concepts in vitamin responsive disorders of folate transport and metabolism. J Inherit Metab Dis 35:665–670
Weisfeld-Adams JD, Bender HA, Miley-Åkerstedt A et al (2013) Neurologic and neurodevelopmental phenotypes in young children with early-treated combined methylmalonic acidemia and homocystinuria, cobalamin C type. Mol Genet Metab 110:241–247
Weisfeld-Adams JD, Mc Court EA, Diaz GA, Oliver SC (2015) Ocular disease in the cobalamin C defect: a review of the literature and a suggested framework for clinical surveillance. Mol Genet Metab 114:537–546
Whitehead VM (2006) Acquired and inherited disorders of cobalamin and folate in children. Br J Haematol 134:125–136
Wong D, Tortorelli S, Bishop L et al (2016) Outcomes of four patients with homocysteine remethylation disorders detected by newborn screening. Genet Med 18:162–167
Wu S, Gonzalez-Gomez I, Coates T, Yano S (2005) Cobalamin C disease presenting with hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol 22:717–721
Yaghmai R, Kashani AH, Geraghty MT et al (2002) Progressive cerebral edema associated with high methionine levels and betaine therapy in a patient with cystathionine beta-synthase (CBS) deficiency. Am J Med Genet 108:57–63
Yamada K, Gravel RA, Toraya T, Matthews RG (2006) Human methionine synthase reductase is a molecular chaperone for human methionine synthase. Proc Natl Acad Sci U S A 103(25):9476–9481
Yu HC, Sloan JL, Scharer G et al (2013) An X-linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. Am J Hum Genet 93:506–514
Zavadáková P, Fowler B, Suormala T et al (2005) cblE type of homocystinuria due to methionine synthase reductase deficiency: functional correction by minigene expression. Hum Mutat 25:239–247
Zsengellér ZK, Aljinovic N, Teot LA et al (2014) Methylmalonic acidemia: a megamitochondrial disorder affecting the kidney. Pediatr Nephrol 29:2139–2146
Acknowledgments
We gratefully acknowledge the support from Prof. Brian Fowler who critically revised the manuscript. We thank Marike Gronendijk for her ongoing support of our work.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors of this guideline declare no competing interests but disclose the following: MRB has received financial support for attending E-HOD steering committee meetings from Orphan Europe. MRB and MH have received support from Milupa Metabolics for travel grants to develop a quality of life assessment tool for patients with intoxication type metabolic diseases. Charles University in Prague-First Faculty of Medicine received support from the Recordati Rare Disease Foundation for organizing an educational course on homocystinurias and methylation defects. MH, MRB, CDV, HB received speakers honoraria for their contribution to this educational course. The development of this guideline was part of the “European network and registry for homocystinurias and methylation defects (EHOD) project” (No.2012_12_02) which has received funding from the European Union in the framework of the Health Program. This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Commnunicated by: Sylvia Stoeckler-Ipsiroglu
Martina Huemer, Daria Diodato, Matthias R. Baumgartner and Carlo Dionisi-Vici contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 32 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Huemer, M., Diodato, D., Schwahn, B. et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis 40, 21–48 (2017). https://doi.org/10.1007/s10545-016-9991-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-016-9991-4