
Abstract
Hymenoscyphus fraxineus, causal agent of Ash Dieback, has posed a threat to Fraxinus excelsior (common ash) in Europe since the 1990s. In south-western Europe, optimal climatic conditions for H. fraxineus become scattered and host density decreases, reducing disease spread rates. To date, the Ash Dieback agent has not been reported from southern and most of central Italy, where native F. excelsior is present as small fragmented populations. This study examines the expansion of Ash Dieback into central Italy, and it considers the consequences of further local spread with regards to the loss of F. excelsior genetic resource. Symptomatic F. excelsior were sampled from sixteen sites in northern and central Italy during 2020. Specimens were analyzed with a culturomics and a quantitative PCR approach. A bibliographic search of F. excelsior floristic reports was conducted for the creation of a detailed range map. The combined use of both techniques confirmed the presence of H. fraxineus in all the sites of central Italy where host plants were symptomatic. These new records represent the southern limit of the current known distribution of this pathogen in Italy, and together with Montenegro, in Europe. The characterization of the F. excelsior scattered range suggests that further spread of Ash Dieback across southern Italy is a realistic scenario. This presents a threat not just to the southern European proveniences of F. excelsior, but to the species as a whole, should Ash Dieback lead to the loss of warm climate adapted genetic material, which may become increasingly valuable under climate change.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hymenoscyphus fraxineus (T. Kowalski) Baral, Queloz & Hosoya, the causal agent of Ash Dieback disease, represents a serious threat for the common ash tree (Fraxinus excelsior) in the European continent (Kowalski 2006; Enderle et al. 2019). Wind-dispersed ascospores of Hymenoscyphus fraxineus form in apothecia on infected leaf petioles in the leaf litter (Kowalski and Holdenrieder 2009). The pathogen enters the host through leaves, young stalks and epicormic shoots and progresses from there to reach the woody tissue (Chandelier et al. 2013). Symptoms occur during spring in in the xylem and the bark of leaves, twigs, shoots and stems (Kowalski et al. 2010), causing wilting of leaves, shoot dieback, necrotic lesions in the bark and wood discoloration (Timmermann et al. 2011). In young ash trees, stem symptoms typically vary from a necrotic bark lesion with a small dead twig in the center to extensive bark necrosis (Kirisits et al. 2012). In this last case, apex dieback may occur in a few weeks. On older trees, ash dieback often becomes chronic. This leads to a progressive deterioration of the plant, predisposing it to other damaging factors, and eventually results in death (Timmermann et al. 2011).
The disease was reported for the first time in north-eastern Poland in 1993 (Kowalski 2006; Gil et al. 2017) and today is present in most parts of the ash distribution range in Europe. Anthropogenic factors, i.e. mainly the trade of infected plant material, and airborne dispersal of ascospores are the main pathways for spread of the pathogen to many countries (Sansford 2013; Drenkhan et al. 2017; Gil et al. 2017; Longauerová et al. 2017). The disease spread is fast, estimated to be in the range of 40–75 km a year on a continental/national scale (Gross 2013; Laivins et al. 2016). This value is likely the combined effect of human mediated and natural dispersal, since natural aerial spread for ascospores dispersal from inoculum sources has been reported to be as high as 1.4–2.6 km (Grosdidier et al. 2018a). More recently, extraordinary colonization rates of 50 km a year, driven by wind turbulences, were observed along the disease front of Northern Italy (Giongo et al. 2017) and France (Grosdidier et al. 2018b). Such impressive and fast long-distance movement is supported by the results of Grosdidier et al. (2018a), who detected aerial ascospores of H. fraxineus as far as 100 km beyond the disease front in France.
Moving south and west in the European continent, the spread of H. fraxineus rapidly decreases. The suitable environmental conditions for disease development are represented by abundant summer precipitation, high soil moisture and low air temperature (Dal Maso and Montecchio 2014). These conditions are common in central and northern European countries, where the pathogen spread fast from the early years of its establishment in the continent (Pautasso et al. 2013). As reported by Ghelardini et al. (2017) such environmental traits become scattered and geographically isolated in the European Mediterranean countries, limiting the potential presence of H. fraxineus to the main mountain ranges over a certain altitude. In addition, the diminishing host density and increasing tree cover fragmentation are limiting factors to disease establishment (Grosdidier et al. 2020).
The southern and western limits of H. fraxineus in Europe are currently represented by Montenegro in the Balkans (Milenković et al. 2017), Central Italy (Ghelardini et al. 2017) and Central France (Grosdidier et al. 2018a). It is believed that beyond these areas, Mediterranean climatic conditions render unlikely the spread and establishment of Ash Dieback by natural dispersal (Dal Maso and Montecchio 2014). However, in southern Italy, sites suitable for the establishment of the fungus are present, especially along the Apennines chain (Dal Maso and Montecchio 2014; Ghelardini et al. 2017). In this area F. excelsior, even if rare, is diffused as small fragmented populations or as singular trees, across the entire mountain chain (Caudullo et al. 2017 and citations reported in Table 3). Consequently, human mediated or natural, localized introductions of H. fraxineus cannot be excluded.
The Apennines seem to have held a glacial refugium for at least one F. excelsior haplotype, which is now occurring throughout Italy and in south western France (Heuertz et al. 2004a). It is widely accepted that refugium populations may accumulate higher diversity and/or unique haplotypes owing to their persistence and relative stability over the glacial era (Hewitt 1996; Newton et al. 1999; Tzedakis et al. 2002). These populations are probably better suited to warmer climates and could be an important source of adaptive genes to survive a changing climate characterized by increasing temperatures. For these reasons, the spread of disease in these areas could represent a threat to the conservation of genetic diversity and destroy a useful source of genes for adaptation to climate change.
The aim of this study is to describe the present spread of the Ash Dieback epidemic in central Italy, and to draw attention to its possible threat for F. excelsior in the stands where the pathogen is not yet present.
Material and methods
Plant material and genomic DNA extraction
In August–September 2020, symptomatic trees of F. excelsior were sampled from 16 stands in northern and central Italy, along the Apennine Mountains (Table 1). Samples from sites 2 to 13 were collected in the area of the 2015 first report from the Apennines (Ghelardini et al. 2017; Luchi et al. 2016) in order to verify the consistency of local disease spread during the last years. Samples from sites 14 to 18 were collected across the three most relevant mountain massifs of central Italy, following the host natural range: Monti Sibillini National Park (14), Gran Sasso and Monti della Laga National Park (15 and 16) and Abruzzo, Lazio and Molise National Park (17 and 18). In this last location, F. excelsior was present but no symptoms of the disease were detected, therefore no samples were collected. (see Results). Symptoms included blackish discoloration of leaves; necrotic leaf rachises; wilting and leaf dropping; dieback of shoots, twigs and branches; necrosis of bark tissue.
Isolations were made from symptomatic branches and rachises after surface sterilization with sodium hypochlorite and ethanol according to EPPO protocol (Chandelier et al. 2013). Fragments of necrotic tissue were placed on 2% malt extract agar (MEA, DIFCO, Detroit, Michigan, USA) amended with streptomycin (0.050 gr/l) and incubated at 5 °C in the dark. After 2–4 weeks, small, dull white to fulvous brown colonies were observed and sub-cultured on MEA and maintained at 20 °C.
DNA was extracted from a portion of branches and rachises from each sampling site and from one apothecium collected in site 16 (Table 1). About 100 mg fresh weight each (the entire apothecium for the sample of site 16) was transferred to 2-ml microfuge tubes with two tungsten beads (3 mm) (Qiagen, Venlo, Netherlands) and 0.6-ml lysis buffer (EZNA Plant DNA Kit—Omega Bio-tek, Norcross, Georgia, USA), and ground with a Mixer Mill 300 (Qiagen) (2 min; 20 Hz). DNA was extracted from all samples using the EZNA Plant DNA Kit (Omega Bio-tek), following the manufacturer’s protocol.
Isolate sequencing and taxonomical analysis
Twelve representative isolates obtained in this study (Table 2) were used for phylogenetic analysis. Each isolate was grown on 300PT cellophane discs (Celsa, Varese, Italy) on MEA in 90-mm Petri dishes and maintained in the dark at 20 °C. After 15 days, mycelium was scraped from the cellophane surface, placed into 1.5 ml microfuge tubes (Sarstedt, Verona, Italy) and stored at -20 °C until use. DNA was extracted from all samples using the EZNA Plant DNA Kit (Omega Bio-tek), as previously described. The DNA concentration was measured using a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, Delaware, USA). Amplification of the internal transcribed spacer ITS region (including spacers ITS1 and ITS2 and the 5.8S gene of the rDNA) was carried out by using ITS1 and ITS4 primers (White et al. 1990).
PCR reaction mixtures (25 µl) contained 1 µl of genomic DNA, 2.5 µl of each 10 µM primer, 0.5 µl of 10 mM dNTPs (GeneSpin, Milan, Italy), 2.5 µl of 10X PCR Buffer (GeneSpin), 3 µl of 25 nM MgCl2 and 0.5 µl (5 U/µl) of Taq polymerase (TaqPol, GeneSpin). Amplifications were carried out in a Mastercycler (Eppendorf, Hamburg, Germany) using the following PCR conditions: initial denaturation at 95 °C for 5 min; followed by 35 cycles of denaturation at 94 °C for 90 s, annealing at 56 °C for 1 min, and extension at 72 °C for 2 min; and a final elongation step at 72 °C for 10 min. (Migliorini et al. 2020). Amplification products were separated by electrophoresis on gels containing 1% (w/v) of agarose LE (GeneSpin). The approximate length (bp) of the amplification products was determined using the 100-bp DNA ladder Ready to Load (Genespin). PCR amplicons were purified with a miPCR Purification Kit (Metabion International, Planegg, Germany) and sequenced in both directions at Macrogen (Seoul, South Korea). The quality of amplified nucleotide sequences was checked with the software package Geneious version 10.0.9 (Biomatters, Auckland, New Zealand). Generated sequences were submitted to NCBI GenBank (Table 2) and the new obtained cultures were stored in the Fungal Collection of the Institute for Sustainable Plant Protection-National Research Council (IPSP-CNR, Sesto Fiorentino, Italy).
Database for phylogeny
Genbank sequences used in this study were selected from the available literature according to the isolate’s provenience in the H. fraxinus whole range and the nucleotides polymorphisms present in each country (Table 2). This method provided a proper diagnostic output and, at the same time, it allowed the taxonomical placement of our isolates according to their ITS gene region. An accurate search and collection of numerous H. fraxineus ITS sequences available in Genbank was conducted (data not shown). The database used in this study was then built selecting at maximum two sequences per each identical nucleotide sequence per country within those with a robust bibliographic reference. Belgium, Serbia and Ukraine represented exceptions, with only one available sequence each.
Phylogenetic analysis
BLAST searches of the generated sequences were done against the NCBI GenBank database (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to identify the most closely-related sequences. Sequences were compared to those of the Hymenoscyphus fraxineus isolates collected across the entire Eurasian range of this fungus (Table 2). The software package Geneious (Auckland, New Zealand) was used for manual optimisation and alignment (ClustalW) of the sequences. Gaps were treated as missing data. Phylogenetic analyses of all obtained sequences were performed using MEGA X (Kumar et al. 2018).
The evolutionary history was inferred by using the Maximum Likelihood method and Tamura-Nei model (Tamura and Nei 1993). The tree with the highest log likelihood (-774.51) was shown. The percentage of trees in which the associated taxa clustered together was shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura-Nei model, and then selecting the topology with superior log likelihood value. This analysis involved 74 nucleotide sequences. There was a total of 423 positions in the final dataset. Evolutionary analyses were conducted in MEGA X (Kumar et al. 2018; Stecher et al. 2020).
qPCR analysis
DNA samples extracted from ash branches, rachises (including samples from Ash Dieback free area, used as negative control) and apothecia were amplified in MicroAmp Fast 96-well Reaction Plates (0.1 mL) closed with optical adhesive, and using the StepOnePlus™ Real-Time PCR System (Applied Biosystems, Life Science, Foster City, CA, USA). Real time PCR was assayed by using the H. fraxineus primers and probe designed by (Ioos et al. 2009), according to the EPPO protocol (Chandelier et al. 2013). Each DNA sample was assayed in duplicate. Two wells, each containing 5 μl of sterile water, were used as the no-template control.
Results were analyzed using an SDS 1.9 sequence detection system (Applied Biosystems) after manual adjustment of the baseline and fluorescence threshold. Quantification of Hymenoscyphus fraxineus DNA in the samples was made by interpolation from a standard curve generated with a H. fraxineus DNA standard that was amplified in the same PCR run. The standard curve was generated from ten fivefold serial dilutions (ranging from 10 ng/tube to 0.128 pg/tube) of a known concentration of H. fraxineus DNA (strain CBS 122,191) and analyzed in duplicate. The amount of H. fraxineus DNA from plant samples was indicated as pg of fungal DNA/µg of total DNA extracted.
Mapping Fraxinus excelsior in the southern Apennines
The databases Google Scholar, Scopus and Web of Science were searched using the searchstring ‘Fraxinus excelsior AND Apennines’. This search included both natural and planted nuclei. In addition, relevant studies were selected from among the papers that had cited the articles found in the search. A profuse search in grey literature was also done. We also contacted scientists and researchers to find upcoming but still unpublished studies of interest.
The results of this search were uploaded in the Free and Open Source QGis format map to implement the Common ash distribution map in southern Italy. The linear distance between the different cores of common ash were calculated to verify the possibility of being infected by the aerial spread of the fungus and the timeframe for infection under a constant spread rate.
Results
Plant material
In sites 1 to 16 we found symptoms of the disease on mature and young plants as leaves wilting, bark necrosis, dieback of shoots and branches. Young plants (dbh about 4 cm) also presented symptoms of stem dieback. Hymenoscyphus fraxineus-like sporocarps were present on litter petioles in all sites except Site 13 (data not shown). Common ash plants in Site 17 and Site 18 did not present any disease symptoms, therefore, no specimens were collected.
Phylogenetic analysis
BLASTn search in GenBank of sequences from all the isolates obtained in this survey revealed a 100% homology with Hymenoscyphus fraxineus. The ITS sequences (strains CNRHfraxGRUVB, CNRHfraxDNOV, CNRHfraxLST, CNRHfraxABT, CNRHfraxPDNOV2, CNRHfraxPDNOV2.2, CNRHfraxPOPTR, CNRHfraxINF13, CNRHfraxINF10RCHD, CNRHfraxVVAC, CNRHfraxLDP) have been deposited in Genbank under Accession Numbers MW238530, MW238533, MW238528, MW238534, MW238532, MW238529, MW238525, MW238527, MW238531, MW238523, MW357546 (Table 2).
The ITS gene phylogenetic tree included 74 ingroup and no outgroup sequences, comprising 423 alignment characters, including gaps (Fig. 1). Phylogenetic analysis showed that the isolates obtained in the sample collection are H. fraxineus.

Phylogenetic tree, based on an ML analysis of DNA dataset of ITS sequences for Hymenoscyphus fraxineus species. Numbers above the branches indicate ML bootstraps (MLBS ≥ 50%). Isolates from the current study are signed with a rhombus. The scale bar indicates the number of substitutions per site
Fungal isolates CNRHfraxVVAC (MW238523), CNRHfraxPOPTR (MW238525), CNRHfraxINF13 (MW238527), CNRHfraxLST (MW238528), CNRHfraxPDNOV2.2 (MW238529), CNRHfraxDNOV (MW238533) and CNRHfraxABT (MW238534) differed from the rest of the sequences for a nucleotide deletion at position 395 (*2 in Fig. 2). Isolates CNRHfraxGRUVB (MW238530), CNRHfraxINF10RCHD (MW238531), CNRHfraxLDP (MW357546) and CNRHfraxPDNOV2 (MW238532) did not present nucleotide differences with the larger group of H. fraxineus sequences retrieved in Genbank (*1 in Fig. 2).

Nucleotide polymorphism from aligned sequence data of ITS gene showing the variation between specimens of Hymenoscyphus fraxineus included in this study. Different colors demark base variations found in the sequences. Position of nucleotide variations are reported in the upper line. Genbank sequences with nucleotide variations per position (in bold): 2 -KJ511193; 18 -KJ511216; 29 -KJ511216, KJ511214; 31 -KJ511216, KP068053, KP068080, KP068073, KU323588, KJ511184, KJ511193, AB705220, KF188727, KP068075, KP403806, KU323576, KJ511214, AB705218, KF188725, KJ511195, KJ511197, KP068055; 32 -KC576535; 33 -KC576535; 44 -KP068053, KP068080; 46 -MG182858; 51 -KP068073, KU323588; 63 -MG182858; 76 -MG182858; 78 -KJ511216, KP068053, KP068080, KP068073, KU323588, FJ429386, GU586921, KC576530, KJ413058, KJ780084, KJ511184, KJ511193, AB705220, KF188727, KP068075, KP403806, KU323576, KJ511214, KJ820505, AB705218, KF188725, KJ511195, KJ511197, KP068055; 83 -MG182857; 91 -MG182858, MG182857, MG182859; 111 -AB705220; 162 -MG182858, MG182857; 167 -MG182858; 172 -MG182858; 206 -MG182858, MG182859; 320 -KP068053, KP068080; 323 -KJ511216 JP; 329 -MT053856, MT053857; 331 -MT053856, MT053857; 333 -KP068073, KU323588; 334 -MT053856, MT053857; 339 -FJ429386, GU586921, KC576530, KJ413058, KJ780084; 365 -MG182858; 373 -MK491651; 376 -MK491651; 377 -MK491651; 384 -MK491651; 395 -MW238534, MW238533, MW238527, MW238528, MW238529, MW238525, MW238523; 400 -MG182858; 406 -KF188727, KP068075, KP403806, KU323576; 414 -MK491649.
Sequences MW238530 (CNRHfraxGRUVB), MW238531 (CNRHfraxINF10RCHD), MW357546 (CNRHfraxLDP), MW238532 (CNRHfraxPDNOV2) did not present nucleotide deformities with the larger group of H. fraxineus sequences included in this study and they are represented by the nucleotide description of the first line (*1). Sequences MW238523 (CNRHfraxVVAC), MW238525 (CNRHfraxPOPTR), MW238527 (CNRHfraxINF13), MW238528 (CNRHfraxLST), MW238529 (CNRHfraxPDNOV2.2), MW238533 (CNRHfraxDNOV) and MW238534 (CNRHfraxABT) differed from the rest of the sequences for a nucleotide deletion at position 395 (line marked with *2).
qPCR analysis
Almost all samples from symptomatic plants were positive to the H. fraxineus-specific qPCR assay (0,001218—28830,11 pg H. fraxineus DNA/µg total DNA) (Table 1). The two samples from asymptomatic plants included as negative controls did not show any amplification signals, as expected. DNA from branches in Site 10 was negative, while rachises DNA from this site revealed the presence of H. fraxineus in the site. The apothecia and the rachides collected in Site 16 did not produce a positive amplification signal. Indeed, ITS gene sequencing revealed that the apothecia were from Hymenoscyphus albidus, (sample CNRHalbLDPRO) Hymenoscyphus fraxineus was, however, confirmed from Site 16, with the fungus both isolated and detected with qPCR in branch samples. No quantitative data could be provided for Site 14 because the plant samples were lost before the quantitative analysis.
Mapping Fraxinus excelsior in the southern Apennines
The results of this search are reported in Table 3 and synthetized in Fig. 3.

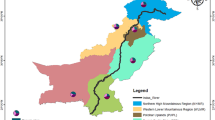
Spatial range map of Fraxinus excelsior (see Table 3 for references) and location of 18 sites visited in Italy during 2020. Orange dots are sites where Hymenoscyphus fraxineus was detected during the 2020 surveys. Yellow dots are sites were no samples were collected because plants did not present ash dieback symptoms. Red dots are the sites of previous H. fraxineus reports in Italy
Common ash scattered nuclei together with the closest limit on the main range have an average linear distance of 57 (min. 3- max. 110) km from one another (excluding the locations in Sardinia and Corsica islands).
The linear distance between the most southern report of the disease in 2015 (Castellaro di Galeata (FC) (Ghelardini et al. 2017), Fig. 3) and in 2020 (Lago di Provvidenza (TE), Site 16) is 201 km. The distance between the present report of H. fraxineus (Site 16) and the southern border of the host’s natural range (Longi, Alcara Li Fusi, Tassita (ME) (Schicchi and Marino 2011; Caudullo et al. 2017); Table 3) is about 330 km (this last measure is not linear and It corresponds to the sum of linear distances between common ash scattered nuclei).
Discussions
To the best of our knowledge, this is the first report of Hymenoscyphus fraxineus in Marche and Abruzzo regions along the central Apennines, an area characterized by mountain Mediterranean climatic conditions, with cold and snowy winters and cool summers without drought (Santini et al. 2010; Pesaresi et al. 2014). Detection of H. fraxineus in branches and rachises proved that the pathogen is diffused across all central Italy; it has moved from its previous southernmost distribution limit (Ghelardini et al. 2017; Luchi et al. 2016, this study), where it established (sites 1 to 12), to approximately 1.5 degrees of latitude further south at sites 14 (159 km), 15 (197 km) and 16 (201 km) (Table 1, Fig. 3). Together with the report of Ash Dieback in Montenegro by Milenković et al. (2017), this latitude represents the current known southern limit of Ash Dieback in Europe.
The taxonomical identification of the ITS gene region placed the analyzed isolates in the H. fraxineus most common sequence variant distributed in Europe. None of the isolates belong to the East Asian strains found in Czech Republic, Germany, Lithuania and Switzerland (Drenkhan et al. 2017), suggesting a central European origin for all the strains collected in this survey. Future research should focus on the origin of the pathogen we found in peninsular Italy following the population genetics approaches proposed by Burokiene et al. (2015) and Orton et al. (2018).
We combined the qPCR detection technique with the use of culture-based isolations to produce the most complete sampling results possible. Moreover, the detection of H. fraxineus in symptomatic specimens using both tools is always recommended, due to unpredictable probability of isolation success with some types of tissues and the high presence of false negative results due to the slow growth of the pathogen in culture (Bakys et al. 2009; Chandelier et al. 2011).
In our survey, successful H. fraxineus isolations were consistent with qPCR detection results, confirming the presence of active mycelium in all the sites except Site 13 where no fungal cultures were obtained, despite the positive qPCR result for this site. In this locality, symptomatic plants were at low altitude (590 m a.s.l.), which lies outside of the optimum ash growing conditions. The samples were collected at the end of the summer, and consisted of seedlings used to establish a new forest plantation during spring 2020. Studies comparing the efficiency of qPCR and isolation methods for Ash Dieback are lacking, and possible explanations for the inconsistent results for Site 13 can only be hypothesized based on biological traits of the fungus. To date, significant effects of high temperature on H. fraxineus have been demonstrated in lab conditions, but not yet in the field (Hauptman et al. 2013; Grosdidier et al. 2018b). This suggests that the mycelium can persist in the host tissues exposed to heat across the southern areas of its geographic diffusion in a summer-latent form. Such low viability may negatively affect isolation success, but not the qPCR detection, which can detect very low quantities of target DNA, as low as 20 fg of H. fraxineus DNA (Ioos et al. 2009). This may explain our failure to isolate H. fraxineus from symptomatic tissue of Site 13.
So far, symptoms of H. fraxineus have never been reported before at latitude below 43° 57'N (Ghelardini et al. 2017). This, together with our observations of initial symptoms during sampling, seems to suggest that the disease in site 14, 15 and 16 has begun very recently, possibly in 2019. This also indicates that H. fraxineus has likely moved about 201 km in the past four-five years (40–50 km/year). No symptomatic adult or young trees were found at Site 17 and Site 18, therefore we should consider Site 16 as the southernmost front of the disease spread. Further diffusion is still possible, however, along the natural range and also in the scattered range areas of the host plant, as despite the discontinuous nature of the populations, the distances are within the spread ability of the disease (Giongo et al. 2017; Grosdidier et al. 2018b) or of the ascospores aerial transport (Grosdidier et al. 2018a).
If the pathogen continues to spread at the same rate as observed in the present study, it can reach the southern border of the F. excelsior natural range in five more years and spread over the scattered populations in the entire Italian peninsula and Sicily Island. The scenario is likely to accelerate in case of unintentional human-aided spread, by the introduction of infected propagating material in areas still free from the disease.
As reported by Marigo et al. (2000), drawing the limits of F. excelsior in southern Europe is difficult, especially because its distribution is scattered in small groups of trees. However, following the literature review (Table 3) this study demonstrates that the natural occurrence of common ash, whose presence in the southern Apennines was generally neglected, is quite common, at least in sites where environmental conditions are favorable. We also observed that some common ash plantations were established, but data are not available, due to their occasionally and lack of financial value, or because they were planted not as forest but as ornamental trees. The role of planted trees should not be underestimated because they can act as aerial spore "bridges" between already infected and uninfected sites.
A significant additional risk factor in the disease diffusion may come from the natural widespread presence in central and southern Italy of Fraxinus angustifolia, a species also susceptible to Hymenoscyphus fraxineus. Infections similar to those caused on common ash have been reported in Austria and Hungary (Kirisits et al. 2010), Croatia (Barić et al. 2012), Serbia (Keča et al. 2017) and Slovakia (Kádasi-Horáková et al. 2017).
Although F. angustifolia is more thermophilic than F. excelsior, they are nevertheless sympatric along river-banks and in very humid valleys (Heuertz et al. 2006). For this reason it is reasonable to assume that where their presence intersects with the optimal environmental conditions for pathogen growth e.g. abundant summer rainfall, high soil moisture and low air temperature (Dal Maso and Montecchio 2014) F. angustifolia can act as a bridgehead for Ash Dieback. Identifying these sites and overseeing the possible arrival of the disease should be a priority to protect common ash biodiversity. It must be said that, unlike F. excelsior, the disease would not threaten the survival of this species, since its range covers also areas with a Mediterranean climate with summer aridity, where H. fraxineus cannot thrive.
Even if poorly studied, southeastern Europe, including south Italy, is considered a putative ice refuge for common ash (Heuertz et al. 2004b, 2006). In southeastern Europe, common ash populations are characterized by a strong genetic structure and by a marked genetic differentiation between neighboring populations (Heuertz et al. 2004b). In this area, the arrival of the pathogen by natural aerial spread, as well as the introduction of common ash from disease infected nurseries, may threaten near to extinction not the species itself, but the many F. excelsior small scattered marginal populations that probably conserve rare forms, harbor a wide genetic diversity and hold rare genes that could be particularly useful to improve the plasticity of this species to a changing climate. There are real risks that repeated localized tree losses and a reduction in the number of genets that effectively contribute to offspring can lead to long-term genetic depletion. This scenario is likely to be realistic in the environmental conditions typical of the central and southern Apennines, where the limiting environmental conditions for H. fraxineus (35°-36 °C) reported by Hauptman et al. (2013) and Grosdidier et al. (2018b) occur with a very low frequency.
In order to preserve the genetic heritage of common ash populations in this marginal portion of the range,
the implementation of disease monitoring for H. fraxineus spread in the remaining southern stands will be crucial. According to Pautasso et al. (2013), common ash conservation strategies will need a total mapping of F. excelsior distribution along the southern Apennines, subsequent plant phenotypic and genomic characterization and a disease resistance screening within all the proveniences.
Genotype x environment interaction is known to play a primary role in modulating the host response to pathogens (Bruck and Manion 1980; Manion 1986; Santini et al. 1997). Results from other pathosystems (Santini et al. 2005; Ahrens et al. 2019) provide evidence that plants have evolved different adaptive growth and defense strategies according to the environmental conditions in the area of origin and that these traits are stable over the years and sites (Ghelardini et al. 2006). This stability gives hope for setting up successful management strategies to provide greater resilience for natural ecosystems to epidemics caused by an introduced pathogen.
References
Ahrens CW, Mazanec RA, Paap T et al (2019) Adaptive variation for growth and resistance to a novel pathogen along climatic gradients in a foundation tree. Evol Appl 12:1178–1190. https://doi.org/10.1111/eva.12796
Bakys R, Vasaitis R, Barklund P et al (2009) Investigations concerning the role of chalara fraxinea in declining fraxinus excelsior. Plant Pathol 58:284–292. https://doi.org/10.1111/j.1365-3059.2008.01977.x
Barberis G, Nepi C, Peccenini S, Peruzzi L (2014) Notulae alla checklist della Flora vascolare Italiana 18 (2071–2099). Inf Bot Ital 46:267–279
Barić L, Županić M, Pernek M, Diminić D (2012) Prvi nalazi patogene gljive Chalara fraxinea u Hrvatskoj—Novog uzročnika odumiranja jasena (Fraxinus spp.). Sumar List 136:461–469
Bruck RI, Manion PD (1980) Interacting environmental factors associated with the incidence of Hypoxylon canker on trembling aspen. Can J for Res 10:17–24. https://doi.org/10.1139/x80-004
Burokiene D, Prospero S, Jung E et al (2015) Genetic population structure of the invasive ash dieback pathogen Hymenoscyphus fraxineus in its expanding range. Biol Invasions 17:2743–2756. https://doi.org/10.1007/s10530-015-0911-6
Caudullo G, Welk E, San-Miguel-Ayanz J (2017) Chorological maps for the main European woody species. Data Br 12:662–666. https://doi.org/10.1016/j.dib.2017.05.007
Chandelier A, Delhaye N, Helson M (2011) First report of the ash dieback pathogen Hymenoscyphus pseudoalbidus (Anamorph Chalara fraxinea) on Fraxinus excelsior in Belgium. Plant Dis 95:220. https://doi.org/10.1094/PDIS-07-10-0540
Chandelier A, Cech T, Ioos R (2013) Normes OEPP-Diagnostic/EPPO standards-diagnostics. PM 7/117 1 hymenoscyphus pseudoalbidus. Bull OEPP EPPO B 43:449–461. https://doi.org/10.1111/epp.12061
Dal Maso E, Montecchio L (2014) Risk of natural spread of hymenoscyphus fraxineus with environmental niche modelling and ensemble forecasting technique. For Res. https://doi.org/10.4172/2168-9776.1000131
Davydenko K, Vasaitis R, Stenlid J, Menkis A (2013) Fungi in foliage and shoots of Fraxinus excelsior in eastern Ukraine: A first report on Hymenoscyphus pseudoalbidus. For Pathol 43:462–467. https://doi.org/10.1111/efp.12055
De Natale A (2004) Note di floristica per il Parco Nazionale del Cilento e Vallo di Diano ( Salerno, Campania ). Inf Bot Ital 36:29–33
Di ZH, Zhuang WY (2014) Hymenoscyphus albidoides sp. nov. and H. pseudoalbidus from China. Mycol Prog 13:625–638. https://doi.org/10.1007/s11557-013-0945-z
Drenkhan R, Adamson K, Hanso M (2015) Fraxinus sogdiana, a central asian ash species, is susceptible to hymenoscyphus fraxineus. Plant Prot Sci 51:150–152. https://doi.org/10.17221/89/2014-PPS
Drenkhan R, Solheim H, Bogacheva A et al (2017) Hymenoscyphus fraxineus is a leaf pathogen of local Fraxinus species in the Russian Far East. Plant Pathol 66:490–500. https://doi.org/10.1111/ppa.12588
Enderle R, Stenlid J, Vasaitis R (2019) An overview of ash (Fraxinus spp.) and the ash dieback disease in Europe. CAB Rev Perspect Agric Vet Sci Nutr Nat Resour. https://doi.org/10.1079/PAVSNNR201914025
Ghelardini L, Falusi M, Santini A (2006) Variation in timing of bud-burst of Ulmus minor clones from different geographical origins. Can J for Res 36:1982–1991. https://doi.org/10.1139/x06-092
Ghelardini L, Migliorini D, Santini A, et al (2017) From the Alps to the Apennines: possible spread of ash dieback in Mediterranean areas. In: Dieback of European Ash (Fraxinus spp.)—consequences and guidelines for sustainable management. Uppsala, Sweden: Swedish University of Agricultural Science. pp 140–149
Gil W, Kowalski T, Kraj W, et al (2017) Ash Dieback in Poland–history of the phenomenon and possibilities of its limitation. In: Die-back of European ash (Fraxinus spp.). Consequences and guidelines for sustainable management (eds.: R. Vasaitis, R. Enderle). Uppsala, Sweden: Swedish University of Agricultural Science. pp 176–184
Giongo S, Longa CMO, Dal Maso E et al (2017) Evaluating the impact of hymenoscyphus fraxineus in trentino (Alps, Northern Italy): first investigations. Iforest 10:871–878. https://doi.org/10.3832/ifor2486-010
Grosdidier M, Ioos R, Husson C et al (2018a) Tracking the invasion: dispersal of Hymenoscyphus fraxineus airborne inoculum at different scales. FEMS Microbiol Ecol 94:1–11. https://doi.org/10.1093/femsec/fiy049
Grosdidier M, Ioos R, Marçais B (2018b) Do higher summer temperatures restrict the dissemination of Hymenoscyphus fraxineus in France? For Pathol 48:1–8. https://doi.org/10.1111/efp.12426
Grosdidier M, Scordia T, Ioos R, Marçais B (2020) Landscape epidemiology of ash dieback. J Ecol 108:1789–1799. https://doi.org/10.1111/1365-2745.13383
Gross A, Han JG (2015) Hymenoscyphus fraxineus and two new Hymenoscyphus species identified in Korea. Mycol Prog. https://doi.org/10.1007/s11557-015-1035-1
Gross A, Hosoya T, Queloz V (2014) Population structure of the invasive forest pathogen Hymenoscyphus pseudoalbidus. Mol Ecol 23:2943–2960. https://doi.org/10.1111/mec.12792
Gross A (2013) Reproduction system and population structure of Hymenoscyphus pseudoalbidus, causal agent of ash dieback. Zurich
Han JG, Shrestha B, Hosoya T et al (2014) First report of the ash dieback pathogen Hymenoscyphus fraxineus in Korea. Mycobiology 42:391–396. https://doi.org/10.5941/MYCO.2014.42.4.391
Hauptman T, Piškur B, de Groot M et al (2013) Temperature effect on Chalara fraxinea: heat treatment of saplings as a possible disease control method. For Pathol 43:360–370. https://doi.org/10.1111/efp.12038
Heuertz M, Fineschi S, Anzidei M et al (2004a) Chloroplast DNA variation and postglacial recolonization of common ash (Fraxinus excelsior L.) in Europe. Mol Ecol 13:3437–3452. https://doi.org/10.1111/j.1365-294X.2004.02333.x
Heuertz M, Hausman JF, Hardy OJ et al (2004b) Nuclear microsatellites reveal contrasting patterns of genetic structure between western and southeastern European populations of the common ash (Fraxinus excelsior L.). Evolution (n Y) 58:976–988. https://doi.org/10.1111/j.0014-3820.2004.tb00432.x
Heuertz M, Carnevale S, Fineschi S et al (2006) Chloroplast DNA phylogeography of European ashes, Fraxinus sp. (Oleaceae): Roles of hybridization and life history traits. Mol Ecol 15:2131–2140. https://doi.org/10.1111/j.1365-294X.2006.02897.x
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276. https://doi.org/10.1006/bijl.1996.0035
Husson C, Scala B, Caël O et al (2011) Chalara fraxinea is an invasive pathogen in France Claude. Eur J Plant Pathol 130:311–324. https://doi.org/10.1007/s10658-011-9755-9
Ioos R, Kowalski T, Husson C, Holdenrieder O (2009) Rapid in planta detection of Chalara fraxinea by a real-time PCR assay using a dual-labelled probe. Eur J Plant Pathol 125:329–335. https://doi.org/10.1007/s10658-009-9471-x
Junker C, Mandey F, Pais A et al (2014) Hymenoscyphus pseudoalbidus and Hymenoscyphus albidus: Viridiol concentration and virulence do not correlate. For Pathol 44:39–44. https://doi.org/10.1111/efp.12066
Kádasi-Horáková M, Adamčíková K, Pastirčáková K et al (2017) Natural infection of Fraxinus angustifolia by Hymenoscyphus fraxineus in Slovakia. Balt for 23:52–55
Keča N, Kirisits T, Menkis A (2017) First report of the invasive ash dieback pathogen Hymenoscyphus fraxineus on Fraxinus excelsior and F. angustifolia in Serbia. Balt for 23:56–59. https://doi.org/10.5281/zenodo.556518
Kirisits T, Matlakova M, Mottinger-Kroupa S et al (2010) Chalara fraxinea associated with dieback of narrow-leafed ash (Fraxinus angustifolia). Plant Pathol 59:411. https://doi.org/10.1111/j.1365-3059.2009.02162.x
Kirisits T, Kritsch P, Kräutler K et al (2012) Ash dieback associated with Hymenoscyphus pseudoalbidus in forest nurseries in Austria. J Agric Ext Rural Dev 4:230–235. https://doi.org/10.5897/jaerd12.057
Kowalski T (2006) Chalara fraxinea sp. nov. associated with dieback of ash (Fraxinus excelsior) in Poland. For Pathol 36:264–270. https://doi.org/10.1111/j.1439-0329.2006.00453.x
Kowalski T, Holdenrieder O (2009) Pathogenicity of Chalara Fraxinea for Pathol. Forest Pathol 39:1–7. https://doi.org/10.1111/j.1439-0329.2008.00565.x
Kowalski T, Schumacher J, Kehr R (2010) Das Eschensterben in Europa—Symptome , Erreger und Empfehlungen für die Praxis. Ash dieback in Europe—symptoms, causes and prognosis. Dujesiefken D (ed.) Jahrbuch der Baumpflege, Haymarket Media Braunschweig 184–195
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Laivins M, Priede A, Puspure I (2016) Spread of Hymenoscyphus fraxineus in Latvia: analysis based on dynamics of young Ash stands. Proc Latv Acad Sci Sect B Nat Exact Appl Sci 70:124–130. https://doi.org/10.1515/prolas-2016-0020
Longauerová V, Kunca A, Longauer R, et al (2017) The ash and Ash Dieback in Slovakia. In: Dieback of European Ash (Fraxinus spp.)- consequences and guidelines for sustainable management. Uppsala, Sweden: Swedish University of Agricultural Science. pp 209–219
Luchi N, Ghelardini L, Santini A et al (2016) First record of Ash Dieback caused by Hymenoscyphus Fraxineus on Fraxinus excelsior in the Apennines (Tuscany, Italy). Plant Dis 100:535. https://doi.org/10.1094/PDIS-09-15-0975-PDN
Manion PD (1986) Sixty-five years of research on hypoxylon Canker of Aspen. Plant Dis 70:803–808
Marigo G, Peltier JP, Girel J, Pautou G (2000) Success in the demographic expansion of Fraxinus excelsior L. Trees - Struct Funct 15:1–13. https://doi.org/10.1007/s004680000061
Migliorini D, Luchi N, Pepori AL et al (2020) Caliciopsis moriondi, a new species for a fungus long confused with the pine pathogen C. pinea. MycoKeys 73:87–108. https://doi.org/10.3897/MYCOKEYS.73.53028
Milenković I, Jung T, Stanivuković Z, Karadžić D (2017) First report of hymenoscyphus fraxineus on fraxinus excelsior in montenegro. For Pathol. https://doi.org/10.1111/efp.12359
Newton MA, Mau B, Larget B (1999) Markov chain monte carlo for the bayesian analysis of evolutionary trees from aligned molecular sequences. Lecture Notes-Monograph Series 33:143–162
Orton ES, Brasier CM, Bilham LJ et al (2018) Population structure of the ash dieback pathogen, Hymenoscyphus fraxineus, in relation to its mode of arrival in the UK. Plant Pathol 67:255–264. https://doi.org/10.1111/ppa.12762
Pastircáková K, Adamcíková K, Barta M et al (2020) Host range of hymenoscyphus fraxineus in Slovak Arboreta. Forests 11:1–18. https://doi.org/10.3390/F11050596
Pautasso M, Aas G, Queloz V, Holdenrieder O (2013) European ash (Fraxinus excelsior) dieback—A conservation biology challenge. Biol Conserv 158:37–49. https://doi.org/10.1016/j.biocon.2012.08.026
Pesaresi S, Galdenzi D, Biondi E, Casavecchia S (2014) Bioclimate of Italy: application of the worldwide bioclimatic classification system. J Maps 10:538–553. https://doi.org/10.1080/17445647.2014.891472
Przybylski P, Sikora K, Mohytych V, Włostowski M (2020) Effect of agrotechnical treatment on the health condition of the clonal seed ash plantation (Fraxinus excelsior L.) in the context of its infection by Hymenoscyphus fraxineus (T. Kowalski). Sylwan 164:404–413. https://doi.org/10.26202/sylwan.2020036
Queloz V, Grünig CR, Berndt R et al (2011) Cryptic speciation in Hymenoscyphus albidus. For Pathol 41:133–142. https://doi.org/10.1111/j.1439-0329.2010.00645.x
Rosati L, Filibeck G, De Lorenzis A et al (2010) La vegetazione forestale dei Monti Alburni, nel Parco Nazionale del Cilento e Vallo di Diano (Campania): analisi fitosociologica e significato fitogeografico. Fitosociologia 47:17–55
Rytkönen A, Lilja A, Drenkhan R et al (2011) First record of Chalara fraxinea in Finland and genetic variation among isolates sampled from Åland, mainland Finland, Estonia and Latvia. For Pathol 41:169–174. https://doi.org/10.1111/j.1439-0329.2010.00647.x
Sansford CE (2013) Pest Risk Analysis for Hymenoscyphus pseudoalbidus (anamorph Chalara fraxinea) for the UK and the Republic of Ireland
Santini A, Casini N, Di Lonardo V, Raddi P (1997) Canker resistance stability of some Cupressus sempervirens clones to Seiridium cardinale. J Genet Breed 51:269–277
Santini A, Fagnani A, Ferrini F et al (2005) Variation among Italian and French elm clones in their response to Ophiostoma novo-ulmi inoculation. For Pathol 35:183–193. https://doi.org/10.1111/j.1439-0329.2005.00401.x
Santini A, Pecori F, Pepori AL et al (2010) Genotype×environment interaction and growth stability of several elm clones resistant to Dutch elm disease. For Ecol Manage 260:1017–1025. https://doi.org/10.1016/j.foreco.2010.06.025
Schicchi R, Marino P (2011) Taxa relitti della flora forestale siciliana e problemi di conservazione. Biogeogr J Integr Biogeogr. https://doi.org/10.21426/b630110595
Schoebel CN, Zoller S, Rigling D (2014) Detection and genetic characterisation of a novel mycovirus in Hymenoscyphus fraxineus, the causal agent of ash dieback. Infect Genet Evol 28:78–86. https://doi.org/10.1016/j.meegid.2014.09.001
Stecher G, Tamura K, Kumar S (2020) Molecular evolutionary genetics analysis (MEGA) for macOS. Mol Biol Evol 37:1237–1239. https://doi.org/10.1093/molbev/msz312
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526. https://doi.org/10.1093/oxfordjournals.molbev.a040023
Timmermann V, Børja I, Hietala AM et al (2011) Ash dieback: pathogen spread and diurnal patterns of ascospore dispersal, with special emphasis on Norway. EPPO Bull 41:14–20. https://doi.org/10.1111/j.1365-2338.2010.02429.x
Tzedakis PC, Lawson IT, Frogley MR et al (2002) Buffered tree population changes in a quaternary refugium: evolutionary implications. Science 297:2044–2047. https://doi.org/10.1126/science.1073083
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Gelfland DH, Sninsky JJ, White TJ (eds) PCR Protocols: A Guide to Methods and Applications (Innis MA. Academic Press, San Diego, CA, pp 315–322
Acknowledgements
This study was funded by European Commission Horizon 2020 Research and Innovation Programme ‘HOlistic Management of Emerging forest pests and Diseases’ (HOMED) (grant No 771271). We wish to thank Gruppo Carabinieri Forestali, Teramo and the Administration or the Parco Nazionale d’Abruzzo, Lazio e Molise for providing us logistic support during the specimens' collection. We are grateful to Claudia Alessandrelli for her assistance and support in the field activity and we warmly thank our friend and colleague Trudy Paap for the precious assistance during the manuscript preparation.
Funding
This study was funded by European Commission Horizon 2020 Research and Innovation Programme ‘HOlistic Management of Emerging forest pests and Diseases’ (HOMED) (Grant No 771271).
Author information
Authors and Affiliations
Contributions
DM and AS conceived the study. DM, NL, EN, ALP, FP and AS collected and prepared the samples, and analysed data. DM wrote the first draft and all the others contributed to the following versions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The author declares their no conflict of interest.
Data availability
Fungal sequences generated in this study were submitted to NCBI GenBank (sequence codes are reported in the text) and the new obtained cultures were stored in the Fungal Collection of the Institute for Sustainable Plant Protection-National Research Council (IPSP-CNR, Sesto Fiorentino, Italy).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Migliorini, D., Luchi, N., Nigrone, E. et al. Expansion of Ash Dieback towards the scattered Fraxinus excelsior range of the Italian peninsula. Biol Invasions 24, 1359–1373 (2022). https://doi.org/10.1007/s10530-021-02716-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-021-02716-z