
Abstract
Cellular adaptation to various types of stress requires a complex network of steps that altogether lead to reconstitution of redox balance, degradation of damaged macromolecules and restoration of cellular metabolism. Advances in our understanding of the interplay between cellular signalling and signal translation paint a complex picture of multi-layered paths of regulation. In this review we explore the link between cellular adaptation to metabolic and oxidative stresses by activation of autophagy, a crucial cellular catabolic pathway. Metabolic stress can lead to changes in the redox state of nicotinamide adenine dinucleotide (NAD), a co-factor in a variety of enzymatic reactions and thus trigger autophagy that acts to sequester intracellular components for recycling to support cellular growth. Likewise, autophagy is activated by oxidative stress to selectively recycle damaged macromolecules and organelles and thus maintain cellular viability. Multiple proteins that help regulate or execute autophagy are targets of post-translational modifications (PTMs) that have an effect on their localization, binding affinity or enzymatic activity. These PTMs include acetylation, a reversible enzymatic modification of a protein’s lysine residues, and oxidation, a set of reversible and irreversible modifications by free radicals. Here we highlight the latest findings and outstanding questions on the interplay of autophagy with metabolic stress, presenting as changes in NAD levels, and oxidative stress, with a focus on autophagy proteins that are regulated by both, oxidation and acetylation. We further explore the relevance of this multi-layered signalling to healthy human ageing and their potential role in human disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
NAD depletion, oxidative stress and loss of macroautophagy (from herein referred to as autophagy) efficiency have all been linked to healthy, pathological and premature ageing (Kubben and Misteli 2017; López-Otín et al. 2013, 2016). Individually, these alterations may underlie seven of the nine outlined hallmarks of ageing including genomic instability (all), telomere attrition (oxidative stress), epigenetic alterations (NAD), loss of proteostasis (autophagy), de-regulated nutrient sensing (NAD), cellular senescence (all) and mitochondrial dysfunction (all) (López-Otín et al. 2013, 2016). Moreover, it is becoming increasingly clear that a significant degree of crossover and interdependence between the three phenomena occur in ageing cells and tissues. Specifically, increased reactive oxygen species (ROS) and depletion of NAD can impact autophagy by influencing post-translational modifications (PTMs) of autophagy proteins (Filomeni et al. 2015; Sedlackova et al. 2020; Zhang et al. 2016a). Furthermore, autophagy impairment may lead to the failure to reconstitute cellular metabolism and detoxify oxidised substrates (Li et al. 2015; Morishita and Mizushima 2019).
Nicotinamide adenine dinucleotide (NAD)
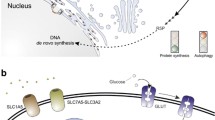
NAD is an essential metabolite that participates in cellular energy generation and signalling. When plentiful, the redox balance and availability of NAD aid cellular adaptation to metabolic stress and help maintain genomic stability, mitochondrial function, detoxification of ROS and cell survival (Fang et al. 2017). Due to its ability to accept or donate electrons, NAD in its reduced (NADH) or oxidised (NAD+) form assists energy metabolism in the cytosol and within mitochondria (Canto et al. 2015). In addition, NAD+ is cleaved into ADP-ribose (ADPR) and nicotinamide (NAM) by three classes of enzymes: sirtuins (SIRTs), poly(ADPR) polymerases (PARPs) and cyclic ADPR synthases (CD38 and CD157) (Fig. 1), which require ADPR for their enzymatic activity (Canto et al. 2015; Fang et al. 2017). Crucially, although SIRT activity depends on NAD+ availability and cannot contribute to uncontrolled NAD+ cleavage, PARPs and CD38 are known for their indiscriminate NAD+ consumption and their role in age- and disease-related NAD depletion (Canto et al. 2015). Homeostasis of intracellular NAD pools is maintained by either local synthesis from NAD+ precursors (nicotinamide (NAM), nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN)) or centralised de novo synthesis from nicotinic acid or L-tryptophan (Canto et al. 2015). Therefore, it is the balance between NAD+ cleavage and synthesis that dictates the total intracellular NAD pool, and by extension, cellular metabolism and protein acetylation status (Strømland et al. 2019).

Molecular outcomes of NAD+ cleavage. The three major groups of NAD+-consuming enzymes include sirtuins (SIRT), poly(ADP-ribose) polymerases (PARPs) and cyclic ADP-ribose synthases (cADPRs, CD38, CD157). a SIRT1-3 are NAD + -dependent deacetylases that bind an acetylated (Ac) protein substrates and transfer the Ac moiety onto ADP-ribose (ADPR) to give rise to O-acetyl-ADP-ribose, a deacetylated protein substrate and a by-product of the reaction, nicotinamide (NAM). b PARPs are indiscriminate NAD+ consumers that use NAD+ as a co-substrate to generate poly(ADP)-ribose (PAR) chains on protein substrates, and generate NAM as a by-product. c cADPRs consume NAD+ to generate cyclic ADP-ribose (cADPR), a second messenger, and a by-product, NAM
Reactive oxygen species (ROS)
ROS are highly reactive molecules of oxygen, which harbour one unpaired electron (superoxide anion (O2•−), hydroxyl radical (OH•)) or an additional electron pair (H2O2) on its valence orbital (Halliwell and Gutteridge 2015). The increased electron content in oxygen molecules makes them more reactive and more likely to participate in one-electron oxidative transfer reactions that lead to macromolecule modification and/or damage (Halliwell and Gutteridge 2015). ROS can also interact with nitric oxide to generate reactive nitrogen species (RNS), including peroxynitrite (ONOO−) (Bartesaghi and Radi 2018). Electrons from ROS/RNS interact with amino acid residues incorporated in proteins and thus translate the cellular redox state into protein activating or inhibiting signals by the modulation of protein enzymatic activity, binding affinity or structural conformation. Particularly sensitive to ROS-mediated redox regulation are cysteine (Cys) residues (Bischoff and Schlüter 2012). Cys is one of the least represented, and yet often highly conserved amino acids that participates in protein structural integrity by formation of covalent disulphide-bridges between two cysteine residues, or protein enzymatic activity, i.e. by thioester bond formation or co-factor stabilisation (Bak et al. 2019; Marino and Gladyshev 2010).
Autophagy
Autophagy is a cytosolic pathway of dynamic membrane rearrangement and cargo sequestration that is assisted and executed by a set of highly conserved autophagy (ATG) proteins (Dikic and Elazar 2018). Autophagy is a catabolic process responsible for cargo recognition, its engulfment in a double membraned vesicle called autophagosome and delivery to the lysosomal lumen for degradation. The subsequent release of amino acids, lipids and nucleosides reconstitutes cellular homeostasis and sustains viability in times of stress (Morishita and Mizushima 2019). The molecular execution of autophagy initiation is mediated by ATG protein association into functional complexes known as the Unc-51-like kinase 1 (ULK1) complex, the class III phosphatidylinositol 3 kinase (PI(3)K) complex, the ATG9-membrane complex, an ATG2–WIPI (WD-repeat protein interacting with phosphoinositides) complex and two conjugation systems consisting of the ATG3-ATG8/LC3 and the ATG5-ATG12:ATG16L complex (Table 1) (Suzuki et al. 2017). The combined action of these complexes is responsible for ER localization of all autophagy components and for the formation and maturation of the autophagic membrane. In addition, a group of autophagy receptors, e.g. sequestosome 1 (SQSTM1/p62), is then responsible for spatially linking the ubiquitylated cargo, including long-lived or aggregated proteins, pathogens and organelles, to the growing autophagosome (Dikic and Elazar 2018; Johansen and Lamark 2019).
The canonical pathway of starvation-induced autophagy was long thought to rely on phosphorylation cascades that are triggered by the loss of nutrient signalling and converge on a small number of regulating kinase complexes (Beurel et al. 2015; Rabanal-Ruiz et al. 2017; Tamargo-Gómez and Mariño 2018). These regulators then either lose function and thus release downstream autophagy components from an inhibitory state, or become activated and promote autophagy initiation. In addition, multiple layers of regulation involved in autophagy initiation, cargo sequestration and degradation, incorporate various stress signals and often improve the efficiency of autophagic flux via PTMs of autophagy proteins or their upstream regulators (Filomeni et al. 2015; Montagna et al. 2016; Sedlackova et al. 2020; Zhang et al. 2016a).
In this review, we explore the current knowledge of how two types of PTMs, lysine (Lys) acetylation and cysteine (Cys) oxidation, regulate the abundance and activity of ATG proteins, and highlight which Lys modifications are subject to NAD+ availability. We then summarize the main concepts of autophagy regulation by oxidative stress and discuss the implications and consequences of age-related changes to NAD+ availability and an increase in oxidative stress on the efficiency of autophagy. We further explore whether autophagy directly influences the homeostasis of cellular NAD levels and outline how aberrations in either of the three phenomena could lead to dysfunction observed in physiological and pathological ageing.
Targets of acetylation in autophagy
Lysine acetylation is a major reversible PTM in eukaryotes that arises by donation of the acetyl moiety from acetyl coenzyme A (Ac-CoA) via its re-direction from mitochondrial energy generation (Drazic et al. 2016). Protein acetylation status is balanced by the activity of multiple lysine acetyl transferases (KATs, historically known as histone acetyl transferases HATs) and lysine deacetylases (KDAC, or HDACs) (Narita et al. 2019). KATs catalyse acetyl moiety transfer from Ac–CoA to a lysine residue of the target protein, while KDACs cleave and release the acetyl moiety (KDAC, classes I, II and IV) or catalyse transfer of the acetyl moiety onto ADPR, a product of NAD+ cleavage (class III KDACs, sirtuins (SIRTs) (Fig. 1). Acetylation status of autophagy proteins is largely controlled by p300 (KAT3B) and 60 kDa Tat-interactive protein (TIP60/KAT5) KATs and SIRT1-3 and HDAC2/6 KDACs (summarized in Table 1). In the next section, we explore how protein acetylation status, generally high in conditions of nutrient abundance and low under nutrient starvation, regulates the activity and localisation of TFs, proteins and receptors involved in autophagy.
Regulation of transcription factors involved in autophagy gene transcription
The loss of lysine acetylation triggers stimulation of several TFs involved in the transcription of ATG genes (Fig. 2a) (Füllgrabe et al. 2016). The strongest link between TF deacetylation and autophagy stimulation comes from studies of transcription factor EB (TFEB), a member of the microphthalmia family of bHLH-LZ transcription factors (Mit/TFE), a group of TFs that stimulate lysosomal biogenesis and expression of autophagy proteins (Yang et al. 2018). Specifically, TFEB is responsible for transcription of multiple autophagy genes (ATG4, ATG9B, MAP1LC3B (LC3B), UVRAG (UV radiation resistance associated gene), WIPI (WD repeat domain phosphoinositide-interacting protein 1), and SQSTM1 (p62)) (Füllgrabe et al. 2016; Settembre et al. 2011). Acetylation of a conserved lysine residue Lys116 was independently identified in three studies as a modifier of TFEB activity in microglia (Bao et al. 2016) and in cancer cells (Wang et al. 2019b; Zhang et al. 2018). In microglia, Lys116 was directly deacetylated by SIRT1 which promoted degradation of fibrillar amyloid β (Bao et al. 2016). In cultured cells, treatment with a KDAC inhibitor, suberoylanilide hydroxamic acid (SAHA), increased the transcriptional activity of TFEB and influenced acetylation of four lysine residues (Lys91, Lys103, Lys116 and Lys430) (Zhang et al. 2018). In addition, authors of this study identified acetyl-coenzyme A acetyltransferase 1 (ACAT1) and HDAC2 as modulators of the overall TFEB acetylation status. Furthermore, a study in a model of chronic kidney disease identified HDAC6 as another KDAC involved in the regulation of TFEB activity (Brijmohan et al. 2018). Importantly, authors of neither of the studies demonstrated a direct interaction between TFEB and HDAC2 or HDAC6, respectively (Brijmohan et al. 2018; Zhang et al. 2018). Overexpression of another KAT, the general control non-repressed protein 5 (GCN5/KAT2A), but not TIP60, p300 or CREB-binding protein (CBP), led to increased TFEB acetylation of Lys116, Lys274 and Lys279 residues (Wang et al. 2019b). Authors further demonstrated that TFEB acetylation at Lys274 and Lys279 mechanistically disrupts TFEB dimerization and its ability to bind DNA, and thus negatively regulates expression of lysosomal and autophagy genes (Fig. 2A) (Wang et al. 2019b). Crucially, Lys116 of TFEB is not conserved in Drosophila melanogaster and Caenorhabditis elegans or in other members of the Mit-TFE family (Wang et al. 2019b), thus SIRT1 and HDAC regulation of TFEB activity is likely to be unique to vertebrates.

Autophagy targets of acetylation and oxidation. Nutrient and oxidative stresses affect proteins that participate in autophagy by lysine (K) acetylation or cysteine (C) oxidation. a Localization of transcriptional factor EB (TFEB), a master regulator of autophagy and lysosomal gene expression, is regulated by oxidative stress. Indirectly, oxidative modification of mucolipin 1 (MCOLN1) leads to TFEB dephosphorylation by Ca2+-sensitive phosphatase, calcineurin and its translocation to the nucleus. Directly, oxidation of TFEBs redox-sensitive residue, C212, promotes rapid nuclear localization. In addition, inhibitory lysine acetylation of K274 and K279 that is regulated by the general control non-repressed protein 5 (GCN5) prevents TFEB dimerization. The molecular and functional outcomes of K116 are not known, but are opposed by nutrient sensitive, NAD + -dependent lysine deacetylase (KDAC), SIRT1. b Acetylation-sensitive lysine residues were detected within members of the ULK1 complex, the class III PI(3)K complex and both ubiquitin-like conjugation systems. Unc-51-like kinase 1 (ULK1, ULK1 complex) contains two lysine residues, K162 and K606 (* in mouse) that are acetylated by TIP60 in response to serum starvation. Vacuolar protein sorting 34 (VPS34, class III PI(3)K complex) contains two acetylation sensitive lysine residues (K29 and K771) that are subject to inhibitory acetylation in fed conditions. Inhibitory acetylation of residues K430 and K437 in Beclin 1 (class III PI(3)K complex) is opposed by nutrient-sensitive SIRT1 KDAC. Within the ubiquitin-like conjugation systems, LC3 (K49, K51), ATG5 (unknown), ATG7 (unknown) and ATG12 (unknown) are subject to inhibitory acetylation by p300 (not shown) in fed conditions. Acetylation of LC3, ATG5 and ATG7 residues is opposed by SIRT1 deacetylase. ATG3 is subject to activating acetylation by TIP60 in starved conditions. Acetylation of lysine residues K19, K48 and K183 (** in yeast) is necessary for ATG3 enzymatic activity and LC3 binding affinity. ATG3 and ATG7 are also subject to inactivation by oxidative stress due to oxidation of their catalytic thiols, C264 and C572, respectively. c Selective autophagy receptor p62 is a target of both, acetylation and oxidation. TIP60-dependent activating acetylation of K420 and K435 residues within the ubiquitin-associated (UBA) domain prevent UBA dimerization and enhance ubiquitin (Ub) binding affinity. Oxidation of C105 and C113 promotes p62 oligomerization and stimulates autophagy by intermolecular disulphide bond formation
Additionally, two members of the forkhead box class O (FoxO) TF family, FoxO1 and FoxO3a, recognized for their role in autophagy/mitophagy (ATG4, ATG5, ATG12, ATG14, BECN1 (beclin 1), BNIP3 (BCL2 interacting protein 3), LC3B, ULK1, VPS34 (vacuolar protein sorting 34) GABARAPL1 (gamma-aminobutyric acid receptor-associated protein-like1), and PARK6/PINK1 (PTEN-induced kinase 1)) gene transcription, are regulated by acetylation PTMs (Fang et al. 2019; Füllgrabe et al. 2016; Requejo-Aguilar et al. 2015). It was first demonstrated that FoxO1 acetylation on Lys242, Lys245 and Lys262 residues (in mice) by CBP is opposed by SIRT1 in response to serum (Daitoku et al. 2004) and glucose starvation (Hariharan et al. 2010). Mechanistically, acetylation of the three Lys residues within FoxO1 interferes with its DNA binding and inhibits its transcriptional activity (Matsuzaki et al. 2005). Furthermore, FoxO1 acetylation permits access for upstream kinases to phosphorylate its Ser253 residue that is otherwise shielded by FoxO1-DNA complex formation (Matsuzaki et al. 2005). FoxO1 phosphorylation sites have since became known to act as docking or shielding sites for 14–3-3 protein binding, and the heterodimer exit into and retention within the cytoplasm (Brunet et al. 1999; Saline et al. 2019).
Similarly to FoxO1, the transcriptional activity of FoxO3 is modulated by SIRT1-3 deacetylases, though the Lys residues susceptible to acetylation remain unknown. First, caloric restriction and oxidative stress increase SIRT2 expression, decrease FoxO3 acetylation and improve gene transcription (Wang et al. 2007). In mitochondria, SIRT3 mediated deacetylation of the mitochondrial FoxO3 pool and led to cellular detoxification of oxidative stress by increased expression of the mitochondrial superoxide dismutase (Jacobs et al. 2008). However, some controversy exists in the perceived outcome of FoxO acetylation. A few articles published in early 2000s reported entirely opposite findings, demonstrating that FoxO acetylation improves transcription of its target genes (Motta et al. 2004; Yang et al. 2005). The pitfall of the majority of FoxO studies centres on the lack of distinction between CBP-mediated acetylation of FoxO, which may increase its DNA binding, and acetylation of histones, which would relax the chromatin condensation and promote gene transcription (discussed in detail in (Daitoku et al. 2011)).
Overall, transcriptional activity of TFEB, a member of the Mit/TFE family, and two FoxO isoforms, FoxO1 and FoxO3, is regulated by acetylation. Three classes of KDACs, SIRT1, HDAC2/6 and GCN5, are thought to deacetylate multiple and variable Lys residues in TFEB, of which the SIRT1 target, Lys116, is unique to vertebrates, and Lys274 and Lys279 deacetylation regulates TFEB-DNA complex formation (Wang et al. 2019b). In the FoxO family, FoxO1 is the better studied isoform, with known target Lys residues in the mouse (Table 1), though both FoxO1 and FoxO3a are likely activated by SIRT-mediated deacetylation. Thus, transcriptional regulation of autophagy/mitophagy genes by TFEB and FoxO1/3a is, at least in part, responsive to intracellular NAD+ levels that influence SIRT activity.
Regulation of autophagy protein complexes
Several autophagy proteins involved in autophagosome formation, growth and maturation may be modified by acetylation (Table 1). Studies from the last decade, summarized below, identify lysine residues sensitive to acetylation in members of the ULK1 kinase complex, the class III PI(3)K kinase complex and the two conjugation systems, ATG12 (ATG7, ATG10, ATG5) and LC3 (ATG4, ATG7, ATG3), as well as ATG12 and LC3 themselves (Fig. 2B).
In the ULK1 complex, ULK1 itself is a target of acetylation by TIP60 (Lin et al. 2012). In serum starved cells, ULK1 was shown to be a target of GSK3-dependent and TIP60-mediated acetylation of two crucial residues, Lys162 and Lys606 (in mouse; likely Lys162 and Lys607 in human), that together stimulate its kinase activity and promote autophagy initiation (Lin et al. 2012). Furthermore, oxidative stress that induces ER stress, was also shown to stimulate ULK1 acetylation by a GSK3-TIP60-dependent mechanism (Nie et al. 2016). These studies together support the idea that ULK1 kinase activity can be modulated by oxidative and metabolic stress via an upstream signalling cascade that results in TIP60 activation and ULK1 acetylation.
Within the Class III PI(3)K complex, VPS34 kinase acetylation by p300 occurs on residues Lys29, Lys771 and Lys781 and inhibits its lipid kinase activity and PI(3)P production (Su et al. 2017). It was further determined that acetylation of Lys29 residue prevents VPS34 association with Beclin 1 that is required for the formation of a complex involved in autophagy progression. Another layer of VPS34 activity regulation occurs upon acetylation of the Lys771 residue located within its catalytic site. In a manner similar to the level of regulation at the Lys29 residue, acetylation of Lys771 disrupts binding between VPS34 and its substrate, PI (Su et al. 2017). However, the KDAC responsible for Lys29 and Lys771 deacetylation remains unknown. In addition to VPS34, Beclin 1 of the Class III PI(3)K complex is also a target of inhibitory acetylation on Lys430 and Lys437 residues by p300 (Sun et al. 2015). Beclin 1 acetylation was demonstrated to promote its binding to Rubicon, and thus shown to result in the loss of autophagosome maturation (Ohashi et al. 2019; Sun et al. 2015). Furthermore, in vitro acetylation analysis revealed that SIRT1 is preferentially responsible for Beclin 1 deacetylation (Sun et al. 2015).
Next, SIRT1-mediated deacetylation of nuclear LC3 at Lys49 and Lys51 residues initiates LC3 translocation to the cytoplasm via a diabetes and obesity regulated (DOR/TP53INP2)-dependent interaction with deacetylated LC3 (Huang et al. 2015). DOR then further assists in LC3 localization to nascent autophagosomes thanks to its ATG7-binding affinity (You et al. 2019b). Furthermore, DOR also contains a ubiquitin-interacting motif and is thus likely to promote LC3-ATG7 formation in the vicinity of ubiquitylated cargo (Xu and Wan 2019; You et al. 2019b). Upon relocation to the cytoplasm, LC3 Lys49 and Lys51 acetylation, that is lost upon nutrient starvation, was recently shown to completely abolish p62 binding (Song et al. 2019). Due to the location and conservation of the two critical lysine residues in the hydrophobic binding grooves of LC3 (Huang et al. 2015; Song et al. 2019), it stands to reason that Lys49 and Lys51 acetylation could disrupt LC3 interaction with multiple binding partners including, but not limited to DOR and p62. Altogether, LC3 deacetylation in response to nutrient starvation not only promotes its exit from the nucleus, but also determines substrate binding specificity of protein partners via their LC3-interacting regions (LIRs).
Cytoplasmic LC3 targeting to and docking on the nascent autophagosomes requires covalent conjugation of LC3 to phosphatidylethanolamine (PE). In a ubiquitin-like conjugation system, ATG7 (and E1-like enzyme), ATG3 (an E2-like enzyme) and an ATG5-ATG12:ATG16L complex (an E3-like enzyme) assist LC3 conjugation to PE (Dikic and Elazar 2018). Nutrient starvation in yeast was first reported to decrease or not change acetylation levels of ATG proteins, with the notable exception of ATG3, in which Lys19, Lys48 and Lys183 acetylation increased (Yi et al. 2012). Authors of this study had further shown that while acetylation of Lys183 is crucial for the enzymatic activity of ATG3, Lys19 and Lys48 acetylation was crucial for autophagy progression by improving interaction between ATG3 and ATG8 (LC3 in mammals), and was regulated by the opposing activities of the yeast histone acetyltransferase Esa1 (TIP60/KAT5 orthologue)) and a histone deacetylase Rpd3 (HDAC1/2 orthologue) enzymes. Furthermore, ATG3 acetylation on Lys19 and Lys48 was shown to enhance its ER membrane localization and binding in vitro (Li et al. 2017).
Other members of the ubiquitin-like conjugation system, ATG7, ATG5 and ATG12, are targets of p300-mediated acetylation (Lee and Finkel 2009) and SIRT1-dependent deacetylation (Lee et al. 2008). In direct contrast to ULK1 and ATG3, acetylation of these ATG proteins generally inhibits their function. However, the specific residues, their location and effect of acetylation on the structure or function of ATG proteins remains unknown. Structural studies of the ATG12-ATG5:ATG16 complex (Otomo et al. 2013) and the nature of interaction between ATG12 and ATG3 (Metlagel et al. 2013) point towards several key lysine residues that could be targets of acetylation in ATG12. First, lysine residues 60, 69, 71 and 128 located on the surface of ATG12 (Metlagel et al. 2013) could contribute to binding affinity between ATG12 (E3-like) and ATG3 (E2-like) that is required for the spatiotemporal regulation of LC3 lipidation. Furthermore, ATG5 contains multiple lysine residues, of which Lys53, Lys130, Lys171 are conserved (Matsushita et al. 2007). Although Lys130 is the known catalytic site for conjugation between ATG5 and ATG12 (Mizushima et al. 1998), the function and acetylation-sensitivity of Lys53 and Lys171 remain unknown. Lastly, no published study followed-up reports of ATG7 acetylation-sensitivity (Lee et al. 2008; Lee and Finkel 2009). However, a high resolution mass spectrometry study of global protein acetylation identified Lys306 of the human ATG7 protein as a residue that might be relevant for further study (Choudhary et al. 2009). Thus, although ATG5, ATG7 and ATG12 have been known substrates of p300 and SIRT for almost a decade, the lysine residues sensitive to acetylation, or indeed the nature of protein inhibition by acetylation have not been elucidated.
Selective cargo recognition
Autophagy receptors modulate the selectivity and specificity of cargo recognition in the autophagy pathway. Although the current knowledge of about PTMs that affect the structure, function and localisation of the canonical autophagy receptors is fairly limited, phosphorylation and ubiquitylation sites were identified in all canonical receptors (THANATOS, https://thanatos.biocuckoo.org) (Deng et al. 2018). The best characterization of acetylation-dependent regulation of autophagy receptors concerns the p62 protein and its affinity for ubiquitin (Fig. 2c). Binding between ubiquitin and p62 to spatially link cargo to the forming autophagosome is, in fed condition, restricted due to the low binding activity of the ubiquitin associated (UBA) domain of p62 and further restricted by UBA homodimerisation (Long et al. 2010). Briefly, Lys420 monoubiquitylation (Lee et al. 2017; Peng et al. 2017), and Ser403 and Ser407 (in humans; Ser405 and Ser409 in mice) phosphorylation (Matsumoto et al. 2015) strengthen the interaction and binding affinity between p62 and ubiquitin. In addition, acetylation of p62 Lys420 and Lys435 residues, regulated by TIP60 and opposed by HDAC6 upon serum and amino acid starvation, interferes with UBA dimerization (Lys420 and Lys435) and enhances ubiquitin-binding affinity (Lys435) (You et al. 2019a). Moreover, spatial proximity between p62 and HDAC6 at sites of protein aggregation promotes their interaction and regulation of HDAC6 deacetylase activity and, by extension, protein aggregate recycling by p62 (Yan et al. 2013).
Moreover, ubiquitin (Ub) itself is a target of lysine acetylation (Ohtake et al. 2015). Formation of stable polyubiquitin chains by covalent linkages of single Ub moieties via homotypic Lys63 linkage (also known as K63) promotes autophagy (Grumati and Dikic 2018). Although the KAT(s) and KDAC(s) involved and the physiological relevance of Ub acetylation remain unknown, acetylation of Lys6 and Lys48 residues was shown to interfere with poly-Ub chain formation (Lys11-, Lys48- and Lys63-linked) in vitro (Ohtake et al. 2015).
Demystification of the acetylation riddle in autophagy regulation
Recent advances in our understanding of which KATs and KDACs are involved in the regulation of autophagy protein acetylation highlight a few interesting phenomena. Overall, autophagy protein acetylation status is mainly regulated by p300, CREB binding protein (CBP) and TIP60 KATs, and HDAC2/6 and SIRT1 KDACs (Table 1). Upon a closer look, targets of p300-mediated acetylation are generally opposed by SIRT1-dependent deacetylation, while the targets of TIP60 may be opposed by HDACs but remain largely unknown. Following this train of thought, targets of p300/SIRT are activated by the loss of acetylation, whereas it is the addition of acetyl group to TIP60 targets that triggers their activation (summarized in Table 1, shown in Fig. 2).
Autophagy is stimulated by the depletion of key nutrients including amino acids, growth factors and glucose. Recognition of nutrient availability by multiple intracellular sensors converges on a handful of regulators that integrate nutrient signals into several key responses. These include mammalian target of rapamycin complex 1 (mTORC1) (Rabanal-Ruiz et al. 2017), glycogen synthase kinase 3 (GSK3) (Mancinelli et al. 2017), and adenine monophosphate-activated protein kinase (AMPK) (Tamargo-Gómez and Mariño 2018). Perhaps unsurprisingly, these three kinases have also been directly linked to the regulation of KATs and KDACs that influence the acetylation status of autophagy proteins. mTORC1 was recently shown to activate the acetyl-transferase activity of p300 by serine phosphorylation that was lost upon amino acid starvation (Wan et al. 2017). Activation of GSK3β by the loss of growth factor signalling is known to phosphorylate and thus activate TIP60 (Lin et al. 2012). Finally, AMPK activation releases SIRT1 inhibition in a GAPDH-dependent manner in response to glucose starvation (Chang et al. 2015). Thus, three potential axes regulate autophagy stimulation in response to nutrient stress by (a) loss of FoxO, VPS34, Beclin1, ATG7, ATG5 and ATG12 acetylation (amino acids/growth factors-mTORC1-p300), (b) increased ULK1 and possible ATG3 and p62 acetylation (serum/ER stress-GSK3β-TIP60) (Lin et al. 2012; Nie et al. 2016; Yi et al. 2012; You et al. 2019a), and (c) FoxO, Beclin1, ATG7, ATG5 and ATG12 deacetylation (glucose–AMPK–GAPDH–SIRT1) that could explain the conundrum of the varied nature of autophagy protein acetylation status upon nutrient starvation and its link to autophagy stimulation.
Targets of cysteine oxidative PTMs in autophagy
Protein modification by ROS and RNS constitutes a covalent modification of amino acid residues by the reactive species directly, or as a secondary interaction in an oxidative relay. Briefly, irreversible (carbonylation, nitration) oxidative modifications affect a variety of amino acids including cysteine (Cys), threonine and tyrosine (Ahmad et al. 2017; Cai and Yan 2013; Xie et al. 2018). In contrast, reversible amino acid oxidation involves modification of the thiol group (-SH) of Cys protein residues that are first modified to sulfenic acid (–SOH) (Cai and Yan 2013). Sulfenic acid can then undergo nitrosylation (-SNO) by reacting with RNS, or disulphide bond formation (R–S–S–R) by intra-/inter-molecular bond formation between two cysteine residues. A specialised form of disulphide bond formation, glutathionylation (R–S–S–G) arises as a mixed disulphide bond formation between a target protein Cys residue and the non-enzymatic antioxidant, glutathione (GSH) (Cai and Yan 2013). Further oxidation of –SOH results in an irreversible Cys oxidation by the formation of sulfinic (–SO2H) and sulfonic (–SO3H) acids (Ahmad et al. 2017; Cai and Yan 2013; Murray and Van Eyk 2012). Autophagy regulation by ROS is linked to the reversible oxidative Cys modification of (a) transcription factors (TFs) that regulate expression of proteins involved in the autophagy process, (b) upstream regulators of autophagy initiation, (c) autophagy proteins themselves and (d) receptors that mediate autophagy substrate selectivity (Filomeni et al. 2015; Montagna et al. 2016; Sedlackova et al. 2020).
The most substantial link between ROS and autophagy TF activation was established in the studies of the Mit/TFE family of transcription factors (Yang et al. 2018). Three members of the Mit/TFE protein family were recently shown to contain redox sensitive Cys residues (TFEB Cys212, TFE3 Cys322, MITF Cys281) that mediate a rapid response to increased intracellular oxidative stress by promoting their nuclear translocation (Wang et al. 2019a). Another layer of regulation by oxidative stress was previously uncovered for TFEB that regulates expression of several autophagy proteins including, ATG4, ATG9, LC3B and p62 (Settembre et al. 2011). Increased intracellular oxidative stress is sensed by the lysosomal cation channel, mucolipin 1 (MCOLN1/TRPML1) in a manner that is not yet understood (Zhang et al. 2016c). What is known is that MCOLN1 oxidation promotes channel opening, Ca2+ release from the lysosomal lumen and activation of a Ca2+ dependent phosphatase, calcineurin (Medina et al. 2015; Zhang et al. 2016c). Calcineurin-dependent TFEB phosphorylation then promotes TFEB translocation to the nucleus and autophagy stimulation (Fig. 2A).
At the stage of autophagy execution, redox-sensitive Cys residues were identified in proteins involved in LC3 processing (ATG4B) and LC3-PE conjugation (ATG7 and ATG3). ATG4B is a Cys-dependent protease that cleaves pro-LC3 at a C-terminal glycine residue prior to LC3-PE conjugation (Kirisako et al. 2000). Its protease activity is also involved in correcting the amount of LC3–PE formation on non-autophagic membranes by the hydrolysis of the LC3–PE bond, and presumably on the outer membrane leaflet of the growing autophagosome. In human cells, the hydrolysing (deconjugating) activity of ATG4B is inhibited by the oxidation of one of two Cys residues (Cys74 or Cys78) and leads to improved stability of LC3–PE and increased formation of autophagosomes (Scherz‐Shouval et al. 2007). Similarly, oxidation of the catalytic thiols in ATG3 (Cys264) and ATG7 (Cys572) inhibits their activity in LC3–PE conjugation and results in the loss of autophagic flux (Fig. 2b) (Frudd et al. 2018). Interestingly, oxidation of these Cys residues can only occur when the thiols are not shielded by their interaction with LC3.
Oxidative stress influences the selectivity of the autophagic process via p62, a known redox sensitive autophagy receptor protein (Fig. 2c). Intermolecular disulphide formation in p62 was first observed in studies of its involvement in the N-end rule pathway of substrate degradation, where Cys113-dependent oligomerisation promoted substrate clearance via autophagy (Cha-Molstad et al. 2017). Subsequently, we have demonstrated that elevated ROS levels promote the formation of disulphide-linked conjugates, intermolecular Cys bonds, that assist p62 oligomer assembly (Carroll et al. 2018). Crucially, we have identified two Cys residues (Cys105 and Cys113) located within the regulatory linker region of the p62 protein, that are necessary and sufficient for the activation of pro-survival autophagy triggered by increased ROS (Carroll et al. 2018).
Reversible oxidation of Cys residues in redox-sensitive autophagy proteins thus appears to have a dual role of pathway stimulation by autophagy gene expression (TFEB), increased autophagosome formation (ATG4B) and substrate selectivity (p62), and autophagy inhibition upon depletion of available LC3 substrate (ATG3, ATG7). However, due to the novelty of these findings, the physiological role of ATG3 and ATG7 inhibition and possible downstream signalling events remain unknown. We propose a regulatory feedback loop whereby sensing depletion of local LC3 pools results in inactivation of ATG3 and ATG7 that serves to prevent indiscriminate autophagy activation. We envision that this inactivation would persist until such a time that the antioxidant defences decrease the oxidative stress load and resolve the ATG3-ATG7 heterodimer, and the expression of autophagy genes restores the available pools of ATG proteins to sustain further autophagy.
The interrelatedness of target oxidation and acetylation in autophagy
Protein deacetylation and oxidation appear to be individually sufficient to regulate the initiation, promotion, efficiency and selectivity of autophagy. However, an interesting crosstalk between oxidative and acetyl-linked PTMs of autophagy proteins arises due to the dual control of several proteins including TFEB, ATG3, ATG7 and p62, which appear to be regulated by both, oxidation and acetylation status (Fig. 2a–c). First, upstream oxidation of MCOLN1 regulates TFEB localization by calcineurin-dependent dephosphorylation (Medina et al. 2015; Zhang et al. 2016c) and direct oxidation of its Cys212 residue (Wang et al. 2019a). Further, TFEB deacetylation at residues Lys274 and Lys279 promotes its dimerization and increases its binding affinity for DNA (Wang et al. 2019b). It would be interesting to study whether oxidation and acetylation PTMs act in concert to establish the optimal TFEB activity and whether TFEB oxidation promotes rapid expression of its target genes in the absence of Lys residue deacetylation.
Second, ATG3 acetylation at residues Lys19 and Lys48 by TIP60, increased in conditions of nutrient starvation, improves interaction between ATG3 and LC3 and promotes autophagy (Yi et al. 2012). Not much is known regarding the functional effect of deacetylation in ATG7, except that it promotes autophagy and Lys306 residue may be the target (Choudhary et al. 2009). In contrast to TFEB, a recently published study suggests that upon loss of LC3 binding, oxidation of ATG3 (Cys264) and ATG7 (Cys572) catalytic cysteine residues inhibits their enzymatic activity and blocks their further interaction with LC3 (Frudd et al. 2018).
Lastly, oxidation and acetylation of p62 could act in concert to achieve optimal selectivity of its interaction with cargo and oligomerization to stimulate autophagy. First, TIP60-dependent acetylation of Lys420 and Lys435 within the UBA domain interferes with its inter-protein dimerization and enhances the ubiquitin binding affinity of p62 upon serum and amino acid starvation (You et al. 2019a). Second, oxidation of Cys105 and Cys113 residues within the regulatory linker region promotes intermolecular p62 disulphide bond formation and thus assist in autophagy stimulation (Carroll et al. 2018).
In addition, activity of NAD+-dependent KDACs, or SIRTs, is directly or indirectly regulated by both, oxidative and metabolic stress stimuli. First, a shift in the NAD redox balance towards oxidation, suggestive of metabolic stress, leads to an increased pool of available NAD+ and thus stimulates SIRT activity (Imai and Guarente 2016). Second, SIRT regulation by oxidative stress was demonstrated in multiple cell culture experiments (reviewed in (Santos et al. 2016)), in which a mild oxidative environment promotes SIRT1 expression and activation by upstream kinases. In contrast, study of SIRT1 oxidation, specifically nitrosylation (–SNO+), suggests that this reversible oxidative PTM of Cys371, Cys374, Cys395 and Cys398 residues within a tetrathiolate formation results in loss of Zn2+ binding, structural destabilization and loss of NAD+ and acetyl-lysine binding ability (Kalous et al. 2016). Thus, SIRT1 activity can be stimulated by both, nutrient starvation, and oxidative stress. However, persistent ROS release may lead to SIRT1 destabilization, loss of its deacetylase activity and might contribute to its degradation by the proteasome (Caito et al. 2010).
NAD depletion, oxidative stress, and autophagy in physiological and pathological ageing
The NAD nucleotide is an important redox molecule required for fundamental molecular processes of energy generation via glycolysis, tricarboxylic acid cycle, oxidative phosphorylation and β-oxidation, and a co-factor to enzymes involved in cellular signalling and longevity. Age-related depletion of available NAD+ pools was, in human disease, animal models and in vitro studies, reported as a result of increased PARP activity due to an elevation in oxidative stress and levels of DNA damage (Pacher and Szabo 2008) and increased CD38 expression and activity (Camacho-Pereira et al. 2016; Polzonetti et al. 2012). Combined with the age-dependent reduction in the enzymatic activity of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of the NAM-based NAD+ salvage pathway (Stein and Imai 2014), these conditions perpetuate the perfect storm of total NAD depletion, loss of NAM recycling and reduction in SIRT activity in physiological ageing.
Study of human skin tissue from volunteers of different ages partially supports these findings (Massudi et al. 2012). In this study, an age-dependent increase in DNA damage correlated with an increase in PARP activity, NAD+ depletion and, in the elderly, a reduction in SIRT1 activity. Notably, these associations with age were strong only in the male participants and it would be interesting to see whether these findings can be reproduced in females and other accessible human tissues, including muscle or post-mortem brain tissues. In a more recent study carried out on human skeletal muscle samples, authors demonstrate that levels of NAMPT negatively correlate with age, body mass index and body fat percentage (de Guia et al. 2019). Another study utilised the power of magnetic resonance-based non-invasive in vivo imaging of the human brain and revealed an age-dependent decrease in total NAD levels, concomitant with an increase in NADH/NAD+ ratio, indicative of metabolic dysfunction (Zhu et al. 2015). While these studies were carried out on healthy human volunteers and suggest that a decline in NAD levels occurs in physiological ageing, multiple studies of accelerated human ageing (progeria) syndromes and patients suffering from metabolic and neurodegenerative diseases strongly link NAD decline to age-related pathology (Kubben and Misteli 2017; Lautrup et al. 2019; Okabe et al. 2019).
Furthermore, studies of two age-related conditions, sarcopenia and frailty, as well as a variety of progeria, neurodegenerative, metabolic and cardiac diseases, demonstrate a strong link between pathology and increased oxidative stress (Derbré et al. 2014; Inglés et al. 2014; Kubben and Misteli 2017; Liguori et al. 2018; Massudi et al. 2012; Soysal et al. 2017). Not only does lipid peroxidation, a proxy measurement for increased oxidative stress, correlate with age (Massudi et al. 2012), a systematic review of available literature suggests that long lived humans (centenarians) have lower levels of oxidative protein damage and lipid peroxidation compared to other elderly individuals (Belenguer-Varea et al. 2019). Given the number and severity of clinical conditions related to healthy ageing, and age-related diseases that are associated with an increase in oxidative stress, it is necessary to design interventions that prevent production of free radicals, boost cellular antioxidant systems, or understand and target the processes downstream of ROS-mediated protein, lipid or nucleotide damage.
Importantly, molecular studies of free radical generation, NAD+-dependent enzymatic processes and disease pathology suggest a link between ROS accumulation, NAD depletion and compromised mitochondrial recycling by autophagy, mitophagy. Mitochondria are energy-generating organelles that act as hubs of pro-survival or pro-apoptotic signalling (Sedlackova and Korolchuk 2019). Although mitochondrial health is maintained by a complex net of quality control mechanisms, whole organelle recycling of damaged and ROS-producing mitochondria is only achieved by selective autophagy. A causal link between NAD+ depletion and mitochondrial dysfunction due to loss of mitophagy was established in studies of premature ageing syndromes including Xeroderma Pigmentosum, Cockayne syndrome and Ataxia-telangiectasia (Fang et al. 2016, 2014; Scheibye-Knudsen et al. 2014, 2012; Valentin-Vega et al. 2012). In these studies, loss of SIRT activity and autophagy abnormalities occur as a result of PARP1 hyperactivation due to unresolved DNA damage. In addition to SIRT inactivation, uncontrolled NAD+ cleavage and protein PAR-ylation by PARPs results in loss of ATP availability and, if persistent, in cell death (Andrabi et al. 2014; Bai et al. 2011; Fouquerel et al. 2014; Pillai et al. 2005). Persistent NAD+ depletion was thus shown to compromise mitochondrial function due to loss of energy generation, impairment in mitochondrial recycling through lack of autophagy/mitophagy stimulation, and to initiate cellular death due to energy collapse. An alternative outcome to cell death upon PARP1 activation was linked to autophagy initiation in independent cell culture experiments (Jiang et al. 2018; Muñoz-Gámez et al. 2009). In the earlier study, authors demonstrated that PARP-dependent stimulation of autophagy due to short-lived energy crisis had a cytoprotective effect as genetic or pharmacological inhibition of autophagy led to increased level of necrotic death (Muñoz-Gámez et al. 2009). In the latter study, authors aimed to mimic constant ROS production in vivo by glucose oxidase (GO) treatment, which led to PARP-induced cell death, parthanatos (Jiang et al. 2018). In this study, inhibition of autophagy led to a significant collapse in mitochondrial polarization and an approximately 50% increase in cell death within four hours of GO treatment. Taken together with the role of SIRT-mediated autophagy stimulation, we wonder whether convergence of these signalling pathways on autophagy suggests a conserved role of this catabolic pathway in healthy ageing by preservation of cellular NAD pools.
An exciting development in the field of ageing and NAD metabolism is the ‘druggability’ of NAD metabolism by exogenous addition of natural, or synthetic, bioavailable NAD+ precursors. This universal approach of NAD+ precursor supplementation is known to increase NAD biosynthesis and alleviate the symptoms of pathological states including metabolic, cardiac and neurodegenerative disorders (Kane and Sinclair 2018; Lautrup et al. 2019). Additionally, evidence from NAD+ supplementation studies in cell culture and in animal models suggests that boosting NAD levels is sufficient to not only improve mitochondrial function, but also stimulate SIRT-dependent mitochondrial recycling via increased TFEB- and FoxO-dependent expression of autophagy/mitophagy genes and PTMs of autophagy proteins, and thus promote clearance of dysfunctional organelles and protein aggregates (Fang et al. 2019, 2016; Hou et al. 2018; Schöndorf et al. 2018; Vannini et al. 2019; Zhang et al. 2016b). Altogether, this ‘silver bullet’ approach might serve as an intervention to the vicious cycle of damage and NAD depletion and thus not only combat the depletion itself, but also support resolution of the underlying stresses and promote long-term cellular health.
Following the success of NAD+-boosting strategies in cell and animal models, NAD+ precursors, and predominantly nicotinamide riboside (NR), are now subjects of multiple clinical trials. Precursors have so far been reported as safe, well tolerated and capable of increasing NAD levels in healthy volunteers (Conze et al. 2019; Martens et al. 2018; Minto et al. 2017; Stea et al. 2017). However, challenges remain in translation of laboratory findings into the design of clinical trials (Gilmour et al. 2020). While some early success was found in disease outcomes of amyotrophic lateral sclerosis (ALS) (NCT03489200) (de la Rubia et al. 2019), others found no benefit in patients with Alzheimer’s disease (NCT00580931) (Phelan et al. 2017), or studies of metabolic disorders or mitochondrial bioenergetics in men (NCT02303483) (Dollerup et al. 2018, 2019a, 2019b). Although only a limited number of trials testing NAM, NMN and NR have been recently completed (> 10) or are currently ongoing (− 3), many are actively recruiting (− 21) (https://clinicaltrials.gov/) (Lautrup et al. 2019) and it will be interesting to see what lessons can be learned about precursor dosage, NAD+/NADH detection methods and bioavailability in the coming years. Considering that the pathological role of NAD depletion in many metabolic and neurodegenerative diseases is not yet firmly established, reporting of relevant disease outcomes is eagerly awaited as they will inform about the feasibility of translating success from the laboratory to human age- and disease-related interventions.
Concluding Remarks
Autophagy is a highly conserved catabolic process that is controlled by multiple nutritional and stress-related cues by reversible protein PTMs. In this review, we first explored the latest findings on how two PTMs, Lys acetylation and Cys oxidation, regulate the localization and function of autophagy proteins. Collectively, novel findings published in 2015–2020 identify TFEB, ULK1, VPS34, ATG3, LC3 and p62 as targets of acetylation PTMs which, in response to metabolic cues, stimulate the expression and enzymatic activity of autophagy proteins and improve pathway selectivity. Furthermore, Mit/TFE family of TFs (including TFEB), ATG3, ATG7 and p62 are also known to contain redox-sensitive Cys residues the oxidation of which influences autophagy outcomes. The dual control of protein localization/enzymatic activity by acetylation and oxidation links the efficiency of autophagy outcomes to nutrient loss and metabolic dysfunction and thus contributes to cellular homeostasis and healthy ageing.
Crucially, studies of the molecular mechanisms of NAD function in cellular physiology and ageing suggest a central role of autophagy in first, preventing increases in DNA damage and NAD+ consumption via mitochondrial recycling and second, by alleviating nutritional crisis via recycling amino acids, lipids and nucleosides. Autophagy thus appears to be necessary in supporting cellular survival upon either nutritional stress that changes NAD redox ratio towards the oxidised form (NAD+) and stimulates SIRTs, or upon DNA damage followed by NAD+ depletion due to PARP1 hyperactivation. Thus, although short term insults to cellular heath are resolved by autophagy stimulation and cellular detoxification, we wonder whether persistent oxidation and NAD+ loss in aged tissues result in stalled autophagy, and due to lack of stress resolution, ultimately in loss of cell viability and tissue dysfunction.
References
Ahmad S et al (2017) Protein oxidation: an overview of metabolism of sulphur containing amino acid, cysteine. Front Biosci (Schol Ed) 9:71–87
Andrabi SA et al (2014) Poly (ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Nat Acad Sci 111:10209–10214
Bai P et al (2011) PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13:461–468
Bak DW, Bechtel TJ, Falco JA, Weerapana E (2019) Cysteine reactivity across the subcellular universe. Curr Opin Chem Biol 48:96–105. https://doi.org/10.1016/j.cbpa.2018.11.002
Bao J et al (2016) Deacetylation of TFEB promotes fibrillar Aβ degradation by upregulating lysosomal biogenesis in microglia. Protein & cell 7:417–433
Bartesaghi S, Radi R (2018) Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol 14:618–625. https://doi.org/10.1016/j.redox.2017.09.009
Belenguer-Varea Á, Tarazona-Santabalbina FJ, Avellana-Zaragoza JA, Martínez-Reig M, Mas-Bargues C, Inglés M (2019) Oxidative stress and exceptional human longevity: systematic review. Free Radic Biol Med. https://doi.org/10.1016/j.freeradbiomed.2019.09.019
Beurel E, Grieco SF, Jope RS (2015) Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148:114–131. https://doi.org/10.1016/j.pharmthera.2014.11.016
Bischoff R, Schlüter H (2012) Amino acids: chemistry, functionality and selected non-enzymatic post-translational modifications. J Prot 75:2275–2296. https://doi.org/10.1016/j.jprot.2012.01.041
Brijmohan AS et al (2018) HDAC6 inhibition promotes transcription factor eb activation and is protective in experimental kidney disease. Front Pharmacol 9:34
Brunet A et al (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription. Fact Cell 96:857–868
Cai Z, Yan L-J (2013) Protein oxidative modifications: beneficial roles in disease and health. J Biochem Pharmacol Res 1:15–26
Caito S et al (2010) SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J 24:3145–3159
Camacho-Pereira J et al (2016) CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab 23:1127–1139
Canto C, Menzies KJ, Auwerx J (2015) NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22:31–53
Carroll B et al (2018) Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat Commun 9:256
Cha-Molstad H et al (2017) p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat Commun 8:102. https://doi.org/10.1038/s41467-017-00085-7
Chang C et al (2015) AMPK-dependent phosphorylation of GAPDH triggers sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell 60:930–940. https://doi.org/10.1016/j.molcel.2015.10.037
Choudhary C et al (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840
Conze D, Brenner C, Kruger CL (2019) Safety and metabolism of long-term administration of NIAGEN (Nicotinamide Riboside Chloride) in a randomized, double-blind, placebo-controlled clinical trial of healthy overweight adults. Sci Rep 9:9772. https://doi.org/10.1038/s41598-019-46120-z
Daitoku H et al (2004) Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Nat Acad Sci 101:10042–10047
Daitoku H, Sakamaki J-I, Fukamizu A (2011) Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochimica et Biophysica Acta (BBA)—Molecular Cell Research 1813:1954–1960 doi:10.1016/j.bbamcr.2011.03.001
de Guia RM et al (2019) Aerobic and resistance exercise training reverses age-dependent decline in NAD+ salvage capacity in human skeletal muscle. Physiol Rep 7:e14139
de la Rubia JE et al (2019) Efficacy and tolerability of EH301 for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled human pilot study. Amyotroph Lateral Scler Frontotemporal Degener 20:115–122
Deng W, Ma L, Zhang Y, Zhou J, Wang Y, Liu Z, Xue Y (2018) THANATOS: an integrative data resource of proteins and post-translational modifications in the regulation of autophagy. Autophagy 14:296–310
Derbré F, Gratas-Delamarche A, Gómez-Cabrera MC, Viña J (2014) Inactivity-induced oxidative stress: a central role in age-related sarcopenia? Eur J Sport Sci 14:S98–S108. https://doi.org/10.1080/17461391.2011.654268
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19:349–364. https://doi.org/10.1038/s41580-018-0003-4
Dollerup OL et al (2018) A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am J Clin Nutr 108:343–353. https://doi.org/10.1093/ajcn/nqy132
Dollerup OL et al (2019a) Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J Physiol 598(4):731–754
Dollerup OL et al (2019b) Effects of nicotinamide riboside on endocrine pancreatic function and incretin hormones in nondiabetic men with obesity. J Clin Endocrinol Metabol 104:5703–5714
Drazic A, Myklebust LM, Ree R, Arnesen T (2016) The world of protein acetylation. Biochimica et Biophysica Acta (BBA)—Proteins and Proteomics 1864:1372–1401. https://doi.org/10.1016/j.bbapap.2016.06.007
Fang EF et al (2019) Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22:401
Fang EF et al (2016) NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab 24:566–581
Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, Bohr VA (2017) NAD(+) in aging: molecular mechanisms and translational implications. Trends Mol Med 23:899–916. https://doi.org/10.1016/j.molmed.2017.08.001
Fang EF et al (2014) Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction. Cell 157:882–896
Filomeni G, De Zio D, Cecconi F (2015) Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22:377
Fouquerel E et al (2014) ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep 8:1819–1831
Frudd K, Burgoyne T, Burgoyne JR (2018) Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat Commun 9:95
Füllgrabe J, Ghislat G, Cho D-H, Rubinsztein DC (2016) Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci 129:3059–3066
Gilmour BC et al (2020) Targeting NAD+ in translational research to relieve diseases and conditions of metabolic stress and ageing. Mech Ageing Dev 186:111208. https://doi.org/10.1016/j.mad.2020.111208
Grumati P, Dikic I (2018) Ubiquitin signaling and autophagy. J Biol Chem 293:5404–5413
Halliwell B, Gutteridge JMC (2015) Oxygen: boon yet bane—introducing oxygen toxicity and reactive species: In: Free radicals in biology and medicine. 5 edn. Oxford University Press, Oxford. doi:10.1093/acprof:oso/9780198717478.003.0001
Hariharan N, Maejima Y, Nakae J, Paik J, DePinho RA, Sadoshima J (2010) Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res 107:1470–1482
Hou Y et al (2018) NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Nat Acad Sci 115:E1876–E1885
Huang R et al (2015) Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell 57:456–466. https://doi.org/10.1016/j.molcel.2014.12.013
Imai S-i, Guarente L (2016) It takes two to tango: NAD+ and sirtuins in aging/longevity control npj. Aging Mech Dis 2:16017. https://doi.org/10.1038/npjamd.2016.17
Inglés M et al (2014) Oxidative stress is related to frailty, not to age or sex, in a geriatric population: lipid and protein oxidation as biomarkers of frailty. J Am Geriatr Soc 62:1324–1328
Jacobs KM et al (2008) SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression International. J Biol Sci 4:291
Jiang H-Y, Yang Y, Zhang Y-Y, Xie Z, Zhao X-Y, Sun Y, Kong W-J (2018) The dual role of poly(ADP-ribose) polymerase-1 in modulating parthanatos and autophagy under oxidative stress in rat cochlear marginal cells of the stria vascularis. Redox Biol 14:361–370. https://doi.org/10.1016/j.redox.2017.10.002
Johansen T, Lamark T (2019) Selective autophagy: atg8 family proteins, LIR motifs and cargo receptors. J Mol Biol
Kalous KS, Wynia-Smith SL, Olp MD, Smith BC (2016) Mechanism of Sirt1 NAD+-dependent protein deacetylase inhibition by cysteine S-nitrosation. J Biol Chem 291:25398–25410. https://doi.org/10.1074/jbc.M116.754655
Kane AE, Sinclair DA (2018) Sirtuins and NAD+ in the development and treatment of metabolic and cardiovascular diseases. Circ Res 123:868–885
Kirisako T et al (2000) The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 151:263–276
Kubben N, Misteli T (2017) Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat Rev Mol Cell Biol 18:595–609. https://doi.org/10.1038/nrm.2017.68
Lautrup S, Sinclair DA, Mattson MP, Fang EF (2019) NAD+ in brain aging and neurodegenerative disorders. Cell Metab 30:630–655. https://doi.org/10.1016/j.cmet.2019.09.001
Lee IH et al (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA 105:3374–3379. https://doi.org/10.1073/pnas.0712145105
Lee IH, Finkel T (2009) Regulation of autophagy by the p300 acetyltransferase. J Biol Chem 284:6322–6328. https://doi.org/10.1074/jbc.M807135200
Lee Y, Chou T-F, Pittman SK, Keith AL, Razani B, Weihl CC (2017) Keap1/Cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep 19:188–202
Li L, Tan J, Miao Y, Lei P, Zhang Q (2015) ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol 35:615–621. https://doi.org/10.1007/s10571-015-0166-x
Li Y-T et al (2017) A semisynthetic Atg3 reveals that acetylation promotes Atg3 membrane binding and Atg8 lipidation. Nat Commun 8:14846. https://doi.org/10.1038/ncomms14846
Liguori I et al (2018) Oxidative stress, aging, and diseases. Clin Interv Aging 13:757–772. https://doi.org/10.2147/CIA.S158513
Lin S-Y et al (2012) GSK3-TIP60-ULK1 Signaling pathway links growth factor deprivation to autophagy. Science 336:477–481. https://doi.org/10.1126/science.1217032
Long J et al (2010) Dimerisation of the UBA domain of p62 inhibits ubiquitin binding and regulates NF-κB signalling. J Mol Biol 396:178–194
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
López-Otín C, Galluzzi L, Freije JMP, Madeo F, Kroemer G (2016) Metabolic control of longevity. Cell 166:802–821. https://doi.org/10.1016/j.cell.2016.07.031
Mancinelli R et al. (2017) Multifaceted roles of GSK-3 in cancer and autophagy-related diseases. Oxid Med Cell Longev
Marino SM, Gladyshev VN (2010) Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J Mol Biol 404:902–916. https://doi.org/10.1016/j.jmb.2010.09.027
Martens CR et al (2018) Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat Commun 9:1286. https://doi.org/10.1038/s41467-018-03421-7
Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ (2012) Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7:e42357
Matsumoto G, Shimogori T, Hattori N, Nukina N (2015) TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet 24:4429–4442
Matsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F (2007) Structure of Atg5·Atg16, a complex essential for autophagy. J Biol Chem 282:6763–6772
Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A (2005) Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA 102:11278–11283. https://doi.org/10.1073/pnas.0502738102
Medina DL et al (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17:288
Metlagel Z, Otomo C, Takaesu G, Otomo T (2013) Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc Nat Acad Sci 110:18844–18849. https://doi.org/10.1073/pnas.1314755110
Minto C, Vecchio MG, Lamprecht M, Gregori D (2017) Definition of a tolerable upper intake level of niacin: a systematic review and meta-analysis of the dose-dependent effects of nicotinamide and nicotinic acid supplementation. Nutr Rev 75:471–490. https://doi.org/10.1093/nutrit/nux011
Mizushima N, Sugita H, Yoshimori T, Ohsumi Y (1998) A new protein conjugation system in human: the counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 273:33889–33892. https://doi.org/10.1074/jbc.273.51.33889
Montagna C, Rizza S, Maiani E, Piredda L, Filomeni G, Cecconi F (2016) To eat, or NOt to eat: S-nitrosylation signaling in autophagy. FEBS J 283:3857–3869
Morishita H, Mizushima N (2019) Diverse cellular roles of autophagy. Ann Rev Cell Dev Biol 35:453–475
Motta MC et al (2004) Mammalian SIRT1 represses forkhead transcription factors. Cell 116:551–563. https://doi.org/10.1016/S0092-8674(04)00126-6
Muñoz-Gámez JA et al (2009) PARP-1 is involved in autophagy induced by DNA damage. Autophagy 5:61–74
Murray CI, Van Eyk JE (2012) Chasing cysteine oxidative modifications: proteomic tools for characterizing cysteine redox status. Circ: Cardiovasc Gen 5:591
Narita T, Weinert BT, Choudhary C (2019) Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 20:156–174. https://doi.org/10.1038/s41580-018-0081-3
Nie T et al (2016) Regulation of ER stress-induced autophagy by GSK3β-TIP60-ULK1 pathway. Cell Death Dis 7:e2563–e2563. https://doi.org/10.1038/cddis.2016.423
Ohashi Y, Tremel S, Williams RL (2019) VPS34 complexes from a structural perspective. J Lipid Res 60:229–241
Ohtake F et al (2015) Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep 16:192–201
Okabe K, Yaku K, Tobe K, Nakagawa T (2019) Implications of altered NAD metabolism in metabolic disorders. J Biomed Sci 26:34–34. https://doi.org/10.1186/s12929-019-0527-8
Otomo C, Metlagel Z, Takaesu G, Otomo T (2013) Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 20:59–66. https://doi.org/10.1038/nsmb.2431
Pacher P, Szabo C (2008) Role of the peroxynitrite-poly(ADP-Ribose) polymerase pathway in human disease. Am J Pathol 173:2–13. https://doi.org/10.2353/ajpath.2008.080019
Peng H et al (2017) Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res 27:657
Phelan M, Mulnard R, Gillen D, Schreiber S (2017) Phase II clinical trial of nicotinamide for the treatment of mild to moderate. Alzheimer’s Dis J Geriatr Med Gerontol 3:021
Pillai JB, Isbatan A, Imai S-i, Gupta MP (2005) Poly (ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J Biol Chem 280:43121–43130
Polzonetti V, Carpi FM, Micozzi D, Pucciarelli S, Vincenzetti S, Napolioni V (2012) Population variability in CD38 activity: correlation with age and significant effect of TNF-α −308G%3eA and CD38 184C%3eG SNPs. Mol Gen Metab 105:502–507. https://doi.org/10.1016/j.ymgme.2011.12.016
Rabanal-Ruiz Y, Otten Elsje G, Korolchuk Viktor I (2017) mTORC1 as the main gateway to autophagy. Essays Biochem 61:565–584. https://doi.org/10.1042/EBC20170027
Requejo-Aguilar R, Lopez-Fabuel I, Jimenez-Blasco D, Fernandez E, Almeida A, Bolaños Juan P (2015) DJ1 represses glycolysis and cell proliferation by transcriptionally up-regulating pink1. Biochem J 467:303–310. https://doi.org/10.1042/bj20141025
Saline M et al (2019) AMPK and AKT protein kinases hierarchically phosphorylate the N-terminus of the FOXO1 transcription factor, modulating interactions with 14-3-3 proteins. J Biol Chem 294:13106–13116
Santos L, Escande C, Denicola A (2016) Potential modulation of sirtuins by oxidative stress. Oxid Med Cell Longev 2016
Scheibye-Knudsen M et al (2014) A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 20:840–855
Scheibye-Knudsen M et al (2012) Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med 209:855–869
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26:1749–1760
Schöndorf DC et al (2018) The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep 23:2976–2988. https://doi.org/10.1016/j.celrep.2018.05.009
Sedlackova L, Kelly G, Korolchuk VI (2020) The pROS of autophagy in neuronal health. J Mol Biol
Sedlackova L, Korolchuk VI (2019) Mitochondrial quality control as a key determinant of cell survival. Biochimica et Biophysica Acta (BBA)—Mol Cell Res 1866:575–587
Settembre C et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433
Song T, Su H, Yin W, Wang L, Huang R (2019) Acetylation modulates LC3 stability and cargo recognition FEBS Lett 593:414–422 doi:10.1002/1873–3468.13327
Soysal P et al (2017) Oxidative stress and frailty: a systematic review and synthesis of the best evidence. Maturitas 99:66–72. https://doi.org/10.1016/j.maturitas.2017.01.006
Stea TH et al. (2017) Assessment of dietary intake of nicotinic acid and nicotinamide in relation to tolerable upper intake levels: opinion of the panel on nutrition, dietetic products, novel food and allergy of the Norwegian Scientific Committee for food safety. VKM report 2017: 27 VKM report
Stein LR, Imai S-i (2014) Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J 33:1321–1340. https://doi.org/10.1002/embj.201386917
Strømland Ø, Niere M, Nikiforov AA, VanLinden MR, Heiland I, Ziegler M (2019) Keeping the balance in NAD metabolism. Biochem Soc Trans 47:119–130. https://doi.org/10.1042/BST20180417
Su H et al (2017) VPS34 Acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy. Mol Cell 67:907–921.e907. https://doi.org/10.1016/j.molcel.2017.07.024
Sun T et al (2015) Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun 6:7215
Suzuki H, Osawa T, Fujioka Y, Noda NN (2017) Structural biology of the core autophagy machinery. Curr Opin Struc Biol 43:10–17. https://doi.org/10.1016/j.sbi.2016.09.010
Tamargo-Gómez I, Mariño G (2018) AMPK: Regulation of metabolic dynamics in the context of autophagy. Int J Mol Sci 19:3812
Valentin-Vega YA et al (2012) Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119:1490–1500
Vannini N et al (2019) The NAD-booster nicotinamide riboside potently stimulates hematopoiesis through increased mitochondrial clearance cell. Stem Cell 24(405–418):e407
Wan W et al (2017) mTORC1 Phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis. Mol Cell 68:323–335.e326. https://doi.org/10.1016/j.molcel.2017.09.020
Wang F, Nguyen M, Qin FX-F, Tong Q (2007) SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6:505–514. https://doi.org/10.1111/j.1474-9726.2007.00304.x
Wang H et al. (2019a) Oxidation of multiple MiT/TFE transcription factors links oxidative stress to transcriptional control of autophagy and lysosome biogenesis. Autophagy
Wang Y et al. (2019b) Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep n/a:e48335 doi:10.15252/embr.201948335
Xie Y et al (2018) DeepNitro: prediction of protein nitration and nitrosylation sites by deep learning. Genomics, Proteomics Bioinform 16:294–306. https://doi.org/10.1016/j.gpb.2018.04.007
Xu Y, Wan W (2019) TP53INP2 mediates autophagic degradation of ubiquitinated proteins through its ubiquitin-interacting motif. FEBS Lett 593:1974–1982. https://doi.org/10.1002/1873-3468.13467
Yan J et al (2013) SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS One 8:e76016–e76016. https://doi.org/10.1371/journal.pone.0076016
Yang M et al (2018) Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun Signal 16:31
Yang Y, Hou H, Haller EM, Nicosia SV, Bai W (2005) Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J 24:1021–1032
Yi C et al (2012) Function and molecular mechanism of acetylation in autophagy regulation. Science 336:474. https://doi.org/10.1126/science.1216990
You Z et al (2019a) Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat Commun 10:5792. https://doi.org/10.1038/s41467-019-13718-w
You Z et al (2019b) TP53INP2 contributes to autophagosome formation by promoting LC3-ATG7 interaction. Autophagy 15:1309–1321. https://doi.org/10.1080/15548627.2019.1580510
Zhang D-X, Zhang J-P, Hu J-Y, Huang Y-S (2016a) The potential regulatory roles of NAD+ and its metabolism in autophagy. Metabolism 65:454–462. https://doi.org/10.1016/j.metabol.2015.11.010
Zhang H et al (2016b) NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352:1436–1443
Zhang J et al (2018) Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy 14:1043–1059. https://doi.org/10.1080/15548627.2018.1447290
Zhang X et al (2016c) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7:12109
Zhu X-H, Lu M, Lee B-Y, Ugurbil K, Chen W (2015) In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Nat Acad Sci 112:2876–2881
Acknowledgements
This work was funded by an MRC PhD program (V.I.K.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sedlackova, L., Korolchuk, V.I. The crosstalk of NAD, ROS and autophagy in cellular health and ageing. Biogerontology 21, 381–397 (2020). https://doi.org/10.1007/s10522-020-09864-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-020-09864-0