
Abstract
Nicotinamide adenine dinucleotide (NAD) is an important coenzyme that participates in various energy metabolism pathways, including glycolysis, β-oxidation, and oxidative phosphorylation. Besides, it is a required cofactor for post-translational modifications such as ADP-ribosylation and deacetylation by poly (ADP-ribose) polymerases (PARPs) and sirtuins, respectively. Thus, NAD regulates energy metabolism, DNA damage repair, gene expression, and stress response through these enzymes. Numerous studies have shown that NAD levels decrease with aging and under disturbed nutrient conditions, such as obesity. Additionally, a decline in NAD levels is closely related to the development of various metabolic disorders, including diabetes and fatty liver disease. In addition, many studies have revealed that administration of NAD precursors, such as nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR), efficiently increase NAD levels in various tissues and prevent such metabolic diseases. These NAD precursors are contained in natural foods, such as cow milk, vegetables, and meats. Therefore, altered NAD metabolism can be a practical target for nutritional intervention. Recently, several human clinical trials using NAD precursors have been conducted to investigate the safety, pharmacokinetics, and efficacy against metabolic disorders such as glucose intolerance. In this review, we summarize current knowledge on the implications of NAD metabolism in metabolic diseases and discuss the outcomes of recent human clinical trials.
Similar content being viewed by others
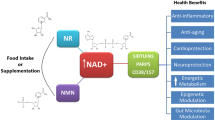

Introduction
Metabolic syndrome is increasing worldwide and is becoming a global health concern because it is a critical risk for various life threatening diseases, including cardiovascular diseases, stroke, and cancer [1]. Its pathophysiology is based on obesity, which consequently causes diabetes, dyslipidemia, and hypertension. Development of metabolic syndrome is closely associated with nutrient status and lifestyle [2]. Excess energy intake and sedentary lifestyle cause obesity and subsequent metabolic disorders. In mammalian cells, energy-sensing pathways are important for maintaining an adequate balance between energy production and expenditure. Disturbance of these pathways results in various metabolic disorders, such as insulin resistance and fatty liver [3]. Endogenous metabolites reflect the nutrient status in cells, and their levels regulate the activity of energy-sensing molecules. For instance, adenosine monophosphate (AMP) and adenosine triphosphate (ATP) levels regulate AMP-activated protein kinase (AMPK) activity and control glucose and lipid metabolism [4]. The mammalian target of rapamycin (mTOR) senses amino acid levels and determines protein synthesis or degradation depending on nutrient availability [3]. Nicotinamide adenine dinucleotide (NAD) is also one of such energy-sensing metabolites and is an essential cofactor that mediates various biological processes, including metabolism, aging, cell death, DNA repair, and gene expression (Fig. 1) [5]. It functions as a coenzyme in various redox reactions in the major energy production pathways, such as glycolysis, tricarboxylic acid (TCA) cycle, and fatty acid oxidation [6]. NAD levels directly influence the activity of metabolic enzymes in these pathways as a coenzyme. In particular, many enzymes in the mitochondrial energy production pathway employ NAD in their redox reactions. Further, NAD acts as a substrate for poly (ADP-ribose) polymerases (PARPs) and class III NAD-dependent deacetylases (sirtuins), regulating their activities [7].

NAD metabolism has a potential protective effect against various metabolic diseases through redox reactions, sirtuins, and possibly PARPs. NAD is a co-enzyme that mediates various redox reactions in glycolysis, the TCA cycle, fatty acid oxidation, and oxidative phosphorylation. It also serves as a substrate for PARPs and sirtuins and regulates various biological pathways, including energy metabolism, gene expression, DNA repair, and cellular stress response
A number of studies have demonstrated that NAD levels decline with age and aberrant nutritional status, such as in obesity (Table 1) [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23, 96]. Decreased NAD levels suppress activities of NAD (H)-dependent enzymes in oxidative phosphorylation, TCA cycle, and glycolysis, which result in lower ATP production [24]. Additionally, decreased NAD levels affect PARPs and sirtuins and lead to the inactivation of downstream molecular pathways, including DNA repair, cellular stress responses, and energy metabolism regulation [5]. Thus, preventing the decline of NAD is suggested as a promising strategy to combat metabolic disorders. Dietary intervention is an ideal way to increase NAD levels in cells and tissues. However, NAD is considered impermeable to the plasma membrane, and NAD administration cannot efficiently increase NAD levels [25]. Therefore, NAD precursors, such as nicotinamide (NAM), nicotinic acid (NA), tryptophan, nicotinamide mononucleotide (NMN), and nicotinamide riboside (NR), are utilized to increase NAD levels in rodents and humans [26]. In particular, NMN and NR administration efficiently boost NAD levels and has beneficial effects for obesity and glucose tolerance in mice [8, 22, 27, 28]. NAM also prevents hepatic steatosis and improves glucose tolerance by reducing oxidative stress and inflammation in diet-induced obese mice [29]. NA improves glucose tolerance and lipid metabolism, and it has already been applied for the treatment of dyslipidemia in humans [30]. In this review, the association of NAD with each metabolic disease and the therapeutic potential of NAD precursors for these diseases are discussed.
NAD synthesis and consuming pathways
There are three NAD synthesis pathways named salvage, de novo, and Preiss-Handler, where NAD is synthesized from NAM, tryptophan, and NA, respectively (Fig. 2) [31]. These NAD precursors are ingested from dietary sources, and their shortage causes pellagra with characteristic symptoms of inflamed skin, diarrhea, dementia, and sores in the mouth [32]. In mammalian cells, NAD is predominantly synthesized through the salvage pathway where nicotinamide phophoribosyltransferase (Nampt) generates NMN from NAM and 5-phosphoribosyl-1-pyrophosphate (PRPP) [33]. Subsequently, NMN is conjugated to ATP and converted to NAD by NMN adenylyltransferase (Nmnat) [34]. In mammals, there are three Nmnat isozymes that are encoded by different genes. Nmnat1, Nmnat2, and Nmnat3 exist in nucleus, Golgi apparatus, and mitochondria, respectively [34]. The salvage pathway is coupled with NAD-consuming enzymes, such as PARPs, sirtuins, CD38 (T10), CD157 (BST1), and SARM1. These enzymes degrade NAD and generate NAM as a by-product [35, 36]. Nampt is a rate-limiting enzyme in the salvage pathway, and the global deletion of Nampt in mice results in embryonic lethality [33, 37]. Furthermore, the tissue-specific deletion of Nampt in murine metabolic tissues, including skeletal muscle, liver, and adipose tissues, decreases NAD levels in each organ [21, 38, 39]. Most tryptophan, a precursor for de novo synthesis pathway, is consumed in the liver, which is the only organ that possesses all synthetic enzymes of this pathway [40]. However, deficiency of quinolinate phosphoribosyltransferase (Qprt), a key enzyme in the de novo pathway, has no effect in the NAD levels in murine tissues, including the liver [41]. These results indicate that NAD synthesis in mammalian cells largely depends on the salvage pathway. However, a recent study has demonstrated that the de novo pathway contributes to synthesis and maintenance of NAD levels in the macrophages, particularly during aging and inflammation [42]. Therefore, it is possible that the NAD synthesis pathway can switch between the de novo and salvage pathways under certain stress conditions.

NAD is synthesized through de novo, Preiss-Handler, and salvage pathways. NAM: nicotinamide, NA; nicotinic acid, NAD: nicotinamide adenine dinucleotide, NMN: nicotinamide mononucleotide, NR: nicotinamide riboside, NAAD: nicotinic acid adenine dinucleotide, Nampt: nicotinamide phophoribosyltransferase, Nmnat: NMN adenylyltransferase, NADS: NAD synthase, NRK: nicotinamide riboside kinase
Although Nampt functions as a NAD synthesis enzyme in cells, it is also found in serum. It was originally reported as a cytokine named pre-B-cell colony-enhancing factor (PBEF) as well as visfatin, a type of adipokine [43, 44]. The extracellular form of Nampt (eNampt) is secreted from several kinds of cells, including mature adipocytes, pancreatic β-cells, myocytes, and hepatocytes [37, 45, 46]. Reportedly, the intracellular form of Nampt (iNampt) is acetylated in the cytoplasm during normal nutrient status. However, once food becomes scarce, iNampt is deacetylated by SIRT1 [47]. In addition, the deacetylation of Nampt enhances its secretion and enzymatic activity [47]. Interestingly, genetic deletion of Nampt in the adipocytes decreases hypothalamic NAD levels [47]. Likewise, eNampt depletion by neutralizing antibodies has the same effect on hypothalamic NAD levels [47]. These results suggest that eNampt may generate NMN in the blood, thus supplying NMN to various tissues, including the hypothalamus. However, another study determined that eNampt did not participate in the generation of extracellular NMN because the physiological concentrations of NAM, ATP, and PRPP in the plasma were insufficient for the catalysis of Nampt [48]. Therefore, the contribution of eNampt to the generation of extracellular NMN is still under debate.
NR is an alternative NAD precursor, and a study using various chemical inhibitors suggested that NR is incorporated into cells using equilibrative nucleoside transporters (ENTs) [49, 50]. Inside the cells, NR is converted to NMN by nicotinamide riboside kinase (NRK), and knockdown of NRK1 in mammalian cells eliminated NAD synthesis from NR. Interestingly, NRK1 also regulates NAD synthesis from NMN [51]. In NRK1 knockout mice, administration of NMN failed to increase NAD levels in the liver, kidney, and brown adipose tissue [51]. Furthermore, a study using stable isotope-labeled NR and NMN revealed that NMN is dephosphorylated into NR extracellularly [51]. These results suggest that NMN is incorporated into cells after extracellular conversion to NR. Meanwhile, a recent study identified Slc12a8 as a NMN transporter [52]. This study demonstrated that Slc12a8 directly transports NMN across the plasma membrane, and deletion of Slc12a8 in the hepatocytes largely diminished the incorporation of NMN. Slc12a8 is strongly expressed in the small intestine and may contribute to oral uptake of NMN. Therefore, it is possible that uptake pathways of NMN vary with tissue types. Therefore, further studies are necessary to reveal the mode and kinetics of uptake of NAD precursors specific to each tissue and/or cell.
Obesity
Obesity is a fundamental pathophysiology for various metabolic diseases, such as diabetes, dyslipidemia, and fatty liver. Several studies have revealed that intracellular NAD levels decreased with obesity in multiple murine tissues, including the adipose tissue, skeletal muscles, liver, and hypothalamus [8, 10, 12, 15]. Further, obesity causes low-grade inflammation, and inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, are induced in various tissues, including adipose tissues, liver, and skeletal muscle [53]. These inflammatory cytokines impair the gene expression of Nampt [8, 54]. In humans, several studies have found reduced Nampt levels in adipose tissue, serum, and liver from obese patients [55,56,57]. However, conflicting results have been reported by several studies [58,59,60,61,62]. It is considered that eNampt is mainly released from adipose tissue [37, 44]. Therefore, it is possible that the increased amount of adipose tissue in obese patients resulted in the enhancement of eNampt secretion. The adipose tissue-specific overexpression of Nampt in mice also shows significant increase in plasma eNampt levels [47]. Reduced iNampt levels correlate with decreased NAD levels in obese tissues; however, the biological significance of increased eNampt in obesity remains unclear. Thus, further studies are warranted to reveal the role of increased eNampt levels in obese patients.
Conversely, NMN or NR administration can prevent the reduction in NAD levels in diet-induced obese mice (Table 2) [27, 28, 65]. Moreover, NR administration partially suppresses weight gain in mice fed a high-fat diet (HFD) by enhancing energy expenditure [8, 28]. Mice with long-term NMN administration exhibit both higher energy expenditure and physical activity, and weight gain during aging is suppressed [27]. Thus, administration of NAD precursors can ameliorate diet- and age-associated weight gain, and nutritional intervention using NMN and NR may be a promising strategy against obesity.
Diabetes
Nampt and insulin secretion
Insulin resistance and subsequent impaired insulin secretion compose the pathophysiology of type 2 diabetes. Both insulin sensitivity and secretion are coordinated by NAD metabolism [26]. Reportedly, NAD levels of islet cells are decreased in heterozygous whole body Nampt knockout mice, and glucose-stimulated insulin secretion (GSIS) is compromised in these mice [37]. Conversely, NMN administration recovers NAD, and ameliorates impaired GSIS in these mice [37]. Although eNampt was reported as a ligand for the insulin receptor (IR) and had an insulin-mimetic effect, the study has been retracted [44]. Later studies also argue that eNampt does not directly activate the insulin-signaling pathway in β-cell lines [37]. However, several studies have suggested positive effects of eNampt on insulin secretion [37, 63, 66]. Reportedly, mice fed a fructose-rich diet (FRD) show significantly reduced eNampt levels, leading to increased islet inflammation and impaired insulin secretion [63]. Islet cells in FRD-fed mice exhibited increased expression of inflammatory cytokines, including TNFα and IL-1β, whereas NMN administration reduced IL-1β expression and restored the decreased insulin secretion in FRD-fed mice, suggesting that eNampt regulates β-cell function through a mechanism of NAD synthesis [63].
Adipocyte Nampt and insulin resistance
Adipocyte-specific deletion of Nampt caused insulin resistance, and this effect is systemic and not restricted to the adipose tissue [67]. Loss of Nampt in adipocytes increases CDK5 and PPARγ phosphorylation, leading to reduce the serum adiponectin levels and conversely increase serum free fatty acid levels [67]. Thus, adipocyte-specific Nampt knockout (FANKO) mice have demonstrated a systemic insulin resistance when fed a normal chow diet. A recent study has demonstrated that FANKO mice are resistant to obesity induced by HFD and lack healthy adipose tissue expansion [68]. Although adipose tissue mitochondria in HFD-fed FANKO mice have a reduced respiratory capacity, the mice exhibit improved glucose tolerance compared with control mice [68]. Furthermore, FANKO mice exhibit reduced food intake [68]. These results suggest that Nampt in adipocytes is necessary for healthy expansion during diet-induced obesity and it is also important for the maintenance of insulin sensitivity in normal nutrient status. Thus, the roles of Nampt in adipose tissues may differ by the nutrient status.
Skeletal muscle Nampt and metabolic disorders
The role of skeletal muscle Nampt in metabolic disorders has been reported using muscle-specific Nampt-overexpressing mice [12, 21, 69]. Although Nampt-overexpressing mice have higher NAD levels in skeletal muscles, there is no significant difference in weight gain between Nampt-overexpressing and control mice fed an NCD or HFD [12, 69]. Nampt-overexpressing mice fed a very HFD are partially protected against body weight gain but not against diet-induced insulin resistance [69]. However, Nampt-overexpressing mice have higher exercise endurance capacity and enhanced mitochondrial gene expression [69]. Furthermore, muscle-specific Nampt knockout mice display progressive muscle degeneration with a significant reduction in NAD levels in muscle [21]. Respiratory capacity also decreases in mitochondria from muscle-specific Nampt knockout mice [21]. It is of further interest to investigate the effect of skeletal muscle-specific deletion of Nampt against glucose tolerance.
Nmnat3 and insulin resistance
Recently, the authors reported that systemic overexpression of Nmnat3 in mice can efficiently increase NAD levels in various tissues and ameliorate the onset of diet- and age-associated insulin resistance [13]. In the skeletal muscles of Nmnat3-overexpressing (Nmnat3 Tg) mice, the increase in TCA cycle intermediates was accompanied by repletion of mitochondrial NAD level, suggesting the activation of the TCA cycle. Additionally, the fuel for energy metabolism was shifted from carbohydrates to fatty acids. Furthermore, overexpression of Nmnat3 modulates the ratio of mitochondrial respiratory chain complexes, which might be associated with lower reactive oxygen species (ROS) generation during aging [13]. Of note, Nmnat3 Tg mice have significantly increased concentrations of nicotinamide guanine dinucleotide (NGD), a NAD analog [13]. However, it is still unclear whether increased NGD levels contribute to the phenotypes in Nmnat3 Tg mice, and further studies are awaited.
CD38 and insulin resistance
A recent study has demonstrated that the decline of NAD levels with aging is largely dependent on CD38 [18]. CD38 has an enzymatic activity catalyzing the degradation of NAD into NAM and ADP-ribose (ADPR). CD38 also has ADPR cyclase activity generating cyclic-ADPR from NAD [70]. Interestingly, the knockout of CD38 increases basal NAD levels in tissue, suggesting the importance of NAD degradation by CD38 in regulating NAD levels. Additionally, expression of CD38 in various tissues is remarkably elevated with aging, and NAD consumption is also accelerated in correlation with CD38 levels. In contrast, CD38 deficiency in mice eliminates NAD decline during aging [18]. Although CD38 has been found in plasma membrane as an ectoenzyme, it has been also detected in mitochondria [71, 72]. Importantly, CD38-deficient mice display improved age-associated glucose intolerance. This benefit should be attributed to the upregulation of mitochondrial function by increasing mitochondrial NAD levels and SIRT3 activity [18]. Recently, a CD38 specific inhibitor, 78c, has been reported to have beneficial effects against age-associated physical declines including glucose tolerance and exercise capacity [73]. Treatment with 78c prevents NAD decline with aging and activates sirtuins, AMPK, and PARPs.
NAD precursors and diabetes
Several research groups used the precursors of NAD, NR or NMN, to increase the level of NAD and showed they could improve insulin resistance due to obesity [8, 28, 64]. The effect of NAD precursors is mainly through the enhancement of sirtuin pathways. The intracellular level of NAD is increased in liver after the administration of NR, leading to activated SIRT1 and SIRT3 [28]. Then SIRT1 promotes the deacetylation of FOXO1, inducing SOD2. SIRT3 promotes the deacetylation of both SOD2 and NDUFA9 [28]. In addition, administered NR contributes to the enrichment of the mitochondrial content in skeletal muscle and brown adipose tissues. Thus, the administration of NR protects mice against obesity and glucose tolerance, increasing fatty acid oxidation and energy expenditure and improving insulin sensitivity [28]. As well as NR, the administration of NMN increases hepatic NAD level and ameliorates the insulin sensitivity in liver. Besides, NMN relieves oxidative stress and the inflammatory response which is induced by diet-induced-obesity and recovers the perturbed circadian rhythm [8]. Long-term NMN administration also ameliorates age-associated insulin resistance and prevents changes in gene expression with aging [27]. Aged mice administered NMN also maintained better mitochondrial respiratory capacity in skeletal muscle, which may contribute to improved glucose tolerance.
Recently, a research group examined the effect of long-term administration of NAM in mice. [29]. Although the level of NAD and the mean or maximum lifespan were unchanged, the administration of NAM restored some aging-related metabolic decline including increased protein carbonylation and the reduction of oxygen consumption rates. Consequently, NAM administration ameliorates glucose tolerance during diet-induced obesity. Thus, NAM promotes a healthy lifespan without obvious adverse effects, and this can be translated into humans. Previous studies have also demonstrated that NAM administration ameliorates sustained hyperglycemia by increasing β-cell proliferation in various diabetic rodent models [74, 75]. In particular, NAM can rescue streptozotocin (STZ)-induced β-cell damage and diabetes (model of type 1 diabetes [T1DM]) [75]. Thus, oral NAM administration is thought to be a therapeutic agent for T1DM. Although small-scale clinical trials have reported the beneficial effects of NAM against T1DM [76], a large-scale randomized controlled trial demonstrated that NAM intervention in patients with confirmed anti-islet cell antibodies failed to prevent the onset of T1DM [77]. For T1DM treatment, NMN and NR are still attractive candidates because NAD-mediated SIRT1 activation augments GSIS in β-cells [78]. Further studies are needed to investigate the effects of NMN or NR in the prevention and/or treatment of T1DM.
Dyslipidemia
Dyslipidemia is caused by both nutritional and genetic factors, and it is associated with various metabolic disorders and cardiovascular diseases. NA was the first therapeutic agent to treat dyslipidemia and has been used to prevent cardiovascular disease clinically for a long time [79]. NA lowers the level of triglycerides and low-density lipoprotein cholesterol (LDL-C) and raises the level of high-density lipoprotein cholesterol (HDL-C) [79]. However, the mechanisms of how NA improves dyslipidemia remain unclear. Several studies have indicated that these effects are due to the activation of the G protein-coupled receptor GPR109A in adipocytes [80,81,82]. Alternatively, a study using Gpr109A-deficient mice and the clinical trials for GPR109 agonists contradict this hypothesis [83]. A recent study demonstrated that NA administration increases NAD levels, and the subsequent activation of sirtuins contributes to improved lipid metabolism [84]. Therefore, NMN and NR, which activate SIRT1 without activating GPR109A, can be potential therapeutic options [28]. Although the beneficial outcome of NA on dyslipidemia has been confirmed by numerous studies, two recent large clinical trials concluded that NA combined with statin therapy did not provide additional benefits over statin monotherapy against cardiovascular incidents [85, 86]. However, the favorable effect of NA on LDL-C, triglycerides, and HDL-C has been clearly shown, and NA administration is still used as an adjuvant therapy to reduce atherogenic lipoprotein burden.
NAFLD and hepatic steatosis
Excess calorie intake causes ectopic lipid accumulation in the liver, known as non-alcoholic fatty liver disease (NAFLD) [87]. The progression of NAFLD leads to hepatic steatosis, hepatitis, liver cirrhosis, and ultimately, liver dysfunction. Further, these hepatic diseases occasionally coincide with hepatocellular carcinoma [87]. In NAFLD, the ectopic lipid accumulation results in increased ROS and the mitochondrial dysfunction [88]. It is reported that NAD levels are decreased in the liver of the diet-induced NAFLD mice model [89]. Along with this, the activity of both SIRT1 and SIRT3 is decreased [89,90,91,92]. Conversely, SIRT1 overexpression restores the diet-induced hepatic steatosis [93, 94]. NR administration also protects against mitochondrial dysfunction with diet-induced NAFLD through NAD elevation and subsequent SIRT1 activation [9, 28]. In humans, patients with NAFLD present lowered Nampt levels in the liver [55]. Altogether, the administration of NAD precursors is considered a potential therapeutic option for the treatment of NAFLD. In contrast, the inhibition of Nampt using FK866 promotes lipid accumulation and hepatic steatosis in HFD-fed mice [95]. FK866 treatment decreases protein levels of SIRT1 and phospho-AMPK, and also increases the gene expression of SREBP1 and fatty acid synthase in the liver of HFD-fed mice [95]. Similarly, dominant-negative Nampt-overexpressing (DN-Nampt Tg) mice display NAFLD-like phenotypes, including lipid accumulation, chronic inflammation and impaired insulin sensitivity, in the liver. NR administration to DN-Nampt Tg mice can rescue the NAFLD-like phenotypes [17]. miR-34a negatively regulates the expression of Nampt and SIRT1 during obesity [96]. Obesity induces the expression of miR-34a, which resulted in the reduction of Nampt levels and subsequent aggravation of hepatic lipid accumulation in vivo. In contrast, reducing miR-34a levels in obese mice restores Nampt and NAD levels and improves inflammation, glucose intolerance, and hepatic steatosis through the Nampt-NAD-SIRT1 axis [96]. Thus, it has been proposed that the Nampt/NAD/SIRT1 axis can suppress hepatic steatosis in HFD-fed mice.
Human clinical trials
In various mouse models of human disease, the benefit of NAD precursors, in particularly NMN and NR, has been demonstrated (Table 2). Currently, there are several ongoing human clinical trials or recently reported trials (Table 3). The first report of oral NR administration revealed that NR could increase NAD levels in plasma and peripheral blood mononuclear cells (PBMC) [97]. In this study, consented healthy volunteers received a single dose of 100, 300, and 1000 mg NR in different sequences with 7-day washout periods between data collection. Two participants reported flushing at the dose of 300 mg, but no other serious adverse side effects were reported. Interestingly, NR administration also increased nicotinic acid adenine dinucleotide (NAAD) levels in PBMC [97]. Another clinical trial of oral NR administration for 8 days was conducted as an open-label, non-randomized study in healthy volunteers [98]. In this study, participants took gradually incremented doses of NR from 250 mg to 2000 mg per day, and NR administration was well tolerated with no unfavorable side effects [98]. Importantly, NR administration in healthy subjects significantly increased plasma NAD levels in correlation with plasma NR levels [97, 98]. Chronic NR administration to healthy-aged volunteers (average age, 65 years) was reported [99]. In this study, participants were orally administered 500 mg NR twice daily for 6 weeks, and there were no serious side effects. NAD levels in the NR treatment group were significantly increased in PBMC by approximately 60% compared with that in the placebo group. Consistent with previous reports, NAAD levels were also significantly increased in the NR treatment group [99]. This study also reported that NR treatment lowered systolic blood pressure and arterial stiffness. Similarly, acute NR supplementation in old individuals increased NADH and NADPH levels and improved exercise performance [100]. Another clinical trial in obese men investigated safety and insulin sensitivity [101]. Men with a body mass index > 30 kg/m2, with an age range of 40–70 years, were randomly assigned to 12 weeks of NR administration (1000 mg twice daily) or placebo. Although no serious adverse events occurred with NR administration, insulin sensitivity, endogenous glucose production, and glucose disposal and oxidation were not improved [101]. Another clinical trial with a combination of NR and pterostilbene (NRPT), a polyphenol found in blueberries, studied healthy volunteer subjects [102]. NR and NPRT at recommended dose (NRPT 1X; 250 mg of NR plus 50 mg of PT), and NRPT at double dose (NRPT 2X; 500 mg of NR plus 100 mg of PT) were orally administrated to participants for 8 weeks. In this study, NAD levels were increased in a dose-dependent manner (approximately 40% in NRPT 1X and 90% in NRPT 2X), and no serious adverse side effects were observed [102]. Clinical trials to examine the safety and pharmacokinetics of NMN have been recently initiated in the United States and Japan [103], and the results of these trials are yet to be seen.
Conclusions
NAD metabolism is spotlighted as a therapeutic target for metabolic disorders, such as obesity, diabetes, dyslipidemia, and fatty liver. The genetic manipulation of NAD synthesis or catabolizing enzymes has established that reduction in NAD levels causes metabolic disorders in mice. Furthermore, mounting evidence has demonstrated that complementing NAD with NAD precursors ameliorates various metabolic diseases. Recently, several human clinical trials have been reported. Overall, NR administration is safe, well tolerated, and can efficiently increase NAD levels in healthy volunteers. However, efficacy in patients with metabolic disorders remains unclear, and further studies are awaited. Moreover, some small molecules boosting NAD levels have been reported [73, 104, 105]. Outcomes of these molecules against metabolic diseases in patients should be clarified in future studies. It is also demonstrated that NMN and NR are contained in natural foods, including cow milk, broccoli, cucumber, avocado, and beef [27, 106, 107]. Thus, NAD metabolism is considered a practical target for a nutritional intervention.
References
Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. 2018;20(2):12. https://doi.org/10.1007/s11906-018-0812-z.
O'Neill S, O'Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1–12. https://doi.org/10.1111/obr.12229.
Chantranupong L, Wolfson RL, Sabatini DM. Nutrient-sensing mechanisms across evolution. Cell. 2015;161(1):67–83. https://doi.org/10.1016/j.cell.2015.02.041.
Burkewitz K, Zhang Y, Mair WB. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20(1):10–25. https://doi.org/10.1016/j.cmet.2014.03.002.
Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, Bohr VA. NAD (+) in aging: molecular mechanisms and translational implications. Trends Mol Med. 2017;23(10):899–916. https://doi.org/10.1016/j.molmed.2017.08.001.
Yaku K, Okabe K, Nakagawa T. NAD metabolism: implications in aging and longevity. Ageing Res Rev. 2018;47:1–17. https://doi.org/10.1016/j.arr.2018.05.006.
Katsyuba E, Auwerx J. Modulating NAD (+) metabolism, from bench to bedside. EMBO J. 2017;36(18):2670–83. https://doi.org/10.15252/embj.201797135.
Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD (+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528–36. https://doi.org/10.1016/j.cmet.2011.08.014.
Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER, Moullan N, Zhang H, et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology. 2016;63(4):1190–204. https://doi.org/10.1002/hep.28245.
Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA, Brenner C. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep. 2016;6:26933. https://doi.org/10.1038/srep26933.
Dall M, Penke M, Sulek K, Matz-Soja M, Holst B, Garten A, Kiess W, Treebak JT. Hepatic NAD (+) levels and NAMPT abundance are unaffected during prolonged high-fat diet consumption in C57BL/6JBomTac mice. Mol Cell Endocrinol. 2018;473:245–56. https://doi.org/10.1016/j.mce.2018.01.025.
Frederick DW, Davis JG, Davila A Jr, Agarwal B, Michan S, Puchowicz MA, Nakamaru-Ogiso E, Baur JA. Increasing NAD synthesis in muscle via nicotinamide phosphoribosyltransferase is not sufficient to promote oxidative metabolism. J Biol Chem. 2015;290(3):1546–58. https://doi.org/10.1074/jbc.M114.579565.
Gulshan M, Yaku K, Okabe K, Mahmood A, Sasaki T, Yamamoto M, Hikosaka K, Usui I, et al. Overexpression of Nmnat3 efficiently increases NAD and NGD levels and ameliorates age-associated insulin resistance. Aging Cell. 2018:e12798. https://doi.org/10.1111/acel.12798.
Liu S, Kim TH, Franklin DA, Zhang Y. Protection against high-fat-diet-induced obesity in MDM2(C305F) mice due to reduced p53 activity and enhanced energy expenditure. Cell Rep. 2017;18(4):1005–18. https://doi.org/10.1016/j.celrep.2016.12.086.
Sasaki T, Kikuchi O, Shimpuku M, Susanti VY, Yokota-Hashimoto H, Taguchi R, Shibusawa N, Sato T, et al. Hypothalamic SIRT1 prevents age-associated weight gain by improving leptin sensitivity in mice. Diabetologia. 2014;57(4):819–31. https://doi.org/10.1007/s00125-013-3140-5.
Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, et al. The NAD (+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154(2):430–41. https://doi.org/10.1016/j.cell.2013.06.016.
Zhou CC, Yang X, Hua X, Liu J, Fan MB, Li GQ, Song J, Xu TY, et al. Hepatic NAD (+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br J Pharmacol. 2016;173(15):2352–68. https://doi.org/10.1111/bph.13513.
Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016;23(6):1127–39. https://doi.org/10.1016/j.cmet.2016.05.006.
Yaku K, Okabe K, Nakagawa T. Simultaneous measurement of NAD metabolome in aged mice tissue using liquid chromatography tandem-mass spectrometry (LC/MS/MS). Biomed Chromatogr. 2018;32:e4205. https://doi.org/10.1002/bmc.4205.
Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, et al. Declining NAD (+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–38. https://doi.org/10.1016/j.cell.2013.11.037.
Frederick DW, Loro E, Liu L, Davila A Jr, Chellappa K, Silverman IM, Quinn WJ 3rd, Gosai SJ, et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 2016;24(2):269–82. https://doi.org/10.1016/j.cmet.2016.07.005.
Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D'Amico D, Ropelle ER, et al. NAD (+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352(6292):1436–43. https://doi.org/10.1126/science.aaf2693.
Clement J, Wong M, Poljak A, Sachdev P, Braidy N. The plasma NAD+ metabolome is dysregulated in ‘normal’ ageing. Rejuvenation Res. 2018. https://doi.org/10.1089/rej.2018.2077.
Hikosaka K, Ikutani M, Shito M, Kazuma K, Gulshan M, Nagai Y, Takatsu K, Konno K, et al. Deficiency of nicotinamide mononucleotide adenylyltransferase 3 (nmnat3) causes hemolytic anemia by altering the glycolytic flow in mature erythrocytes. J Biol Chem. 2014;289(21):14796–811. https://doi.org/10.1074/jbc.M114.554378.
Zocchi E, Usai C, Guida L, Franco L, Bruzzone S, Passalacqua M, De Flora A. Ligand-induced internalization of CD38 results in intracellular Ca2+ mobilization: role of NAD+ transport across cell membranes. FASEB J. 1999;13(2):273–83.
Yoshino J, Baur JA, Imai SI. NAD (+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27(3):513–28. https://doi.org/10.1016/j.cmet.2017.11.002.
Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, et al. Long-term Administration of Nicotinamide Mononucleotide Mitigates age-Associated Physiological Decline in mice. Cell Metab. 2016;24(6):795–806. https://doi.org/10.1016/j.cmet.2016.09.013.
Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, et al. The NAD (+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15(6):838–47. https://doi.org/10.1016/j.cmet.2012.04.022.
Mitchell SJ, Bernier M, Aon MA, Cortassa S, Kim EY, Fang EF, Palacios HH, Ali A, et al. Nicotinamide improves aspects of Healthspan, but not lifespan, in mice. Cell Metab. 2018;27(3):667–76 e4. https://doi.org/10.1016/j.cmet.2018.02.001.
Julius U. Niacin as antidyslipidemic drug. Can J Physiol Pharmacol. 2015;93(12):1043–54. https://doi.org/10.1139/cjpp-2014-0478.
Badawy AA. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res. 2017;10:1–10. https://doi.org/10.1177/1178646917691938.
Cervenka I, Agudelo LZ, Ruas JL. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science. 2017;357(6349). https://doi.org/10.1126/science.aaf9794.
Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279(49):50754–63. https://doi.org/10.1074/jbc.M408388200.
Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280(43):36334–41. https://doi.org/10.1074/jbc.M508660200.
Sauve AA. NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther. 2008;324(3):883–93. https://doi.org/10.1124/jpet.107.120758.
Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, Funaro A, Horenstein AL, Malavasi F. CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin Cytom. 2013;84(4):207–17. https://doi.org/10.1002/cyto.b.21092.
Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6(5):363–75. https://doi.org/10.1016/j.cmet.2007.09.003.
Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, Heiland I, Birringer M, Groth M, et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol. 2013;9(11):693–700. https://doi.org/10.1038/nchembio.1352.
Mukherjee S, Chellappa K, Moffitt A, Ndungu J, Dellinger RW, Davis JG, Agarwal B, Baur JA. Nicotinamide adenine dinucleotide biosynthesis promotes liver regeneration. Hepatology. 2017;65(2):616–30. https://doi.org/10.1002/hep.28912.
Shibata K. Organ co-relationship in tryptophan metabolism and factors that govern the biosynthesis of nicotinamide from tryptophan. J Nutr Sci Vitaminol (Tokyo). 2018;64(2):90–8. https://doi.org/10.3177/jnsv.64.90.
Terakata M, Fukuwatari T, Sano M, Nakao N, Sasaki R, Fukuoka S, Shibata K. Establishment of true niacin deficiency in quinolinic acid phosphoribosyltransferase knockout mice. J Nutr. 2012;142(12):2148–53. https://doi.org/10.3945/jn.112.167569.
Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, Contrepois K, Wang Q, et al. Macrophage de novo NAD (+) synthesis specifies immune function in aging and inflammation. Nat Immunol. 2019;20(1):50–63. https://doi.org/10.1038/s41590-018-0255-3.
Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14(2):1431–7.
Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307(5708):426–30. https://doi.org/10.1126/science.1097243.
Garten A, Petzold S, Barnikol-Oettler A, Korner A, Thasler WE, Kratzsch J, Kiess W, Gebhardt R. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem Biophys Res Commun. 2010;391(1):376–81. https://doi.org/10.1016/j.bbrc.2009.11.066.
Kover K, Tong PY, Watkins D, Clements M, Stehno-Bittel L, Novikova L, Bittel D, Kibiryeva N, et al. Expression and regulation of nampt in human islets. PLoS One. 2013;8(3):e58767. https://doi.org/10.1371/journal.pone.0058767.
Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, Nakagawa T, Yoshino J, Imai S. SIRT1-mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD+ and function in mice. Cell Metab. 2015;21(5):706–17. https://doi.org/10.1016/j.cmet.2015.04.002.
Hara N, Yamada K, Shibata T, Osago H, Tsuchiya M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS One. 2011;6(8):e22781. https://doi.org/10.1371/journal.pone.0022781.
Nikiforov A, Dolle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem. 2011;286(24):21767–78. https://doi.org/10.1074/jbc.M110.213298.
Lu SP, Lin SJ. Phosphate-responsive signaling pathway is a novel component of NAD+ metabolism in Saccharomyces cerevisiae. J Biol Chem. 2011;286(16):14271–81. https://doi.org/10.1074/jbc.M110.217885.
Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun. 2016;7:13103. https://doi.org/10.1038/ncomms13103.
Grozio A, Mills KF, Yoshino J, Bruzzone S, Sociali G, Tokizane K, Lei HC, Cunningham R, et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat Metab. 2019;1(1):47–57. https://doi.org/10.1038/s42255-018-0009-4.
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9. https://doi.org/10.1093/gerona/glu057.
Kralisch S, Klein J, Lossner U, Bluher M, Paschke R, Stumvoll M, Fasshauer M. Interleukin-6 is a negative regulator of visfatin gene expression in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 2005;289(4):E586–90. https://doi.org/10.1152/ajpendo.00090.2005.
Dahl TB, Haukeland JW, Yndestad A, Ranheim T, Gladhaug IP, Damas JK, Haaland T, Loberg EM, et al. Intracellular nicotinamide phosphoribosyltransferase protects against hepatocyte apoptosis and is down-regulated in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2010;95(6):3039–47. https://doi.org/10.1210/jc.2009-2148.
Gaddipati R, Sasikala M, Padaki N, Mukherjee RM, Sekaran A, Jayaraj-Mansard M, Rabella P, Rao-Guduru V, Reddy-Duvvuru N. Visceral adipose tissue visfatin in nonalcoholic fatty liver disease. Ann Hepatol. 2010;9(3):266–70.
Barth S, Klein P, Horbach T, Dotsch J, Rauh M, Rascher W, Knerr I. Expression of neuropeptide Y, omentin and visfatin in visceral and subcutaneous adipose tissues in humans: relation to endocrine and clinical parameters. Obes Facts. 2010;3(4):245–51. https://doi.org/10.1159/000319508.
Auguet T, Terra X, Porras JA, Orellana-Gavalda JM, Martinez S, Aguilar C, Lucas A, Pellitero S, et al. Plasma visfatin levels and gene expression in morbidly obese women with associated fatty liver disease. Clin Biochem. 2013;46(3):202–8. https://doi.org/10.1016/j.clinbiochem.2012.11.006.
Terra X, Auguet T, Quesada I, Aguilar C, Luna AM, Hernandez M, Sabench F, Porras JA, et al. Increased levels and adipose tissue expression of visfatin in morbidly obese women: the relationship with pro-inflammatory cytokines. Clin Endocrinol. 2012;77(5):691–8. https://doi.org/10.1111/j.1365-2265.2011.04327.x.
Kaminska A, Kopczynska E, Bronisz A, Zmudzinska M, Bielinski M, Borkowska A, Tyrakowski T, Junik R. An evaluation of visfatin levels in obese subjects. Endokrynol Pol. 2010;61(2):169–73.
Jaleel A, Aheed B, Jaleel S, Majeed R, Zuberi A, Khan S, Ahmed B, Shoukat F, Hashim H. Association of adipokines with obesity in children and adolescents. Biomark Med. 2013;7(5):731–5. https://doi.org/10.2217/bmm.13.41.
Chen MP, Chung FM, Chang DM, Tsai JC, Huang HF, Shin SJ, Lee YJ. Elevated plasma level of visfatin/pre-B cell colony-enhancing factor in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2006;91(1):295–9. https://doi.org/10.1210/jc.2005-1475.
Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC. Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia. 2011;54(12):3083–92. https://doi.org/10.1007/s00125-011-2288-0.
Uddin GM, Youngson NA, Sinclair DA, Morris MJ. Head to head comparison of short-term treatment with the NAD (+) precursor nicotinamide mononucleotide (NMN) and 6 weeks of exercise in obese female mice. Front Pharmacol. 2016;7:258. https://doi.org/10.3389/fphar.2016.00258.
Shi W, Hegeman MA, van Dartel DAM, Tang J, Suarez M, Swarts H, van der Hee B, Arola L, Keijer J. Effects of a wide range of dietary nicotinamide riboside (NR) concentrations on metabolic flexibility and white adipose tissue (WAT) of mice fed a mildly obesogenic diet. Mol Nutr Food Res. 2017;61(8). https://doi.org/10.1002/mnfr.201600878.
Spinnler R, Gorski T, Stolz K, Schuster S, Garten A, Beck-Sickinger AG, Engelse MA, de Koning EJ, et al. The adipocytokine Nampt and its product NMN have no effect on beta-cell survival but potentiate glucose stimulated insulin secretion. PLoS One. 2013;8(1):e54106. https://doi.org/10.1371/journal.pone.0054106.
Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, Franczyk MP, Kelly SC, Qi N, Imai S, Yoshino J. NAMPT-mediated NAD (+) biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Rep. 2016;16(7):1851–60. https://doi.org/10.1016/j.celrep.2016.07.027.
Nielsen KN, Peics J, Ma T, Karavaeva I, Dall M, Chubanava S, Basse AL, Dmytriyeva O, Treebak JT, Gerhart-Hines Z. NAMPT-mediated NAD (+) biosynthesis is indispensable for adipose tissue plasticity and development of obesity. Mol Metab. 2018;11:178–88. https://doi.org/10.1016/j.molmet.2018.02.014.
Costford SR, Brouwers B, Hopf ME, Sparks LM, Dispagna M, Gomes AP, Cornnell HH, Petucci C, et al. Skeletal muscle overexpression of nicotinamide phosphoribosyl transferase in mice coupled with voluntary exercise augments exercise endurance. Mol Metab. 2018;7:1–11. https://doi.org/10.1016/j.molmet.2017.10.012.
Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88(3):841–86. https://doi.org/10.1152/physrev.00035.2007.
Yamada M, Mizuguchi M, Otsuka N, Ikeda K, Takahashi H. Ultrastructural localization of CD38 immunoreactivity in rat brain. Brain Res. 1997;756(1–2):52–60.
Ceni C, Pochon N, Brun V, Muller-Steffner H, Andrieux A, Grunwald D, Schuber F, De Waard M, et al. CD38-dependent ADP-ribosyl cyclase activity in developing and adult mouse brain. Biochem J. 2003;370(Pt 1):175–83. https://doi.org/10.1042/BJ20020604.
Tarrago MG, Chini CCS, Kanamori KS, Warner GM, Caride A, de Oliveira GC, Rud M, Samani A, et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD (+) decline. Cell Metab. 2018;27(5):1081–95 e10. https://doi.org/10.1016/j.cmet.2018.03.016.
Shima K, Zhu M, Kuwajima M. A role of nicotinamide-induced increase in pancreatic beta-cell mass on blood glucose control after discontinuation of the treatment in partially pancreatectomized OLETF rats. Diabetes Res Clin Pract. 1998;41(1):1–8.
Schein PS, Cooney DA, Vernon ML. The use of nicotinamide to modify the toxicity of streptozotocin diabetes without loss of antitumor activity. Cancer Res. 1967;27(12):2324–32.
Olmos PR, Hodgson MI, Maiz A, Manrique M, De Valdes MD, Foncea R, Acosta AM, Emmerich MV, et al. Nicotinamide protected first-phase insulin response (FPIR) and prevented clinical disease in first-degree relatives of type-1 diabetics. Diabetes Res Clin Pract. 2006;71(3):320–33. https://doi.org/10.1016/j.diabres.2005.07.009.
Cabrera-Rode E, Molina G, Arranz C, Vera M, Gonzalez P, Suarez R, Prieto M, Padron S, et al. Effect of standard nicotinamide in the prevention of type 1 diabetes in first degree relatives of persons with type 1 diabetes. Autoimmunity. 2006;39(4):333–40. https://doi.org/10.1080/08916930600738383.
Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2(2):105–17. https://doi.org/10.1016/j.cmet.2005.07.001.
Altschul R, Hoffer A, Stephen JD. Influence of nicotinic acid on serum cholesterol in man. Arch Biochem Biophys. 1955;54(2):558–9.
Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9(3):352–5. https://doi.org/10.1038/nm824.
Zhang Y, Schmidt RJ, Foxworthy P, Emkey R, Oler JK, Large TH, Wang H, Su EW, et al. Niacin mediates lipolysis in adipose tissue through its G-protein coupled receptor HM74A. Biochem Biophys Res Commun. 2005;334(2):729–32. https://doi.org/10.1016/j.bbrc.2005.06.141.
Li X, Millar JS, Brownell N, Briand F, Rader DJ. Modulation of HDL metabolism by the niacin receptor GPR109A in mouse hepatocytes. Biochem Pharmacol. 2010;80(9):1450–7. https://doi.org/10.1016/j.bcp.2010.07.023.
Lauring B, Taggart AK, Tata JR, Dunbar R, Caro L, Cheng K, Chin J, Colletti SL, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. 2012;4(148):148ra15. https://doi.org/10.1126/scitranslmed.3003877.
Bruckbauer A, Banerjee J, Cao Q, Cui X, Jing J, Zha L, Li F, Xue B, Shi H, Zemel MB. Leucine-nicotinic acid synergy stimulates AMPK/Sirt1 signaling and regulates lipid metabolism and lifespan in Caenorhabditis elegans, and hyperlipidemia and atherosclerosis in mice. Am J Cardiovasc Dis. 2017;7(2):33–47.
Group HTC, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203–12. https://doi.org/10.1056/NEJMoa1300955.
Investigators A-H, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–67. https://doi.org/10.1056/NEJMoa1107579.
Stefan N, Haring HU, Cusi K. Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2018. https://doi.org/10.1016/S2213-8587(18)30154-2.
Satapati S, Kucejova B, Duarte JA, Fletcher JA, Reynolds L, Sunny NE, He T, Nair LA, et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest. 2015;125(12):4447–62. https://doi.org/10.1172/JCI82204.
Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433(3):505–14. https://doi.org/10.1042/BJ20100791.
Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464(7285):121–5. https://doi.org/10.1038/nature08778.
Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, Sanyal AJ. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15(5):665–74. https://doi.org/10.1016/j.cmet.2012.04.004.
Xu F, Gao Z, Zhang J, Rivera CA, Yin J, Weng J, Ye J. Lack of SIRT1 (mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/− mice: a role of lipid mobilization and inflammation. Endocrinology. 2010;151(6):2504–14. https://doi.org/10.1210/en.2009-1013.
Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, Kharitonenkov A, Yang Q, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology. 2014;146(2):539–49 e7. https://doi.org/10.1053/j.gastro.2013.10.059.
Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105(28):9793–8. https://doi.org/10.1073/pnas.0802917105.
Wang LF, Wang XN, Huang CC, Hu L, Xiao YF, Guan XH, Qian YS, Deng KY, Xin HB. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKalpha/SREBP1 signaling pathway. Lipids Health Dis. 2017;16(1):82. https://doi.org/10.1186/s12944-017-0464-z.
Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, Kang Y, Li X, Kemper B, Kemper JK. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell. 2013;12(6):1062–72. https://doi.org/10.1111/acel.12135.
Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. https://doi.org/10.1038/ncomms12948.
Airhart SE, Shireman LM, Risler LJ, Anderson GD, Nagana Gowda GA, Raftery D, Tian R, Shen DD, O'Brien KD. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One. 2017;12(12):e0186459. https://doi.org/10.1371/journal.pone.0186459.
Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, Chonchol M, Seals DR. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD (+) in healthy middle-aged and older adults. Nat Commun. 2018;9(1):1286. https://doi.org/10.1038/s41467-018-03421-7.
Dolopikou CF, Kourtzidis IA, Margaritelis NV, Vrabas IS, Koidou I, Kyparos A, Theodorou AA, Paschalis V, Nikolaidis MG. Acute nicotinamide riboside supplementation improves redox homeostasis and exercise performance in old individuals: a double-blind cross-over study. Eur J Nutr. 2019. https://doi.org/10.1007/s00394-019-01919-4.
Dollerup OL, Christensen B, Svart M, Schmidt MS, Sulek K, Ringgaard S, Stodkilde-Jorgensen H, Moller N, et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am J Clin Nutr. 2018;108(2):343–53. https://doi.org/10.1093/ajcn/nqy132.
Dellinger RW, Santos SR, Morris M, Evans M, Alminana D, Guarente L, Marcotulli E. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD (+) levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. NPJ Aging Mech Dis. 2017;3:17. https://doi.org/10.1038/s41514-017-0016-9.
Tsubota K. The first human clinical study for NMN has started in Japan. NPJ Aging Mech Dis. 2016;2:16021. https://doi.org/10.1038/npjamd.2016.21.
Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014;158(6):1324–34. https://doi.org/10.1016/j.cell.2014.07.040.
Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27(3):529–47. https://doi.org/10.1016/j.cmet.2018.02.011.
Trammell SA, Yu L, Redpath P, Migaud ME, Brenner C. Nicotinamide riboside is a major NAD+ precursor vitamin in cow Milk. J Nutr. 2016;146(5):957–63. https://doi.org/10.3945/jn.116.230078.
Ummarino S, Mozzon M, Zamporlini F, Amici A, Mazzola F, Orsomando G, Ruggieri S, Raffaelli N. Simultaneous quantitation of nicotinamide riboside, nicotinamide mononucleotide and nicotinamide adenine dinucleotide in milk by a novel enzyme-coupled assay. Food Chem. 2017;221:161–8. https://doi.org/10.1016/j.foodchem.2016.10.032.
Acknowledgements
Not applicable.
Funding
This work is partially supported by JSPS KAKENHI (Grant Number 17 K08653 to TN, 18 K17921 to KY, and 18 K16193 to KO). The grant from Takeda Science Foundation to TN also supported this work.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
OK collected literature, designed and wrote the manuscript. KY edited and revised the manuscript. KT revised the manuscript. TN designed, wrote, edited and prepared manuscript for submission. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Okabe, K., Yaku, K., Tobe, K. et al. Implications of altered NAD metabolism in metabolic disorders. J Biomed Sci 26, 34 (2019). https://doi.org/10.1186/s12929-019-0527-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-019-0527-8