
Abstract
The MiT/TFE transcription factors play a pivotal role in the regulation of autophagy and lysosomal biogenesis. The subcellular localization and activity of MiT/TFE proteins are primarily regulated through phosphorylation. And the phosphorylated protein is retained in the cytoplasm and subsequently translocates to the nucleus upon dephosphorylation, where it stimulates the expression of hundreds of genes, leading to lysosomal biogenesis and autophagy induction. The transcription factor-mediated lysosome-to-nucleus signaling can be directly controlled by several signaling molecules involved in the mTORC1, PKC, and AKT pathways. MiT/TFE family members have attracted much attention owing to their intracellular clearance of pathogenic factors in numerous diseases. Recently, multiple studies have also revealed the MiT/TFE proteins as master regulators of cellular metabolic reprogramming, converging on autophagic and lysosomal function and playing a critical role in cancer, suggesting that novel therapeutic strategies could be based on the modulation of MiT/TFE family member activity. Here, we present an overview of the latest research on MiT/TFE transcriptional factors and their potential mechanisms in cancer.
Similar content being viewed by others


Background
The Microphthalmia family of bHLH-LZ transcription factors (MiT/TFE) is composed of four members. Previous studies reveal that the MiT/TFE transcriptional factors play important roles in the regulation of cellular processes. In particular, recent evidence suggests that the MiT/TFE family members might function as critical factors in cancer. In this review, we focus on the novel mechanism involved in the regulation of the activation of MiT/TFE transcriptional factors and their role in lysosomal homeostasis and autophagy induction. We also describe the participation of the aberrant activation of MiT/TFE members in cancer and discuss the potential therapeutic strategy in cancer.
MiT/TFE family of transcription factors
The Microphthalmia family of bHLH-LZ transcription factors (MiT/TFE) is composed of four members: MITF, TFEB, TFE3 and TFEC [1, 2]. All members of the MiTF/TFE family share a similar structure that includes three critically important regions. The basic motif, which is required for DNA binding, and highly similar Helix-loop-helix (HLH) and leucine-zipper (Zip) regions are important for their dimerization; however, outside of these regions, these proteins are quite different [3]. Besides, TFEB, TFE3 and MITF also contain a conserved activation domain that is important for their transcriptional activation [1], while the activation domain is missing in TFEC, which is the most divergent member of the family and appears to inhibit, rather than activate, transcription [4].
Homodimerization and heterodimerization within the members of the MiTF/TFE family is critical for binding to DNA and the transcriptional activation of target genes [5]. MiT/TFE members bind the palindromic CACGTG E-box, a motif also recognized by other bHLH-Zip transcription factors, such as MYC, MAX and MAD proteins [6]. Unlike other bHLH-Zip transcription factors, MiT/TFE proteins also specifically bind the asymmetric TCATGTG M-box response elements present in the promoter region of their downstream target genes [7]. However, MiT/TFE proteins do not heterodimerize with other bHLH-Zip-containing proteins directly, such as MYC and USF [5, 6]. A previous study demonstrated that a conserved three-residue shift within the Zip domain of the MiT/TFE members generates an unusual out-of-register leucine zipper that allows for specific heterodimerization among MiT/TFE members, while preventing binding to other bHLH-Zip transcription factors [5]. However, the functional relevance of MiT/TFE homodimers compared to heterodimers remains unknown.
All four MiT/TFE members are conserved in vertebrates [8]. The MITF gene is predominantly expressed in the retinal pigment epithelium (RPE), macrophages, osteoclasts, mast cells, melanocytes and natural killer cells [2], while TFEC expression is restricted to cells of myeloid origin [9]. In contrast, TFE3 and TFEB show a more ubiquitous pattern of expression and have been detected in multiple cell types [10]. A large body of evidence suggests that the expression of MITF isoforms (this expression is different in the amino-terminal regions) is due to alternative splicing and posttranslational modifications [11]. Many of these isoforms show a tissue-dependent manner due to the usage of alternative promoters. For example, MITF-M is mainly expressed in melanoblasts and melanocytes, MITF-D is preferentially expressed in monocyte lineages [12], and MITF-A and MITF-H can be detected in several cells [3]. Analyzing their molecular structures, we conclude that TFEB and TFEC contain multiple alternative first exons with restricted and differential tissue distributions, whereas the TFE3 gene may be regulated by a single promoter [13].
Numerous studies have provided evidence suggesting that MiT/TFE transcription factors are important for the maintenance of cellular physiological and pathological processes. Among all the four members of the MiT/TFE family, TFEC is the least studied, and its function has not been widely investigated [14]. While MITF is critical for proliferation, survival and differentiation of melanocytes [3, 15]. And Mutations in MITF are also linked to the pigment and deafness disorder, Waardenburg syndrome type 2A [9]. And the presentation of TFE3 and TFEB is correlated with the development of osteoclasts [16], mast cell differentiation [17, 18], regulation of the expression of genes encoding critical metabolic regulators [19], activation of immune system [20] and control of allergic diseases [18, 21]. Additionally, TFEB is also essential for placental vascularization [22]. Furthermore, previous studies have revealed that the aberrant expression of several MiT/TFE family members is associated with different types of human cancers, such as renal carcinomas [23], alveolar sarcomas [24], and melanomas [25].
Role of MiT/TFE family in lysosome and autophagy biogenesis
Role of MiT/TFE in the transcriptional regulation of lysosome biogenesis
Lysosomes are crucial components of the cellular degradation and recycling system, and their correct function is required to maintain proper cell homeostasis [26]. These organelles are indeed involved in a number of essential cellular processes, including autophagy and lysosomal exocytosis [27]. Autophagosome clearance is initiated upon fusion with lysosomes and/or late endosomes, which introduce into the resulting autolysosome dozens of lysosomal acidic hydrolases and the acidification machinery necessary for enzyme activation and substrate digestion [28].
Lysosomes have long been considered as static organelles devoted to the terminal degradation of waste material; however, this concept has been challenged by subsequent discoveries that lysosomal biogenesis and function are subject to transcriptional regulation [1, 29]. The promoter analysis of lysosomal genes revealed that these molecules share a common 10-base E-box-like palindromic sequence (GTCACGTGAC), in most cases, localized within 200 base pairs of the transcription initiation site. This motif was named Coordinated Lysosomal Expression and Regulation (CLEAR) element [30]. TFE3 and TFEB directly bind to CLEAR elements on the promoters of several autophagic lysosomal genes to promote their expression [31, 32]. Accordingly, TFEB and TFE3 overexpression increases the number of lysosomes and levels of lysosomal enzymes, thus promoting lysosomal catabolic activity [2, 30], while the depletion of these genes abolishes the enhanced expression of lysosomal genes [2, 33]. Moreover, previous studies have also shown that TFEB can induce lysosomal exocytosis [34], a process by which lysosomes fuse to the plasma membrane and secrete their contents into the extracellular space, suggesting the transcriptional regulation of lysosome function which promotes intracellular clearance by TFEB. In a recent study, the role of MITF in the regulation of lysosomal biogenesis has been established in multiple cell types [35, 36]. MITF-A is localized at the lysosomal membrane, mimicking the transcriptional regulation of TFEB and TFE3 [37], and MITF-M drives the transcription of lysosomal markers and activates the CLEAR motif reporter in melanoma cells [36].
Notably, TFEB overexpression results in the enhanced degradation of bulk autophagy substrates, such as long-lived proteins [32], as well as the clearance of lipid droplets and damaged mitochondria [38, 39], suggesting that this transcription factor also plays a role in modulating organelle-specific autophagy, such as lipophagy and mitophagy. Previous studies have revealed that TFEB regulated peroxisome proliferator activated receptor gamma coactivator-1 alpha (PGC1α) expression, a regulator of mitochondrial biogenesis and oxidative stress, by directly binding to the PGC1α promoters in the liver [39] and cardiomyocyte [40]. While, absence of TFEB could affect mitochondrial complex II activity, increased oxidative stress, and decreased ATP production in skeletal muscle [41]. These results reveal an integrated view of how lysosomal signaling affects mitochondria. Interestingly, other recent studies suggested that the reciprocal positive feedforward TFEB-PGC1α signaling pathway plays a crucial role in the clearance of damaged mitochondria and mitochondrial biogenesis in skeletal muscle [42], and in mice with a Q311X Parikin mutation [43], as well as Huntington’s disease mouse model [44, 45]. Besides, the TFEB-dependent lysosomal alterations were also detected in others mitochondrial dysfunction, including the deletion of TFAM (loss of mtDNA), and PINK1 (mitophagy), and STUB1 (reduce mitochondrial protein degradation) and GCN5L1 (mitophagy), and inhibition of mitochondrial fission protein Drp1 (mitophagy), as well as mitochondrial complex I inhibition in various cell types and organs (T lymphocytes [46], neurons [38, 47, 48], heart [49], and mouse embryonic fibroblasts [50, 51]). All those experimental findings support that the biogenesis of mitochondria and lysosomes is regulated by the same MiT/TFE transcriptional factor. Similarly, TFE3 was also observed directly in regulation PGC1α expression in mitochondrial biogenesis. TFE3 ectopic expression induces PGC1α, while silencing TFE3 suppresses PGC1α expression in myotubes [52] and in liver [53]. In line with TFEB, MITF also regulates the expression of PGC1α in retinal pigment epithelium [54] and melanomas [55,56,57], further confirming the role of those transcription factors in energy metabolism (Table 1).
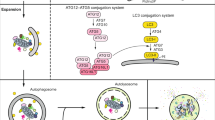
These finding suggest that lysosomal biogenesis and function are coordinated by the transcriptional regulation of MiT/TFE family members (Fig. 1). And by modulating transcription factor activities, the cells can monitor lysosomal function and adapt to environmental signals.

The MiT/TFE family of transcription factors regulates cellular clearance. The members of the MiT/TFE family participate in the regulation of cellular clearance via autophagy and lysosome biogenesis
MiT/TFE family transcription factors participate in the regulation of autophagosome biogenesis
Autophagy is a critical catabolic pathway that is essential for the maintenance of cellular homeostasis via enabling the degradation of cellular components and recycling of important molecules under nutrient deprivation conditions [58, 59]. Autophagy is a complicated process that requires numerous autophagy-related genes (ATGs) to act at multiple stages of autophagy, including initiation, nucleation, and elongation steps [60]. Under normal conditions, the levels of ATG proteins are typically high, but some key autophagy regulators could be depleted under certain pathological conditions. For this reason, increasing the transcription of autophagy genes plays a pivotal role in cells under certain conditions, such as nutrient scarcity [9].
Subsequent studies have shown that TFEB is also a key transcriptional regulator of autophagy biogenesis, in addition to its crucial role in lysosomal function [32, 61]. In particular, TFEB directly binds to the promoter regions of numerous autophagy-related genes and promotes their expression [32]. These autophagy-related genes include LC3B, P62, VPS11, VPS18, UVRAG, WIPI and ATG9B. The overexpression of TFEB increases the number of autophagosomes in numerous cells, while the suppression of TFEB expression reduces autophagosome formation. [9]. At the same time, similar results are also obtained in the liver of GFP-LC3 transgenic mice, where activation of autophagy is observed following TFEB overexpression [32]. Therefore, by regulating lysosomal and autophagy biogenesis, TFEB coordinates a transcriptional program to control the main cellular degradative pathways and promote intracellular clearance. Importantly, TFEB does not regulate the basal transcription of its targets but rather enhances their transcriptional levels in response to environmental cues [1] and additional regulators of autophagic-lysosomal function have also been identified with the identification of TFEB [62].
Similar to TFEB, the overexpression of TFE3 in ARPE-19 cells promotes the transcriptional regulation of several critical autophagy-related genes, such as WIPI and ATG16L, suggesting a key role for TFE3 in the regulation of cellular processes [2]. Previous studies have shown that autophagy-related genes are not significantly correlated with MITF expression, while microarray analysis revealed that although many other autophagosomal genes were negatively associated with MITF, some autophagosomal genes were positively correlated with MITF, including UVRAG, VPS11, ATG3, SNCA, and AMBRA1 [32]. In GSEA analyses, many autophagy-related genes did not show a significant relationship with MITF, whereas lysosomal genes were highly correlated with this protein [36]. Furthermore, the overexpression of MITF-A, but not lysosomal genes, in RPE cells enhanced the activation of the autophagic process, adding an additional layer of mystery concerning the role of MITF in autophagy.
Taken together, these results suggested that MiT/TFE transcription factors play a pivotal role in the regulation of the autophagic process (Fig. 1), but the contribution of each member or certain MITF isoforms to autophagy biogenesis may depend on nutritional conditions, cell types and signaling inputs [14].
Signaling involved in regulation of MiT/TFE family
Regulation of MiT/TFE family member activities in mTORC1 signaling
The activity of TFEB is primarily modulated by post-translational modifications, protein-protein interactions and spatial organization [1]. Under normal conditions, TFEB is largely in an inactive state and remains in the cytoplasm [32, 63]. Under starvation or other extreme conditions, nuclear translocation of TFEB occurs rapidly with the transcriptional activation of its target genes.
The localization and activity of TFEB is mainly manipulated by phosphorylation. Two serine residues, that is, Ser142 [31] and Ser211 [31, 64], determine the subcellular localization of the TFEB protein. When both residues are phosphorylated, TFEB remains inactive in the cytosol, while variants of TFEB carrying Ser-to-Ala mutations of either Ser142 or Ser211 consistently show nuclear translocation and constitutive activation [64, 65]. Notably, the phosphorylation of Ser211 serves as a docking site for the chaperone 14–3-3 protein, which retains TFEB in the cytosol and prevents its nuclear translocation, likely by masking its nuclear localization signal [1, 65].
Previous studies have demonstrated that the Mechanistic target of rapamycin complex 1 (mTORC1) is the main protein kinase regulating TFEB phosphorylation in most cell types [65, 66]. Remarkably, mTORC1 activation occurs at the lysosomal membrane. Under nutrition-full conditions, a mechanism involving the v-ATPase complex promotes the activation of the small Rag (Rag-related GTP-binding) GTPases, which recruit mTORC1 to the lysosomal membrane, thus promoting its activation through the small GTPase Rheb and inhibiting TFEB nuclear translocation [67, 68]. Intriguingly, active Rag GTPase can also bind TFEB and recruit it to the lysosomal membrane, thus enhancing TFEB phosphorylation by mTORC1 [37]. Upon starvation or lysosomal stress, mTORC1 is released from the lysosomal membrane and becomes inactive, and unphosphorylated TFEB accumulates in the nucleus, where it binds to CLEAR sequences and promotes subsequent gene expression [69] (Fig. 2). Interestingly, nutrient deprivation induces the release of lysosomal Ca2+ through Ca2+ channel mucolipin 1 (MCOLNI), thus activating the phosphatase calcineurin, which in turn dephosphorylates and promotes the nuclear translocation of TFEB [70]. The suppression of MCOLN1 expression inhibits lysosomal Ca2+ release and calcineurin activation, thus preventing TFEB activation and autophagy induction under nutrient insufficient conditions [70]. These results suggested that mTORC1 signaling was involved in the regulation of TFEB phosphorylation at the lysosomal membrane. However, it is unclear whether substrate phosphorylation by mTORC1 also occurs in lysosomes or whether active mTORC1 is released into the cytosol to exert its kinase activity [1].

The mTORC1 signaling pathway is involved in the autophagy-lysosomal pathway via MiT/TFE transcriptional factors. Under normal nutrition conditions, the MiT/TFE members were phosphorylated by mTORC1, which were sequestered in the cytoplasm by 14–3-3 proteins. Upon starvation, mTORC1 was inactivated, leading to the dephosphorylation of MiT/TFE transcription factors, which resulted in the dissociation of the binding with 14–3-3 proteins that subsequently freely translocate to the nucleus where their transcriptional activation occurs in the autophagy-lysosome pathway [69]
A recent study also revealed that Ser122, another critical residue of TFEB, is important for TFEB subcellular localization by mTORC1 regulation [66]. These authors reported that Ser122 of TFEB was phosphorylated in an mTORC1-dependent manner. Specifically, TFEB nuclear localization following mTORC1 inhibition is blocked by Ser122 mutation of TFEB. And this mutation of TFEB also inhibits lysosomal biogenesis induced by Torin1, an inhibitor of mTORC1 [66]. These data reveal a novel mechanism of TFEB regulation by mTORC1, which is essential for lysosomal biogenesis.
Similar to TFEB, the activity of other MiT/TFE members can also be controlled by mTORC1 signaling. In the case of TFE3, its activity is also dictated by its nuclear localization, which is regulated by mTORC1 and nutrient levels. When nutrition is sufficient, the mTORC1 is recruited to the lysosomal membrane and become active, leading to phosphorylation of TFE3 at critical serine residues, such as Ser321, which cause TFE3 cytosolic retention; under nutrition-deprived conditions, TFE3 nuclear translocation occurs and activates the transcription of genes containing CLEAR promoter elements with mTORC1 signaling inhibition [2]. Additionally, a similar activation mechanism was also implicated in regulation of the localization of several MITF isoforms [37].
Taken together, these data demonstrated that the mTORC1 activation controls the activity and function of MiT/TFE transcription factors.
Role of PKC in the regulation of MiT/TFE family members
Protein kinase C (PKC) signaling has been correlated with MITF in human melanogenesis [71, 72]. In osteoclasts, TFEB is directly phosphorylated at three serine residues located in its C-terminal region by PKCβ upon stimulation with receptor activator of nuclear factor κB ligand (RANKL) to promote lysosomal biogenesis and maintain its stability [73]. Additionally, recent evidence suggests that another PKC isoform can control lysosomal biogenesis without compromising mTORC1. These authors reported that PKC can couple the activation of the TFEB transcription factor, but not TFE3, with the inactivation of ZKSCAN3, a zinc-finger transcriptional factor, through two parallel signaling cascades. In the TFEB activation pathway, activated PKCδ phosphorylates and inactivates GSK3β, leading to the reduced phosphorylation of TFEB at Ser134 and Ser138 residues; this reduced phosphorylation is critical for the cytoplasmic sequestration of TFEB. Additionally, PKC further activates JNK and p38 MAPK, which in turn phosphorylate ZKSCAN3, leading to ZKSCAN3 translocation to the cytoplasm, which consequently alleviates the transcriptional repression of TFEB, with reduced the accumulation of polyQ aggregates and lipid droplets in HeLa cells. Moreover, injection of PKC inhibitor significantly increases Aβplaques in APP/PS1 mice, an animal model of Alzheimer’s disease, which further identify the evidence that PKC signaling is involved in regulation TFEB, thereby facilitating lysosomal clearance and mediating cellular adaptation to many extracellular cues [74, 75] (Fig. 3). Altogether, these data suggested that PKC is a master switch that controls two protein phosphorylation cascades to activate TFEB-mediated lysosomal gene expression without compromising mTORC1.

PKC triggered the transcriptional regulation of the TFEB-dependent autophagy-lysosome pathway. Under normal conditions, the PKC isoforms PKCα and PKCδ are inactivated. During hormone, neurotransmitter and bacterial lipopolysaccharide (LPS) stimulation, PKCα and PKCδ are activated, leading to their phosphorylation and inhibition of GSK3β, inhibiting the phosphorylation of TFEB serine sites needed for binding to cytoplasmic 14–3-3 proteins. TFEB nuclear translocation occurred and activated the expression of autophagy-lysosomal related genes. PKCδ activates JNK and P38, leading to the export of the repressor ZKSCAN3 from the nucleus to the cytoplasm,consequently alleviating transcriptional repression [74, 75]
AKT/PKB regulates the autophagy-lysosome pathway involved in MiT/TFE transcriptional factors
Upon nutrition sufficiency, the nuclear translocation and activity of TFEB is inhibited by mTORC1 [64]. The mTORC1 is mainly controlled by several signal transduction pathways, such as Wnt and phosphatidylinositol 3(PI3K)-serine/threonine kinase AKT (also known as protein kinase B, PKB) signaling. The mTORC1 is activated downstream of AKT and PI3K kinases, and growth factor receptor signaling and inhibits autophagy under those growth promoting conditions [76, 77]. The canonical PI3K-AKT-mTORC1 pathway has been reported in several studies on TFEB and TFE3 activation [78,79,80]. However, mTORC1-independent TFEB activation via AKT inhibition, which promotes lysosome-mediated cellular clearance, has also been reported in recent years. Palmieri M et al. [81, 82] reported that TFEB activity is modulated by AKT phosphorylation at Ser467, and both AKT knockdown and the pharmacological inhibition of AKT promoted the nuclear translocation and stability of TFEB, as well as increased its ability to activate downstream target genes, thus promoting cellular clearance in a variety of models of genetic diseases, including patient-derived primary fibroblasts defective for PPT1, and TPP1M, and MFSD8, presenting with an impairment of lysosomal pathways (Fig. 4). In addition, a similar consequence was also observed for the TFEB paralogs, MITF and TFE3. Pharmacological inhibition of AKT promoted the nuclear translocation of these two transcriptional factors, suggesting the potential conservation of this regulatory pathway [81, 82]. Taken together, these findings provide novel perspectives for directly regulating cellular clearance by MiT/TFE transcriptional factors via AKT, without mTORC1 participation.

Signaling mechanism that regulates the TFEB nuclear translocation and activation involved AKT/PKB. Under normal conditions, AKT/PKB is activated, leading to phosphorylation, decreased nuclear translocation and TFEB activation. The inhibition of AKT activity with pharmacological inhibitors promotes TFEB nuclear translocation, which enhanced the transcriptional regulation of the autophagy-lysosome pathway and increased cellular clearance [81, 82]
The MiT/TFE family in cancer
The tight connection of TFEB and TFE3 with clear cell renal cell carcinoma (RCC) has been reported [83, 84]. MiT/TFE family translocation renal cell carcinoma (tRCC) comprises Xp11 tRCC and t(6:11)RCC, which were characterized by the rearrangement of the MiT transcription factors TFE3 and TFEB, respectively [85]. The fusion partner of TFEB in t(6:11) RCC is MALAT1 (also known as Alpha) [86, 87]. MALAT1 is a well-known long non-coding RNA (lncRNA) that fuses to TFEB upstream of the translation initiation codon ATG in exon 3, and this fusion promotes the translation of the encoded full-length TFEB protein [88]. In the case of TFE3, several RCC-associated fusion events have also been reported to occur between the TFE3 genes and various other genes, such as CLTC [89] and ASPL [90]. Intriguingly, the TFE3-ASPL fusion has also been reported in alveolar soft part sarcoma [90]. Moreover, multiple lines of evidence have shown that MITF plays an important role in melanoma biogenesis [15, 36]. Taken together, these results revealed that the upregulation of the MiT/TFE transcriptional network can drive tumorigenesis in many tissues.
Growing evidence has established the acceleration of autophagic and/or lysosomal dysfunction, with protein aggregates accumulation, as the major pathogenesis underlying these diseases [91, 92]. In recent years, researchers have paid more attention to whether the MiT/TFE family-mediated autophagic or lysosomal dysfunction also plays a critical role in cancers [93]. In human pancreatic ductal adenocarcinoma (PDA) cells, studies have revealed that autophagy induction occurs in PDA, relying on autophagy-lysosome function regulated by the MiT/TFE proteins, MITF, TFE3, and TFEB. Escape of those factors from inhibition by mTOR which control their cytoplasmic retention enables nuclear import increase, in turn, drives the expression of a coherent network of genes that facilitate the survival of PDA in response to nutrient stress [94]. Besides, to further extend this findings in primary patient-derived samples, Perera RM et al. also examined a series of early passage PDA cultures. Obviously, these cells have showed high level of autophagy, nuclear localization of MiT/TFE proteins as well as its mediated autophagy-lysosome gene expression. Furthermore, knockdown of TFE3 and MITF can abolish xenograft tumor growth of PDA cells, while MITF overexpression in Kras G12D mouse PanIN cells promoted tumorigenesis upon orthotopic injection [94]. Thus, those results suggested that MiT/TFE proteins are potent drivers of PDA pathogenesis with its requirement for increasing autophagy-lysosome function.
Similarly, up-regulation of MiT/TFE genes in cells and tissues from patients and mouse models of renal cell carcinoma, and melanoma enables cellular adaptation to nutrient availability and support the cellular metabolism of cancer cells, resulting in cell hyperproliferation and cancer growth [58]. Previous studies have revealed that mTORC1 can negatively regulate the activity of those MiT/TFE transcription factors, leading to their cytoplasmic retention. Here, Malta CD and his colleagues [58] reported that the MiT/TFE transcription factors can also in turn influence mTORC1 activity via RagD GTPase. They observed that hyperactivation and recruitment to lysosome of mTORC1 signaling and increased RagD levels in MiT/TFE overexpression cell and mouse model, whereas an opposite effect was also observed by MiT/TFE depletion, which was rescued by RagD overexpression. Notably, silencing of either MiT/TFE or RagD reduced cell proliferation in MiT/TFE overexpression tumor both in vivo and in vitro, and similar result was also obtained by mTORC1 inhibitor treatment. All those results suggested an MiT/TFE-RagD-mTORC1-MiT/TFE feedback circuit is critical for promoting tumor growth.
In parallel, the overexpression of TFEB was significantly correlated with poor prognosis in non-small cell lung cancer (NSCLC), Silencing of TFEB with siRNA could reduce the migration, but not proliferation, of NSCLC cells [95]. Furthermore, the enhancement of the autophagy flux by TFEB could rescue MITA (mediator of IRF3 activation)-induced cell death in breast cancer cells [96]. Taken together, these studies have demonstrated the MiT/TFE-controlled autophagy-lysosome function for the maintenance of intracellular homeostasis is critical for cancer.
Conclusion
The identification of MiT/TFE transcriptional factors as master regulators of intracellular clearance and energy metabolism by orchestrating the genes involved in lysosomal-autophagic pathways has emphasized a mechanism by which the cell responds to environmental cues, such as nutrient deprivation. However, the mechanisms involved in the integration of multiple growth-stimulating and inhibitory signals to modulate an appropriate response and regulate a wide array of key cellular functions requires further investigation. Moreover, it remains unclear that how different signals can activate particular MiT/TFE members and promote responses to different environmental stimuli.
So far, we recognize that the TFEB and other MiT/TFE members, associated with autophagic or lysosomal dysfunction and the accumulation of toxic aggregates, present more and more close connection with cancers. This regulation mechanism enables cancer cells to maintain robust activation of anabolic pathways, thus promote cellular adaptation to stress, evidenced by the increase activation of MiT/TFE members in several cancers. However, the detail mechanism of MiT/TFE activation in cancer is still unclear and need further investigation. Therefore, understanding the specific functions of MiT/TFE transcriptional factors and the mechanism of their activation will certainly raise our cognizance of how cancer cell response so that they can survive under stress condition. And modulation of the activity of MiT/TFE transcriptional factors targeting aberrant autophagic and lysosomal functions may have a potential therapeutic benefit in cancer.
Abbreviations
- AKT:
-
Serine/threonine kinase
- ATG:
-
Autophagy-related genes
- CLEAR:
-
Coordinated lysosomal expression and regulation
- GSK3β:
-
Glycogen synthase kinase 3 beta
- HLH:
-
Helix-loop-helix
- IRF3:
-
Interferon regulating factor 3
- JNK:
-
c-Jun N-terminal kinase
- LDSs:
-
Lysosomal storage disorders
- MAPK:
-
Mitogen activated kinase-like protein
- MCOLNI:
-
Ca2+ channel mucolipin 1
- MiT/TFE:
-
the Microphthalmia family of transcription factors
- MITF:
-
Melanogenesis associated transcription factor
- mTORC1:
-
the Mechanistic target of rapamycin complex 1
- NSCLC:
-
Non-small cell lung cancer
- PDA:
-
Pancreatic ductal adenocarcinoma
- PKC:
-
Protein kinase C
- RANKL:
-
Nuclear factor κB ligand
- RPE:
-
Retinal pigment epithelium
- TFE3:
-
Transcription factor E3
- TFEB:
-
Transcription factor EB
- TFEC:
-
Transcription factor EC
- Zip:
-
Leucine-zipper
- ZKSCAN3:
-
Zinc finger with KRAB and SCAN domains 3
References
Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129(13):2475–81.
Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014;7(309):ra9.
Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet. 2014;38:365–411.
Zhao GQ, Zhao Q, Zhou X, Mattei MG, de Crombrugghe B. TFEC, a basic helix-loop-helix protein, forms heterodimers with TFE3 and inhibits TFE3-dependent transcription activation. Mol Cell Biol. 1993;13(8):4505–12.
Pogenberg V, Ogmundsdottir MH, Bergsteinsdottir K, Schepsky A, Phung B, Deineko V, Milewski M, Steingrimsson E, Wilmanns M. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012;26(23):2647–58.
Hemesath TJ, Steingrimsson E, McGill G, Hansen MJ, Vaught J, Hodgkinson CA, Arnheiter H, Copeland NG, Jenkins NA, Fisher DE. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8(22):2770–80.
Aksan I, Goding CR. Targeting the microphthalmia basic helix-loop-helix-leucine zipper transcription factor to a subset of E-box elements in vitro and in vivo. Mol Cell Biol. 1998;18(12):6930–8.
Bouche V, Espinosa AP, Leone L, Sardiello M, Ballabio A, Botas J. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy. 2016;12(3):484–98.
Martina JA, Diab HI, Li H, Puertollano R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell Mol Life Sci. 2014;71(13):2483–97.
Jebbink JM, Boot RG, Keijser R, Moerland PD, Aten J, Veenboer GJ, van Wely M, Buimer M, Ver Loren van Themaat E, Aerts JM, van der Post JA, Afink GB, Ris-Stalpers C. Increased glucocerebrosidase expression and activity in preeclamptic placenta. Placenta. 2015;36(2):160–9.
Bharti K, Liu W, Csermely T, Bertuzzi S, Arnheiter H. Alternative promoter use in eye development: the complex role and regulation of the transcription factor MITF. Development. 2008;135(6):1169–78.
Takeda K, Yasumoto K, Kawaguchi N, Udono T, Watanabe K, Saito H, Takahashi K, Noda M, Shibahara S. Mitf-D, a newly identified isoform, expressed in the retinal pigment epithelium and monocyte-lineage cells affected by Mitf mutations. Biochim Biophys Acta. 2002;1574(1):15–23.
Kuiper RP, Schepens M, Thijssen J, Schoenmakers EF, van Kessel AG. Regulation of the MiTF/TFE bHLH-LZ transcription factors through restricted spatial expression and alternative splicing of functional domains. Nucleic Acids Res. 2004;32(8):2315–22.
Ploper D, De Robertis EM. The MITF family of transcription factors: role in endolysosomal biogenesis, Wnt signaling, and oncogenesis. Pharmacol Res. 2015;99:36–43.
Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12(9):406–14.
Steingrimsson E, Tessarollo L, Pathak B, Hou L, Arnheiter H, Copeland NG, Jenkins NA. Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc Natl Acad Sci U S A. 2002;99(7):4477–82.
Kitamura Y, Oboki K, Ito A. Molecular mechanisms of mast cell development. Immunol Allergy Clin N Am. 2006;26(3):387–405.
Yagil Z, Hadad Erlich T, Ofir-Birin Y, Tshori S, Kay G, Yekhtin Z, Fisher DE, Cheng C, Wong WS, Hartmann K, Razin E, Nechushtan H. Transcription factor E3, a major regulator of mast cell-mediated allergic response. J Allergy Clin Immunol. 2012;129(5):1357–1366.e5.
Nakagawa Y, Shimano H, Yoshikawa T, Ide T, Tamura M, Furusawa M, Yamamoto T, Inoue N, Matsuzaka T, Takahashi A, Hasty AH, Suzuki H, Sone H, Toyoshima H, Yahagi N, Yamada N. TFE3 transcriptionally activates hepatic IRS-2, participates in insulin signaling and ameliorates diabetes. Nat Med. 2006;12(1):107–13.
Huan C, Kelly ML, Steele R, Shapira I, Gottesman SR, Roman CA. Transcription factors TFE3 and TFEB are critical for CD40 ligand expression and thymus-dependent humoral immunity. Nat Immunol. 2006;7(10):1082–91.
Tsicopoulos A, Joseph M. The role of CD23 in allergic disease. Clin Exp Allergy. 2000;30(5):602–5.
Steingrimsson E, Tessarollo L, Reid SW, Jenkins NA, Copeland NG. The bHLH-zip transcription factor Tfeb is essential for placental vascularization. Development. 1998;125(23):4607–16.
Cardili L, Wrublevsky Pereira G, Viana CR. A rare case of TFE-related pigmented renal tumor with overlapping features between melanotic Xp11 translocation renal cancer and Xp11 renal cell carcinoma with melanotic features. Pathol Int. 2017;67(4):208–13.
Tanaka M, Homme M, Yamazaki Y, Shimizu R, Takazawa Y, Nakamura T. Modeling alveolar soft part sarcoma unveils novel mechanisms of metastasis. Cancer Res. 2017;77(4):897–907.
Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, Golub TR, Rimm DL, Meyerson ML, Fisher DE, Sellers WR. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–22.
Ballabio A. The awesome lysosome. EMBO Mol Med. 2016 Feb 1;8(2):73–6.
Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control Centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–96.
Kondratskyi A, Kondratska K, Skryma R, Klionsky DJ, Prevarskaya N. Ion channels in the regulation of autophagy. Autophagy. 2018;14(1):3–21.
Helip-Wooley A, Thoene JG. Sucrose-induced vacuolation results in increased expression of cholesterol biosynthesis and lysosomal genes. Exp Cell Res. 2004;292(1):89–100.
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–7.
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–108.
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–33.
Li M, Pi H, Yang Z, Reiter RJ, Xu S, Chen X, Chen C, Zhang L, Yang M, Li Y, Guo P, Li G, Tu M, Tian L, Xie J, He M, Lu Y, Zhong M, Zhang Y, Yu Z, Zhou Z. Melatonin antagonizes cadmium-induced neurotoxicity by activating the transcription factor EB-dependent autophagy-lysosome machinery in mouse neuroblastoma cells. J Pineal Res. 2016;61(3):353–69.
Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, Palmieri M, Polishchuk R, Puertollano R, Ballabio A. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21(3):421–30.
Cheli Y, Ohanna M, Ballotti R, Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2011;23(1):27–40.
Ploper D, Taelman VF, Robert L, Perez BS, Titz B, Chen HW, Graeber TG, von Euw E, Ribas A, De Robertis EM. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc Natl Acad Sci U S A. 2015;112(5):E420–9.
Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013;200(4):475–91.
Nezich CL, Wang C, Fogel AI, Youle RJ. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol. 2015;210(3):435–50.
Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15(6):647–58.
Ma X, Liu H, Murphy JT, Foyil SR, Godar RJ, Abuirqeba H, Weinheimer CJ, Barger PM, Diwan A. Regulation of the transcription factor EB-PGC1alpha axis by beclin-1 controls mitochondrial quality and cardiomyocyte death under stress. Mol Cell Biol. 2015;35(6):956–76.
Mansueto G, Armani A, Viscomi C, D'Orsi L, De Cegli R, Polishchuk EV, Lamperti C, Di Meo I, Romanello V, Marchet S, Saha PK, Zong H, Blaauw B, Solagna F, Tezze C, Grumati P, Bonaldo P, Pessin JE, Zeviani M, Sandri M, Ballabio A. Transcription factor EB controls metabolic flexibility during exercise. Cell Metab. 2017;25(1):182–96.
Vainshtein A, Desjardins EM, Armani A, Sandri M, Hood DA. PGC-1alpha modulates denervation-induced mitophagy in skeletal muscle. Skelet Muscle. 2015;5:9.
Siddiqui A, Bhaumik D, Chinta SJ, Rane A, Rajagopalan S, Lieu CA, Lithgow GJ, Andersen JK. Mitochondrial quality control via the PGC1alpha-TFEB signaling pathway is compromised by Parkin Q311X mutation but independently restored by rapamycin. J Neurosci. 2015;35(37):12833–44.
Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4(142):142ra197.
La Spada AR. PPARGC1A/PGC-1alpha, TFEB and enhanced proteostasis in Huntington disease: defining regulatory linkages between energy production and protein-organelle quality control. Autophagy. 2012;8(12):1845–7.
Baixauli F, Acin-Perez R, Villarroya-Beltri C, Mazzeo C, Nunez-Andrade N, Gabande-Rodriguez E, Ledesma MD, Blazquez A, Martin MA, Falcon-Perez JM, Redondo JM, Enriquez JA, Mittelbrunn M. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015;22(3):485–98.
Ivankovic D, Chau KY, Schapira AH, Gegg ME. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem. 2016;136(2):388–402.
Rao L, Sha Y, Eissa NT. The E3 ubiquitin ligase STUB1 regulates autophagy and mitochondrial biogenesis by modulating TFEB activity. Mol Cell Oncol. 2017;4(6):e1372867.
Chen Z, Li Y, Wang Y, Qian J, Ma H, Wang X, Jiang G, Liu M, An Y, Ma L, Kang L, Jia J, Yang C, Zhang G, Chen Y, Gao W, Fu M, Huang Z, Tang H, Zhu Y, Ge J, Gong H, Zou Y. Cardiomyocyte-restricted low density lipoprotein receptor-related protein 6 (LRP6) deletion leads to lethal dilated cardiomyopathy partly through Drp1 signaling. Theranostics. 2018;8(3):627–43.
Fernandez-Mosquera L, Diogo CV, Yambire KF, Santos GL, Luna Sanchez M, Benit P, Rustin P, Lopez LC, Milosevic I, Raimundo N. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci Rep. 2017;7:45076.
Scott I, Webster BR, Chan CK, Okonkwo JU, Han K, Sack MN. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J Biol Chem. 2014;289(5):2864–72.
Salma N, Song JS, Arany Z, Fisher DE. Transcription factor Tfe3 directly regulates Pgc-1alpha in muscle. J Cell Physiol. 2015;230(10):2330–6.
Xiong J, Wang K, He J, Zhang G, Zhang D, Chen F. TFE3 alleviates hepatic steatosis through autophagy-induced Lipophagy and PGC1alpha-mediated fatty acid beta-oxidation. Int J Mol Sci. 2016;17(3):387.
Hua J, Chen H, Chen Y, Zheng G, Li F, Qu J, Ma X, Hou L. MITF acts as an anti-oxidant transcription factor to regulate mitochondrial biogenesis and redox signaling in retinal pigment epithelial cells. Exp Eye Res. 2018;170:138–47.
Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23(3):302–15.
Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23(3):287–301.
Abbate F, Badal B, Mendelson K, Aydin IT, Serasinghe MN, Iqbal R, Mohammed J, Solovyov A, Greenbaum BD, Chipuk JE, Celebi JT. FBXW7 regulates a mitochondrial transcription program by modulating MITF. Pigment Cell Melanoma Res. 2018; https://doi.org/10.1111/pcmr.12704. [Epub ahead of print]
Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, Eastes AN, Davis O, De Cegli R, Zampelli A, Di Giovannantonio LG, Nusco E, Platt N, Guida A, Ogmundsdottir MH, Lanfrancone L, Perera RM, Zoncu R, Pelicci PG, Settembre C, Ballabio A. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356(6343):1188–92.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–42.
Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12(2):245–60.
Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20(19):3852–66.
Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25(6):354–63.
Sakamaki JI, Long JS, New M, Van Acker T, Tooze SA, Ryan KM. Emerging roles of transcriptional programs in autophagy regulation. Transcription. 2018;9(2):131–6.
Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5(228):ra42.
Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–14.
Vega-Rubin-de-Celis S, Pena-Llopis S, Konda M, Brugarolas J. Multistep regulation of TFEB by MTORC1. Autophagy. 2017;13(3):464–72.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303.
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334(6056):678–83.
Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, Delling M, Marugan J, Ferrer M, Xu H. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109.
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17(3):288–99.
Liu LH, Fan X, Xia ZK, An XX, Yang RY. Angiotensin II stimulates melanogenesis via the protein kinase C pathway. Exp Ther Med. 2015;10(4):1528–32.
Kim DS, Cha SB, Park MC, Park SA, Kim HS, Woo WH, Mun YJ. Scopoletin stimulates melanogenesis via cAMP/PKA pathway and partially p38 activation. Biol Pharm Bull. 2017;40(12):2068–74.
Ferron M, Settembre C, Shimazu J, Lacombe J, Kato S, Rawlings DJ, Ballabio A, Karsenty G. A RANKL-PKCbeta-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013;27(8):955–69.
Li Y, Xu M, Ding X, Yan C, Song Z, Chen L, Huang X, Wang X, Jian Y, Tang G, Tang C, Di Y, Mu S, Liu X, Liu K, Li T, Wang Y, Miao L, Guo W, Hao X, Yang C. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol. 2016;18(10):1065–77.
Saftig P, Haas A. Turn up the lysosome. Nat Cell Biol. 2016;18(10):1025–7.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017 Apr 20;169(3):381–405.
Lina TT, Luo T, Velayutham TS, Das S, McBride JW. Ehrlichia activation of Wnt-PI3K-mTOR signaling inhibits autolysosome generation and autophagic destruction by the mononuclear phagocyte. Infect Immun. 2017;85(12):e00690–17.
Fang Y, Bao W, Rao Q, Wang X, Xia Q, Shen Q, Zhou X, Yao B. TFE3 regulates renal adenocarcinoma cell proliferation via activation of the mTOR pathway. Mol Med Rep. 2017;16(3):2721–5.
Zhang J, Wang J, Xu J, Lu Y, Jiang J, Wang L, Shen HM, Xia D. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget. 2016;7(46):75659–71.
Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, Chaudhury A, Bajaj L, Bondar VV, Bremner L, Saleem U, Tse DY, Sanagasetti D, Wu SM, Neilson JR, Pereira FA, Pautler RG, Rodney GG, Cooper JD, Sardiello M. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun. 2017;8:14338.
Palmieri M, Pal R, Sardiello M. AKT modulates the autophagy-lysosome pathway via TFEB. Cell Cycle. 2017;16(13):1237–8.
Inamura K. Translocation renal cell carcinoma: an update on Clinicopathological and molecular features. Cancers (Basel). 2017;9(9). https://doi.org/10.3390/cancers9090111.
Williamson SR, Eble JN, Palanisamy N. Sclerosing TFEB-rearrangement renal cell carcinoma: a recurring histologic pattern. Hum Pathol. 2017;62:175–9.
Humphrey PA, Moch H, Cubilla AL, Ulbright TM, Reuter VE. The 2016 WHO classification of Tumours of the urinary system and male genital organs-part B: prostate and bladder Tumours. Eur Urol. 2016;70(1):106–19.
Davis IJ, Hsi BL, Arroyo JD, Vargas SO, Yeh YA, Motyckova G, Valencia P, Perez-Atayde AR, Argani P, Ladanyi M, Fletcher JA, Fisher DE. Cloning of an alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci U S A. 2003;100(10):6051–6.
Kuiper RP, Schepens M, Thijssen J, van Asseldonk M, van den Berg E, Bridge J, Schuuring E, Schoenmakers EF, van Kessel AG. Upregulation of the transcription factor TFEB in t(6;11)(p21;q13)-positive renal cell carcinomas due to promoter substitution. Hum Mol Genet. 2003;12(14):1661–9.
Inamura K, Fujiwara M, Togashi Y, Nomura K, Mukai H, Fujii Y, Yamamoto S, Yonese J, Fukui I, Ishikawa Y. Diverse fusion patterns and heterogeneous clinicopathologic features of renal cell carcinoma with t(6;11) translocation. Am J Surg Pathol. 2012;36(1):35–42.
Argani P, Lui MY, Couturier J, Bouvier R, Fournet JC, Ladanyi M. A novel CLTC-TFE3 gene fusion in pediatric renal adenocarcinoma with t(X;17)(p11.2;q23). Oncogene. 2003;22(34):5374–8.
Ladanyi M, Lui MY, Antonescu CR, Krause-Boehm A, Meindl A, Argani P, Healey JH, Ueda T, Yoshikawa H, Meloni-Ehrig A, Sorensen PH, Mertens F, Mandahl N, van den Berghe H, Sciot R, Dal Cin P, Bridge J. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20(1):48–57.
Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16(6):345–57.
Medina DL, Settembre C, Ballabio A. Methods to monitor and manipulate TFEB activity during autophagy. Methods Enzymol. 2017;588:61–78.
Slade L, Pulinilkunnil T. The MiTF/TFE family of transcription factors: master regulators of organelle signaling, metabolism, and stress adaptation. Mol Cancer Res. 2017;15(12):1637–43.
Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, Settleman J, Stephanopoulos G, Dyson NJ, Zoncu R, Ramaswamy S, Haas W, Bardeesy N. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524(7565):361–5.
Giatromanolaki A, Kalamida D, Sivridis E, Karagounis IV, Gatter KC, Harris AL, Koukourakis MI. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer. 2015;90(1):98–105.
Bhatelia K, Singh K, Prajapati P, Sripada L, Roy M, Singh R. MITA modulated autophagy flux promotes cell death in breast cancer cells. Cell Signal. 2017;35:73–83.
Acknowledgments
Work performed in the author’s laboratory on the topic was supported by grant from the Joint NSFC-ISF Research Grant (No.81661148050) and grant from National Natural Science Foundation of China (NO.81602577).
Funding
This work was supported by the Joint NSFC-ISF Research Grant (No.81661148050) and National Natural Science Foundation of China (NO.81602577).
Author’s contributions
MY and EL collected all the data and wrote the manuscript. LT, YL, XS and JH prepared the figure. HD and SMY provided the important guidance for paper writing. And MG and BT reviewed and revised the paper. All authors approved the final version.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, M., Liu, E., Tang, L. et al. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun Signal 16, 31 (2018). https://doi.org/10.1186/s12964-018-0242-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-018-0242-1