
Abstract
Childhood general cognitive ability (g) is important for a wide range of outcomes in later life, from school achievement to occupational success and life expectancy. Large-scale association studies will be essential in the quest to identify variants that make up the substantial genetic component implicated by quantitative genetic studies. We conducted a three-stage genome-wide association study for general cognitive ability using over 350,000 single nucleotide polymorphisms (SNPs) in the quantitative extremes of a population sample of 7,900 7-year-old children from the UK Twins Early Development Study. Using two DNA pooling stages to enrich true positives, each of around 1,000 children selected from the extremes of the distribution, and a third individual genotyping stage of over 3,000 children to test for quantitative associations across the normal range, we aimed to home in on genes of small effect. Genome-wide results suggested that our approach was successful in enriching true associations and 28 SNPs were taken forward to individual genotyping in an unselected population sample. However, although we found an enrichment of low P values and identified nine SNPs nominally associated with g (P < 0.05) that show interesting characteristics for follow-up, further replication will be necessary to meet rigorous standards of association. These replications may take advantage of SNP sets to overcome limitations of statistical power. Despite our large sample size and three-stage design, the genes associated with childhood g remain tantalizingly beyond our current reach, providing further evidence for the small effect sizes of individual loci. Larger samples, denser arrays and multiple replications will be necessary in the hunt for the genetic variants that influence human cognitive ability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To date, most studies addressing the genetics of g have opted to explore genome-wide linkage or candidate gene associations. Although linkage analyses have met with some success (Houlihan et al. 2009; Dick et al. 2006; Luciano et al. 2006; Posthuma et al. 2005), these studies lack power to detect common variation and have difficulty pinpointing the variant of interest (Balding 2006). Similarly, although candidate gene studies provide useful hypothesis-driven exploration of particular genetic variants, they lack the systematic approach of genome-wide association and are likely to miss real associations that do not involve biologically obvious candidate genes. In contrast, genome-wide association studies (GWAS) have the potential to provide high-resolution mapping of common genetic variants associated with g, even where the associations come from unexpected quarters of the genome. For example, the recent spate of publications implicating non-coding RNA in neurodevelopmental pathways (Kosik 2006; Cao et al. 2006; Berezikov et al. 2006) highlights areas of the genome that would until lately have remained unexplored by candidate gene studies searching for genetic influences on human cognition. A genome-wide association study stands a better chance of detecting such effects, opening up new areas of the genome for detailed exploration.
Our recent genome-wide association study identified four SNPs of small effect associated with g at 7 years of age (Butcher et al. 2008). This study concluded, in common with genome-wide association studies for common medical disorders (Wellcome Trust Case Control Consortium 2007), that there are unlikely to be any common SNPs of large effect influencing variation of g in the normal range. Rather, many genes of small effect make up the 60% heritable variation in the trait. However, by selecting 47 SNPs out of half a million for replication we potentially missed the opportunity to replicate real effects that by chance fell lower down the list; replicating a greater number of the top SNP associations from this first stage in a second population would increase our chances of winnowing real associations from the thousands of possibilities.
Our aim in this new study was to greatly expand the scope of our analysis using our sample of over 7,900 twins, replicating the top several thousand associations from an initial GWAS in a second sample before narrowing in to validate and extend the associations across the normal distribution of g scores in the population.
Methods
Participants
The Twins Early Development Study (TEDS) recruited over 11,000 families of twins born in England and Wales in 1994, 1995 and 1996 (Oliver and Plomin 2007). Since then, the sample has remained representative of the UK population (ascertained by comparison with census data from the Office of National Statistics; Kovas et al. 2007). Informed consent is obtained by post or online consent forms, and a test administrator is then assigned who telephones the family to assist or encourage. Ethical approval was provided by the Institute of Psychiatry ethics committee (05/Q0706/228).
We excluded from the analyses children with severe current medical problems and children who had suffered severe problems at birth or whose mothers had suffered severe problems during pregnancy. We also excluded twins whose zygosity was unknown or uncertain or whose first language was other than English. Finally, we included only twins whose parents reported their ethnicity as ‘white’ (93% of this UK sample) to mitigate false positive associations attributable to ethnic stratification. 7,924 Children from 4,039 families assessed at 7 years of age also provided us with DNA samples.
Measures
Verbal and nonverbal tests were administered by telephone (Petrill et al. 2002). Prior to the telephone call, parents were sent a booklet of test items along with instructions. The booklet contained two tests of verbal abilities and two nonverbal tests. The verbal tests consisted of the Similarities subtest and the Vocabulary subtest from the Wechsler Intelligence Scale for Children (WISC-III-UK; Wechsler 1992). The nonverbal tests were the Picture Completion subtest from the WISC-III-UK and Conceptual Grouping from the McCarthy Scales of Children’s Abilities (McCarthy 1972). A g score was derived from the test battery as the standardized sum of the standardized subtest scores. A score derived as the first principal component of the battery correlates .99 with this score (Davis et al. 2008).
Genome-wide association
860 children (only one per family) were selected from the quantitative extremes of the g distribution for the first stage of the GWAS. The individuals selected all scored one standard deviation above or below the mean. Although DNA from only 860 children was pooled, selecting from the extremes of the distribution retains much of the power of the original quantitative distribution (Sham et al. 2002). All genotyping was carried out at the MRC SGDP. Genomic DNA for each individual, extracted from buccal swabs (Freeman et al. 2003) and suspended in Tris-ethylenediaminetetraacetic acid (TE) buffer (0.01 m Tris–HCl, 0.001 m EDTA, pH 8.0), was quantified in triplicate using PicoGreen™ double-stranded DNA quantification reagent (Invitrogen, Carlsbad, CA, USA). Upon obtaining reliable triplicate readings, each individual contributed the same amount of DNA to their respective pool. DNA samples were assigned to one of 10 biological replicate low pools or one of 10 high pools to make 20 independent DNA pools in all (ten high g, ten low g). Each pool was hybridized to an Affymetrix GeneChip Human Mapping 500K array set using the standard protocol (Butcher et al. 2008, 2004; Kirov et al. 2006; Pearson et al. 2007). Power analyses suggest that a pooling study of this kind retains around two-thirds of the power of the equivalent sample individually genotyped (Barratt et al. 2002), which implies 80% power to detect an additive SNP accounting for 1.7% of the variance where the causal allele is typed and alpha is 5 × 10−7. Microarrays were scanned using a GeneChip® Scanner 3000 and Cell intensity (CEL) files were exported for analysis.
Probe intensities were derived from the CEL files, quantile normalized and combined to produce relative allele scores (RAS) using the SNPMaP package (Davis et al. 2009) for the R statistical computing environment (R Development Core Team 2008). X chromosome SNPs and SNPs with minor allele frequencies lower than 5% were removed from the analysis at this stage because of limited statistical power. In addition, SNPs that were excluded from analysis in the recent WTCCC study using the same arrays (WTCCC 2007) were removed. This left 358,948 autosomal SNPs. The RAS scores from each probe quartet for these SNPs were analyzed for association with high/low status using linear mixed effects models implemented in R with the lme4 package. High/low pool status, the variable of interest, was modelled as a fixed effect; array and assay strand were modelled as random effects to account for the additional variation they introduce to the analysis. The 3,000 top-ranked SNPs from the first stage were taken forward to the second stage, where a second sample of 1,000 children (only one per family) was selected from the extremes of the same distribution (500 low and 500 high). The selection of 3,000 SNPs was intended to balance taking forward a larger number of SNPs than in our previous study (Butcher et al. 2008) with requiring a relatively low P value in the first stage; although this cast the net wider than before, this threshold was largely arbitrary, intended only as a screen to enrich true positive associations in the second stage. As before, 20 pools were constructed and hybridized to 20 Affymetrix 500K array sets. The same analysis in R tested for replication of association in the 3,000 SNPs of interest from the first stage.
Quantitative trait association
The 32 top-ranking SNPs from the second stage were selected for validation using individual genotyping in a sample of 3,297 children from the full population (maximum of one per family). Although the current study delayed the arbitrary selection of a small number of top-ranking SNPs by introducing a second pooling stage, the selection of 32 SNPs for individual genotyping was a pragmatic decision based on the capacity of our genotyping platform and the funds available to follow up SNPs. Because g is inherently a quantitative trait, it was important at this stage of the analysis to test whether the SNPs identified in the first two stages using extreme samples would also show association across the full normal range of g. Where possible, assays were designed for the Sequenom MassARRAY iPlex Gold® system and 28 SNPs were genotyped using the standard protocols. The remaining 4 SNPs were genotyped using Applied Biosystems’ TaqMan® assay. 678 individuals overlapped with the pooling stages. Although removing these individuals from the third stage would provide an additional independent replication, these 678 individuals correspond to the extremes of the third sample, so removing them would severely compromise the statistical power of the third stage. On the other hand, the third stage provides additional information in the form of individual genotypes rather than pooled estimates of allele frequency. It also uses quantitative estimates of g rather than a dichotomous trait and includes individuals from the full range of the normal distribution, extending the analysis to accommodate the quantitative trait locus model (Plomin et al. 2008). For these reasons, it is a validation and extension of the analysis for high-ranking SNPs from the first two stages. Individuals calling on fewer than 70% of the SNPs were re-genotyped, as were SNPs with a call rate lower than 95%. Individuals and SNPs with persistently low call rates were removed from the analysis. The 28 remaining SNPs were assessed for Hardy–Weinberg equilibrium and analyzed using linear models in R, fitting an additive model to test for association with g, then testing for evidence of non-additivity by likelihood ratio test comparison of nested models (Balding 2006). The mean sample size of 3,236 gives us 99.5% power to detect a SNP accounting for 1% of the variance at α = 0.001786 (controlling family-wise error rate at 0.05 for 28 tests), or 82% power to detect a SNP accounting for 0.5%.
Results
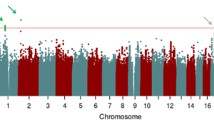
Figure 1 shows genome-wide signal plots for the two pooling replication stages. Figure 2 shows that even though no SNPs stand out in a quantile–quantile plot of approximate P values from stage 1 of the study, selecting the top 3,000 SNPs for the second stage in a new sample results in SNPs enriched for lower P values.

Genome-wide signal plots. Negative log base 10 P values from a mixed-effects model are plotted against genomic position for samples 1 and 2. Only SNPs associated in the same direction in both samples are displayed. The selected SNPs from Table 1 that were successfully genotyped in the full sample are plotted as darker dots. The dotted line represents P = 5 × 10−7. Chromosome-by-chromosome signal plots are presented in supplementary materials

Quantile–quantile plots for samples 1 and 2. In each panel negative log base 10 P values from a mixed-effects model are plotted against theoretical quantiles from the null distribution. The straight line at x = y represents the null distribution and the gray areas represent 95% bootstrapped confidence intervals on the null. The left-hand plot represents genome-wide SNPs passing quality control in sample 1; the right-hand plot represents the top 3,000 SNPs from sample 1 tested in sample 2 (one-tailed). Although the left-hand plot shows no associations greater than chance, the right-hand plot shows that the SNPs tested in the second sample are enriched for associations; this can be seen by the deviation of the SNPs from the x = y line. The best-performing SNPs from this second stage were individually genotyped in a large sample of TEDS individuals across the distribution of g and tested for quantitative association; the results are presented in Table 1
Of the 32 SNPs selected from the second stage, 28 were successfully genotyped in the unselected population sample (Table 1). After quality control, all reported SNPs showed Hardy–Weinberg equilibrium P values > 0.05 and call rates above 95%. Assessed quantitatively across the distribution and with individual genotyping, these SNPs retain the enrichment of P values above that expected by chance. For example, by chance we would expect one or two SNPs to be significant at the nominal P value of 0.05, whereas we find nine (32%), although this enrichment is not independent of the previous stages. Despite enrichment of low P values, none of these SNPs survive correction for multiple testing using either Bonferroni or false discovery rate (Benjamini and Hochberg 1995).
Discussion
Despite our three-stage study demonstrating enrichment of associations for general cognitive ability, the genetic variants that make up the heritable component of intelligence remain elusive. Our findings suggest that the hunt for these genes will require even larger sample sizes and more powerful methodologies.
Although our study attempts as comprehensive a screen as possible of SNPs across the genome and uses large sample sizes, there may be other explanations for why the specific genetic variants remain beyond our grasp. For example, we were unable to assess SNPs on the X chromosome because of relatively low power, and the combination of males and females in the pools. In addition, although over 350,000 SNPs passed our stringent quality control to be included in our analysis, this provides us with less than 60% coverage of the remaining autosomes at r 2 of 0.8 (Pe’er et al. 2006). This coverage will improve as newer arrays provide a greater density of SNPs. In addition, future studies that model interactions among polymorphisms, or between polymorphisms and measured environmental variables, may detect larger effects. Despite these limitations of the current study, assuming the SNPs we typed represent a random sample of autosomal SNPs, it seems unlikely that a few loci of large effect size directly account for the heritable component of g.
Bearing in mind these caveats, the enrichment of low P values in the second (and third) stage suggest that at least some of the SNPs we selected are likely to show true association with g. One tool that may assist in exploring and replicating these weak associations is the SNP set. Creating a SNP set involves coding a group of putatively associated SNPs in the same direction by counting the number of high-g-associated alleles carried by each individual at each locus and summing the score across loci. To demonstrate, we created a SNP set using the 9 SNPs from the third stage of the analysis that were associated with g at the nominal significance level of P < 0.05 (Fig. 3). This SNP set correlates r = 0.11 with g, accounting for 1.2% of the variance. Or, to put it another way, each additional high-g associated allele increases g by 0.06 standard deviations or 0.92 IQ points. However, these calculations involve some members of the discovery population, so effect size estimates are likely to be smaller in an independent sample because of the winner’s curse (Lohmueller et al. 2003). A SNP set score may be used for attempted replication in samples that, because of the expected small effect sizes, do not have sufficient power to replicate individual SNP associations. For example, a sample of over 4,500 would be required to replicate the individual SNPs within the SNP set (accounting for a mean 0.136% of the variance) with 80% power at α = 0.05. However, a sample of just over 500 is required to attempt replication of the SNP set. SNP set scores may also be used for behavioural genomic analyses that explore developmental, multivariate and gene-environment issues (Harlaar et al. 2005; Haworth et al. 2007).

A SNP set for g at 7 years of age. The SNP set is formed from the nominally associated SNPs from Table 1 by counting the number of alleles associated with high g in each individual. Because rs10997145 is significantly non-additive, it was scored 0, 2 and 2 instead. The points represent mean g scores and the line represents the regression of the g score on the SNP set score. The underlying bar chart shows the number of individuals with each SNP set score. The graph runs from 6 to 16 rather than from 0 to 18, because there were no individuals with SNP set scores of 0 to 5 or 17 to 18
Although the slight associations suggested here require replication to meet rigorous standards, it is informative to explore the regions surrounding our selected SNPs. In some cases the nearby gene has previously been implicated in association with linked phenotypes. For example, Fig. 4 shows a signal plot for the region surrounding CTNNA3, the gene linked to rs10997145 (P = 0.011) in Affymetrix’s annotation for the array set. Variants of this gene have previously been reported to be associated with Alzheimer’s disease (Bertram et al. 2007). Although we have studied children who are at the other end of the lifespan and genotyped a different marker, this suggests an interesting pattern for follow-up. Other genes shown in Table 1 such as NR2F1, a nuclear receptor, are highly expressed in the foetal brain and post-natal prefrontal cortex. Intriguingly, MAP3K7 is implicated in cellular response to environmental stress, which may suggest a candidate for gene-environment interplay in relation to g. Finally, although they narrowly miss out on nominal significance in the third sample, the presence of non-coding RNAs in this list derived from the first two stages suggests that the hypothesis-free genome-wide association approach is a valuable complement to candidate gene studies because it includes genomic regions that would previously have been disregarded as “junk DNA”. Plots of the regions surrounding each of the SNPs in Table 1 are presented in supplementary materials.

Signal plot for the region surrounding rs10997145 on Chromosome 10. The top panel shows negative log base 10 P values from a mixed-effects model in Sample 1 plotted against physical position; the darker dot represents the target SNP. The second panel shows the same for Sample 2. The third panel shows genes downloaded from Ensembl (release 49; http://mar2008.archive.ensembl.org/index.html), plus and minus strands. The bottom panel shows recombination rate in cM per Mb (darker line and left axis) and genetic distance from the target SNP in cM (lighter line and right axis), both extracted from HapMap CEPH data. Regional signal plots for all the SNPs in Table 1 are presented in supplementary materials
Two of the SNPs individually genotyped in Stage 3 here overlap with those selected for individual genotyping in Butcher et al. (2008), demonstrating robust associations even with an additional pooling stage; rs959922, re-typed in a sample almost 500 individuals larger than the previous study narrowly misses out on nominal significance at P = 0.091; its proximity to NR2F1 suggests a possible role in the regulation of other genes. rs6794128 was selected for genotyping in Butcher et al. but the assay failed. Using Sequenom technology the SNP was successfully genotyped, with a low but non-significant P value (P = 0.161). Nevertheless, its appearance in both studies and its position in the third intron of PDZRN3, which is highly expressed in the foetal brain (Su et al. 2004), make it an interesting candidate for future study.
Despite its three-stage design incorporating over 350,000 SNPs and its large sample size, our study has not identified strong associations between genetic variants and childhood general cognitive ability. Although there are some interesting regions nominated for future investigation, the small effect sizes mean that they will require replication in multiple other samples before drawing strong conclusions. This study provides further evidence that although we know that intelligence is among the most heritable of traits (Deary et al. 2006), we should not expect to find a few genes of large effect. Instead, we should be seeking and replicating many genes of small effect in our hunt for the specific genetic polymorphisms that account for the heritable component of variation in human cognitive ability.
References
Balding DJ (2006) A tutorial on statistical methods for population association studies. Nat Rev Genet 7(10):781–791
Barratt BJ, Payne F, Rance HE, Nutland S, Todd JA, Clayton DG (2002) Identification of the sources of error in allele frequency estimations from pooled DNA indicates an optimal experimental design. Annu Hum Genet 66:393–405
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, Plasterk RH (2006) Diversity of microRNAs in human and chimpanzee brain. Nat Genet 38:1375–1377
Bertram L, Mullin K, Parkinson M, Hsiao M, Moscarillo TJ, Wagner SL, Becker KD, Velicelebi G, Blacker D, Tanzi RE (2007) Is a-T catenin (VR22) an Alzheimer’s disease risk gene? Br Med J 44:63
Butcher LM, Meaburn E, Liu L, Hill L, Al-Chalabi A, Plomin R, Schalkwyk L, Craig IW (2004) Genotyping pooled DNA on microarrays: a systematic genome screen of thousands of SNPs in large samples to detect QTLs for complex traits. Behav Genet 34:549–555
Butcher LM, Davis OSP, Craig IW, Plomin R (2008) Genome-wide quantitative trait locus association scan of general cognitive ability using pooled DNA and 500K single nucleotide polymorphism microarrays. Genes Brain Behav 7:435–446
Cao X, Yeo G, Muotri AR, Kuwabara T, Gage FH (2006) Noncoding RNAs in the mammalian central nervous system. Annu Rev Neurosci 29:77–103
Crabbe JC (2002) Alcohol and genetics: new models. Am J Med Genet 114(8):969–974
Davis OSP, Arden R, Plomin R (2008) g in middle childhood: moderate genetic and shared environmental influence using diverse measures of general cognitive ability at 7, 9 and 10 years in a large population sample of twins. Intelligence 36:68–80
Davis OSP, Plomin R, Schalkwyk LC (2009) The SNPMaP package for R: a framework for genome-wide association using DNA pooling on microarrays. Bioinformatics 25:281–283
Deary IJ, Spinath FM, Bates TC (2006) Genetics of intelligence. Eur J Hum Genet 14:690–700
Dick DM, Aliev F, Bierut L, Goate A, Rice J, Hinrichs A, Bertelsen S, Wang JC, Dunn G, Kuperman S (2006) Linkage analyses of IQ in the collaborative study on the genetics of alcoholism (COGA) sample. Behav Genet 36:77–86
Freeman B, Smith N, Curtis C, Huckett L, Mill J, Craig I (2003) DNA from buccal swabs recruited by mail: evaluation of storage effects on long-term stability and suitability for multiplex polymerase chain reaction genotyping. Behav Genet 33:67–72
Harlaar N, Butcher L, Meaburn E, Sham P, Craig IW, Plomin R (2005) A behavioural genomic analysis of DNA markers associated with general cognitive ability in 7-year-olds. J Child Psychol Psychiatry 46(10):1097–1107
Haworth CMA, Meaburn EL, Harlaar N, Plomin R (2007) Reading and generalist genes. Mind, Brain, Educ 1(4):173–180
Houlihan LM, Harris SE, Luciano M, Gow AJ, Starr JM, Visscher PM, Deary IJ (2009) Replication study of candidate genes for cognitive abilities: the Lothian Birth Cohort 1936. Genes Brain Behav 8(2):238–247
Kirov G, Nikolov I, Georgieva L, Moskvina V, Owen MJ, O’Donovan MC (2006) Pooled DNA genotyping on Affymetrix SNP genotyping arrays. BMC Genomics 7:27
Kosik KS (2006) The neuronal microRNA system. Nat Rev Neurosci 7:911–920
Kovas Y, Haworth CM, Dale PS, Plomin R (2007) The genetic and environmental origins of learning abilities and disabilities in the early school years. Monogr Soc Res Child Dev 72(3):1–144
Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN (2003) Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 33:177–182
Luciano M, Wright MJ, Duffy DL, Wainwright MA, Zhu G, Evans DM, Geffen GM, Montgomery GW, Martin NG (2006) Genome-wide scan of IQ finds significant linkage to a quantitative trait locus on 2q. Behav Genet 36:45–55
McCarthy D (1972) McCarthy scales of children’s abilities. The Psychological Corporation, New York
McClearn GE (2006) Contextual genetics. Trends Genet 22(6):314–319
McClearn GE, Kakihana R (1973) Selective breeding for ethanol sensitivity in mice. Behav Genet 3:409–410
McClearn GE, Rodgers DA (1959) Differences in alcohol preference among inbred strains of mice. Q J Stud Alcohol 20(4):691–695
McClearn GE, Plomin R, Gora-Maslak G, Crabbe JC (1991) The gene chase in behavioral science. Psychol Sci 2(4):222–229
Oliver BR, Plomin R (2007) Twins early development study (TEDS): a multivariate, longitudinal genetic investigation of language, cognition and behavior problems from childhood through adolescence. Twin Res Hum Genet 10:96–105
Pe’er I, de Bakker PIW, Maller J, Yelensky R, Altshuler D, Daly MJ (2006) Evaluating and improving power in whole-genome association studies using fixed marker sets. Nat Genet 38:663–667
Pearson JV, Huentelman MJ, Halperin RF, Tembe WD, Melquist S, Homer N, Brun M, Szelinger S, Coon KD, Zismann VL et al (2007) Identification of the genetic basis for complex disorders by use of pooling-based genome wide single-nucleotide-polymorphism association studies. Am J Hum Genet 80:126–139
Petrill SA, Rempell J, Oliver B, Plomin R (2002) Testing cognitive abilities by telephone in a sample of 6- to 8-year olds. Intelligence 30:353–360
Plomin R, DeFries JC, McClearn GE, McGuffin P (2008) Behavioral genetics, 5th edn. Worth, New York
Posthuma D, Luciano M, Geus EJ, Wright MJ, Slagboom PE, Montgomery GW, Boomsma DI, Martin NG (2005) A genome wide scan for intelligence identifies quantitative trait loci on 2q and 6p. Am J Hum Genet 77:318–326
R Development Core Team (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Sham PC, Bader JS, Craig I, O’Donovan M, Owen M (2002) DNA pooling: a tool for large-scale association studies. Nat Rev Genet 3:862–871
Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci 101:6062–6067
Wechsler D (1992) Wechsler intelligence scale for children—third edition UK (WISC-IIIUK) Manual. The Psychological Corporation, London
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678
Acknowledgments
Our genome-wide association scans for g were supported by a grant from the Wellcome Trust (GR75492). The Twins Early Development Study (TEDS) has been funded since 1995 by a program grant from the UK Medical Research Council (G9424799, now G0500079). We are grateful to the TEDS families for their participation and support for more than a decade.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by George Vogler.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Davis, O.S.P., Butcher, L.M., Docherty, S.J. et al. A Three-Stage Genome-Wide Association Study of General Cognitive Ability: Hunting the Small Effects. Behav Genet 40, 759–767 (2010). https://doi.org/10.1007/s10519-010-9350-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-010-9350-4