
Abstract
In many areas of Africa, recent studies highlighted the great impact of ticks on animal and human health throughout the continent. On the other hand, very limited information on the bacterial endosymbionts of the African ticks and their pattern of co-infections with other bacteria are found in literature, notwithstanding their pivotal role in tick survival and vector efficiency. Thus, we investigated the distribution of selected pathogenic and symbiotic bacteria in hard ticks collected from wild, domestic animals and from vegetation in various ecological zones in Africa and their co-occurrence in the same tick host. Overall, 339 hard ticks were morphologically identified as belonging to the genera Amblyomma, Dermacentor, Hyalomma, Haemaphysalis, Ixodes and Rhipicephalus. Molecular screening provided information on pathogens circulation in Africa, detecting spotted fever group rickettsiae, Anaplasma spp., Ehrlichia ruminantium, Borrelia garinii, Babesia spp., Theileria spp. and Coxiella burnetii. Furthermore, our work provides insights on the African scenario of tick-symbiont associations, revealing the presence of Coxiella, Francisella and Midichloria across multiple tick populations. Coxiella endosymbionts were the most prevalent microorganisms, and that with the broadest spectrum of hosts, being detected in 16 tick species. Francisella was highly prevalent among the Hyalomma species tested and correlated negatively with the presence of Coxiella, showing a potential competitive interaction. Interestingly, we detected a positive association of Francisella with Rickettsia in specimens of Hy. rufipes, suggesting a synergistic interaction between them. Finally, Midichloria was the most prevalent symbiont in Rhipicephalus sanguineus sensu lato from Egypt.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the last few decades, a growing number of studies have focused on exploring the composition of microbial communities harboured by blood-feeding arthropods, such as ticks (Acari: Ixodidae), reporting a mixture of commensal, mutualistic and pathogenic microorganisms (Andreotti et al. 2011; Narasimhan and Fikrig 2015; Bonnet et al. 2017; Duron et al. 2017). As a result of these efforts, a number of new tick-borne pathogens (TBPs) and new microbial associations have been described (Vayssier-Taussat et al. 2013; Greay et al. 2018). Many of these microorganisms can coexist simultaneously within the same host and synergistic or antagonistic interactions have been hypothesized (Vautrin and Vavre 2009; Moutailler et al. 2016; Díaz-Sánchez et al. 2019) and also proven in specific cases (Paddock et al. 2015; Budachetri et al. 2018).
In many areas of Africa, recent studies highlighted the great impact of ticks on animal and human health throughout the continent (Jongejan and Uilenberg 2004; Maina et al. 2014; Lorusso et al. 2016; Kamani et al. 2018; Asante et al. 2019). The local environmental conditions together with the close contact of wildlife animals with domestic animals and humans provide the opportunities for colonizing multiple niches, driving the spread of TBPs. The most common zoonotic bacteria reported in Africa are the spotted fever group (SFG) rickettsiae, mainly represented by Rickettsia africae, R. aeschlimannii, R. conorii and R. massiliae (Macaluso et al. 2003; Parola et al. 2005). The circulation of pathogens of veterinary importance have also been commonly reported, including Ehrlichia ruminantium, Anaplasma marginale, A. phagocytophilum, and A. centrale, widespread among ruminants (Bekker et al. 2002; Ikwap et al. 2010; Allsopp 2015), and piroplasms (Babesia spp. and Theileria spp.), which infect ruminants and equids (Gebrekidan et al. 2014; Hawkins et al. 2015).
Whereas studies on TBPs in Africa are flourishing, to date there is very limited information regarding the bacterial endosymbionts of the African ticks and their pattern of co-infections with other bacteria. Endosymbionts, intracellular bacteria with high prevalence and load that are generally transovarially transmitted, have been proven to be fundamental in the survival of hematophagous arthropods, ticks included, and thus warrant extensive investigation. The main bacterial endosymbionts of ticks are Coxiella (order Legionellales), Francisella (order Thiotrichales), ‘Candidatus Midichloria’ and Rickettsia (order Rickettsiales) (Duron et al. 2017). The most common tick endosymbiont is Coxiella, detected in most individuals of numerous tick species (Clay et al. 2008; Lalzar et al. 2012; Machado-Ferreira et al. 2016; Duron et al. 2017). Recent studies focused on the intricate interaction of this symbiont in ticks showed that Coxiella endosymbionts possess the typical hallmarks of an obligate symbiont from a physiological point of view. For example, their pronounced tropism to the host ovary is indicative of the predominantly maternal transmission (typical of bacterial intracellular symbionts), and the negative effect on the hosts physiology caused by a reduction of the symbiont load is consistent with a mutualistic role (Zhong et al. 2007; Guizzo et al. 2017; Zhang et al. 2017). Such role is thought to be the provisioning of essential nutrients. Indeed, the presence of B vitamins and cofactors biosynthesis pathways in genomes of different strains of Coxiella endosymbionts suggest their capability of supplementing the unbalanced blood diet of the hosts (Gottlieb et al. 2015; Smith et al. 2015). Coxiella is believed to be the bacterium with the oldest symbiotic association with tick hosts, but other endosymbiotic bacteria, especially Francisella, have been reported to have a similar role, possibly having replaced Coxiella in some tick species (Duron et al. 2017).
Indeed, Francisella endosymbionts have been commonly reported in Coxiella-free ticks, belonging to the genera Dermacentor, Amblyomma, Hyalomma and Ornithodorus. Genome comparison of selected Francisella symbionts together with physiological experiments strongly suggest their important role in conferring advantages for the tick fitness, mainly providing B vitamins (Gerhart et al. 2016; Duron et al. 2018).
Multiple essential roles, including B vitamins provision, were also hypothesized for the symbiont ‘Candidatus Midichloria mitochondrii’ (hereafter M. mitochondrii) (Sassera et al. 2011; Olivieri et al. 2019). This bacterial endosymbiont was originally described in one of the most widespread ticks in Europe, Ixodes ricinus, and later reported in several other tick species from different continents (Beninati et al. 2004, 2009; Sacchi et al. 2004; Epis et al. 2008; Cafiso et al. 2016).
Interestingly, recent phylogenetic investigations revealed the occurrence of regular transitions between endosymbiotic and pathogenic forms during the course of evolution, such as Coxiella burnetii that seems to have recently evolved from a Coxiella endosymbiont ancestor (Duron et al. 2015) or conversely Francisella endosymbionts that probably originated from a pathogenic ancestor (Gerhart et al. 2016, 2018).
The well-known relevance of symbionts of arthropods on the host physiology and the nested interactions that can develop among symbionts and pathogens call for further investigation. For these reasons, the aims of this work were: (i) to update the knowledge on the prevalence, distribution and molecular characterization of selected TBPs and symbionts in different ecological zones in Africa, and (ii) to evaluate the patterns of co-infections detecting eventual competitive or facilitative interactions.
Materials and methods
Study sites, tick collection and identification
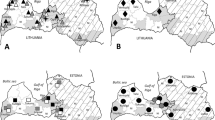
From 2009 to 2017 ticks were collected in various locations in Kenya from sympatric wild (African elephant, African buffalo, black and white rhinoceros, bongo antelope, dromedary camel, giraffe, hyena, lion, leopard, zebra and Grévy’s zebra) and domestic animals (cattle, sheep). Most of the samples were collected during routine veterinary surveillances of the Kenya Wildlife Service (KWS) performed in national parks, reserves, game reserves and from the vegetation (Fig. 1). Ticks were additionally collected in two districts in Ethiopia from cattle and sheep, where animals are managed under an extensive farming system at communal grazing land shared among small scale farmers. An additional portion of the dataset was collected from dogs living in close proximity with domestic ruminants in a single location in Egypt (Fig. 1). For each sampling point, the ecological zone values were extracted from the African ecological zones layer (AEZs; HarvestChoice 2011), by using the QGIS 3. According to this database, the samples were located in six agro-ecological zones. Collected ticks were preserved in vials containing 70% ethanol and morphologically identified using standard taxonomic keys (Theiler and Salisbury 1959; Walker et al. 2003).

Political map of Africa indicating the countries where ticks were collected (i.e., Egypt in pink, Ethiopia in light-blue and Kenya in light-green). Insets show the localities where the ticks were collected. (Color figure online)
Molecular analyses
Genomic DNA was extracted individually from 339 ticks using the NucleoSpin® Tissue Kit (Macherey Nagel, Duren, Germany), according to the manufacturer’s instructions. The DNA quality was tested on a random subset of 68 samples (20%) using PCR amplification of tick mitochondrial ribosomal small RNA gene (12S rRNA) using a previously described protocol (Beati and Keirans 2001) (Additional file 1: Table S1).
The DNA samples were then tested by PCR for the presence of Rickettsia spp., Anaplasma spp./Ehrlichia spp., Borrelia burgdorferi (s.l.), Babesia spp./Theileria spp., Coxiella spp., Midichloria and Francisella using primers and conditions previously described (Additional file 1: Table S1). Positive PCR products of the expected size were extracted from agarose gel, purified using the QIAquick® Gel Extraction Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Purified DNA was sequenced with forward and reverse amplification primers (Eurofins Genomics, Ebersberg, Germany). Sequences were manually verified with Chromas Lite (Technelysium, Australia) and compared with those available in GenBank database using Basic Local Alignment Search Tool (BLAST: http://www.ncbi.nlm. nih.gov/BLAST). All the consensus sequences obtained in this study were deposited in GenBank database under the accession numbers given in the Additional file 2.
Phylogenetic analyses
For the phylogenetic analyses on the sequences obtained in this study, two approaches were employed depending on the kind of sequence amplified. For each of the assays targeting SSU rRNA gene (Anaplasma/Ehrlichia, Coxiella, Midichloria, Babesia/Theileria), the newly obtained sequences were aligned on the SSU rRNA SILVA 128 Ref NR 99 database (Quast et al. 2012) with the ARB software package (Westram et al. 2011). After selection of similar sequences, the alignments were manually edited to optimize base-pairing in the predicted stems of the rRNA, and trimmed at both ends to the length of the amplicon sequences (i.e., excluding flanking regions present only in the database-derived sequences). For all other non-SSU assays (the three Rickettsia genes, Borrelia and Francisella), the sequences were directly aligned with selected database sequences using MUSCLE (Edgar 2004), and polished with Gblocks (Talavera and Castresana 2007).
For each final alignment thereby obtained, nucleotide substitution models were ranked according to the Akaike’s Information Criterion with jModeltest (Darriba et al. 2012). After model selection, maximum likelihood phylogenetic analyses were performed using phyML (Guindon and Gascuel 2003) with 100 bootstrap pseudo-replicates.
Microorganisms co‐presence and ecological network inference
An ad-hoc script in R (R Core Team 2019) has been developed (available at https://github.com/MontagnaLab/co-presence_test) for testing whether the co-presence/co-absence of two microorganisms in the same tick individual is due to chance. This hypothesis has been tested simulating a null model (representing the hypothesis that co-presence of the same microorganism in individuals is due to chance) developed permuting the columns of a presence/absence matrix obtained for each couple of microorganisms based on PCR assays results (21 matrices in total). Each matrix was permuted 9999 times and the number of co-presences of each couple of microorganisms estimated for each permuted matrix. A two-tailed test with α/2 = 2.5% was performed for testing the null hypothesis. The values corresponding to the 2.5th and 97.5th percentiles of the simulated distribution were estimated. The number of co-presences observed for each couple of microorganisms in the total number of screened ticks was than calculated from the real presence/absence matrix. The null hypothesis is accepted when the observed value of co-presence was included between the values corresponding to the 2.5th and the 97.5th percentiles of the simulated distribution. In the event that the null hypothesis was rejected a p-value was calculated.
The relation between each tick-borne microorganism, tick species and vertebrate host was analysed and visualized by constructing a bipartite ecological network. Nodes of the network represent the vertebrate host and the tick species, whereas the edges represent the presence of individuals of the tick species on the vertebrate host. In addition, the information of the percentage of carried microorganisms was plotted as pie charts for each tick species. The network visualization was carried out using Cytoscape v.3.7.1 by importing the nodes and edges data mentioned above (Shannon et al. 2003).
Results
In total, 339 ticks belonging to the Ixodidae family were collected. The ticks were morphologically identified as belonging to six genera: Amblyomma, Dermacentor, Haemaphysalis, Hyalomma, Ixodes, Rhipicephalus, for a total of 30 tick species. Additional information on tick species identification, number, gender, collection sites and hosts is listed in Table 1.
Molecular screening revealed the presence of pathogens belonging to the genera Rickettsia, Anaplasma, Ehrlichia, Borrelia, Babesia, and Theileria, with Rickettsia bacteria being the most widespread. Indeed, Rickettsia spp. were found in 18 out of 339 ticks tested (5.3%). Subsequent analyses of the gltA gene sequences revealed that 10 out of 18 rickettsias share high identity with Rickettsia aeschlimannii (detected in 9/30 Hy. rufipes and 1/7 Hy. impeltatum). The gltA marker did not allow to discriminate the remaining eight Rickettsia sequences at the species level (Additional file 2: Fig. S1a). Thus, additional sequencing of ompA and ompB genes of a representative subset of positive samples was performed, and, besides confirming that the most prevalent species was R. aeschlimannii (n = 10), allowed to identify other rickettsial species: R. africae (n = 5) detected in Am. gemma (n = 2), Am. variegatum (n = 2) and Hy. impeltatum (n = 1); R. massiliae (n = 2) in Rh. praetextatus; and one R. rhipicephali in Am. cohaerens (Additional file 2: Fig. S1b,c).
Anaplasma spp. DNA was detected in 2.1% (7/339) of the ticks. The phylogenetic analysis based on 16S rRNA sequences did not provide sufficient discriminatory power to clarify the species assignment. However, the obtained sequences formed two distinct clusters: the sequences from three Rh. pravus and two Rh. decoloratus ticks clustered with A. marginale, A. centrale and A. ovis sequences downloaded from NCBI with 100% bootstrap support; whereas two sequences, from Am. variegatum and Rh. decoloratus, clustered with A. platys (74% bootstrap support) (Additional file 2: Fig. S2).
Ehrlichia bacteria were detected in three ticks only (0.9% of the total, one Am. variegatum, one Am. lepidum, one Hy. impeltatum), collected from cattle and Grevy’s zebra. The sequences showed 100% of identity with E. ruminantium (GenBank: NR074155), supported with 100% bootstrap in the phylogeny (Additional file 2: Fig. S2).
Different Theileria spp. were detected in seven out of 339 ticks (2.1%). Among these, three were clearly identified as T. taurotragi (detected in two Rh. appendiculatus collected from antelope) and as T. velifera (detected in an Am. cohaerens collected from Black rhinoceros). Two sequences, from Am. cohaerens and Am. gemma collected from white rhinoceros, clustered together with an unknown Theileria species detected in cheetahs in the same area in 2009 (Githaka et al. 2012). One Theileria sequence detected in Rh. pulchellus collected from black rhinoceros clustered together with another unknown Theileria sp. detected in blood samples from giraffes in the same area in 2011 (GenBank AB650504). Finally, a Theileria sequence detected in Am. cohaerens collected from white rhinoceros likely represents a new species, showing only 89.97% nucleotide identity with T. mutans (GenBank: JN572694). However, additional characterization would be required as it is not possible to establish new variants of piroplasms based only on the use the 18S rRNA gene (Chae et al. 1999; Allsopp and Allsopp 2006) (see Additional file 2: Fig. S3 for the phylogeny of the Theileria).
Babesia was detected in two ticks (0.6%). The sequence obtained from Am. variegatum shows 98% of identity with B. caballi (GenBank: MH424325) and the one from Hy. rufipes shows 100% identity with B. occultans (GenBank: MH899757). Both identifications were highly supported in the phylogeny (Additional file 2: Fig. S3).
Borrelia positivity was detected in only one tick (Hy. rufipes). The obtained ITS sequence shows 100% identity with B. garinii from an Ixodes ricinus sample in Finland (GenBank: MG356954). Consistently, the novel sequence results embedded in a clade of B. garinii in a phylogenetic analysis (Additional file 2: Fig. S4).
Molecular screening of bacterial symbionts revealed the presence of Coxiella, Francisella and Midichloria across the tick populations. The most prevalent endosymbionts were Coxiella spp., successfully amplified from 95 of the 339 ticks tested (28%). Putative Coxiella endosymbionts were found among 16 tick species, whereas only one Coxiella strain identical to the pathogenic Coxiella burnetii was detected in one specimen of Rh. pulchellus. (Table 2). Phylogenetic analysis based on the 16S rRNA gene showed, in most cases, that closely related Coxiella strains are found in closely related tick species (Additional file 2: Fig. S5).
Francisella spp. were detected in 32 ticks out of 339 tested (9.4%). Francisella positive ticks belonged to seven tick species, mainly within the Hyalomma genus, in which the prevalence was high, ranging from 20 to 50% (Table 2). Although the phylogenetic analysis of the rpoB gene was poorly informative in terms of species determination, it still allowed to identify the detected organisms as members of the Francisella-like endosymbionts (FLE) clade, and genetically distant from strains of pathogenic Francisella species and subspecies. In addition, all of the sequences of FLE detected in Hyalomma ticks were closely related, whereas FLE detected in De. rhinocerinus and Rh. praetextatus clustered together with a distinct, long branch, probably due to higher sequence divergence (Additional file 2: Fig. S6).
A total of 24 ticks out of 339 (7.1%) were positive for Midichloria. The rate of infection among specimens of the positive tick species was generally lower compared to Coxiella and Francisella endosymbionts. However, Midichloria resulted the most prevalent symbiont of Rh. sanguineus s.l., reaching an infection rate of 33.3% versus 11.1% of Coxiella endosymbionts, and the two symbionts were never detected in the same individual (Table 2). On the other hand, the phylogenetic tree clearly showed that similar sequences of Midichloria are found in genetically distant tick species, with the most diverging Midichloria member identified in Am. lepidum (Additional file 2: Fig. S7).
Interestingly, co-infections were spotted: 21 ticks resulted infected with more than one microorganism, including 16 double infections with seven combinations and five triple infections, mainly involving Midichloria, Francisella and Rickettsia (Table 3). Co-infection between tick-borne microorganisms occurred more frequently in generalist tick species with a broad host spectrum, such as Hy. rufipes and Am. variegatum, whereas ticks with a pronounced host specificity, such as Am. tholloni and Rh. carnivoralis, resulted mainly bearing single microorganisms, especially vertically transmitted endosymbionts (Fig. 2).

Bipartite ecological network showing the relation among tick-borne microorganism, tick species and its vertebrate host. The vertebrate host and the tick species, the nodes of the network, are represented in the form of squares and circles, respectively; whereas edges represent the associations between the tick species and their vertebrate host. For each tick species, the relative abundance (expressed as percentage) of microorganisms detected through the PCR screening of individuals is reported in pie charts. A.co, Amblyomma cohaerens; A.eb, Amblyomma eburneum; A.ge, Amblyomma gemma; A.le, Amblyomma lepidum; A.nu, Amblyomma nuttalli; A.pe, Amblyomma personatum; A.th, Amblyomma tholloni; A.va, Amblyomma variegatum; D.rh, Dermacentor rhinocerinus; Ha.sp, Haemaphysalis sp; Hy.al, Hyalomma albiparmatum; Hy.dr, Hyalomma dromedarii; Hy.im, Hyalomma impeltatum; Hy.ru, Hyalomma rufipes; Hy.tr, Hyalomma truncatum; I.sp, Ixodes sp.; R.ap, Rhipicephalus appendiculatus; R.cam, Rhipicephalus camicasi; R.car, Rhipicephalus carnivoralis; R.com, Rhipicephalus compositus; R.de, Rhipicephalus decoloratus; R.ev, Rhipicephalus evertsievertsi; R.hu, Rhipicephalus humeralis; R.mu, Rhipicephalus muelensi; R.prae, Rhipicephalus praetextatus; R.prav, Rhipicephalus pravus; R.pu, Rhipicephalus pulchellus; R.sa, Rhipicephalus sanguineus; R.sp., Rhipicephalus sp
Furthermore, comparing the null model distribution with the observed values of co-presence, the association between Rickettsia and Francisella in the same host tick resulted positively significant (p < 0.01) (Additional file 3: Fig. S8 A), this association was often observed in Hy. rufipes individuals (Fig. 2). Through the same analysis, Francisella and Coxiella association was found to be negatively significant (p < 0.001) (Additional file 3: Fig. S8 B).
Discussion
Spotted fever group (SFG) rickettsioses are the most frequently tick-borne diseases recognised among travellers returning from sub-Saharan Africa with acute febrile illness, this is indicative of the endemicity of rickettsial diseases in African countries and their impact on public health (Freedman et al. 2006; Parola et al. 2013). Rickettsia africae, R. aeschlimannii and R. massiliae, all detected in this investigation, are considered among the main pathogenic SFG rickettsiae (Parola 2006). Our results are in agreement with those of previous studies, which identified several tick species as potential vectors for these rickettsiae in multiple sub-Saharan African countries. Indeed Hyalomma ticks frequently harbour R. aeschlimanni, especially Hy. rufipes and Hy. marginatum (Mura et al. 2008; Kumsa et al. 2015; Azagi et al. 2017). On the other side, our finding of R. africae in multiple Amblyomma species, with higher prevalence in Am. variegatum and Am. gemma, confirms previous findings (Jensenius et al. 2003; Macaluso et al. 2003; Mediannikov et al. 2010; Mutai et al. 2013; Vanegas et al. 2018). The geographical distribution of these SFG rickettsiae strongly overlaps with the distribution of their respective tick vectors.
In the last years several SFG rickettsial species that are pathogenic for the vertebrate hosts have also been identified as secondary tick symbionts, reaching a high frequency of infection in some tick populations, enhancing the host fitness and being transovarially transmitted to the offspring, e.g., Rickettsia parkeri or R. monacensis (Ahantarig et al. 2013). Whether the three rickettsial species detected here play a similar role in their host remains an open question.
A noteworthy finding for human health is the unusual detection of B. garinii DNA in a Hy. rufipes tick collected from a Giraffe in Kenya. Borrelia garinii is one of the predominant genospecies of the B. burgdorferi sensu lato complex, known to cause Lyme disease in Europe, and is considered the most neurotropic Borrelia spirochete (Benredjem et al. 2014; Stanek and Strle 2018). Borrelia garinii is usually vectored by Ixodes ticks in Europe and Asia, but was also reported in North Africa (Tunisia and Morocco) in association with Ixodes species (Bouattour et al. 2004), identified as I. ricinus by Bouattour, but possibly belonging to the subsequently described species I. inopinatus (Estrada-Pena et al. 2014). Birds are considered the main reservoirs and biological carriers of B. garinii (Comstedt et al. 2011; Pajoro et al. 2018). The role of migratory birds in the spread of this spirochete can explain the novel finding of the positivity of Hy. rufipes, a tick species that has been reported infesting various migratory birds worldwide (England et al. 2016). Based on this evidence, and on previous reports of Borrelia lusitaniae in Hy. marginatum (Michelis et al. 2000), these findings represent uncommon cases of B. burgdorferi sensu lato species associated with metastriate ticks (Margos et al. 2020). Considering that ticks can be infected following an infected blood meal, only further studies can confirm the vectorial competence of Hyalomma ticks for Borrelia species focusing on the acquisition, maintenance, and subsequent transmission into a vertebrate host during blood feeding.
Additionally, a high diversity of tick-borne pathogens relevant for domestic and wild animal health were here detected in the tick populations tested, although with low prevalence. Among others, we detected E. ruminantium, a bacterium mainly transmitted by ticks of the genus Amblyomma, causing heartwater disease affecting wild and domestic ruminants (Uilenberg 1997; Allsopp 2010). The occurrence of several piroplasm species, such as B. caballi, B. occultans, T. taurotragi and T. velifera, considered mildly to severely pathogenic with significant impact on animal health, is here reported, in accordance with previous surveys (de la Fuente et al. 2008; Sivakumar et al. 2014; Omondi et al. 2017).
The most retrieved symbiont was Coxiella, found in representatives of four out of six tick genera analysed, reaching high prevalence in many of the analysed species, especially within the Rhipicephalus and Amblyomma genera (Table 2). According to phylogenetic analysis based on the 16S rRNA gene sequence, most of the novel sequences result closely related to other Coxiella associated to tick species of the same genus. Moreover, although not fully supported, the deeper tree topology is overall consistent with the four Coxiella clades identified by Duron and colleagues through multilocus sequence typing (MLST) (Duron et al. 2015). Accordingly, whereas a great diversity exists within the genus, our results confirm the overall co-cladogenesis of Coxiella symbionts with their hosts, but, at the same time, presence of highly related Coxiella in unrelated ticks suggest relatively frequent host species shifts (Duron et al. 2015). These features likely reflect a long mutualistic coevolution, conferring significant advantages to both organisms, and with a certain degree of flexibility with respect to host/symbiont species.
The second most widespread symbiont is Francisella. Consistently with previous studies (Ivanov et al. 2011; Szigeti et al. 2014; Azagi et al. 2017; Duron et al. 2017), Francisella resulted highly prevalent among the Hyalomma species tested, but we additionally detected this bacterium in species in which it was never reported before (Hy. impeltatum and Hy. albiparmatum). The nutritional mutualism of Francisella can explain the negative correlation we found with Coxiella endosymbionts, since they provide the same benefit for the host (Duron et al. 2017, 2018). Indeed, in recent studies Francisella was defined as an alternative obligate symbiont to Coxiella, which appeared to be replaced by Francisella in multiple tick species (Duron et al. 2017). In our dataset Francisella was found to significantly co-occur with Rickettsia, as frequently reported previously across tick taxa (Scoles 2004; Ahantarig et al. 2013; Budachetri et al. 2015; Azagi et al. 2017), whereas Coxiella endosymbionts were often reported as single infections. Taken together, these data allow to hypothesize that Francisella is less competitive than the Coxiella primary symbiont, or that multiple co-occurring symbionts can act in conjunction or even synergistically.
Noteworthy, Midichloria is the most prevalent (33%) symbiont in Rh. sanguineus s.l. with Coxiella as second (11%). This finding is interesting when compared with a recent study on the microbial communities of various Rh. sanguineus s.l. populations in France, Arizona (USA) and Senegal, which indicated Coxiella and Rickettsia as the predominant endosymbionts, with strong geographical clustering (René-Martellet et al. 2017). In particular, René-Martellet and colleagues concluded that the relative abundance of these endosymbionts varies depending on the geographical origin and the lineage of the tick, with Coxiella strongly associated with Senegal ticks. We can add to the complex landscape of the symbionts of Rh. sanguineus s.l. the notion that in Egypt the predominant symbiont is neither Coxiella, nor Rickettsia, but Midichloria. These results confirm the lability of the bacterial community structure hosted by this tick species, much differently that what seen in most other species (Duron et al. 2017). There are various possible explanations, such as the influence of multiple ecological and geographical factors (Lalzar et al. 2014; Abraham et al. 2017; Bonnet et al. 2017) as well as the host-feeding behavior of the ticks, the host’s immune system and the direct interaction of protozoan or bacterial pathogens (Adegoke et al. 2020; Aivelo et al. 2019; Hawley and Altizer 2011). Alternatively, or in conjunction, the possibility that the analysed individuals belong to different sibling species of the Rh. sanguineus s.l. group must be considered (Dantas-Torres and Otranto 2015; Coimbra-Dores et al. 2020).
Despite the low prevalence of Midichloria symbionts in African ticks, the detection of similar sequences of Midichloria in genetically distant tick species provides additional support to the hypothesis of frequent horizontal transfers of these bacteria (Skarphédinsson et al. 2005; Bazzocchi et al. 2013; Cafiso et al. 2018; Di Lecce et al. 2018; Serra et al. 2018). Low genetic variation of Midichloria was commonly reported in surveys based on phylogenetic analysis of 16S rRNA gene sequences (Cafiso et al. 2016; Duron et al. 2017), whereas recent MLST-based studies provide evidence of co-evolution of Midichloria in some tick populations (Buysse and Duron 2018; Al-khafaji et al. 2019).
Additionally, we report a frequent albeit not statistically significant co-occurrence of Midichloria with Rickettsia in specimens of Hy. rufipes. We can draw a parallel with what recently reported in the tick A. maculatum, in which R. parkeri infection was found to promote Midichloria colonization in the midgut, salivary glands, and ovarian tissues of fed and unfed ticks, indicating a synergistic relationship between them (Budachetri et al. 2018).
Conclusions
This study brings further attention to the complexity of ticks’ microbial communities and calls for an in-depth analysis of the interactions among the tick-borne microorganisms. Further multidisciplinary investigations involving metagenomics, genomics, and ecology are pivotal to better understand these dynamics, with possible important consequences on human and animal health, economy, and on the preservation of endangered species.
References
Abraham NM, Liu L, Jutras BL et al (2017) Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc Natl Acad Sci 114:E781–E790. https://doi.org/10.1073/pnas.1613422114
Adegoke A, Kumar D, Bobo C et al (2020) Tick-borne pathogens shape the native microbiome within tick vectors. Microorganisms 8:1–16. https://doi.org/10.3390/microorganisms8091299
Ahantarig A, Trinachartvanit W, Baimai V, Grubhoffer L (2013) Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia Microbiol (Praha) 58:419–428. https://doi.org/10.1007/s12223-013-0222-1
Aivelo T, Norberg A, Tschirren B (2019) Bacterial microbiota composition of Ixodes ricinus ticks: the role of environmental variation, tick characteristics and microbial interactions. PeerJ 7:e8217. https://doi.org/10.7717/peerj.8217
Al-khafaji AM, Clegg SR, Pinder AC et al (2019) Multi-locus sequence typing of Ixodes ricinus and its symbiont Candidatus Midichloria mitochondrii across Europe reveals evidence of local co- cladogenesis in Scotland. Ticks Tick Borne Dis 10:52–62. https://doi.org/10.1016/j.ttbdis.2018.08.016
Allsopp BA (2010) Natural history of Ehrlichia ruminantium. Vet Parasitol 167:123–135. https://doi.org/10.1016/j.vetpar.2009.09.014
Allsopp BA (2015) Heartwater-Ehrlichia ruminantium infection. Rev Sci Tech 34:557–568
Allsopp MTEP, Allsopp BA (2006) Molecular sequence evidence for the reclassification of some Babesia species. Ann N Y Acad Sci 1081:509–517. https://doi.org/10.1196/annals.1373.076
Andreotti R, De León AAP, Dowd SE et al (2011) Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol 11:6. https://doi.org/10.1186/1471-2180-11-6
Asante J, Noreddin A, El Zowalaty ME (2019) Systematic review of important bacterial zoonoses in africa in the last decade in light of the “One Health” concept. Pathogens 8:50. https://doi.org/10.3390/pathogens8020050
Azagi T, Klement E, Perlman G et al (2017) Francisella-like endosymbionts and Rickettsia species in local and imported Hyalomma ticks. Appl Environ Microbiol 83:eo01302–e17. https://doi.org/10.1128/AEM.01302-17
Bazzocchi C, Mariconti M, Sassera D et al (2013) Molecular and serological evidence for the circulation of the tick symbiont Midichloria (Rickettsiales: Midichloriaceae) in different mammalian species. Parasites Vectors 6:1–7. https://doi.org/10.1186/1756-3305-6-350
Beati L, Keirans JE (2001) Analysis of the systematic relationships among ticks of the genera Rhipicephalus and Boophilus (Acari: Ixodidae) based on mitochondrial 12S ribosomal DNA gene sequences and morphological characters. J Parasitol 87:32–48. https://doi.org/10.1645/0022-3395(2001)087[0032:AOTSRA]2.0.CO;2
Bekker CPJ, de Vos S, Taoufik A et al (2002) Simultaneous detection of Anaplasma and Ehrlichia species in ruminants and detection of Ehrlichia ruminantium in Amblyomma variegatum ticks by reverse line blot hybridization. Vet Microbiol 89:223–238
Beninati T, Lo N, Sacchi L et al (2004) A novel alpha-Proteobacterium resides in the mitochondria of ovarian cells of the tick Ixodes ricinus. Appl Environ Microbiol 70:2596–2602
Beninati T, Riegler M, Vilcins IME et al (2009) Absence of the symbiont Candidatus midichloria mitochondrii in the mitochondria of the tick Ixodes holocyclus. FEMS Microbiol Lett 299:241–247. https://doi.org/10.1111/j.1574-6968.2009.01757.x
Benredjem W, Leulmi H, Bitam I et al (2014) Borrelia garinii and Rickettsia monacensis in Ixodes ricinus ticks, Algeria. Emerg Infect Dis 20:1776–1777. https://doi.org/10.3201/eid2010.140265
Bonnet SI, Binetruy F, Hernández-Jarguín AM, Duron O (2017) The tick microbiome: why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front Cell Infect Microbiol 7:1–14. https://doi.org/10.3389/fcimb.2017.00236
Bouattour A, Ghorbel A, Chabchoub A, Postic D (2004) Lyme borreliosis situation in North Africa. Arch Inst Pasteur Tunis 81:13–20
Budachetri K, Browning RE, Adamson SW et al (2015) An insight into the Microbiome of the Amblyomma maculatum (Acari: Ixodidae). J Med Entomol 51:119–129
Budachetri K, Kumar D, Crispell G et al (2018) The tick endosymbiont Candidatus Midichloria mitochondrii and selenoproteins are essential for the growth of Rickettsia parkeri in the Gulf Coast tick vector. Microbiome 6:1–15. https://doi.org/10.1186/s40168-018-0524-2
Buysse M, Duron O (2018) Multi-locus phylogenetics of the Midichloria endosymbionts reveals variable specificity of association with ticks. Parasitology 1–10
Cafiso A, Bazzocchi C, De Marco L et al (2016) Molecular screening for Midichloria in hard and soft ticks reveals variable prevalence levels and bacterial loads in different tick species. Ticks Tick Borne Dis 7:1186–1192. https://doi.org/10.1016/j.ttbdis.2016.07.017
Cafiso A, Sassera D, Romeo C et al (2018) Midichloria mitochondrii, endosymbiont of Ixodes ricinus: evidence for the transmission to the vertebrate host during the tick blood meal. Ticks Tick Borne Dis 10:5–12. https://doi.org/10.1016/j.ttbdis.2018.08.008
Chae JS, Allsopp BA, Waghela SD et al (1999) A study of the systematics of Theileria spp. based upon small-subunit ribosomal RNA gene sequences. Parasitol Res 85:877–883. https://doi.org/10.1007/s004360050651
Clay K, Klyachko O, Grindle N et al (2008) Microbial communities and interactions in the lone star tick, Amblyomma americanum. Mol Ecol 17:4371–4381. https://doi.org/10.1111/j.1365-294X.2008.03914.x
Coimbra-Dores MJ, Jaarsma RI, Carmo AO et al (2020) Mitochondrial sequences of Rhipicephalus and Coxiella endosymbiont reveal evidence of lineages co-cladogenesis. FEMS Microbiol Ecol 96:1–18. https://doi.org/10.1093/femsec/fiaa072
Comstedt P, Jakobsson T, Bergström S (2011) Global ecology and epidemiology of Borrelia garinii spirochetes. Infect Ecol Epidemiol 1:9545. https://doi.org/10.3402/iee.v1i0.9545
Dantas-Torres F, Otranto D (2015) Further thoughts on the taxonomy and vector role of Rhipicephalus sanguineus group ticks. Vet Parasitol 208:9–13. https://doi.org/10.1016/j.vetpar.2014.12.014
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Meth 9:772
de la Fuente J, Estrada-Pena A, Venzal JM et al (2008) Overview: ticks as vectors of pathogens that cause disease in humans and animals. Front Biosci 13:6938–6946. https://doi.org/10.2741/3200
Di Lecce I, Bazzocchi C, Cecere JG et al (2018) Patterns of Midichloria infection in avian- borne African ticks and their trans-Saharan migratory hosts. Parasit Vectors 11:1–11. https://doi.org/10.1186/s13071-018-2669-z
Díaz-Sánchez S, Estrada-Peña A, Cabezas-Cruz A, de la Fuente J (2019) Evolutionary insights into the tick hologenome. Trends Parasitol. https://doi.org/10.1016/j.pt.2019.06.014
Duron O, Noël V, McCoy KD et al (2015) The recent evolution of a maternally-inherited endosymbiont of ticks led to the emergence of the Q fever pathogen, Coxiella burnetii. PLoS Pathog 11:1–23. https://doi.org/10.1371/journal.ppat.1004892
Duron O, Binetruy F, Noël V et al (2017) Evolutionary changes in symbiont community structure in ticks. Mol Ecol 26:2905–2921. https://doi.org/10.1111/mec.14094
Duron O, Morel O, Erie Noë V et al (2018) Tick-bacteria mutualism depends on B vitamin synthesis pathways. Curr Biol 28:1–7. https://doi.org/10.1016/j.cub.2018.04.038
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
England ME, Phipps P, Medlock JM et al (2016) Hyalomma ticks on northward migrating birds in southern Spain: Implications for the risk of entry of Crimean-Congo haemorrhagic fever virus to Great Britain. J Vector Ecol 41:128–134. https://doi.org/10.1111/jvec.12204
Epis S, Sassera D, Beninati T et al (2008) Midichloria mitochondrii is widespread in hard ticks (Ixodidae) and resides in the mitochondria of phylogenetically diverse species. Parasitology 135:485–494. https://doi.org/10.1017/S0031182007004052
Estrada-Pena A, Nava S, Petney T (2014) Description of all the stages of Ixodes inopinatus n. sp. (Acari: Ixodidae). Ticks Tick Borne Dis 5:734–743. https://doi.org/10.1016/j.ttbdis.2014.05.003
Freedman DO, Weld LH, Kozarsky PE et al (2006) Spectrum of disease and relation to place of exposure among ill returned travelers. N Engl J Med 354:119–130
Gebrekidan H, Hailu A, Kassahun A et al (2014) Theileria infection in domestic ruminants in northern Ethiopia. Vet Parasitol 200:31–38. https://doi.org/10.1016/j.vetpar.2013.11.017
Gerhart JG, Moses AS, Raghavan R (2016) A Francisella-like endosymbiont in the Gulf Coast tick evolved from a mammalian pathogen. Sci Rep 6:1–6. https://doi.org/10.1038/srep33670
Gerhart JG, Auguste Dutcher H, Brenner AE et al (2018) Multiple Acquisitions of Pathogen-Derived Francisella Endosymbionts in Soft Ticks. Genome Biol Evol 10:607–615. https://doi.org/10.1093/gbe/evy021
Githaka N, Konnai S, Kariuki E et al (2012) Molecular detection and characterization of potentially new Babesia and Theileria species / variants in wild felids from Kenya. Acta Trop 124:71–78. https://doi.org/10.1016/j.actatropica.2012.06.013
Gottlieb Y, Lalzar I, Klasson L (2015) Distinctive genome reduction rates revealed by genomic analyses of two Coxiella-like endosymbionts in ticks. Genome Biol Evol 7:1779–1796. https://doi.org/10.1093/gbe/evv108
Greay TL, Gofton AW, Paparini A et al (2018) Recent insights into the tick microbiome gained through next-generation sequencing. Parasit Vectors 1–14. https://doi.org/10.1186/s13071-017-2550-5
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Guizzo MG, Parizi LF, Nunes RD et al (2017) A Coxiella mutualist symbiont is essential to the development of Rhipicephalus microplus. Sci Rep 7:17554. https://doi.org/10.1038/s41598-017-17309-x
HarvestChoice (2011) AEZ Tropical (8-class). International Food Policy Research Institute, Washington, DC., and University of Minnesota, St. Paul, MN. Available online at http://harvestchoice.org/node/4997
Hawkins E, Kock R, Mckeever D et al (2015) Prevalence of Theileria equi and Babesia caballi as well as the identification of associated ticks in sympatric grevy’ s zebras (Equus grevyi) and donkeys (Equus africanus asinus) in northern Kenya. J Wildl Dis 51:137–147. https://doi.org/10.7589/2013-11-316
Hawley DM, Altizer SM (2011) Disease ecology meets ecological immunology: Understanding the links between organismal immunity and infection dynamics in natural populations. Funct Ecol 25:48–60. https://doi.org/10.1111/j.1365-2435.2010.01753.x
Ikwap K, Picozzi K, Waiswa C (2010) Molecular characterization of Anaplasma and Ehrlichia species in different cattle breeds and age groups in Mbarara District (Western Uganda). Int J Anim Vet Adv 2:76–88
Ivanov IN, Mitkova N, Reye AL et al (2011) Detection of new Francisella-like tick endosymbionts in Hyalomma spp. and Rhipicephalus spp. (Acari: Ixodidae) from Bulgaria. Appl Environ Microbiol 77:5562–5565. https://doi.org/10.1128/AEM.02934-10
Jensenius M, Fournier P-E, Kelly P et al (2003) African tick bite fever. Lancet Infect Dis 3:557–564
Jongejan F, Uilenberg G (2004) The global importance of ticks. Parasitology 129:S3–S14. https://doi.org/10.1017/S0031182004005967
Kamani J, Baneth G, Gutiérrez R et al (2018) Coxiella burnetii and Rickettsia conorii: two zoonotic pathogens in peridomestic rodents and their ectoparasites in Nigeria. Ticks Tick Borne Dis 9:86–92. https://doi.org/10.1016/j.ttbdis.2017.10.004
Kumsa B, Socolovschi C, Raoult D, Parola P (2015) Spotted fever group rickettsiae in ixodid ticks in oromia, Ethiopia. Ticks Tick Borne Dis 6:8–15. https://doi.org/10.1016/j.ttbdis.2014.08.001
Lalzar I, Harrus S, Mumcuoglu KY, Gottlieb Y (2012) Composition and seasonal variation of Rhipicephalus turanicus and Rhipicephalus sanguineus bacterial communities. Appl Environ Microbiol 78:4110–4116. https://doi.org/10.1128/AEM.00323-12
Lalzar I, Friedmann Y, Gottlieb Y (2014) Tissue tropism and vertical transmission of Coxiella in Rhipicephalus sanguineus and Rhipicephalus turanicus ticks. Environ Microbiol 16:3657–3668. https://doi.org/10.1111/1462-2920.12455
Lorusso V, Wijnveld M, Majekodunmi AO et al (2016) Tick-borne pathogens of zoonotic and veterinary importance in Nigerian cattle. Parasit Vectors 9:217. https://doi.org/10.1186/s13071-016-1504-7
Macaluso KR, Davis J, Alam U et al (2003) Spotted fever group rickettsiae in ticks from the Masai Mara region of Kenya. Am J Trop Med Hyg 68:551–553. https://doi.org/10.4269/ajtmh.2003.68.551
Machado-Ferreira E, Vizzoni VF, Balsemão-Pires E et al (2016) Coxiella symbionts are widespread into hard ticks. Parasitol Res 115:4691–4699. https://doi.org/10.1007/s00436-016-5230-z
Maina AN, Jiang J, Omulo SA et al (2014) High prevalence of Rickettsia africae variants in Amblyomma variegatum ticks from domestic mammals in rural western Kenya: implications for human health. Vector-Borne Zoonotic Dis 14:693–702. https://doi.org/10.1089/vbz.2014.1578
Margos G, Fingerle V, Cutler S et al (2020) Controversies in bacterial taxonomy: the example of the genus Borrelia. Ticks Tick Borne Dis 11:101335. https://doi.org/10.1016/j.ttbdis.2019.101335
Mediannikov O, Trape J-F, Diatta G et al (2010) Rickettsia africae, Western Africa. Emerg Infect Dis 16:571–573
Michelis SDE, Sewell H, Collares-pereira M et al (2000) Genetic diversity of Borrelia burgdorferi sensu lato in ticks from mainland Portugal. J Clin Microbiol 38:2128–2133
Moutailler S, Valiente Moro C, Vaumourin E et al (2016) Co-infection of ticks: the rule rather than the exception. PLoS Negl Trop Dis 10:1–17. https://doi.org/10.1371/journal.pntd.0004539
Mura A, Socolovschi C, Ginesta J et al (2008) Molecular detection of spotted fever group rickettsiae in ticks from Ethiopia and Chad. Trans R Soc Trop Med Hyg 102:945–949
Mutai BK, Wainaina JM, Magiri CG et al (2013) Zoonotic surveillance for rickettsiae in domestic animals in Kenya. Vector-Borne Zoonotic Dis 13:360–366. https://doi.org/10.1089/vbz.2012.0977
Narasimhan S, Fikrig E (2015) Tick microbiome: the force within. Trends Parasitol 31:315–323. https://doi.org/10.1016/j.pt.2015.03.010
Olivieri E, Epis S, Castelli M et al (2019) Tissue tropism and metabolic pathways of Midichloria mitochondrii suggest tissue-specific functions in the symbiosis with Ixodes ricinus. Ticks Tick Borne Dis 10:1070–1077. https://doi.org/10.1016/j.ttbdis.2019.05.019
Omondi D, Masiga DK, Fielding BC et al (2017) Molecular detection of tick-borne pathogen diversities in ticks from livestock and reptiles along the shores and adjacent Islands of Lake Victoria and Lake Baringo, Kenya. Front Vet Sci 4:1–15. https://doi.org/10.3389/fvets.2017.00073
Paddock CD, Denison AM, Dryden MW et al (2015) High prevalence of “Candidatus Rickettsia andeanae” and apparent exclusion of Rickettsia parkeri in adult Amblyomma maculatum (Acari: Ixodidae) from Kansas and Oklahoma. Ticks Tick Borne Dis 6:297–302
Pajoro M, Pistone D, Boccazzi IV et al (2018) Molecular screening for bacterial pathogens in ticks (Ixodes ricinus) collected on migratory birds captured in northern Italy. Folia Parasitol (Praha) 65:4–9. https://doi.org/10.14411/fp.2018.008
Parola P (2006) Rickettsioses in sub-Saharan Africa. Ann N Y Acad Sci 1078:42–47
Parola P, Paddock CD, Raoult D (2005) Tick-borne rickettsioses around the world: emerging diseases challenging old concepts. Clin Microbiol Rev 18:719–756. https://doi.org/10.1128/CMR.18.4.719-756.2005
Parola P, Paddock CD, Socolovschi C et al (2013) Update on tick-borne rickettsioses around the world: A geographic approach. Clin Microbiol Rev 26:657–702. https://doi.org/10.1128/CMR.00032-13
Quast C, Pruesse E, Yilmaz P et al (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
R Core Team (2019) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
René-Martellet M, Minard G, Massot R et al (2017) Bacterial microbiota associated with Rhipicephalus sanguineus (s.l.) ticks from France, Senegal and Arizona. Parasites Vectors 10:1–10. https://doi.org/10.1186/s13071-017-2352-9
Sacchi L, Bigliardi E, Corona S et al (2004) A symbiont of the tick Ixodes ricinus invades and consumes mitochondria in a mode similar to that of the parasitic bacterium Bdellovibrio bacteriovorus. Tissue Cell 36:43–53. https://doi.org/10.1016/j.tice.2003.08.004
Sassera D, Lo N, Epis S et al (2011) Phylogenomic evidence for the presence of a flagellum and cbb 3 oxidase in the free-living mitochondrial ancestor. Mol Biol Evol 28:3285–3296. https://doi.org/10.1093/molbev/msr159
Scoles GA (2004) Phylogenetic analysis of the Francisella-like endosymbionts of Dermacentor ticks. J Med Entomol 41:277–286. https://doi.org/10.1603/0022-2585-41.3.277
Serra V, Cafiso A, Formenti N et al (2018) Molecular and serological evidence of the presence of Midichloria mitochondrii in Roe Deer (Capreolus capreolus) in France. J Wildl Dis
Shannon P, Markiel A, Ozier O et al (2003) Cytoscape: a software environment for integrated models. Genome Biol Evol 13:2498–2504. https://doi.org/10.1101/gr.1239303.metabolite
Sivakumar T, Hayashida K, Sugimoto C, Yokoyama N (2014) Evolution and genetic diversity of Theileria. Infect Genet Evol 27:250–263. https://doi.org/10.1016/j.meegid.2014.07.013
Skarphédinsson S, Jensen PM, Kristiansen K (2005) Survey of tick borne infections in Denmark. Emerg Infect Dis 11:1055–1061. https://doi.org/10.3201/eid1107.041265
Smith TA, Driscoll T, Gillespie JJ, Raghavan R (2015) A Coxiella-like endosymbiontis a potential vitamin source for the lone star tick. Genome Biol Evol 7:831–838. https://doi.org/10.1093/gbe/evv016
Stanek G, Strle F (2018) Lyme borreliosis-from tick bite to diagnosis and treatment. FEMS Microbiol Rev 42:233–258. https://doi.org/10.1093/femsre/fux047
Szigeti A, Kreizinger Z, Hornok S et al (2014) Detection of Francisella-like endosymbiont in Hyalomma rufipes from Ethiopia. Ticks Tick Borne Dis 5:818–820. https://doi.org/10.1016/j.ttbdis.2014.06.002
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577
Theiler G, Salisbury LE (1959) Ticks in the South African zoological survey collection-Part IX-“The Amblyomma marmoreum group”. Onderstepoor J Vet Res 28:47–124
Uilenberg G (1997) General review of tick-borne diseases of sheep and goats world-wide. Parassitologia 39:161–165
Vanegas A, Keller C, Krüger A et al (2018) Molecular detection of spotted fever group rickettsiae in ticks from Cameroon. Ticks Tick Borne Dis 9:1049–1056. https://doi.org/10.1016/j.ttbdis.2018.03.022
Vautrin E, Vavre F (2009) Interactions between vertically transmitted symbionts: cooperation or conflict? Trends Microbiol 17:95–99. https://doi.org/10.1016/j.tim.2008.12.002
Vayssier-Taussat M, Moutailler S, Michelet L et al (2013) Next generation sequencing uncovers unexpected bacterial pathogens in ticks in western Europe. PLoS One 8. https://doi.org/10.1371/journal.pone.0081439
Walker AR, Bouattour A, Camicas J-L et al (2003) Ticks of domestic animals in Africa: a guide to identification of species. Bioscience Reports, Edinburgh
Westram R, Bader K, Prüsse E et al (2011) ARB: a software environment for sequence data. Handb Mol Microb Ecol I metagenomics Complement approaches 399–406
Zhang CM, Li NX, Zhang TT et al (2017) Endosymbiont CLS-HI plays a role in reproduction and development of Haemaphysalis longicornis. Exp Appl Acarol 73:429–438. https://doi.org/10.1007/s10493-017-0194-y
Zhong J, Jasinskas A, Barbour AG (2007) Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS ONE 2:1–7. https://doi.org/10.1371/journal.pone.0000405
Acknowledgements
This research was supported by the Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022) - Dept. of Biology and Biotechnology “L. Spallanzani”, University of Pavia (to DS).
Funding
Open access funding provided by Università degli Studi di Pavia within the CRUI-CARE Agreement. This work was supported by Human Frontiers Science Program grant RGY0075-2017 to DS and by CARIPLO Regione Lombardia rafforzamento ERC grant (2016 − 1012) to DS.
Author information
Authors and Affiliations
Contributions
The study was conceived and designed by DS and EO. EK and BK performed field studies. EO, YMT, EK performed the molecular studies. GM and MM performed the statistical analysis. MC and AMF performed the phylogenetic analysis. EO and DS drafted the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have nothing to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Olivieri, E., Kariuki, E., Floriano, A.M. et al. Multi‐country investigation of the diversity and associated microorganisms isolated from tick species from domestic animals, wildlife and vegetation in selected african countries. Exp Appl Acarol 83, 427–448 (2021). https://doi.org/10.1007/s10493-021-00598-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-021-00598-3