
Abstract
Background
Ticks are regarded as the most relevant vectors of disease-causing pathogens in domestic and wild animals. The cattle tick, Rhipicephalus (Boophilus) microplus, hinders livestock production in tropical and subtropical parts of the world where it is endemic. Tick microbiomes remain largely unexplored. The objective of this study was to explore the R. microplus microbiome by applying the bacterial 16S tag-encoded FLX-titanium amplicon pyrosequencing (bTEFAP) technique to characterize its bacterial diversity. Pyrosequencing was performed on adult males and females, eggs, and gut and ovary tissues from adult females derived from samples of R. microplus collected during outbreaks in southern Texas.
Results
Raw data from bTEFAP were screened and trimmed based upon quality scores and binned into individual sample collections. Bacteria identified to the species level include Staphylococcus aureus, Staphylococcus chromogenes, Streptococcus dysgalactiae, Staphylococcus sciuri, Serratia marcescens, Corynebacterium glutamicum, and Finegoldia magna. One hundred twenty-one bacterial genera were detected in all the life stages and tissues sampled. The total number of genera identified by tick sample comprised: 53 in adult males, 61 in adult females, 11 in gut tissue, 7 in ovarian tissue, and 54 in the eggs. Notable genera detected in the cattle tick include Wolbachia, Coxiella, and Borrelia. The molecular approach applied in this study allowed us to assess the relative abundance of the microbiota associated with R. microplus.
Conclusions
This report represents the first survey of the bacteriome in the cattle tick using non-culture based molecular approaches. Comparisons of our results with previous bacterial surveys provide an indication of geographic variation in the assemblages of bacteria associated with R. microplus. Additional reports on the identification of new bacterial species maintained in nature by R. microplus that may be pathogenic to its vertebrate hosts are expected as our understanding of its microbiota expands. Increased awareness of the role R. microplus can play in the transmission of pathogenic bacteria will enhance our ability to mitigate its economic impact on animal agriculture globally. This recognition should be included as part of analyses to assess the risk for re-invasion of areas like the United States of America where R. microplus was eradicated.
Similar content being viewed by others


Background
Ticks are considered to be second only to mosquitoes as worldwide vectors of human diseases, but they are regarded as the most relevant vectors of disease-causing pathogens in domestic and wild animals [1]. The cattle tick, Rhipicephalus (Boophilus) microplus, hinders livestock production in tropical and subtropical parts of the world where it is endemic. For example, the economic impact on the cattle industry in Brazil by the cattle tick R. microplus is estimated to be two billion U.S. dollars annually [2]. In addition to direct economic loss associated with blood feeding by R. microplus during infestation, indirect effects are also significant due to the transmission of diseases like bovine babesiosis and anaplasmosis caused by the apicomplexan protozoans Babesia bovis and Babesia bigemina, and the bacterium Anaplasma marginale, respectively. The vector competency of R. microplus for A. marginale suggests that other microbial associations with this tick host may exist. However, quantitative and qualitative information on the composition of bacterial communities in R. microplus is scarce.
Seminal studies by Smith and Kilbourne at the end of the 19th century demonstrating that Rhipicephalus annulatus transmitted B. bigemina triggered research on other microorganisms harbored by ticks [3, 4]. Currently, our understanding of ticks as vectors of infectious agents has advanced to the point where some tick-borne bacterial diseases are considered an emerging infectious threat globally [5, 6]. It is estimated that the number of described tick-borne pathogens affecting humans and animals will increase as research on tick biology and ecology progresses [7]. In some cases, species related to pathogenic bacteria were detected and identified in ticks before their effect on human health was fully determined [8]; but our knowledge of bacterial communities in ticks beyond pathogenic species is limited, even though the association between non-pathogenic bacteria and ticks was documented at the beginning of the 20th century [9].
Bacteria are ubiquitous microorganisms and some have evolved symbioses with ticks. In addition to transmitting pathogenic bacteria that include species in the genera Borrelia, Rickettsia, Francisella, Ehrlichia, Anaplasma, and Coxiella, ticks also harbor bacterial endosymbionts which can have commensal, mutualistic, or parasitic relationships with their tick hosts [10–12]. The study of bacterial communities in ticks that transmit disease-causing agents has revealed new microbial associations including previously unknown tick-borne pathogens or vector competencies [13–15]. Elucidating the taxonomic composition of symbiotic bacteria facilitates our understanding of phylogenetic relationships between symbionts and the evolutionary biology of their association with tick hosts [16]. Microbial interactions within the tick host may influence pathogen characteristics and dynamics including transmission [17, 18]. Additionally, the functional and genomic characterization of endosymbionts could provide opportunities for genetic engineering whereby transformants could be developed for use as microbial acaricides.
Molecular methods offer an expedient and efficient opportunity to analyze bacterial communities in ticks avoiding the need for intensive culture-based techniques, and furthermore, allow the identification of species which are not amenable to culturing. Specifically, pyrosequencing of partially amplified 16S rRNA sequences has been applied to study the composition of bacteria associated with biological systems including insect vectors [19–21]. Here, we evaluated bacterial diversity associated with R. microplus using bTEFAP. Bacterial composition was investigated in the egg, adult male and female life stages, and ovary and gut tissues from adult female cattle ticks. This report represents the first comprehensive survey of bacterial communities associated with the cattle tick using a culture-independent method.
Results
Estimated richness and diversity of bacterial communities
The application of bTEFAP reported here enabled us to explore the genome of bacterial symbionts, i.e. the microbiome, living inside and outside the cattle tick R. microplus as a means to initiate the characterization of the microbiota associated with this tick species of economic significance in animal agriculture worldwide. A total of 183,626 sequences were generated and a total of 130,019 sequences utilized for analyses of the 18 samples. Thus, an average of 7200 sequences > 350 bp (avg length 450 bp) per sample were analyzed after all quality control and screening steps. Indices of bacterial richness and diversity, based on Operational Taxonomic Unit (OTU) estimated through Rarefaction curve, Ace, and Chao1 procedures, are summarized in Table 1. Rarefaction and Richards maximum predicted curve modeling indicated that > 98% of OTUs at the 5% divergence were achieved for each sample [22], which suggests adequate depth of coverage (data not shown). Although results are presented at the 1, 3, and 5% dissimilarity levels, attention is focused on OTUs at 5% dissimilarity since it has been reported that reasonable genus-level richness can be achieved using that degree of discrimination [22]. By rarefaction analysis estimates, the trend for genera richness at 5% dissimilarity was: egg>gut > adult male > adult female > ovary.
Identification and quantification of bacterial taxa
In addition to surveying bacterial diversity across tick life stages and tissues, pyrosequencing also allowed assessment of the relative abundance of the taxonomic levels of bacteria detected (Figure 1). Tracebacks entered as "sp" in Figure 1 indicate that the characterization required for identification at the species level exists, but consensus on the particular nomenclature was lacking at the time groupings were done. However, tracebacks with the suffix "_genus" indicate that they may represent novel bacterial species. Genera that may include previously undescribed species of bacteria associated with the cattle tick include Coxiella, Achromobacter, Corynebacterium, Staphyloccocus, Anaerobiospirillum, Roseburia, Prevotella, Nocardioides, and Vagoccocus.

Heat map depicting bacterial diversity and relative abundance in life stages and tissue samples from R. microplus. * Letters (A-C) used to identify individual life stage samples where applicable. Double hierarchical dendogram shows different bacteria distribution between taxonomic levels based on complete linkage clustering, and Manhattan distance methods with no scaling. Dendrogram linkages and distance of the bacterial taxa or traceback groups are not phylogenetic, but based upon relative abundance of the taxa within the samples. Traceback means bacterial classifications were based upon the percent identity of the sample sequence to known sequences, the percent divergence was then used to adjust identifications to the taxonomic level with the highest degree of confidence (e.g. a percent divergence < 3% can be expected to provide confidence at the species level, > 3% but < 5% at the genera level, etc.). Classifications were compiled after traceback. Legend and scale shown in upper left corner of the figure represent colors in heat map associated with the relative percentage of each traceback grouping of bacteria (cluster of variables in Y-axis) within each tick sample (X-axis clustering). Tick samples along the X-axis with Manhattan distances are indicated by branch length and associated with the scale located at the upper right corner of the figure. Bacterial traceback groups along the Y-axis are also clustered according to Manhattan distances; the respective scale is indicated in the figure's lower left corner.
Bacteria identified to the species level include Staphylococcus aureus, Staphylococcus chromogenes, Streptococcus dysgalactiae, Staphylococcus sciuri, Serratia marcescens, Corynebacterium glutamicum, and Finegoldia magna. Staphylococcus aureus was present in adult males, eggs, and the gut of adult female cattle ticks. Similar findings were reported for the closely related tick species Rhipicephalus decoloratus and Rhipicephalus geigyi in Africa where S . aureus was isolated from the hemolymph of adult females and their eggs [23]. However, there was no evidence of transovarial transmission for S . aureus in those tick species. We detected S . chromogenes in adult male and female ticks. Staphylococcus chromogenes was isolated previously from R . microplus collected in Australia using a culture-dependent approach after the ticks had been surface-sterilized [24]. Staphylococcus chromogenes is part of the natural skin flora of cattle but can cause mastitis, and in pigs it may provoke exudative epidermitis [25, 26].
The other five bacterial species represent previously unreported associations for R. microplus. Whereas C. glutamicum and S. marcescens were detected in eggs only, S. sciuri was present in male and female ticks, F. magna in eggs and female ticks, and S. dysgalactiae in eggs, male ticks, and female ticks. Because of our permissive approach to assess bacterial diversity, e.g., the ticks used in this study were not disinfected prior to DNA extraction, the prevalence of these new bacterial associations with R. microplus needs to be confirmed. However, it is relevant to note that S. dysgalactiae and S. marcescens are known to cause bovine mastitis, whereas F. magna was detected in papillomatous digital dermatitis lesions of cattle [27–29]. Staphylococcus sciuris is commonly found in the skin of cattle and other animals, while the natural habitats of C. glutamicum include soil, soils contaminated with bird feces, sewage and manure, and vegetables and fruits [30, 31]. In their natural environment, R. microplus eggs may be exposed to C. glutamicum after oviposition by gravid female ticks.
Clustering analysis showed that the microbial biota detected in the ovary tissue of adult female ticks was the most dissimilar tissue of all the tick samples tested (Figure 1). Additionally, the least diverse microbial biota was detected in this tissue. Members of the Coxiellaceae family were the most prevalent bacteria in cattle tick ovary. Consistent with this finding, the Coxiellaceae were also found in the egg and adult female samples (Figure 1).
Relative abundance of bacterial genera by tick life stage and tissue sample
One hundred twenty-one bacterial genera were detected in all the life stages and tissues sampled in this study (see Additional File 1 Table S1). Among the genera found in our study, Arthrobacter, Bacillus, Curtobacterium, Enterobacter, Microbacterium, Paenibacillus, Pantoea, Pseudomonas, Rhodococcus, Serratia, Staphylococcus, and Stenotrophomonas are genera previously reported to be harbored by R. microplus isolated from ticks collected in Australia [24]. Enterobacter, Pseudomonas, and Staphylococcus, found in both our study and the Australian study, were also cultured from homogenates of R. microplus in Bangladesh that were produced following surface sterilization and dissections using sterile technique [32]. Infection with Achromobacter and Escherichia was previously reported for cattle ticks from the Bangladesh study but not the Australian study.
Among the life stages sampled, the total number of bacterial genera detected in the egg, adult male, and adult female ticks was 54, 53, and 61, respectively (Additional File 1 Table S1). Of those numbers, 25, 25, and 27 genera were unique to the egg, adult male, and adult female life stages, respectively. By comparison, only 7 bacterial genera were identified in tick ovary, whereas 11 genera were found in the tick gut. Cryobacterium, Rhodococcus, and Veillonella were identified only in the ovary, whereas Anaerobiospirillum was the only genera unique to the gut.
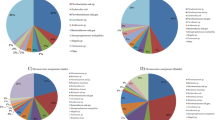
The molecular approach applied in this study allowed us to assess the relative abundance of the microbiota associated with R. microplus. The predominant genera in the bacterial communities of the tick samples analyzed based on an abundance cutoff of 1.0% are shown for each sample in Figure 2. Staphylococcus was relatively abundant ( > 18%) in adult males and eggs, but not in adult female ticks. Other prevalent genera were Corynebacterium ( > 13%) in eggs and adult males, and Coxiella ( > 13%) in tick eggs. Achromobacter (27.7%), Pseudomonas (12.6%), and Sinorhizobium (7.7%) were the predominant genera found in adult female ticks. Among the tissues sampled, Coxiella was the most abundant (98.2%) genus in ovary, whereas Anaerobiospirillum (29.5%) and Brachybacterium (21.9%) predominated in the tick gut. Other relatively less abundant genera, but worth noting, include Borrelia (7.9%) in the tick gut; Clostridium (3.9%) in adult female ticks; Escherichia (1.5%) in the tick gut; Klebsiella (1.3%) in adult female ticks; Streptococcus in eggs (2.9%) and adult males (1.%); Enterococcus in adult male ticks (1.4%), adult female ticks (2.2%), and tick gut (11.4%); and Wolbachia in adult female ticks (1.8%).

Relative abundance of bacterial genera in life stages and tissue samples from R. microplus as detected by bTEFAP pyrosequencing. a) Adult female cattle tick. Mean percentages (n = 2). Values below 1% were grouped as "Other" with total value of 9.5%. "Other" group includes: Staphylococcus (0.7%), Bacillus (0.5%), Streptococcus (0.7%), Vagococcus (0.3%), Pseudobutyrivibrio (0.7%), Nocardioides (0.2%), Asteroleplasma (0.9%), Ruminococcus (0.4%), Escherichia (0.9%), Acetivibrio (0.3%), Erwinia (0.1%), Pedobacter (0.2%), Dermabacter (0.1%), Ornithinicoccus (0.2%), Oribacterium (0.7%), Alkaliflexus (0.2%), Paludibacter (0.5%), Pantoea (0.2%), Cytophaga (0.1%), Mitsuokella (0.1%), Enterobacter (0.1%), Paucisalibacillus (0.4%), Lachnobacterium (0.1%), Caldithrix (0.2%), Shigella (0.1%), Solirubrobacter (0.1%), Rhodobacter (0.1%), Desulfosporosinus (0.1%). b) Adult male cattle tick. Mean percentages (n = 2). Values below 1% were grouped as "Other" with total value of 3.8%. "Other" group includes: Coxiella (0.1%), Prevotella (0.3%), Rikenella (0.1%), Pseudomonas (0.2%), Escherichia (0.3%), Hallella (0.3%), Pantoea (0.1%), Moraxella (0.7%), Arthrobacter (0.1%), Enhydrobacter (0.1%), Mogibacterium (0.1%), Kocuria (0.5%), Enterobacter (0.1%), Exiguobacterium (0.2%), Lysinibacillus (0.1%), Belnapia (0.1%). c) Cattle tick egg. Mean percentages (n = 3). Values below 1% were grouped as "Other" with total value of 6.9%. "Other" group includes: Achromobacter (0.3%), Enterococcus (0.1%), Clostridium (0.1%), Serratia (0.7%), Ruminococcus (0.3%), Propionibacterium (0.4%), Klebsiella (0.2%), Acetivibrio (0.9%), Pedobacter (0.6%), Alkaliflexus (0.4%), Centipeda (0.5%), Pantoea (0.1%), Brevibacterium (0.2%), Rubrivivax (0.4%), Enhydrobacter (0.2%), Rhodoferax (0.3%), Sporocytophaga (0.1%), Alkanindiges (0.2%), Sphingopyxis (0.1%), Caulobacter (0.1%), Trichococcus (0.1%), Comamonas (0.1%), Anaerotruncus (0.1%), Akkermansia (0.1%), Legionella (0.1%). d) Adult female cattle tick gut. Pool of tissue from five ticks tested. Values below 1% were grouped as "Other" with total value of 0.3%. "Other" group includes: Corynebacterium (0.3%). e) Adult cattle tick ovary. Pool of tissue from five ticks tested. Values below 1% were grouped as "Other" with total value of 1.8%. "Other" group includes: Borrelia (0.9%), Cryobacterium (0.9%).
Discussion
To our knowledge, this study represents the first exploration of the diversity of the bacterial biota associated with distinct life stages and tissues of the cattle tick, R. microplus using a nonculturable method. Previous surveys of bacterial diversity in R. microplus employed culture methods, and for the most part, those studies focused on the isolation of bacteria pathogenic to the tick and vertebrate hosts [24, 32–34]. The tag-encoded pyrosequencing approach reported here allowed us to detect and identify bacteria that otherwise might be fastidious, obligate intracellular, or noncultivable. Surveys of bacteria based on 16S rRNA gene sequences have proven useful to analyze the microbiome of bacterial communities in different habitats on and inside the host's body [35]. Our understanding of the ecology and eco-pathogenic relevance of tick-bacterial relationships is expanding as new associations are revealed through 16S rRNA gene-based analyses [14, 36, 37]. We probed deeply into the cattle tick microbiome using the 16S-bTEFAP technique. One hundred seven bacterial genera reported here represent new microbial associations for R. microplus. It has been suggested that the analysis of individual ticks could increase the ability to recognize bacteria in low copy numbers whereas the analysis of dissected organs would exclude the detection of external environmental bacteria [36]. We took a mixed approach by sampling ticks individually, without sterilization and prior to DNA isolation, for broad-range analysis of bacterial communities, while the gut and ovary were dissected for testing. Unique bacteria genera associations were detected for each of the tick samples tested. The symbiotic relationships for the bacterial genera associated with R. microplus remain to be characterized.
Although transovarial transmission enables bacterial colonization very early in the tick life cycle, copulation and egg fertilization could augment bacteria-tick associations through possibly infected sperm or the microbiota associated with the female genital tract [38]. It remains to be determined if antimicrobial activity occurs in R. microplus ejaculate, as has been shown for other arthropod species [39, 40]. The environment where eggs are deposited influences the type of bacterial communities they are exposed to, which in some cases can include bacteria pathogenic to engorged females [41]. Ecological factors related to questing behavior facilitate contact with bacteria in the environment and expand the complexity of bacterial communities residing on a tick's exoskeleton. Further investigation of the microbiota in the tick exoskeleton is needed to understand the ecology of that microbial habitat in the context of host-microbe and microbe-microbe interactions. Studies in other biological systems have revealed the complexity of such interactions that offer the opportunity to develop novel diagnostic and therapeutic interventions [42, 43], which in the context of this study could translate into options for tick biological control. Once on the host, ticks come in contact with the skin microbiota and become exposed to infected blood to fulfill their obligate hematophagous habit, or other host body fluids, while searching for and attaching at predilection sites. Systemic infection with bacteria acquired from the host skin, including S. marcescens, was documented in Dermacentor andersoni following a stringent, sterile sample processing protocol prior to tick trituration and media inoculation with the resulting suspension [44]. Here, it is documented that R. microplus harbors S. marcescens. Isolation of the bacterial genera Staphylococcus from R. annulatus and R. decoloratus, and Streptococcus from R. annulatus without specific characterization was reported previously [41, 45, 46]. Thus, systemic infection of R. microplus with S. sciuri and S. dysgalactiae may have occurred through host skin contact. This route of infection could also apply to F. magna because of its presence in the host skin habitat. Since C. glutamicum was detected in eggs laid by females collected in the field, it is possible that the ticks acquired the bacterium from hosts exposed to environmental sources. Given their economic impact on livestock production systems, our results indicate cattle transmission studies are warranted using R. microplus infected with S. dysgalactiae, S. marcescens, and F. magna.
The detection of S. chromogenes in cattle ticks from Australia and outbreaks in the USA, as well as the suite of bacterial genera shared by specimens from Australia, Bangladesh, and the USA noted here suggest that there may be a core microbiome associated with R. microplus. Alternatively, bacteria found in common between R. microplus, R. annulatus, R. decoloratus, and R. geigyi indicates that microbiota composition is influenced by the ecological niche they occupy during the parasitic stage, i.e. cattle. More extensive surveys are required to ascertain the biogeography of the microbiome across time and space as well as among and between R. microplus populations. As it has been shown for other anthropod vector-bacteria systems, these studies will help determine if bacterial communities associated with R. microplus represent random assemblages and define the influence of biotic and abiotic factors [14, 21, 37]. However, special attention is needed to harmonize sampling methods and molecular protocols given the rapid development of massively parallel sequencing technologies to facilitate meaningful comparisons. Additionally, it has been hypothesized that at least two tick species have evolved under the R. microplus designation [47]. The apparent co-evolution of certain bacterial lineages with their hosts warrants the application of that concept to test the hypothesis of genetic and reproductive divergence between geographic strains of R. microplus [12, 47–49].
The Coxiella-type microbe we detected in R. microplus can be presumed to be an endosymbiont. Although more abundant in adult females, ovary, and eggs, a weak signal for the Coxiella microbe was noticed in one male tick. A similar observation was reported for a Coxiella endosymbiont in Amblyomma americanum [14, 37]. Its presence in ovary and eggs indicates that the putative R. microplus-associated Coxiella endosymbiont can be transmitted vertically. Most of the bacterial sequences detected in the ovary were ascribed to the Coxiella microbe. This may result from selective amplification of the Coxiella symbiont associated with the expansion of ovarian tissue that takes place during engorgement since the ovary was collected from replete female R. microplus undergoing active oviposition [37, 50]. The degree of relatedness between the R. microplus-associated Coxiella symbiont, Coxiella endosymbionts in other tick species, and C. burnetii remains to be determined. This will facilitate testing the hypothesis that the R. microplus-associated Coxiella microbe is a primary endosymbiont as documented for the Coxiella spp. infecting A. americanum, which showed a reduced genome in comparison to C. burnetii [50, 51]. Rhipicephalus microplus has been found to harbor C. burnetii in India and China [52, 53]. Our inability to detect C. burnetii in R. microplus from outbreaks in the USA suggests that the pathogen is not circulating in that tick population; alternatively, its presence in very low numbers prevented detection through the method used in this study. The concept of targeting endosymbionts as a means to control ticks and tick-borne diseases has been tested taking the chemotherapeutic approach [54, 55]. Using antibiotics to treat the infection of A. americanum with a Coxiella spp. endosymbiont resulted in reduced reproductive fitness [55]. Novel approaches for endosymbiont isolation and characterization will facilitate in vitro culture to produce reagents for testing of the immunological approach to control ticks targeting their endosymbionts [54, 56].
Our understanding of the associations between R. microplus and members of the genus Borrelia keeps expanding. Borrelia theileri, the etiologic agent of bovine borreliosis, has been shown to be transmitted by R. microplus in many parts of the world [57]. Borrelia burgdorferi was isolated from R. microplus in China [58]. Detection of the R. microplus-associated Borrelia in the gut and ovary reported here parallels the systemic infection with B. theileri where no adverse effects were observed in tick viability [33, 59]. Like the Borrelia DNA sequences detected in this study, specific identification awaits for other Borrelia microbes isolated from R. microplus in diverse geographic locations [60–62]. However, R. microplus may be acting as a bridging vector facilitating the transmission of microbes across vertebrate hosts and possibly influencing ecological and evolutionary aspects of their natural history. The degree of similarity at the nucleotide level between a Mexican isolate of B. theileri and Borrelia spp. infecting A. americanum from the Northeast region of the USA suggests recent divergence [63]. Because white-tailed deer and cattle used to be sympatric throughout the southern USA prior to 1943, which is when cattle ticks were officially eradicated, it has been hypothesized that spirochetes infecting A. americanum may represent a host shift of B. theileri as R. microplus could have transmitted the spirochete to both ungulate hosts [64]. A Borrelia spp. detected in R. microplus from Brazil was shown to be closely related to B. theileri and Borrelia lonestari and the cattle tick-deer relationship was suggested as a natural process for the spread and/or maintenance of Borrelia spp. [65].
Although bacteria in the genus Wolbachia are generally found in reproductive tissues, the R. microplus-associated Wolbachia was not detected in ovarian tissue, but in the two adult female ticks assayed individually. Since ticks from a laboratory colony established in 1999 were the source of the ovarian tissue samples, it is plausible that Wolbachia infection was lost during the colonization process. It is also possible that laboratory rearing conditions allowed the Coxiella strain in the R. microplus ovaries sampled to out-compete pre-existing Wolbachia microbes with the eventual loss of infection in La Minita strain. Detection of the Wolbachia-type microbe in adult female ticks does not necessarily mean that the ovary was the only tissue infected. Disseminated Wolbachia infection has been documented in other arthropod vector species and similar events were reported for a Coxiella endosymbiont infecting A. americanum where the salivary glands were also infected [50, 66]. The possibility for horizontal transmission would exist if Wolbachia infection of the R. microplus salivary glands were to occur. The horizontal transmission of Wolbachia microbes has been documented to occur more often than previously thought [67–69]. However, it has been shown in mosquitoes that the size of Wolbachia symbionts would prevent its free passage through the salivary ducts [70].
The functional relevance of our findings and observations needs to be tested. Proof of active infection is suggested to confirm the physiological significance of bacterial DNA detection in R. microplus by tag-encoded pyrosequencing or any other molecular or non-culturable approach. Rhipicephalus microplus has evolved various defense mechanisms acting in the hemocel if the external physical barrier represented primarily by the exoskeloton is bridged. Antimicrobial peptides form part of the cattle tick immune system [71, 72]. Additionally, at least two types of R. microplus hemocytes exist that effect phagocytosis and production of reactive oxygen species [73]. Other components of the cattle tick immune system are likely to be discovered as additional functions are identified and assigned to the hemocyte transcriptome [74]. Caution must also be exercised in defining the relationship of bacteria found to be associated with R. microplus in this study. Although a particular genus may include pathogenic species, several of the bacterial genera detected and reported here for the first time in association with the cattle tick comprise groups commonly found in soil, on the surface of plants, or considered enteric bacteria. However, similar results from studies where stringent surface-sterilization was performed and negative controls were tested indicate that such bacteria are truly associated with R. microplus [14, 37]. Lastly, blood feeding has been shown to increase bacterial diversity [37]. Thus, comparative analyses of the R. microplus microbiome between immature stages, unfed and blood-fed ticks across life stages, laboratory colony and wild-caught specimens, and additional organs and tissues are warranted [37].
It is worth noting that certain bacteria were detected in R. microplus by investigators in other parts of the world. Rhipicephalus microplus was found to harbor Rickettsia conorii in India [52]. Ehrlichia canis and a new Ehrlichia species closely related to Ehrlichia chaffeensis were detected in cattle ticks in China and the Thai-Myanmar border [53, 58, 75]. Additionally, R. microplus in the Caribbean contained Ehrlichia ruminantium DNA [76]. Our findings suggest that these pathogens of economic importance to livestock production systems are not circulating among outbreak strains of R. microplus in the USA. However, those studies highlight the potential role of R. microplus as vector of zoonotic bacteria. Although it is considered a rare event, R. microplus can parasitize humans [77, 78].
The analysis of our results in the context of previous bacterial surveys provides an indication of geographic variation in the assemblages of bacteria associated with R. microplus. Additional reports on the identification of new bacterial species maintained in nature by R. microplus that may be pathogenic to its vertebrate hosts are expected as our understanding of its microbiota expands. Increased awareness of the role R. microplus can play in the transmission of pathogenic bacteria will enhance our ability to mitigate its economic impact on animal agriculture globally. This recognition should be included as part of analyses to assess the risk for re-invasion of areas where R. microplus was eradicated like the USA.
Conclusion
Tick microbiomes remain largely unexplored. By comparison to the proposed strategy to accomplish the Human Microbiome Project, the work presented here constitutes the initial data acquisition and analysis exercise towards a comprehensive analysis of the R. microplus microbiome. A thorough understanding of the functional, ecological, and evolutionary aspects of the bacterial diversity in communities associated with the cattle tick requires additional investigations. The bacteria we found could have favorable effects on the tick's successful infestation of its cattle host, perhaps with roles in host blood digestion, immunity against infection by competing microbes potentially deleterious to the tick, or effects on population structure and fertility. Cattle ticks have evolved in conjunction with bovine hosts; therefore, bovine-tick interactions have likely influenced the ecology of their microbiomes. Even within the tick itself, there are feedback mechanisms influencing interactions at the host-microbiome interface. Our results further document the co-infection of cattle ticks with several bacteria, even in the presence of antimicrobial factors that are known to be produced by the tick immune system response in their hemolymph and gut tissues. Further investigations on the cattle tick microbiome are likely to enhance our understanding of the roles this cosmopolitan species serves as vector of bacteria that may be pathogenic to its vertebrate hosts.
Methods
Tick samples
Adult male and female ticks were obtained from a R. microplus infestation outbreak on cattle from Starr County, TX. Samples from the infestation were collected by USDA personnel in November, 2008, and shipped to the USDA Cattle Fever Tick Research Laboratory in Moore Field, TX, where the samples were frozen at -80°C. Prior to freezing, eggs were collected from gravid females, mixed together, and pooled and labeled as f1 generation. A portion of these f1 eggs were used to establish a laboratory colony to obtain adult ticks as described previously [79]. Two adult females and two adult males developed from these f1 eggs and three small clumps of approximately 100 f1 eggs each were used for the DNA extraction and pyrosequencing. The gut and ovary samples were obtained from the f20 generation of the La Minita strain of R. microplus that has been maintained Babesia-free at the University of Idaho Holm Research Center since 1999. The founding ticks for this strain were originally collected in Starr County, TX, in 1996. Using standard protocols approved by the University of Idaho Institutional Animal Care and Use Committee, La Minita larvae were placed on a stanchioned calf and replete females collected and dissected under sterile phosphate-buffered saline during the period of active oviposition. The ventral cuticle was excised; the ovaries and gut separately removed, rinsed in sterile saline, and held on ice until 5 ticks were dissected. The tissues were frozen at -80°C. A clump (~5 mm diameter) of the frozen material was broken off and used for pyrosequencing analysis. All samples used in this study were collected under open benchtop conditions. Neither surface sterilization nor sterile dissection techniques were employed during sample preparation steps prior to DNA extractions.
Pyrosequencing and analysis
Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) was conducted as described previously [19, 20]. Our approach was modified slightly to utilize the Titanium sequencing platform rather than FLX (Roche Applied Science, Indianapolis, IN) to take advantage of the longer average read lengths generated by the Titanium methodology. Additionally, we used a single 35 cycle PCR step with Qiagen HotStar Master Mix and addition of 0.5U of HotStar HiFidelity Polymerase in each reaction (Qiagen Inc., Valencia, CA). Finally, sequences used for analysis had average read length of ~450 bp with sequencing extending from the 27F 5' GAG TTT GAT CNT GGC TCA G 3' to 519r 5' GTN TTA CNG CGG CKG CTG 3' in relation to E. coli 16S extending across V1 and into the V3 ribosomal region (Research and Testing Laboratory, Lubbock, TX). Amplicon sequencing was performed based upon the manufacturers protocols (Roche Applied Science, Indianapolis, IN) for Titanium sequencing on FLX-titanium platform. Raw data from bTEFAP was screened and trimmed based upon quality scores of Phred20 average and binned into individual sample collections. Sequence collections were then depleted of chimeras using B2C2 [80].
The resulting files were then depleted of short reads (< 350 bp) and bacterial species identified using BlastN comparison to a quality controlled and manually curated database derived from the NCBI. Data was compiled and relative percentages of each bacterial identification were determined for each individual sample. Data was also compiled at each individual taxonomic level according to the NCBI taxonomy criteria as described previously [19, 20]. Rarefaction, Ace, and Chao 1 analyses to estimate mathematically predicted diversity and richness in tick samples was performed with DOTUR as described elsewhere [22, 81].
References
de la Fuente J, Estrada-Peña A, Venzal JM, Kocan KM, Sonenshine DE: Overview: ticks as vectors of pathogens that cause disease in humans and animals. Front Biosc. 2008, 13: 6938-6946. 10.2741/3200.
Grisi L, Massard CL, Moya-Borja GE, Pereira JB: Impacto econômico das principais ectoparasitoses em bovinos no Brasil. A Hora Veterinária. 2002, 21: 8-10.
Dutton JE, Todd JL: The nature of tick fever in the eastern part of the Congo Free State, with notes on the distribution and bionomics of the tick. Br Med J. 1905, 2: 1259-1260.
Ricketts HT: Some aspects of Rocky Mountain spotted fever as shown by recent investigations. Med Rec. 1909, 76: 843-855.
Hotez PJ, Kamath A: Neglected tropical diseases in sub-Saharan Africa: review of their prevalence, distribution, and disease burden. PloS Negl Trop Dis. 2009, 3: e412-10.1371/journal.pntd.0000412.
Heyman P, Cochez C, Hofhuis A, van der Giessen J, Sprong H, Porter SR, Losson B, Saegerman C, Donoso-Mantke O, Niedrig M, Papa A: A clear and present danger: tick-borne diseases in Europe. Expert Rev Anti Infect Ther. 2010, 8: 33-50. 10.1586/eri.09.118.
Parola P, Raoult D: Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Inf Dis. 2001, 32: 897-928. 10.1086/319347.
Schouls LM, Van De Pol I, Rijpkema SG, Schot CS: Detection and identification of Ehrlichia, Borrelia burgdorferi sensu lato, and Bartonella species in Dutch Ixodes ricinus ticks. J Clin Microbiol. 1999, 37: 2215-2222.
Cowdry EV: A group of microorganisms transmitted hereditarily in ticks and apparently unassociated with disease. J Exp Med. 1925, 41: 817-830. 10.1084/jem.41.6.817.
Noda H, Munderloh UG, Kurtti TJ: Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol. 1997, 63: 3926-3932.
Sacchi L, Bigliardi E, Corona S, Beninati T, Lo N, Franceschi A: A symbiont of the tick Ixodes ricinus invades and consumes mitochondria in a mode similar to that of the parasitic bacterium Bdellovibrio bacteriovorus. Tissue Cell. 2004, 36: 43-53. 10.1016/j.tice.2003.08.004.
Scoles GA: Phylogenetic analysis of the Francisella-like endosymbionts of Dermacentor ticks. J Med Entomol. 2004, 41: 277-286. 10.1603/0022-2585-41.3.277.
Burgdorfer W, Brinton LP, Hughes LE: Isolation and characterization of symbionts from the Rocky Mountain wood tick, Dermacentor andersoni. J Invert Pathol. 1973, 22: 424-434. 10.1016/0022-2011(73)90173-0.
Clay K, Klyachko O, Grindle N, Civitello D, Oleske D, Fuqua C: Microbial communities and interactions in the lone star tick, Amblyomma americanum. Mol Ecol. 2008, 17: 4371-4381. 10.1111/j.1365-294X.2008.03914.x.
Vilcins IE, Fournier P, Old JM, Deane E: Evidence for the presence of Francisella and spotted fever group Rickettsia DNA in the tick Amblyomma fimbriatum (Acari: Ixodidae), Northern territory, Australia. J Med Entomol. 2009, 46: 926-933. 10.1603/033.046.0427.
Rymaszewska A: Symbiotic bacteria in oocyte and ovarian cell mitochondria of the tick Ixodes ricinus: biology and phylogenetic position. Parasitol Res. 2007, 100: 917-920. 10.1007/s00436-006-0412-8.
Macaluso KR, Sonenshine DE, Ceraul SM, Azad AF: Rickettsial infection in Dermacentor variabilis (Acari: Ixodidae) inhibits transovarial transmission of a second Rickettsia. J Med Entomol. 2002, 39: 809-813. 10.1603/0022-2585-39.6.809.
de la Fuente J, Blouin EF, Kocan KM: Infection exclusion of the rickettsial pathogen Anaplasma marginale in the tick vector Dermacentor variabilis. Clin Diagn Lab Immun. 2003, 10: 182-184.
Dowd SE, Sun Y, Wolcott RD, Domingo A, Carroll JA: Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) for microbiome studies: bacterial diversity in the ileum of newly weaned Salmonella-infected pigs. Foodborne Pathog Dis. 2008, 5: 459-472. 10.1089/fpd.2008.0107.
Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS: Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008, 8: 125-10.1186/1471-2180-8-125.
Jones TR, Knight R, Martin AP: Bacterial communities of disease vectors sampled across time, space, and species. ISME J. 2010, 4: 223-231. 10.1038/ismej.2009.111.
Acosta-Martínez V, Dowd S, Sun Y, Allen V: Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol Chem. 2008, 40: 2762-2770.
Amoo AO, Dipeolu OO, Akinboade AO, Adeyemi A: Bacterial isolation from and transmission by Boophilus decoloratus and Boophilus geigyi. Folia Parasitol. 1987, 34: 69-74.
Murrel A, Dobson SJ, Yang X, Lacey E, Barker SC: A survey of bacterial diversity in ticks, lice and fleas from Australia. Parasitol Res. 2003, 89: 326-334.
Devriese LA, Baele M, Vaneechoutte M, Martel A, Haesebrouk F: Identification and antimicrobial susceptibility of Staphylococcus chromogenes isolates from intramammary infections of dairy cows. Vet Microbiol. 2002, 87: 175-182. 10.1016/S0378-1135(02)00047-0.
Andresen LO, Ahrens P, Daugaard L, Bille-Hansen V: Exudative epidermitis in pigs caused by toxigenic Staphylococcus chromogenes. Vet Microbiol. 2005, 105: 291-300. 10.1016/j.vetmic.2004.12.006.
Garvie EI, Farrow JAE, Bramley AJ: Streptococcus dysgalactiae (Diernhofer) nom. rev. Int J Syst Bacteriol. 1983, 33: 404-405. 10.1099/00207713-33-2-404.
Bannerman DD, Paape MJ, Goff JP, Kimura K, Lippolis JD, Hope JC: Innate immune response to intramammary infection with Serratia marcescens and Strepococcus uberis. Vet Res. 2004, 35: 681-700. 10.1051/vetres:2004040.
Yano T, Moe KK, Yamazaki K, Ooka T, Hayashi T, Misawa N: Identification of candidate pathogens of papillomatous digital dermatitis in dairy cattle from quantitative 16S rRNA clonal analysis. Vet Microbiol. 2010, 143: 352-362. 10.1016/j.vetmic.2009.12.009.
Nagase N, Sasaki A, Yamashita K, Shimizu A, Wakita Y, Kitai S, Kawano J: Isolation and species distribution of staphylococci from animal and human skin. J Vet Med Sci. 2002, 64: 245-250. 10.1292/jvms.64.245.
Liebl W: From Corynebacterium Taxonomy. Handbook of Corynebacterium glutamican. Edited by: Eggeling L, Bott M. 2005, Florida: Taylor & Francis Group, 9-36. full_text.
Rahman MH, Rahman MM: Occurrence of some bacterial isolates in ticks found in Madhupur Forest Area. Bang Vet Jour. 1980, 14: 43-47.
Smith RD, Brener J, Osorno M, Ristic M: Pathobiology of Borrelia theileri in the tropical cattle tick, Boophilus microplus. J Invertebr Pathol. 1978, 32: 182-190. 10.1016/0022-2011(78)90028-9.
Brum JGW, Teixeira MO: Acaricidal activity of Cedecea lapagei on engorged females of Boophilus microplus exposed to the environment. Arq Bras Med Vet Zoot. 1992, 44: 543-544.
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI: The human microbiome project. Nature. 2007, 449: 804-810. 10.1038/nature06244.
Schabereiter-Gurtner C, Lubitz W, Rölleke S: Application of broad-range 16S rRNA PCR amplification and DGGE fingerprinting for detection of tick-infecting bacteria. J Microbiol Meth. 2003, 52: 251-260. 10.1016/S0167-7012(02)00186-0.
Heise SR, Elshahed MS, Little SE: Bacterial diversity in Amblyomma americanum (Acari: Ixodidae) with a focus on members of the genus Rickettsia. J Med Entomol. 2010, 47: 258-268. 10.1603/ME09197.
Afzelius BA, Alberti G, Dallai R, Godula J, Witalinski W: Virus- and Rickettsia-infected sperm cells in arthropods. J Invertebr Path. 1989, 53: 365-377. 10.1016/0022-2011(89)90102-X.
Joseph L, Josekumar VS, George PV: Detection of antimicrobial activity in accessory gland secretions of the virgin male red palm weevil, Rhynchophorus ferrugineus. Internet J Microbiol. 2009, 7: 1-
Otti O, Naylor RA, Siva-Jothy MT, Reinhardt K: Bacteriolytic activity in the ejaculate of an insect. Am Nat. 2009, 174: 292-295. 10.1086/600099.
Hendry DA, Rechav Y: Acaricidal bacterial infecting laboratory colonies of the tick Boophilus decoloratus (Acarina: Ixodidae). J Invertebr Pathol. 1981, 38: 149-151. 10.1016/0022-2011(81)90044-6.
Fierer N, Lauber CL, Zhou N, McDonald D, Costello EK, Knight R: Forensic identification using skin bacterial communities. PNAS. 2010, 107: 6477-6481. 10.1073/pnas.1000162107.
Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, Mizunoe Y: Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature. 2010, 456: 346-349. 10.1038/nature09074.
Steinhaus EA: The microbial flora of the Rocky Mountain Wood Tick, Dermacentor andersoni Stiles. J Bacteriol. 1942, 44: 397-404.
Ahmed LS, Dosoky RM: Some bacterial isolates from Boophilus annulatus ticks under natural conditions in Assiut Governorate. Assuit Vet Med J. 1986, 15: 199-202.
El Kammah KM, Oyoun LMI, Abdel-Shafy S: Detection of microogranisms in the saliva and midgut smears of different tick species (Acari: Ixodoidea) in Egypt. J Egypt Soc Parasitol. 2007, 37: 533-539.
Labruna MB, Naranjo V, Mangold AJ, Thompson C, Estrada-Pena A, Guglielmone AA, Jongejan F, de la Fuente J: Allopatric speciation in ticks: gentic and reproductive divergence between geographic strains of Rhipicephalus (Boophilus) microplus. BMC Evol Biol. 2009, 9: 46-10.1186/1471-2148-9-46.
Hongoh Y, Deevong P, Inoue T, Moriya S, Trakulnaleamsai S, Ohkuma M, Vongkaluang C, Noparatnaraporn N, Kudo T: Intra- and interspecific comparsions of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Appl Environ Microb. 2005, 71: 6590-6599. 10.1128/AEM.71.11.6590-6599.2005.
Dethlefsen L, McFall-Ngai M, Relman DA: An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007, 449: 811-818. 10.1038/nature06245.
Klyachko O, Stein BD, Grindle N, Clay K, Fuqua C: Localization and visualization of a Coxiella-type symbiont within the lone star tick, Amblyomma americanum. Appl Environ Microb. 2007, 73: 6584-6594. 10.1128/AEM.00537-07.
Jasinskas A, Zhong J, Barbour AG: Highly prevalent Coxiella sp. bacterium in the tick vector Amblyomma americanum. Appl Environ Microb. 73: 334-336. 10.1128/AEM.02009-06.
Padbidri VS, Rodrigues JJ, Shetty PS, Joshi MV, Rao BL, Shukla RN: Tick-borne rickettsioses in Pune district, Maharashtra, India. Int J Zoonoses. 1984, 11: 45-52.
Wen B, Cao W, Pan H: Ehrlichiae and ehrlichial diseases in China. Ann NY Acad Sci. 2003, 990: 45-53. 10.1111/j.1749-6632.2003.tb07335.x.
Ghosh S, Azhahianambi P, Yadav MP: Upcoming and future strategies of tick control: a review. J Vect Borne Dis. 2007, 44: 79-89.
Zhong J, Jasinskas A, Barbour AG: Antibotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS ONE. 2007, 2: e405-10.1371/journal.pone.0000405.
Mediannikov O, Sekeyová Z, Birg ML, Raoult D: A novel obligate intracellular gamma-proteobacterium associated with Ixodid ticks, Diplorickettsia massiliensis, gen. nov., sp. nov. PLoS ONE. 2010, 5: e11478-10.1371/journal.pone.0011478.
Matton P, Van Melckebeke H: Bovine borreliosis: comparison of simple methods for detection of the spirochaete in the blood. Trop Anim Hlth Prod. 1990, 22: 147-152. 10.1007/BF02241005.
Wen B, Jian R, Zhang Y, Chen R: Simultaneous detection of Anaplasma marginale and a new Ehrlichia species closely related to Ehrlichia chaffeensis by sequences analyses of 16S ribosomal DNA in Boophilus microplus ticks from Tibet. J Clin Microbiol. 2002, 40: 3286-3290. 10.1128/JCM.40.9.3286-3290.2002.
Smith RD, Miranpuri GS, Adams JH, Ahrens EH: Borrelia theileri: isolation from ticks (Boophilus microplus) and tick-borne transmission between splenectomized calves. Am J Vet Res. 1985, 46: 1396-1398.
Callow LL, Hoyte HMD: Transmission experiments using Babesia bigemina, Theileria mutans, Borrelia sp. and the cattle tick, Boophilus microplus. Aust Vet J. 1961, 73: 381-390. 10.1111/j.1751-0813.1961.tb03790.x.
Rodríguez Vivas RI, Cen Aguilar F, Domínguez Alpízar JL, Cob Galera LA, Solís Calderon JJ: Detección de espiroquetas del género Borrelia en hemolinfas de teleoginas de Boophilus microplus en el estado de Yucatán, México. Vet Mex. 1996, 27: 187-188.
Rezende J, Kessler RH, Soares CO, Martins OP: Ocorrência de Borrelia spp. em cultura de células embrionárias do carrapato Boophilus microplus (Acari: Ixodidae) no estado do Mato Grosso do Sul, Brasil. Rev Brasileira Parasitol Veter. 2008, 17: 50-52.
Rich SM, Armstrong PM, Smith RD, Telford SR: Lone star tick-infecting Borrelia are most closely related to the agent of bovine borreliosis. J Clin Microbiol. 2001, 39: 494-497. 10.1128/JCM.39.2.494-497.2001.
Spielman A, Pollack RJ, Telford SR: The origins and course of the present outbreak of Lyme disease. Ecology and environmental management of Lyme Disease. Edited by: Ginsberg HS. 1992, New Jersey: Rutgers University Press, 83-96.
Yparraguirre LA, Machado-Ferreira E, Ullmann AJ, Piesman J, Zeidner NS, Soares CAG: A hard tick relapsing fever group spirochete in a Brazilian Rhipicephalus (Boophilus) microplus. Vector-Borne Zoonot Dis. 2007, 7: 717-721. 10.1089/vbz.2007.0144.
Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu G, Pyke AT, Hedges LM, Rocha BC, Hall-Mendelin S, Day A, Riegler M, Hugo LE, Johnson KN, Kay BH, McGraw EA, van den Hurk AF, Ryan PA, O'Neill SL: A Wolbachia symbiont in Aedes aegypti limits infection with Dengue, Chikungunya, and Plasmodium. Cell. 2009, 139: 1268-1278. 10.1016/j.cell.2009.11.042.
Vavre F, Fleury F, Lepetit D, Fouillet P, Bouletreau M: Phylogenetic evidence for horizontal transmission of Wolbachia in host-parasitoid associations. Mol Biol Evol. 1999, 16: 1711-1723.
Ahrens ME, Shoemaker D: Evolutionary history of Wolbachia infections in the fire ant Solenopsis invicta. BMC Evol Biol. 2005, 5: 35-10.1186/1471-2148-5-35.
Viljakainen L, Reuter M, Pamilo P: Wolbachia tranmission dynamics in Formica wood ants. BMC Evol Biol. 2008, 8: 55-10.1186/1471-2148-8-55.
Moreira LA, Saig E, Turley AP, Ribeiro JMC, O'Neil SL, McGraw EA: Human probing behavior of Aedes aegypti when infected with a life-shortening strain of Wolbachia. PLoS Negl Trop Dis. 2009, 3: e568-10.1371/journal.pntd.0000568.
Fogaça AC, Lorenzini DM, Kaku LM, Esteves E, Bulet P, Daffre S: Cysteine-rich antimicrobial peptides of the cattle tick Boophilus microplus: isolation, structural characterization and tissue expression profile. Dev Comp Immunol. 2004, 28: 191-200.
Fogaça AC, Almeidae IC, Eberlin MN, Tanaka AS, Bulet P, Daffre S: Ixodidin, a novel antimicrobial peptide from the hemocytes of the cattle tick Boophilus microplus with inhibitory activity against serine proteinases. Peptides. 2006, 27: 667-674.
Pereira LS, Oliveira PL, Barja-Fidalgo C, Daffre S: Production of reactive oxygen species by hemocytes from the cattle tick Boophilus microplus. Exp Parasitol. 2001, 99: 66-72. 10.1006/expr.2001.4657.
Santos IK, Valenzuela JG, Ribeiro JM, de Castro M, Costa JN, Costa AM, da Silva ER, Neto OB, Rocha C, Daffre S, Ferreira BR, da Silva JS, Szabó MP, Bechara GH: Gene discovery in Boophilus microplus, the cattle tick. Ann NY Acad Sci. 2006, 1026: 242-246. 10.1196/annals.1307.037.
Parola P, Cornet JP, Sanogo YO, Miller RS, Van Thien H, Gonzalez JP, Raoult D, Telford SR, Wongsrichanalai C: Detection of Ehrlichia spp., Anaplasma spp., Rickettsia spp., and other eubacteria in ticks from the Thai-Mynmar border and Vietnam. J Clin Microbiol. 2003, 41: 1600-1608. 10.1128/JCM.41.4.1600-1608.2003.
Robinson JB, Eremeeva ME, Olson PE, Thornton SA, Medina MJ, Sumner JW, Dasch GA: New approaches to detection and identification of Rickettsia africae and Ehrlichia ruminatium in Amblyomma variegatum (Acari: Ixodidae) Ticks From the Caribbean. J Med Entomol. 2009, 46: 942-951. 10.1603/033.046.0429.
Estrada-Peña A, Jongejan F: Ticks feeding on humans: a review of records on human-biting Ixodoidea with special reference to pathogen transmission. Exp Appl Acarol. 1999, 23: 685-715.
Girotto A, Zangirolando A, Teixeira Y, Vidotto O: Parasitism by Rhipicephalus (Boophilus) microplus (Canestrini, 1887) in humans in the northern part of Parana State, Brazil. 13th International Congress of Acarology Abstract Book: 23-27 August 2010; Brazil. Edited by: de Moraes GJ, Castilho RC, Flechtmann. 2010, 92-93.
Miller RJ, Li AY, Tijerina M, Davey RB, George JE: Differential response to diazinon and coumaphos in a strain of Boophilus microplus (Acari: Ixodidae) collected in Mexico. J Med Entomol. 2008, 45: 905-911. 10.1603/0022-2585(2008)45[905:DRTDAC]2.0.CO;2.
Gontcharova V, Youn E, Wolcott RD, Hollister EB, Gentry TJ, Dowd SE: Black box chimera check (B2C2): a windows-based software for batch depletion of chimeras from bacterial 16S rRNA gene datasets. Open Microbiol J. 2010, 4: 47-52. 10.2174/1874285801004010047.
Schloss PD, Handlesman J: Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol. 2005, 71: 1501-1506. 10.1128/AEM.71.3.1501-1506.2005.
Acknowledgements
We thank Ralph Horn and Sara Davis for technical assistance and Drs. Ludek Zurek and J. Allen Byrd for critically reviewing the manuscript prior to submission. We also acknowledge Sherri Starks for outstanding programmatic support. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. This work was supported by USDA-ARS CRIS project number 6205-32000-031-00 D.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
FDG and GAS conceived and designed the study; KGB and FDG prepared samples and acquired data for sequence analysis; SED performed sequence and bioinformatics analyses; RA and AAPL analyzed and interpreted the data, and drafted the article. All authors read and approved the final manuscript.
Renato Andreotti, Adalberto A Pérez de León contributed equally to this work.
Electronic supplementary material
12866_2010_1293_MOESM1_ESM.PDF
Additional file 1: Table S1 - Bacterial genera detected in R . ( B .) microplus. Bacterial genera detected in R. (B.) microplus samples. (PDF 120 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Andreotti, R., Pérez de León, A.A., Dowd, S.E. et al. Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplusthrough tag-encoded pyrosequencing. BMC Microbiol 11, 6 (2011). https://doi.org/10.1186/1471-2180-11-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-11-6