
Abstract
Lesions of spiral ganglion cells, representing a restricted sector of the auditory nerve array, produce immediate changes in the frequency tuning of inferior colliculus (IC) neurons. There is a loss of excitation at the lesion frequencies, yet responses to adjacent frequencies remain intact and new regions of activity appear. This leads to immediate changes in tuning and in tonotopic progression. Similar effects are seen after different methods of peripheral damage and in auditory neurons in other nuclei. The mechanisms that underlie these postlesion changes are unknown, but the acute effects seen in IC strongly suggest the “unmasking” of latent inputs by the removal of inhibition. In this study, we explore computational models of single neurons with a convergence of excitatory and inhibitory inputs from a range of characteristic frequencies (CFs), which can simulate the narrow prelesion tuning of IC neurons, and account for the changes in CF tuning after a lesion. The models can reproduce the data if inputs are aligned relative to one another in a precise order along the dendrites of model IC neurons. Frequency tuning in these neurons approximates that seen physiologically. Removal of inputs representing a narrow range of frequencies leads to unmasking of previously subthreshold excitatory inputs, which causes changes in CF. Conversely, if all of the inputs converge at the same point on the cell body, receptive fields are broad and unmasking rarely results in CF changes. However, if the inhibition is tonic with no stimulus-driven component, then unmasking can still produce changes in CF.
Similar content being viewed by others

Introduction
Receptive field (RF) changes in central nervous system (CNS) neurons that follow partial losses of sensory input have been widely used as models of CNS plasticity. In the first demonstration of plasticity as a result of damage to the cochlea (Robertson and Irvine 1989), a basal (high frequency) sector of the organ of Corti was ablated mechanically, resulting in a high-frequency hearing loss over a narrow range of frequencies. Some months after the lesion, it was observed that neurons in the high-frequency region of the auditory cortex now responded to lower frequencies. The new characteristic frequencies (CFs) corresponded to the high-frequency edge of the remaining cochlea inputs, and their CF thresholds averaged only 12.5 dB above the prelesion thresholds of neurons tuned to the same low frequencies. These responses did not simply reflect residual activity that was “left over” from the prelesion RFs. There was new activity in responses to stimuli outside the range which had produced responses prior to the lesion. Thus, Robertson and Irvine observed an expansion of the central representation of these lower frequencies, which radically altered the cortical frequency organization.
Subsequent studies using the same chronic lesion method have confirmed and extended these results in the auditory cortex (Rajan et al. 1993; Irvine et al. 2003; Kamke et al. 2003) and down to the level of the inferior colliculus (IC; Irvine et al. 2003; Kamke et al. 2003). Studies in which damage was induced acoustically, whether chronic (Rajan 1998, 2001) or acute (Calford et al. 1993; Wang et al. 1996; Norena et al. 2003), in either IC (Wang et al. 1996) or cortex (Calford et al. 1993, Norena et al. 2003) also showed changes in tuning and expansion of RFs. However, with one exception (Norena et al. 2003), the changes in CF were accompanied by significant increases in thresholds and did not result from activity at new frequency/level combinations. In the cochlear nucleus, all changes in RFs and CFs are explainable as the “residue” of prelesion tuning, whether lesions are acute and acoustically induced or are chronic mechanical ablations (Kaltenbach et al. 1992; Rajan and Irvine 1998). Tonotopic remapping that does reflect new activity was observed in IC immediately after focal lesions of spiral ganglion cells (Snyder and Sinex 1998; Snyder and Sinex 2002; Snyder et al. 2008). Moreover, rapid RF changes have been seen in other sensory modalities (for review, see Calford 2002). Interestingly, immediately following mechanical lesions involving the organ of Corti and basilar membrane, there are only high-threshold residual changes in CF (Robertson and Irvine 1989). However, trauma may obscure any immediate changes in these experiments. It is clear that the method of damage to the cochlea, the time since the damage, and the location in the neural pathway are all important factors in determining the changes seen to neural response properties and that they interact in quite complex ways.
There are several possible mechanisms that could explain postlesion changes in tuning and tonotopy. Anatomical “rewiring” requires a time course of months after damage (Darian-Smith and Gilbert 1994; Tailby et al. 2005) and so cannot explain acute changes. Another more rapid mechanism is an increase in the strength of individual synaptic inputs (Jenison 1997). A third possibility is that the removal of some afferent inputs might alter the balance between excitatory and inhibitory input in an afferent population. Reduction in the number of inhibitory inputs, for example, might “unmask” or release previously suppressed or subthreshold excitatory inputs, allowing them to evoke excitatory responses at frequency–sound level combinations outside the prelesion excitatory response area. RF changes in the auditory system (Rajan 1998, 2001) have been associated with a loss of surround inhibition, and a wide variety of changes across different sensory modalities can be interpreted as having unmasking as their underlying mechanism (see Calford 2002). This explanation of plasticity implies something about the way sensory systems are hardwired. Specifically, neurons are receiving a far wider range of inputs than is normally measured under acoustic stimulation. It has been suggested that tonic inhibition might be necessary (Calford and Tweedale 1991a) to account for unmasking. Indeed, there is experimental support for this in the somatosensory cortex (Calford and Tweedale 1991b). Yet, there has been no quantitative theoretical demonstration of unmasking and how it can lead directly to changes in RF organization without elevations in threshold.
As a first step in establishing such a quantitative theoretical account in the auditory system, we have explored neural mechanisms in two model neurons that could underlie the immediate changes in frequency tuning seen in IC neurons following spiral ganglion lesions (Snyder et al. 2000; Snyder and Sinex 2002). These cannot be attributed to slow, plastic processes. There are a variety of such immediate changes, but we will focus on those in which new, low-threshold responses are observed, since they result in changes in CFs and frequency organization that cannot be explained by residual responses (pseudoplasticity). The models consist of populations of single neurons receiving convergent afferent excitatory and inhibitory inputs, and they compute only steady-state firing rates. The models ignore any temporal processing and any processing that occurs between the auditory nerve and the IC. They do not, therefore, reproduce the wide range of complex response patterns seen in IC. Nevertheless, these models show that there are at least two mechanisms by which unmasking of latent inputs could account for the immediate shifts in CF observed in IC neurons. Moreover, they indicate some of the constraints on the mechanisms producing these changes.
Methods
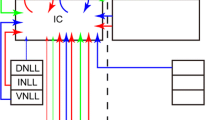
Two stage models were used to simulate the steady-state RF properties (firing rate as a function of tone frequency and level) of neurons in the IC. Figure 1A shows the overall architecture of one of the models. The first stage was the simulated responses of an array of auditory nerve (AN) fibers across the length of the cochlea (Sumner et al. 2003b). The outputs from this array formed both the excitatory and inhibitory inputs to stage 2, a steady-state solution of the membrane potential in a single neuron. There are two different kinds of neuron model. The first and most complex is shown in Figure 1A.

A Schematic of a compartmental model of an IC neuron. A model of the auditory periphery (bottom) to the neuron is based on a previously published model. Its inputs are pure tones, filtered by a bandpass model of the middle ear. The mechanical filtering at fixed points along the basilar membrane is modeled by DRNL filters, which drive a model of transduction by the IHC, which in turn drives 20 Poisson spike generators. The discharge rates of the 20 generators, simulating 20 HSR auditory nerve fibers, are summed to represent a single input channel to the IC model neuron, in spikes per second. Representative FTCs for five (of the 60) input channels from the peripheral model are shown. The CF of each channel is separated by 1/12 of an octave and CFs extend from 1 to 30 kHz. Each channel acts as the input to one neural cell compartment at the top. IC neurons were simulated using an electrical circuit analog equivalent of the membrane potential at different points (compartments) on the cell body and dendrites. The output of each compartment is simulated by its membrane potential (V j ) at discrete points across the cell. Each compartment has a resting (G rj ), excitatory (G ej ), and inhibitory (G ij ) input conductance. The soma is the central compartment of the model and V j at this point drives spike generation. Both excitatory and inhibitory input conductances are derived from the auditory nerve model with different CFs forming synapses tonotopically onto different electrical compartments. The strength of input conductance falls off with distance from the soma as in C. B A single DRNL filter. C Input weighting functions determining the strength of the conductances are Gaussian functions centered on the somatic compartment. Parameters determine the width (We, Wi) and overall scaling factors (Fe, Fi) of the weighting functions. The black bar indicates the CF extent of the simulated lesion. The figure illustrates the parameters for the model output shown in Figure 3 (We = 10 semitones, Wi = 8 semitones), for a CF of 5 kHz (N11).
Stage 1: the model of the auditory periphery
The AN was simulated with a model of the guinea pig cochlea (Sumner et al. 2003b), which consisted of five substages: a middle ear filter stage, a nonlinear mechanical filter bank stage, an inner hair cell (IHC) transduction stage, a spike generation stage, and a channel output stage. The stimulus waveform was input to a linear bandpass filter (refer to Fig. 1A), reflecting the frequency response of the middle ear. The middle ear stage in turn provided the input to a dual-resonance nonlinear (DRNL; Fig. 1B) filter bank. This simulated the nonlinear mechanical filtering in the ear (Meddis et al. 2001). Each channel of the filter bank represented a single position in the cochlea and consisted of two parallel bandpass branches whose outputs were summed (refer to Fig. 1B). One branch was linear, while the other had a broken stick compression function. The nonlinear pathway contributed the narrow tip of the tuning curves (example tuning curves are shown in Fig. 1A). Since this pathway was compressive, its output grew little with sound level, unlike the broadly tuned linear pathway. So at high intensities, the linear pathway dominated the output. Longitudinal variation in tuning and compression arose from variation in the parameters with characteristic frequency (CF: the frequency at which the threshold of the AN fiber is lowest). This architecture can reproduce many of the characteristics of basilar membrane (BM) measurements (Meddis et al. 2001).
Each channel from the DRNL filter bank drove an IHC stage (Sumner et al. 2002; Sumner et al. 2003b). The IHC stage incorporated fluid–cilia coupling, a simple passive equivalent electrical circuit of the IHC receptor potential, and calcium-controlled release of neurotransmitter into the synaptic cleft. Neurotransmitter was recycled via the cleft at a limited rate resulting in a reduction of the amount available for release. This produced adaptation in firing rate much like that seen in auditory nerve responses. In the previously published model, a key feature was that neurotransmitter was released stochastically. However, for computational efficiency in this study, the synapse output was a continuously varying probability of neurotransmitter release. The output of each IHC was input to 20 Poisson spike generators, modified to have absolute and relative refractoriness (Carney 1993). These generators simulated the responses of 20 AN fibers with the same CF. The complete auditory peripheral model can reproduce many of the effects observed in the AN and their variation with fiber type, including realistic RF shapes with the correct tuning asymmetry, compression, adaptation, and phase-locking (Sumner et al. 2002, 2003a, b; Holmes et al. 2004).
The responses of the peripheral model were calculated for high spontaneous rate (HSR) AN fibers with 60 different CFs varying from 1 to 30 kHz in 12th octave (i.e., one semitone) steps. The stimuli were pure tones which varied from 1 to 30 kHz in 12th octave frequency steps and 5 dB steps in level from 0 to 100 dB sound pressure level (SPL). From this, the mean spike rate was calculated from the 20 AN fibers at each CF and stimulus condition. All fibers had a spontaneous firing rate of approximately 100s−1. Some neuron models presented were also tested with peripheral inputs that were not spontaneously active. A value of 125s−1 was subtracted to ensure there was no spontaneous activity, since actual values are variable owing to the stochastic nature of firing. The remainder were then rescaled to produce the original maximum firing rate. Examples of frequency threshold tuning curves (FTCs) are shown in Figure 1A. Channel 30 has a CF of 5 kHz, and each of the illustrated FTCs differs in CF by ten semitones.
Stage 2: the compartmental model of a central auditory neuron
The first neural model presented in this study considered the convergence of excitatory and inhibitory inputs onto a central auditory neuron when the inputs were distributed tonotopically across the cell body and dendrites. All the inputs, excitatory and inhibitory, were taken directly from the peripheral model in which the only variations were CF-related. Figure 1A stage 2 shows the architecture of this model and how it was connected to stage 1, the model AN inputs.
We simulated neurons using an electrical circuit analog equivalent of the membrane potential at different points on the cell body and dendrites (e.g., Banks and Sachs 1991). The top of stage 1 shows the electrical circuit, divided into coupled “compartments” (divided by dashed vertical lines) representing different points along the neuron’s dendritic surface. One central compartment was labeled the “soma” and its membrane potential drove action potential generation in the cell axon. Two dendrites stretched out from the soma in opposite directions. Each compartment received different excitatory and inhibitory inputs.
We were interested in the possibilities for static processing in such a neuron. Therefore, we analytically solved the steady-state solution for the neuron, i.e., as if the inputs were constant and the membrane potential at the various points in the neuron had settled to a steady state. This was much faster than simulating the evolution of the membrane potential in time and allowed us to explore a large parameter space. For a compartment at a single location, j, in our neuron (see Fig. 1A), we applied Kirchoff’s current law, giving a steady-state solution of the form:
where V j represented the membrane potential at a single location along the dendrite or cell body with V j−1 and V j+1 being the membrane potential at the neighboring locations. G ej and E e were the excitatory conductance and excitatory reversal potential, respectively; G ij and E i were the inhibitory conductance and inhibitory reversal potential; E r and G rj were the resting potential and resting (or leakage) conductance of the membrane; and G dj , was the coupling conductance between compartment j and j + 1. None of the conductances in the model were dependent on membrane potential. For all models presented in this study, E e was set to 0mV and E i was set to −80mV, which are both physiologically common values (Johnston and Wu 1995). E r was set to −60 mV, determining the resting potential of the cell with no inputs. G dj was a parameter in the model, but in most instances, it was set to 36 × 10−3S and it was the same for all j within a single model. For n compartments, we thus derived n equations linking each compartment with its two neighbors. This produced n simultaneous equations with n unknowns (the membrane potential at each point) which were solved using Gaussian elimination.
The inputs to the neuron were the weighted outputs from stage 1. Each input channel had a different CF and provided the input to a different compartment in the neuron model in tonotopic order. Thus each adjacent compartment received an input differing in CF by one semitone (1/12 octave) relative to the neighboring compartments with filter bank channel j forming the input to compartment j in the model neuron. Although every neuron had 60 compartments, it was possible for many of these compartments to receive no input. The CF range and strength of the excitatory and inhibitory inputs to a neuron were determined by Gaussian weighting functions. Multiplying the firing rate for the peripheral input by the excitatory weight for a given channel gave the excitatory conductance at the corresponding compartment. For the excitatory inputs:
where G ej was the excitatory input to compartment j, x j was the total firing rate of the 20 AN model fibers from filter bank channel j, and c was the compartment number which was defined as the soma. W e determined the range of stage 1 channel CFs which provided input onto the stage 2 neuron, F e was the maximum synaptic strength (which was at the soma), and c was the input channel which provided the input to the soma. In order to produce a finite range of inputs, if the weight applied to x j was less than 0.1, G ej was set to 0. Notice that because c is both the center of the Gaussian weighting function and the somatic compartment, the strongest input to the neuron is always at the soma and will define the CF of the neuron. The inhibitory inputs were subject to a similar weighting function with different parameters (W i and F i). There was no inhibitory input to the soma (i.e., G ij = 0 when j = c). This was necessary because inhibition at this compartment was often strong enough to suppress all excitatory inputs and completely suppressed any excitatory responses (examination of which was the purpose of the model). There is some experimental support that such an arrangement exists in the avian forebrain (Muller and Scheich 1988). We would like to stress that this does not rule out the existence of other weaker, differently tuned, or higher threshold inhibition at the soma. For simplicity, such inhibition was not considered in this model.
Figure 1 illustrates how, for an example model neuron (the responses of which are shown in Fig. 3), the model parameters determine the inputs to the neuron. The Gaussian weighting function in Figure 1C is centered on j = 30 (i.e., c = 30) with W e = 10 and W i = 8. This produced nonzero excitatory input weights for 41 channels of the filter bank with the maximum weight at j = 30, for which the CF of the input is 5 kHz. Figure 1A shows the FTCs for five of the CF channels of the AN inputs to the neuron (j = 10, 20, 30, 40, 50) and Figure 1C indicates the input weights (colored points) for each of these channels. The weighting functions meant that when j = c ± W e input strengths dropped to 0.6F e. At 20 semitones away from the center of the function, only the excitatory input was nonzero.
The firing rate of the neuron was modeled as an exponential function of the steady-state membrane potential at the soma:
where R max is the maximum firing rate, V c is membrane voltage at the soma, V thr is the threshold membrane voltage for the neuron to discharge, and λ is a constant. We set the neurons’ firing threshold V thr to −50 mV, the maximum firing rate R max to 300s−1, and λ to 15. This produced a threshold effect and a realistic saturating firing rate.
Stage 2: the point neuron model of a central auditory neuron
The second kind of model we considered was slightly different to that shown in Figure 1A, in that it had only one compartment at the soma and no dendrites at all. This allowed us to investigate the necessity of the dendrites. The membrane potential of the cell was given by:
where V m was the membrane potential for the compartment, G e and E e were the excitatory conductance and reversal potential, respectively, G i and E i were the inhibitory conductance and reversal potential, respectively, and G r and E r were the resting conductance and reversal potential, respectively. Effectively, this was a simplified version of Eq. 1 with G dj = 0. All the reversal potentials and the resting conductance took the same values as for the compartmental model.
All the peripheral inputs were summed to form a single excitatory and a single inhibitory input. The same form of Gaussian weighting function was applied to these inputs. Thus, the total excitatory input conductance was:
where j was the peripheral input channel, N was the number of input CFs (i.e., 60), x j was the input firing rate from peripheral channel j, c was the peripheral channel where the weighting function was maximum and thus determined the CF of the model neuron, and F e and W e were the maximum synaptic strength and width of the weighting function, respectively. The inhibitory conductance was determined similarly to the excitatory input. The firing rate of the neuron was determined from V m by Eq. 3.
In one set of point neuron model simulations, we employed a tonic inhibition which was active for each channel providing the input was intact (i.e., not lesioned). Each tonic inhibitory input (x j ) had a constant firing rate of 100s−1 (to make it roughly equivalent to the spontaneous rate in the other inputs used), and these were scaled by the input weighting function (Eq. 5) in the same way as any other inputs.
The simulation process and analysis
For a given neuron model (compartmental or point) and a given set of parameters to describe the weights of the inputs, the center of the input weighting functions, c (Eqs. 2, 3, and 5), was shifted from 20 to 40 across the peripheral inputs, yielding an array of 21 different model neurons with CFs from ∼3 to 9 kHz. Except for the CFs, this array of model neurons had the same parameters and thus similar response properties.
In a given model neuron, the evoked firing rate for each pure tone frequency and level combination was calculated to yield a RF. FTCs were derived from the RF using an algorithm which at each stimulus frequency looked for the level at which firing rate increased by 20% of the difference between spontaneous and maximum driven rate (Moshitch et al. 2006). From the FTC, CF, threshold at CF, and filter Q (center frequency/bandwidth) at 10 dB above threshold were found. Two CFs were recorded if there was an increase in threshold between the FTC minima of more than 20 dB above two thresholds (as used by Moshitch et al. 2006). Some parameter sets produced arrays of neurons with weak and patchy responses or neurons with tuning that was very wide, producing an array that was not tonotopically ordered. All the model neurons associated with a parameter set were rejected if the prelesion CF of any neuron in the tonotopic array differed by more than 20% from the CF of that neuron’s somatic input (i.e., input channel), c. The parameter set was also rejected if one or more neurons did not produce any suprathreshold activity.
To simulate a lesion in the model, we set the firing rates in the peripheral AN model to zero for all stimulus conditions in channels tuned to a specific range of CFs. This procedure removed all spontaneous as well as all stimulus-driven activity for channels tuned to that range. In most cases, the simulated lesion was seven semitones (7/12 octave) wide centered on the AN input having a CF of 5 kHz (channel 30). The width of this lesion is shown as a black bar in Figure 1C, for comparison with an example weighting function. We looked for changes in stimulus-driven activity between prelesion and postlesion RFs, FTCs, CFs, and thresholds.
The responses of models before and after a simulated lesion were explored for a very wide range of parameters and for the different model configurations. Sometimes this was done manually, running a single-parameter set for an array of neurons before and after the simulated lesions. In other instances, the effects of parameters were explored by generating large populations of models run across a range of values for the four parameters (W e, F e, W i, and F i) and for all combinations of those parameters. Each model neuron was then assessed for different kinds of response changes as a result of the simulated lesion. A neuron was counted as having “new” unmasked activity, if there was more than a 10% increase in the area of suprathreshold activity. A CF was judged to have been changed following a lesion, if the shift was more than one semitone (1/12 octave). We judged unmasking leading to CF changes to have occurred if there was new suprathreshold activity at the frequency of the new CF. Other changes in CF were judged to be residual as they did not arise from unmasking of new activity. For a given “family” of model neurons, we examined both the overall frequency of occurrence of different kinds of change and also how those changes varied with the parameter values. The parameter values used and the frequency of changes are given in Table 1.
A summary of observed lesion effects on receptive fields
Figure 2 illustrates the principal changes observed in IC RFs following restricted spiral ganglion lesions. In Figure 2A, the six panels in the top and middle rows illustrate an example of a set of RFs (firing rates as a function of pure tone frequency and level) recorded in the IC before and after a small spiral ganglion lesion. The prelesion CFs were from left to right 13.5, 14.7, 16.0, 17.5, 17.5, and 20.8 kHz; prelesion thresholds were 30, 30, 30, 35, 40, and 55 dB SPL. The lesion was centered at about 19 kHz, thus the prelesion CFs for these neurons span the lesion center frequency. The postlesion responses (middle row) at the three middle sites (sites 10, 11, and 12) had double response tips and are assigned two CFs. Postlesion CFs were 11.3, 13.5 [24.7], 13.5 [26.9], 13.5 [26.9], 29.3, and 26.9 with the second CF indicated in brackets. Postlesion thresholds were 55, 60, 60, 60, 50, and 45 dB SPL. All but the lowest and highest frequency neurons had a notch in their postlesion RF, corresponding to the lesion frequencies, and all had a shift in their CF. Difference response areas (normalized postlesion minus normalized prelesion firing rates) for each pair of responses recorded at each site are illustrated in the bottom row. In these difference RFs, there is a consistent loss in excitation (blue regions) corresponding to the lesion center frequencies and a consistent gain of excitation at frequencies on the high-frequency edge of the lesion (red regions). The effect of the lesion on the compound action potential (measured at the round window) is superimposed as white lines on the RFs and as black lines on the difference plots.

A RFs recorded in the IC before (top row) and after (middle row) a restricted spiral ganglion lesion. Response amplitude differences between the prelesion and postlesion recordings are in the bottom row. Firing rates are normalized relative to the maximum prelesion response at each site and are color-coded according to the scales on the right. These RFs were recorded at six consecutive sites (#9–14) of a 16-site silicon electrode array inserted into the central nucleus of the IC and fixed in place in a normal-hearing cat. The sites were separated by 100 μ and distributed along the tonotopic axis of the IC. In each panel, the abscissa is stimulus frequency; the left ordinate is stimulus level. In the middle and bottom rows, compound action potential (CAP) threshold differences (postlesion minus prelesion) are plotted as a white or black curve, respectively; the horizontal line indicates the zero change level for these differences. The axis on the right indicates the magnitude of the postlesion CAP threshold change. The frequencies with more elevated postlesion thresholds (large positive values) define the lesion frequencies. B Changes to RF. Solid lines indicate the threshold of prelesion RF and dashed lines show postlesion RF. Type 1 postlesion activity is contained within prelesion RF. CF changes are the residue of prelesion RF. Type 2 increases in activity within the prelesion RF. This is shown as the patterned area (dashed lines indicating postlesion RF are offset slightly from the prelesion RF for clarity). Type 3 unmasking of new driven responses that do not result in CF changes. Type 4 unmasking of new driven activity which results in CF changes. C Changes to tonotopic maps seen in the auditory system following damage to the cochlea. Measured CF is plotted against the physical position of an electrode along a tonotopic axis. The dotted line shows smooth progression in normal-hearing animals. The solid line shows the tonotopic progression seen in IC following acute spiral ganglion lesions. The dashed line shows a typical progression seen in various nuclei following a lesion at the basal end of the cochlea (e.g., Robertson and Irvine 1989).
Figure 2B summarizes four different categories of changes that can be observed in RFs of experimental data of the type shown above. Solid lines indicate prelesion RFs and dashed lines show the RF shapes following a lesion. Type 1 illustrates a “residual” RF in which suprathreshold activity is completely contained within the prelesion response area. A type 1 change can also be seen at the lowest CF site in Figure 2A. Changes in CF result from a loss of responsiveness at the prelesion CF. They can be readily explained as a loss in sensitivity, such as that seen in auditory nerve fibers after noise damage (Liberman and Mulroy 1982), or the result of a partial removal of excitatory inputs. Such changes have been reported throughout the auditory pathway: in the cochlear nucleus (Rajan and Irvine 1998; Kaltenbach et al. 1992) and IC (Irvine et al. 2003) following long-term recovery from basal cochlear lesions and immediately after basal cochlea lesions in the auditory cortex (Robertson and Irvine 1989).
In type 2 changes, there is an increase in activity within the boundaries of the prelesion RF. A loss of excitatory input cannot explain the increase in activity (indicated by the cross-hatched area in Fig. 2B), although no previously hidden activity is revealed. Type 2 changes are seen in IC following spiral ganglion lesions (Snyder and Sinex 2002, see lowest CF site, #9, in Fig. 2A) and are widely reported following tone and noise exposure in cochlear nucleus (Boettcher and Salvi 1993), IC (Willott and Lu 1982; Wang et al. 1996), and auditory cortex (Calford et al. 1993; Norena et al. 2003; although this was not reported in Rajan 1998 or Rajan and Irvine 1998). These may or may not be accompanied by other types of changes elsewhere in the RF (as in Fig. 2A).
We define unmasking in this study as the appearance of activity where there was none before. Type 3 changes are characterized by unmasking. It may or may not lead to a change in CF. Unmasking is evident in all except the leftmost panel of Figure 2A. Unmasking has been observed in the IC (Snyder et al. 2000; Snyder and Sinex 2002) and auditory cortex (Snyder and Sinex 1998) immediately following spiral ganglion lesions. It has been observed immediately after tone-induced threshold shifts in the IC (Wang et al. 1996) and the auditory cortex (Calford et al. 1993) and also in the auditory cortex following mild, but permanent, cochlea damage from noise exposure (Rajan 1998).
If unmasking occurs near to the prelesion threshold, it can produce a shift in CF (see Fig. 2A, all except the lowest CF site). We will refer to it as a type 4 change. It is also possible that a V-shaped tuning curve can become W-shaped (Snyder et al. 2000; Fig. 2A), making CF ambiguous. Type 4 changes have been most clearly demonstrated in the IC and cortex following spiral ganglion lesions, when prelesion and postlesion RFs from single recording sites have been compared directly (Snyder and Sinex 2002, Snyder et al. 2000). The changes in tonotopy previously reported by Irvine and colleagues in the thalamus (Kamke et al. 2003), cortex (Robertson and Irvine 1989), and to a lesser extent, in IC (Irvine et al. 2003) after long-term recovery from basal cochlear lesions can only be explained if there is new suprathreshold activity at frequencies near to threshold that were not present in normal animals.
A corollary of changes in CF is that tonotopic progression across the neural tissue, which is usually defined by CF, will also change. Figure 2C shows diagrammatically the progression of CF across a tonotopic axis of the IC of a normal animal (dotted line) and following a large lesion to the basal region of the cochlea (dashed line). From the point along the tonotopic axis corresponding to the edge of the lesion, the CF does not change. The representation of the intact lower lesion edge frequencies expands to replace the representation of higher frequencies lost as a result of the lesion. The figure also illustrates the tonotopic progression of CF (solid line) observed in the IC following a more restricted lesion of the spiral ganglion (Snyder et al. 2000; Snyder and Sinex 2002; Fig. 2A). This more focal lesion produced a step change in CF with two CF plateaus for short ranges above and below the position in the tonotopic array corresponding to the lesion frequency.
As shown in Figure 2A, spiral ganglion lesions lead to all of the above types (1–4) of changes in IC (Snyder et al. 2000; Snyder and Sinex 2002). The most intriguing of the changes are those (type 4 changes) leading to CF shifts, since they cannot be explained as a residue of the prelesion RF. The immediacy of these changes is consistent with a passive, experience-independent shift in the balance of excitatory and inhibitory inputs, providing an ideal set of data against which to compare models of unmasking. Successfully reproducing type 4 changes in a computational model was the main focus of this study.
Model results
Simulating spiral ganglion lesions in single inferior colliculus neurons with dendritic processing
Figure 1A shows the architecture for a model of an IC neuron. This model was, in our simulations, the most successful at producing CF changes that arose from unmasking of new responses (type 4 changes). Its fundamental feature is that excitatory and inhibitory inputs with the same CF formed synapses at matched locations across the cell. These inputs are spread across the cell body and dendrites in a tonotopically organized manner. The discharge rate of the model was dependent on the membrane potential at the soma, but this in turn was a product of local inhibitory–excitatory interactions all along the dendrites. Four parameters determine the CF range and synaptic “strength” of the inputs: W e and F e are parameters of a Gaussian function which determines the CF range (W e) and strength (F e) of all of the excitatory inputs (see the “Methods” section for details). The inhibitory inputs were determined in the same way by the parameters W i and F i, except that the inhibitory input to the soma was omitted. We do not mean to imply that there can be no inhibition on the soma of IC neurons, only that there was no strong, low-threshold inhibition. Such inhibition suppressed all driven activity and would not have allowed the modeling to proceed.
Figure 3 shows the outputs from a set of model neurons, simulating the RF from a tonotopic array of IC neurons and the effects of a spiral ganglion lesion. The top row (Fig. 3A) shows the RF prior to any lesion. Tuning is as narrow as in the auditory nerve model and is tonotopically organized (also shown in Fig. 3D; open circles). All the models in the array had the same parameters, but each was aligned differently relative to the input filter bank, and so varied systematically in CF (the input weights are shown for N11, superimposed on the RF). Figure 3B shows the result of simulating a seven semitone wide spiral ganglion lesion, centered on the peripheral input having a lesion center frequency of 5 kHz (the CF of N11), and Figure 3C shows the changes in firing rate as a result of the lesion. Some model neurons show a shift in their CFs to frequencies above and below the range of lesion frequencies (N8 and N14), while other model neurons having CFs within the range of lesion frequencies have W-shaped tuning curves (N10–N12). The changes in tuning observed in Figure 3B and C are reflected in the tonotopic gradient (Fig. 3D) and occur with shifts in threshold of 10 dB or less (Fig. 3E). This set of model neurons produced three of the four types of changes we outlined in the “A summary of observed lesion effects on receptive fields” section: an increase in activity within the RF (type 2; see N8 and N14), unmasking of new activity (type 3; see N9–N14), and unmasking leading to changes in CF (type 4; see N9–N14). Note, however, that some model neurons (N7 and N15) become almost silent after the lesion. This is rarely seen in the IC, but silent postlesion sites have been observed in other central auditory areas, especially the auditory cortex (e.g., Rajan et al. 1993).

RF changes and tonotopic remapping in the model following simulated spiral ganglion lesions. Model parameters: W e = 10, W i = 8, F e = 80 nS, F i = 300 ns, G d = 36 mS. A RF from an array of neurons differing only in CF with complete auditory nerve inputs. The position in the array is indicated above each plot. The input weight functions for the excitatory (red line) and inhibitory (white line) inputs are superimposed on N11. B RF from the same neurons following a simulated spiral ganglion lesion covering CFs across seven semitones, centered on a 5-kHz CF. C The difference in RF between A and B. D The tonotopic map across an array of model neurons with CFs from ∼3 to 9 kHz. The open circles represent the tonotopic map prior to the lesion and the solid blue line shows the CFs from the same neurons following the lesion. Where two CFs are measurable, the second CF is indicated by a green line. E The thresholds of the model neurons across the neural array before and after the lesion.
A detailed comparison of Figure 3 with the data shown in Figure 2 reveals some discrepancies in the shapes of RFs both before and after a lesion. This is partly because this is a single-parameter set that produces robust effects across a range of CFs. Real neurons are not nearly so homogenous in their characteristics. It also reflects the fact that the model does not attempt to simulate many of the complexities of real neurons such as tuning curves shaped differently to auditory nerve fiber tuning, suprathreshold “best” frequencies, and other significant nonmonotonicities. The features included in this model would be expected to form only part of the many features of real neurons: those sufficient for reproducing the main effects described in the “A summary of observed lesion effects on receptive fields” section.
Examples of more models using the same basic architecture but with different parameter sets are shown in Figure 4A, B, and C. In this figure, each row of panels is a different set of model parameters. Each panel shows, for model neurons with a range of CFs, the prelesion FTC (blue line), postlesion FTC (black line), and any increases in firing rate between the two (graded red shading). If a model neuron received excitatory inputs from a fairly wide frequency range (in Fig. 4A) but no inhibitory inputs, it displayed quite broad prelesion FTCs and only residual (type 1) changes in CF. This is understandable as there is a simple partial removal of excitatory inputs. The presence of inhibitory inputs from a wide CF range and a moderate degree of convergence of excitatory inputs (Fig. 4B) tended to produce a mix of effects. Neurons at the center of the lesion became silent following the lesion. Neurons with CFs nearer the edge of the lesion, which suffered only partial removal of excitatory inputs, displayed residual (type 1) changes. Outside of the lesion frequencies, removal of side band inhibition resulted in increases in firing rate (type 2 changes). When the range of inhibitory inputs was wide and excitatory RFs were narrow (Fig. 4B and C), neurons with CFs around the lesion frequencies became silent, whereas neurons near the lesion edge showed increases (type 2 changes) in firing rate, again due to the removal of off-CF inhibition.

A–C Examples of outputs from the compartmental neuron model for different parameter sets, which reveal the different types of changes which occur following a lesion. The leftmost panel shows the weighting function for excitatory (solid line) and inhibitory (dashed line) inputs. Remaining panels in each row show, for a range of different CFs across the neural array, the FTCs before (blue line) and after (black line) a simulated lesion (blue bar shows the CF extent). Red shading indicates the magnitude of any increases in firing rates as a result of the lesion. A Model showing residual changes (We = 5, Fe = 5 μS, no inhibition). B Model showing residual CF changes and increases in spontaneous activity (We = 2.5, Fe = 40 μS, Wi = 7, Fe = 50 μS). C Model showing within RF increases (We = 0.5, Fe = 80 μS, Wi = 5, Fi = 50 μS). D Range of parameters across which residual CF (type 1; in blue) changes and CF changes from unmasking (type 4; in red) occur. Gray squares indicate no change in CF following a lesion, and open squares are those models with poorly defined tonotopy. E Range of parameters over which there are criterion increases in the firing rate within an RF (type 2; in blue) and unmasking of new activity (type 3; in red). In D and E, the greater the number of RFs showing a given change, the brighter the color is. The range of parameters is given in Table 1.
To investigate these models quantitatively, we systematically varied the model parameters (parameters: W e, F e, W i, F i; ranges are given in Table 1). For all combinations of all parameter values, we determined how many of the neurons in each array showed each type of RF change (see the “Methods” section). In total, 51% of neurons showed some kind of CF shift (type 1 or type 4 changes), 9% of neurons showed increases in firing rate within their RFs (type 2 changes), 11% of neurons showed unmasking of new activity (type 3), and 18% of these neurons showed CF changes resulting directly from unmasking (type 4 changes). These values are summarized in Table 1.
Figure 4D shows the distribution of type 1 and type 4 changes across the range of the four model parameters. Each pixel represents a different set of model parameters. The color shows what types of changes were seen following a lesion and the shade indicates the number of CFs at which they occurred. Parameter sets which produce poor tonotopy before a lesion (see the “Methods” section) have no color. In most parameter sets for which excitatory convergence was small (W e = 0.5), CFs did not change following a lesion (gray squares in Fig. 4D). When excitatory convergence was modest (W e = 2.5), there was a mix of residual CF changes (type 1; blue in Fig. 4D, see also Fig. 4A) and unmasked CF changes (red squares). Type 4 changes tend to occur across more neurons in the array (bright red) when W e > W i. This is a reasonable constraint, since the lesion should shift the relative strengths of the remaining inputs in favor of excitatory inputs in order for there to be any postlesion activity. The mean prelesion Q10 for neurons showing type 4 changes was 4.6 (SD = 0.3). If inhibition does not effectively suppress excitatory inputs, type 1 changes are more likely, as convergent excitatory inputs are not suppressed by inhibition prior to the lesion. Figure 4E shows how within RF increases (type 2; blue shading) and unmasking (type 3; green shading; red shading indicates both types) occur across the range of parameters. Comparing with Figure 4D, it can be seen that both increases and unmasking commonly accompany type 4 changes.
The effects in IC modeled thus far are also reminiscent of experiments (e.g., Robertson and Irvine 1989) in which a fairly large basal region of the cochlear was destroyed. Figure 5A shows the outputs from a set of model neurons for a very large basal lesion which extended from inputs with CFs of 5 kHz. The model showed increases, unmasking, and type 4 CF changes. Most postlesion CFs were near to the lesion edge (N11–N18; shaded blue bar indicates the extent of the lesion) and thresholds remained low. However, there was considerable variation in the change in firing rate across neurons which was not reported in the data (Robertson and Irvine 1989; Kamke et al. 2003; Rajan et al. 1993).

A A set of compartmental model neurons showing type 4 changes following a large basal (high CF) lesion (We = 8, Fe = 80 μS, Wi = 3, Fi = 300 μS). B A set of compartmental model neurons showing type 4 changes, but that have inputs which are not spontaneously active (We = 10, Fe = 80 μS, Wi = 8, Fi = 200 μS). C Another set of compartmental model neurons showing type 4 changes, but that have inputs which are not spontaneously active (We = 10, Fe = 80 μS, Wi = 2, Fi = 200 μS). This example has multipeaked tuning curves prior to the lesion, as a result of inhibition not being effective in suppressing the convergent excitatory inputs. The format of these figures is similar to that in Figure 4A, B, and C.
A component of the inhibition in the models shown in Figures 3, 4, and 5A was tonic (i.e., continuous and spontaneously occurring) because the inputs were spontaneously active. To see if the models would work when inhibition arose from driven activity only, we removed the spontaneous component of the input as described in the “Methods” section. Figure 5B shows an example of the output of a set of model neurons. These neurons showed robust type 4 changes. To determine the robustness of these effects, we ran simulations across the same range of parameters that were used with the spontaneously active inputs. These models showed type 4 changes (46%; the proportions of type 1–3 changes are given in Table 1). However, a proportion of the parameter sets, those with considerably less convergence for inhibitory than for excitatory inputs (i.e., W i < W e), showed spurious excitatory activity. An example of such a model is shown in Figure 5C. Thus, the tonic component of the inhibition was not essential, but it did mean that convergent inputs were more effectively inhibited, producing more physiologically realistic responses.
The role of dendritic processing
The presented model neuron shows robust unmasking and CF changes following simulated lesions. What is not clear is how the model produced these effects. Figure 6 shows a model with only three compartments. Each compartment has an excitatory input with a different CF, and these innervate the cell tonotopically. Additionally, the more proximal dendritic location receives an inhibitory input having exactly the same properties as its excitatory component. Figure 6B shows the RFs of the membrane potential (V m) as measured at the soma (rightmost panel) and at the two dendritic locations. V m at the proximal dendritic location (Fig. 6B, middle panel) is held down by the inhibitory input. The proximal inhibition does not suppress the excitatory inputs at either distal dendrite or at the soma, which show depolarization to their respective inputs, but it prevents either input reaching other compartments. Figure 6C shows the membrane potentials after removal (lesioning) of the inputs to the soma and the more proximal dendrite (a vertical dashed line on the filter bank indicates this lower boundary in Fig. 6A). Now all three locations are depolarized by the remaining distal input and the tuning matches that input (c.f. dashed line showing prelesion CF at the soma). Thus, following the lesion, the input to the distal dendrite causes depolarization at the soma.

A Schematic of a simplified model which has only three compartments: one somatic compartment, which drives spike generation; one proximal; and one distal dendritic compartment. All compartments receive tonotopically aligned excitatory inputs, but only the proximal dendrite receives inhibition. The vertical dashed line in between filters shows the lower limit of the lesion. B Membrane potential RF of each compartment before the lesion. RFs are directly below their respective compartment in A. C Membrane potential RF in each compartment, following the removal of all inputs from the somatic and proximal dendritic compartments to the right of the vertical dashed line in A. The vertical dashed line shows the prelesion CF. Inputs to each compartment have CFs 12% different (one tone) to each neighbor. Gi for middle compartment was 0.75 × 10−4 S; Ge for the three compartments was 0.5, 0.6, and 0.7 × 10−5 S from left to right, respectively; Gd = 6 × 10−3 S.
Using this simplified model, we can also demonstrate how some of the parameters determine the performance of the more elaborate model. Figure 7A shows a model in which inhibition is extended on to the most distal dendrite. Figure 7B and C shows the membrane potential at the soma before and after the lesion, respectively. This time there is no “unmasking” of the distal excitatory inputs because distal inhibition remains. This demonstrates why the model works better when excitatory inputs are more convergent than inhibitory inputs. It also demonstrates how in some model neurons which have CFs close to the edge of a lesion, the firing rate can drop dramatically following a lesion. The somatic and proximal inputs of these neurons are removed by the lesion and the distal inhibition remains strong and largely suppresses the remaining excitatory inputs.

A Schematic of a model like Figure 7, except that the most distal compartment also receives an inhibitory input. B RF showing membrane potential at the soma. C The membrane potentials at the soma following the removal of all inputs from the somatic and proximal dendritic compartments. The vertical dashed line shows the prelesion CF. Model parameters as in Figure 9, except Gi is 4 × 10−5 for the leftmost and middle compartments. D Schematic of a model like A, except that the inputs driving inhibition are offset so that inhibition synapses distal to the excitatory input of the same CF. Only the origin of the inhibition differs from A. E RF showing membrane potential at the soma. F The membrane potentials at the soma following the removal of all inputs from the somatic and proximal dendritic compartments. G–I As above except that the model has inhibition which synapses proximal to the excitatory input with the same CF.
The effects of a slightly different model configuration are shown in Figure 7D–F. In these panels, inhibitory input with a given CF provides the input to a location that was more distal than that of its corresponding excitatory input. Following the simulated lesion, there is an unmasking of the distal input, as in Figure 6. Figure 7G–I shows the reverse situation, i.e., when inhibition at a given CF is located proximal to its excitatory counterpart. In this case, there is virtually no response to sound following the lesion because the inhibition to the middle compartment is still intact. Thus, the relative positions of excitatory and inhibitory inputs tuned to the same frequency have profound effects on processing. In real neurons, one would reasonably expect some degree of variation, which might be another source of diversity not seen in the models.
Simulating spiral ganglion lesions in single IC neurons without any dendritic processing
In the “The role of dendritic processing” section, we demonstrated how dendritic processing produced the model results shown thus far. To investigate whether a model without any local dendritic processing can reproduce the RF changes observed experimentally, we considered the behavior of a point neuron model. This model had one compartment at the soma onto which all excitatory and inhibitory inputs converged. The same Gaussian weighting functions were applied to the AN inputs, but the sum of the weighted excitatory inputs formed a single excitatory conductance. Inhibitory input was derived in the same way (see the “Methods” section).
We again ran several sets of simulations across wide ranges of input parameters (see Table 1 for range). These simulations revealed widespread residual CF changes (type 1; see Table 1), but few other changes. Figure 8A shows an example of a set of parameters that produced type 1 changes. Prelesion tuning is broad (Q10s are around 2 in this example; Q10s for the same CF range in the AN model are 4–5), and the postlesion behavior is very similar to the compartmental example (Fig. 4A). Figure 8E shows the incidence of type 1 and 4 changes across a wide range of parameters (see Table 1). Type 1 changes occurred for a range of values of W e, W i, and F e when F i was low. This figure shows only two incidences of models showing any type 4 changes (in red). Subsequent simulations did reveal two small ranges of parameters that consistently produced type 4 changes (shown in Table 1). Figure 8B shows the tuning curve changes for an example set of model parameters that was typical of those models when W e > W i. In channels 8 and 14, there is unmasking that leads to CF changes. In other channels, the only change is a loss of activity following the lesion. Figure 8C shows an example parameter set when W i > W e. These models showed circumscribed prelesion RFs with considerable unmasking of activity at frequencies above and below the lesion. Occasionally (N4, N18), this unmasking led to subtle CF changes (i.e., type 4). Figure 8F shows the distribution of type 1 (upper panel) and type 4 (lower panel) changes across the neural arrays for all of the models across the three-point neuron parameter sets in Table 1. Type 1 changes can occur across most of the neural array, while type 4 changes tend to occur only beyond the edges of the lesion. Type 2 (within RF increases) and 3 (unmasking) changes occurred for W i > W e (not shown).

A–D Examples of outputs from the point neuron model for different parameter sets. A Model showing residual changes (We = 3, Fe = 0.6 μS, Wi = 3, Fi = 0.2 μS). B Model with Wi < We showing type 4 changes (We = 1.8, Fe = 0.7 μS, Wi = 0.6, Fe = 2.1 μS). C Model with Wi > We showing type 4 changes (We = 3.6, Fe = 2.5 μS, Wi = 6.4, Fi = 2.5 μS). D Model with side band inhibition showing type 4 changes (We = 3.25, Fe = 0.7 μS, Wi = 0.1, Fi = 3 μS). E Range of parameters across which residual CF (type 1; in blue) changes and CF changes from unmasking (type 4; in red) occur. F The distribution of type 1 (upper panel) and type 4 (lower panel) CF changes across the array of neurons, for the three-point neuron models in Table 1.
In order to explore further the possibility for unmasking of (type 4) CF changes in point neuron models, we also explored the effect of inhibition which was restricted to two side bands placed symmetrically about CF. This model was implemented using two separate and very narrow Gaussian functions (see the “Methods” section) with the distance in semitones between the functions being a model parameter. With a gap of zero, the model was identical to the previous point neuron model with the same parameters. Figure 8D shows an example of a point neuron model which was amongst the most successful at producing type 4 changes. Notably, these models were able to reproduce the W-shaped RF at the center of the lesion. Type 4 changes occurred for a range of parameters, when W i ≤ 1, Offi > 2 (in semitones) and was most common for W e < 3.5. In the population of parameters tested (see the “Methods” section and Table 1), these changes were more common than the simple point neuron model, but still comparatively rare (0.6%), and never resembled the results of the compartmental model.
Calford (2002) argued that, conceptually, stimulus-driven inhibition was not likely to result in unmasking. The point neuron model results support this argument. His point was that for one input to inhibit another input, the first input had to be suppressing the second, even if a stimulus was driving the second input more strongly. Tonic (spontaneous) inhibition driven from the first input, on the other hand, would be present even without an effective stimulus. Yet it would be removed if the first input was destroyed. In somatosensory cortex, it was demonstrated experimentally that eliminating C fiber tonic inhibition allowed reorganization (Calford and Tweedale 1991a, b). To explore this in the point neuron model, we modified it so that the inhibitory inputs were entirely tonic. As long as there was an intact AN fiber at a given CF, that input would produce a constant inhibition, equivalent to a discharge rate of 100s−1. Note that these inputs were still subject to the Gaussian weighting function and summed to make the inhibitory conductance. This meant that the strength and degree of convergence of the inhibition could still be controlled parametrically. This time, parametric exploration revealed a considerable percentage of models (6%) which showed type 4 changes. Figure 9A shows an example model set, which changes following a lesion in a similar way to that of the compartmental model. Figure 9B shows how type 1 and type 4 changes depend on the model parameters. It shows that type 1 changes are common in the range 1.5 < W e < 4, while type 4 changes are most common when W i > 1. The mean Q10 for those models showing type 4 changes was 2.6 (SD = 0.9). Figure 9C shows how the type 2 and type 3 changes depend on the choice of model parameters. This suggests that increases can occur for a range of parameters when W e ≠ W i, while unmasking is restricted mostly to conditions when W e > W i.

A A point neuron model showing robust type 4 changes which receives only tonic inhibition (We = 3, Fe = 0.6 μS, Wi = 3, Fi = 0.2 μS). B Range of parameters across which residual CF (type 1; in blue) changes and CF changes from unmasking (type 4; in red) occur for point neuron models with tonic inhibition. C Range of parameters across which increases (type 2; in blue) and unmasking (type 3; in green; red squares indicate both together) occurs for point neuron models with tonic inhibition.
Discussion
We have shown that models of single neurons can exhibit the physiologically observed “unmasking” of low-threshold hidden inputs that lead to “type 4” changes in IC. Since physiologically, it seems clear that inhibition can be responsible for modifying tuning (e.g., (Suga 1995; LeBeau et al. 2001; Xie et al. 2005), it is surprising that robust type 4 changes were restricted to such specific arrangements of model inputs. Currently, we cannot be precise about the exact requirements for these changes to occur.
Type 4 changes may depend on the size of a shift in dominance from inhibitory inputs to excitatory ones and the range of CFs and stimulus conditions across which it occurs. In the basic point neuron models, the total strengths of the excitatory and inhibitory input are both simple linear (Gaussian) weighted sums of the same auditory nerve inputs and are centered on the same input CF. Unless the widths of the two functions differ greatly, the effect of a lesion will tend to be quite similar for excitation and inhibition, and so any shift toward the excitatory inputs will often be small. When the functions are quite different, there are either no excitatory inputs to conceal or there is insufficient inhibition with which to conceal them. In models where the inhibitory inputs form two side bands, shifts in the balance following a lesion can be larger, but still this does not appear to be enough to produce type 4 changes across a range of CFs. The relative effects of lesioning on stimulus-driven excitatory inputs and tonic inhibitory inputs can be quite different and this is enough to produce robust type 4 changes. This is consistent with an argument made by Calford (2002) in favor of tonic inhibition. Finally, in the compartmental model described in this study, one strong proximal inhibitory input can easily suppress more distal excitatory inputs and its removal by a lesion will reveal them.
Residual CF (type 1) changes occurred in all models if a lesion left sufficient excitatory inputs and these were not inhibited. Postlesion increases in activity (type 2) and unmasking (type 3) also occurred in all types of models tested. The incidence varied considerably among different populations, but a given population of models tended to show similar proportions of both type 2 and 3 changes. However, type 3 changes, by definition, occur at suprathreshold stimulus levels. The auditory nerve model inputs in this study all had uniformly low thresholds and narrow dynamic ranges. Inputs with higher thresholds would have expanded the range of effects seen at higher stimulus levels. Interestingly, however, the proportions of the changes seen in the compartmental models are similar to those found in IC (Synder et al. 2008).
The physiological data (cf. Fig. 2) is diverse both in the prelesion responses and the effects of peripheral lesions. In contrast, individual arrays of model neurons showed stereotypical responses across the CF range. Models showed greater diversity across different parameter values and still more variety across the different types of models. More realistic simulation of the physiological data would be achievable, therefore, with a mix of parameters and even model variants. Also, the IC is a point of integration for ascending afferent pathways bilaterally (Cant 2005; Schofield 2005). As a result, neurons can vary in their tuning (Le Beau et al. 1996; Wang et al. 1996; Ramachandran et al. 2000), rate–level functions (Ehret and Merzenich 1988; Rees and Palmer 1988), temporal coding (Rees and Moller 1983, 1987; Schreiner and Langner 1988), binaural properties (Palmer and Kuwada 2005), and intrinsic currents (Wu 2005). Simulation of these would have made for more diverse and realistic models and might have affected the simulation results.
The models that show CF changes (type 1 or type 4) require a convergence of inputs from a wide range of CFs. The central IC is thought to be divided into discrete laminae (<200 μm wide) within which afferent and neuronal CFs vary by approximately 0.3 octaves (Oliver 2005). Only a minority of cells, stellate type cells, have dendrites (and axons) extending across these laminae. Nevertheless, blocking inhibition reveals excitatory activity beyond the normal excitatory RF (LeBeau et al. 2001; Xie et al. 2005) and intracellular recordings reveal subthreshold tuning of much broader extracellular excitatory RF (Yang et al. 1992; Suga et al. 1997; LeBeau et al. 2001; Xie et al. 2007). Thus, it appears that our current knowledge of anatomy within the IC cannot explain a range of physiological data, including the data that was the focus of this study.
The compartmental models make specific predictions about the relative degree of convergence of excitatory and inhibitory inputs. The most robust type 4 changes occurred when the excitatory inputs had a wider range of CFs than inhibitory inputs. Neurons receiving excitatory inputs from a wider range of CFs than inhibitory inputs would seem unlikely to display lateral inhibitory side bands, and side bands are impossible with tonic inhibition. This is at apparent odds with a role for side band inhibition in unmasking and also in the ubiquity of side band inhibition in IC (and elsewhere). Admittedly, the creation of inhibitory side bands on the flanks of the excitatory response areas was not one of our goals. It is possible that different inhibitory inputs might be responsible for mediating unmasking and inhibitory side bands, and this might be the case if there were a mix of stimulus-driven wideband inhibition and, as proposed by Calford, tonic inhibition. This would be consistent with the effects of mild hearing loss on cortical tuning, which produces a loss of side band inhibition but is insufficient to produce tonotopic reorganization (Rajan 1998). Also, local bicuculline application does not block inhibitory side bands in IC (LeBeau et al. 2001), suggesting that they are created by subcollicular nuclei. Of course, none of these considerations rules out the possibility that some other configuration of inhibition which produces inhibitory side bands might also produce robust type 4 changes.
We have focused comparisons of our model results with the effects of spiral ganglion lesions on responses in IC. This data set was convenient as it was limited to acute CF changes resulting from unmasked responses, and spiral ganglion lesions were straightforward to simulate. However, the same mechanisms may contribute to the effects seen in other experiments. Nonresidual type 4 changes were seen in a minority of IC neurons following large chronic lesions to basal regions of the cochlea (Irvine et al. 2003). These lesions were very different to the spiral ganglion lesions. The compartmental model was able to show type 4 changes following large basal lesions, albeit with a wide variation in firing rates which was not reported for the data. Interestingly, Irvine et al. (2003) also observed an increase in the incidence of excitatory ipsilateral responses. Our model did not include any ipsilateral inputs. We suspect that for the removal of contralateral inhibition to affect ipsilateral responses, the inhibition would need to have a tonic component. Furthermore, it would have to innervate either the cell body or the same dendrites as the ipsilateral excitatory inputs in order to affect ipsilateral excitatory responses. The models may also be relevant to the acute effects of pure tone trauma in the IC (Wang et al. 1996). Type 4 changes do not follow pure tone trauma, but it is possible that the mechanisms modeled in this study, perhaps acting on higher threshold inputs, are responsible for some of the type 3 unmasking seen. However, although these experiments using acoustic trauma rule out slower plastic changes, the effects on auditory nerve responses can be complex and different to both spiral ganglion lesions and larger chronic lesions.
The models may also be of relevance to numerous cortical lesion studies (e.g., Robertson and Irvine 1989; Rajan et al. 1993). Studies of A1 responses following cochlear lesions (e.g., Rajan et al. 1993) report cortical locations that no longer respond to sound. This is seen in our compartmental models, but has not been observed in IC following spiral ganglion lesions. In the thalamus and cortex, tonotopic reorganization is restricted to the side of the lesion (Kamke et al. 2003; Rajan et al. 1993), unlike in the IC where excitatory ipsilateral responses are more common. We believe this would be the case in the model if the constraints for reproducing ipsilateral effects in IC, outlined above, were not met. Unmasking of latent inputs to cortical neurons has also been shown in various experiments (Rajan 1998, 2001; Calford et al. 1993; Norena et al. 2003) using acoustic trauma and has been associated with a loss of side band inhibition (Rajan 1998). As in the IC (e.g., Wang et al. 1996), the models may be broadly consistent with these results. Interestingly, however, chronic mild hearing loss does not result in the within RF (type 2) increases which are seen in the models (Rajan 1998; Rajan and Irvine 1998). Clearly, no one model is likely to account for the range of results seen across many experiments and numerous auditory nuclei.
We have demonstrated that rapid RF changes can be modeled by “unmasking”: the removal of inhibitory inputs to reveal previously subthreshold excitatory inputs. Acute changes in RF might also be explained by other forms of synaptic plasticity. Hebbian synaptic plasticity (Hebb 1949) has been proposed as a mechanism for the maintenance of cortical RFs in several modalities (Pearson et al. 1987; Armentrout et al. 1994; Benuskova et al. 1994; Jenison 1997; Benuskova et al. 1999) and models of it can reproduce (Jenison 1997) the normal development of tonotopy (Reale et al. 1987) and the changes following lesions reported by Robertson and Irvine (1989). Another possibility is that multineuron circuitry could result in processing that is functionally similar to that which we see in dendrites in this model. Most IC cells have extensive local projections. Across frequency integration may be a local, multisynaptic process arising from a network of neurons with overlapping arborizations and interactions. It remains an empirical question what mechanisms are at work and how much different mechanisms contribute to the changes seen in the responsiveness of IC neurons and elsewhere.
References
Armentrout SL, Reggia JA, Weinrich M. A neural model of cortical map reorganization following a focal lesion. Artif. Intell. Med. 6:383–400, 1994.
Banks MI, Sachs MB. Regularity analysis in a compartmental model of chopper units in the anteroventral cochlear nucleus. J. Neurophysiol. 65:606–629, 1991.
Benuskova L, Diamond ME, Ebner FF. Dynamic synaptic modification threshold: computational model of experience-dependent plasticity in adult rat barrel cortex. Proc. Natl. Acad. Sci. U. S. A. 91:4791–4795, 1994.
Benuskova L, Ebner FF, Diamond ME, Armstrong-James M. Computational study of experience-dependent plasticity in adult rat cortical barrel-column. Network 10:303–323, 1999.
Boettcher FA, Salvi RJ. Functional-changes in the ventral cochlear nucleus following acute acoustic overstimulation. J. Acoust. Soc. Am. 94:2123–2134, 1993.
Calford MB. Dynamic representational plasticity in sensory cortex. Neuroscience. 111(4):709–738, 2002.
Calford MB, Tweedale R. Acute changes in cutaneous receptive fields in primary somatosensory cortex after digit denervation in adult flying fox. J. Neurophysiol. 65:178–187, 1991a.
Calford MB, Tweedale R. C-fibres provide a source of masking inhibition to primary somatosensory cortex. Proc. Biol. Sci. 243:269–275, 1991b.
Calford MB, Rajan R, Irvine DR. Rapid changes in the frequency tuning of neurons in cat auditory cortex resulting from pure-tone-induced temporary threshold shift. Neuroscience 55:953–964, 1993.
Cant NB. Projectections from the cochlear nucleus complex to the inferior colliculus. In: Winer JA, Schreiner C (eds) The inferior colliculus. New York, Springer, pp. 115–131, 2005.
Carney LH. A model for the responses of low-frequency auditory-nerve fibers in cat. J. Acoust. Soc. Am. 93:401–417, 1993.
Darian-Smith C, Gilbert CD. Axonal sprouting accompanies functional reorganization in adult cat striate cortex. Nature 368:737–740, 1994.
Ehret G, Merzenich MM. Neuronal discharge rate is unsuitable for encoding sound intensity at the inferior-colliculus level. Hear. Res. 35:1–7, 1988.
Hebb DO. The organization of behaviour. New York, Wiley, 1949.
Holmes SD, Sumner CJ, O’Mard LP, Meddis R. The temporal representation of speech in a nonlinear model of the guinea pig cochlea. J. Acoust. Soc. Am. 116:3534–3545, 2004.
Irvine DR, Rajan R, Smith S. Effects of restricted cochlear lesions in adult cats on the frequency organization of the inferior colliculus. J. Comp. Neurol. 467:354–374, 2003.
Jenison RL. A computational model of reorganization in auditory cortex in response to cochlear lesions. In: Jesteadt W (ed) Modelling sensorineural hearing loss. Hillsdale, Erlbaum, 1997.
Johnston D, Wu S. Foundations of cellular neurophysiology. MIT Press, Cambridge 1995.
Kaltenbach JA, Czaja JM, Kaplan CR. Changes in the tonotopic map of the dorsal cochlear nucleus following induction of cochlear lesions by exposure to intense sound. Hear. Res. 59:213–223, 1992.
Kamke MR, Brown M, Irvine DR. Plasticity in the tonotopic organization of the medial geniculate body in adult cats following restricted unilateral cochlear lesions. J. Comp. Neurol. 459:355–367, 2003.
Le Beau FE, Rees A, Malmierca MS. Contribution of GABA- and glycine-mediated inhibition to the monaural temporal response properties of neurons in the inferior colliculus. J. Neurophysiol. 75:902–919, 1996.
LeBeau FE, Malmierca MS, Rees A. Iontophoresis in vivo demonstrates a key role for GABA(A) and glycinergic inhibition in shaping frequency response areas in the inferior colliculus of guinea pig. J. Neurosci. 21:7303–7312, 2001.
Liberman MC, Mulroy MJ. Acute and chronic effects of acoustic trauma: cochlear pathology and auditory-nerve pathophysiology. In: Hamernik RP, Henderson D, Salvi R (eds) New perspectives on noise-induced hearing loss. Raven Press, New York, pp. 105–135, 1982.
Meddis R, O’Mard LP, Lopez-Poveda EA. A computational algorithm for computing nonlinear auditory frequency selectivity. J. Acoust. Soc. Am. 109:2852–2861, 2001.
Moshitch D, Las L, Ulanovsky N, Bar-Yosef O, Nelken I. Responses of neurons in primary auditory cortex (A1) to pure tones in the halothane-anesthetized cat. J. Neurophysiol. 95:3756–3769, 2006.
Muller CM, Scheich H. Contribution of GABAergic inhibition to the response characteristics of auditory units in the avian forebrain. J. Neurophysiol. 59:1673–1689, 1988.
Norena AJ, Tomita M, Eggermont JJ. Neural changes in cat auditory cortex after a transient pure-tone trauma. J. Neurophysiol. 90:2387–2401, 2003.
Oliver DL. Neuronal organization in the inferior colliculus. In: Winer JA, Schreiner C (eds) The inferior colliculus. New York, Springer, pp. 69–114, 2005.
Palmer A, Kuwada S. Binaural and spatial coding in the inferior colliculus. In: Winer JA, Schreiner C (eds) The inferior colliculus. New York, Springer, pp. 377–410, 2005.
Pearson JC, Finkel LH, Edelman GM. Plasticity in the organization of adult cerebral cortical maps: a computer simulation based on neuronal group selection. J. Neurosci. 7:4209–4223, 1987.
Rajan R. Receptor organ damage causes loss of cortical surround inhibition without topographic map plasticity. Nat. Neurosci. 1:138–143, 1998.
Rajan R. Plasticity of excitation and inhibition in the receptive field of primary auditory cortical neurons after limited receptor organ damage. Cereb. Cortex 11:171–182, 2001.
Rajan R, Irvine DR. Absence of plasticity of the frequency map in dorsal cochlear nucleus of adult cats after unilateral partial cochlear lesions. J. Comp. Neurol. 399:35–46, 1998.
Rajan R, Irvine DR, Wise LZ, Heil P. Effect of unilateral partial cochlear lesions in adult cats on the representation of lesioned and unlesioned cochleas in primary auditory cortex. J. Comp. Neurol. 338:17–49, 1993.
Ramachandran R, Davis KA, May BJ. Rate representation of tones in noise in the inferior colliculus of decerebrate cats. J. Assoc. Res. Otolaryngol. 1:144–160, 2000.
Reale RA, Brugge JF, Chan JC. Maps of auditory cortex in cats reared after unilateral cochlear ablation in the neonatal period. Brain Res. 431:281–290, 1987.
Rees A, Moller AR. Responses of neurons in the inferior colliculus of the rat to AM and FM tones. Hear. Res. 10:301–330, 1983.
Rees A, Moller AR. Stimulus properties influencing the responses of inferior colliculus neurons to amplitude-modulated sounds. Hear. Res. 27:129–143, 1987.
Rees A, Palmer AR. Rate-intensity functions and their modification by broadband noise for neurons in the guinea pig inferior colliculus. J. Acoust. Soc. Am. 83:1488–1498, 1988.
Robertson D, Irvine DR. Plasticity of frequency organization in auditory cortex of guinea pigs with partial unilateral deafness. J. Comp. Neurol. 282:456–471, 1989.
Schofield BR. Superior olivary complex and lateral lemniscal connections of the auditory midbrain. In: Winer JA, Schreiner C (eds) The inferior colliculus. New York, Springer, pp. 132–154, 2005.
Schreiner CE, Langner G. Periodicity coding in the inferior colliculus of the cat. II. Topographical organization. J. Neurophysiol. 60:1823–1840, 1988.
Snyder RL, Sinex DG. Tonotopic reorganization of cat primary auditory cortex (AI) after acute lesions of restricted sectors of the spiral ganglion. In: Annual Meeting of the Society for Neuroscience, 1998.
Snyder RL, Sinex DG. Immediate changes in tuning of inferior colliculus neurons following acute lesions of cat spiral ganglion. J. Neurophysiol. 87:434–452, 2002.
Snyder RL, Sinex DG, McGee JD, Walsh EW. Acute spiral ganglion lesions change the tuning and tonotopic organization of cat inferior colliculus neurons. Hear. Res. 147(1–2):200–220, 2000.
Snyder RL, Bonham BH, Sinex DG. Acute changes in frequency responses of inferior colliculus central nucleus (ICC) neurons following progressively enlarge restricted spiral ganglion lesions. Hear. Res. 2008. doi: 10.1016/j.hears.2008.09.010.
Suga N. Sharpening of frequency tuning by inhibition in the central auditory system: tribute to Yasuji Katsuki. Neurosci. Res. 21:287–299, 1995.
Suga N, Zhang Y, Yan J. Sharpening of frequency tuning by inhibition in the thalamic auditory nucleus of the mustached bat. J. Neurophysiol. 77:2098–2114, 1997.
Sumner CJ, Lopez-Poveda EA, O’Mard LP, Meddis R. A revised model of the inner-hair cell and auditory-nerve complex. J. Acoust. Soc. Am. 111:2178–2188, 2002.
Sumner CJ, Lopez-Poveda EA, O’Mard LP, Meddis R. Adaptation in a revised inner-hair cell model. J. Acoust. Soc. Am. 113:893–901, 2003a.
Sumner CJ, O’Mard LP, Lopez-Poveda EA, Meddis R. A nonlinear filter-bank model of the guinea-pig cochlear nerve: rate responses. J. Acoust. Soc. Am. 113:3264–3274, 2003b.
Tailby C, Wright LL, Metha AB, Calford MB. Activity-dependent maintenance and growth of dendrites in adult cortex. Proc. Natl. Acad. Sci. U. S. A. 102:4631–4636, 2005.
Wang J, Salvi RJ, Powers N. Plasticity of response properties of inferior colliculus neurons following acute cochlear damage. J. Neurophysiol. 75:171–183, 1996.
Willott JF, Lu SM. Noise-induced hearing loss can alter neural coding and increase excitability in the central nervous system. Science 216:1331–1334, 1982.
Wu SH. Biolphysical properties of inferior colliculus neurons. In: Winer JA, Schreiner C (eds) The inferior colliculus. New York, Springer, pp. 282–311, 2005.
Xie R, Meitzen J, Pollak GD. Differing roles of inhibition in hierarchical processing of species-specific calls in auditory brainstem nuclei. J. Neurophysiol. 94:4019–4037, 2005.
Xie R, Gittelman JX, Pollak GD. Rethinking tuning: in vivo whole-cell recordings of the inferior colliculus in awake bats. J. Neurosci. 27:9469–9481, 2007.
Yang L, Pollak GD, Resler C. GABAergic circuits sharpen tuning curves and modify response properties in the mustache bat inferior colliculus. J. Neurophysiol. 68:1760–1774, 1992.
Acknowledgements
This work was supported by the Medical Research Council and by the NIDCD grant DC03549. The authors would like to thank Alan Palmer and Stephen Coombes for their comments on an earlier version of this manuscript, and also thank Steve Colburn and two anonymous reviewers for the high quality of their reviews.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sumner, C.J., Scholes, C. & Snyder, R.L. Retuning of Inferior Colliculus Neurons Following Spiral Ganglion Lesions: A Single-Neuron Model of Converging Inputs. JARO 10, 111–130 (2009). https://doi.org/10.1007/s10162-008-0139-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-008-0139-6