
Abstract
Dysfunction of slit diaphragm, a cell–cell junction of glomerular podocytes, is involved in the development of proteinuria in several glomerular diseases. Slit diaphragm should be a target of a novel therapy for proteinuria. Nephrin, NEPH1, P-cadherin, FAT, and ephrin-B1 were reported to be extracellular components forming a molecular sieve of the slit diaphragm. Several cytoplasmic proteins such as ZO-1, podocin, CD2AP, MAGI proteins and Par-complex molecules were identified as scaffold proteins linking the slit diaphragm to the cytoskeleton. In this article, new insights into these molecules and the pathogenic roles of the dysfunction of these molecules were introduced. The slit diaphragm functions not only as a barrier but also as a signaling platform transfer the signal to the inside of the cell. For maintaining the slit diaphragm function properly, the phosphorylation level of nephrin is strictly regulated. The recent studies on the signaling pathway from nephrin, NEPH1, and ephrin-B1 were reviewed. Although the mechanism regulating the function of the slit diaphragm had remained unclear, recent studies revealed TRPC6 and angiotensin II-regulating mechanisms play a critical role in regulating the barrier function of the slit diaphragm. In this review, recent investigations on the regulation of the slit diaphragm function were reviewed, and a strategy for the establishment of a novel therapy for proteinuria was proposed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proteinuria is one of the most important symptoms of kidney diseases and is reported to be one of the most important risk factors of stroke and cardiovascular diseases [1]. However, the etiology, pathogenesis, and clinical significance of proteinuria were not fully understood yet. The development of more effective selective therapy for proteinuria is awaited. Glomerular proteinuria is associated with pathological damage of the glomerular filtration barrier, which is composed of three layers: glomerular endothelial cells, glomerular basement membrane (GBM), and glomerular visceral epithelial cells (podocytes). The studies in the past two decades revealed that the third layer, podocyte functions as the final barrier [2, 3]. It is now widely accepted that slit diaphragm, a cell–cell junction of podocytes, plays a critical role in preventing the leak of plasma proteins into primary urine and that dysfunction of the slit diaphragm is involved in the development of proteinuria in several glomerular diseases. The functional molecules of the slit diaphragm could be novel therapeutic targets for proteinuria.
The authors have previously reviewed the role of the slit diaphragm [4, 5]. In this review, first, we will overview a unique, specialized property of the slit diaphragm, review new insight into the structural and functional properties of the slit diaphragm, and discuss the future line of the investigations for establishing a novel therapy for proteinuria.
Overview: slit diaphragm, a specialized cell–cell junction of podocyte, shares common characteristics with synapse
Foot processes of podocytes cover the outer side of GBM. The neighboring foot processes were derived from different cell bodies, and they were connected by a continuous membrane-like structure, which is called “slit diaphragm”. The early study with electron microscopy showed that the slit diaphragm exhibited a zipper-like substructure with alternating, periodic cross-bridges extending from the opposite podocyte plasma membranes [6]. Arakawa demonstrated that neighboring foot processes were interdigitated with the scanning electron microscopic analysis [7]. Developmental analyses showed the slit diaphragms appear during the capillary loop stage and gradually replace tight junctions [8, 9]. The slit diaphragm is reported to be a specialized adherens junction, because it contains typical adherens junction proteins such as P-cadherin, β-catenin [10], and FAT [11], while Farquhar’s group reported that slit diaphragm is a highly specialized variant of a tight junction, because the slit diaphragm contains several tight junction proteins including ZO-1, JAM-A, occludin, and cingulin [12].
Podocyte shares common characteristics with neuron: both of them are final differentiated cells, which have no ability of proliferation, and they possess unique processes and share common functional molecules such as synaptopodin [13], densin [14], drebrin [15], and dendrin [16]. These molecules are restrictively expressed in podocyte foot processes and neuronal dendric spines. Slit diaphragm connects foot processes of podocytes. Synapse is a structure between axon terminal and dendrite of neurons. Both are highly specialized cell–cell junctions connecting unique processes. Rastaldi et al. reported that Rab3, a synaptic vesicle surface protein, is expressed at the slit diaphragm and that the vesicles locate at the edge of the foot process [17]. Synaptic vesicle-associated protein 2B (SV2B) is also expressed at slit diaphragm [18]. A recent study showed that the deletion of SV2B results in the mislocalization of the slit diaphragm components including nephrin and CD2AP and that several synaptic vesicle-associated molecules including SNARE molecules were expressed in podocyte [19]. Soda et al. reported that dynamin, synaptojanin, and endophilin, which are functional partners in synaptic vesicle recycling, are essential for the formation and maintenance of the podocyte foot process structure [20]. It is reported that neurexin-1, a presynaptic adhesion molecule, is expressed in podocyte [21], and neurexin-1 interacts with CD2AP and SV2B. It is plausible that dysfunction of these neuron-associated molecules in podocyte is involved in the development of slit diaphragm dysfunction and that the pharmacological reagent targeting these molecules could be candidates for a novel therapy protecting podocyte.
Extracellular components forming a molecular sieve of the slit diaphragm
The first molecule identified as an extracellular component of the slit diaphragm is nephrin [22]. Nephrin is accepted to be the main body of the extracellular portion of the slit diaphragm. Nephrin has a long extracellular domain containing eight Ig-like modules and a single fibronectin type III module. Ruotsalainen et al. proposed that nephrin molecules extending from two adjacent foot processes are likely to interact with each other in the slit through homophilic interactions [23], as has been shown for other Ig cell adhesion molecules such as N-CAM [24], C-CAM [25], and L1 [26]. Nephrin was identified as a product of the mutated gene in patients with Finnish-type nephrotic syndrome [22], and it was demonstrated that the antibody against nephrin is capable of inducing massive proteinuria [27,28,29]. Based on these findings, nephrin is accepted to be a key molecule forming a molecular sieve of the slit diaphragm. Downregulation of nephrin is observed in experimental nephrotic models [28, 30,31,32] and in clinical cases of several types of glomerular disease such as minimal change disease [33], membranous nephropathy, membranoproliferative glomerulonephritis, IgA nephropathy, lupus nephritis [34], diabetic kidney disease [35, 36], and preeclampsia [37]. These studies indicated that nephrin dysfunction is one of the common pathogenic mechanism of proteinuria in human glomerular diseases.
Another critical molecule of the extracellular components is NEPH1. NEPH1 was identified as a nephrin-related protein by the gene trapping screen [38]. NEPH1, a transmembrane protein, contains five extracellular immunoglobulin-like domains [38]. NEPH1 interacted with nephrin in a cis form [39]. Glomeruli of NEPH1 knock-out mice showed the effacement of podocyte foot processes and proteinuria, and all mice died at 3–8 weeks of age [38], which indicates that NEPH1 is also essential for maintaining the barrier function of the slit diaphragm. A recent study using high-resolution ultrastructural imaging showed that slit diaphragm is multilayered and that the NEPH1 molecule spanning is in the lower part of the junction, closer to GBM with a width of 23 nm, while nephrin contributes to the apical region of the slit diaphragm with a width of 45 nm [40]. The study also reported that nephrin is not required to build a functional filtration barrier in birds, and discussed that NEPH1 plays a critical role in birds.
Some of other transmembrane proteins were reported to be accumulated at the slit diaphragm. Reiser et al. reported that P-cadherin, a member of cadherin superfamily, was localized at the slit diaphragm [10]. Cadherin forms homophilic Ca2+-dependent cell–cell adhesion. FAT, a large transmembrane protein of 34 tandem cadherin-like repeats, was localized at the slit diaphragm and was costained with nephrin [11]. FAT KO mice showed severe proteinuria, which indicates that FAT is also essential for maintaining the filtration barrier of the slit diaphragm [41]. It was also reported that neurexin [21] and ephrin-B1 [42] are accumulated at the slit diaphragm. Neurexin is a presynaptic adhesion molecule and is known to have multiple splicing variants. It was shown that a unique variant of neurexin is expressed in the podocyte. Immunoprecipitation assay with rat glomerular lysate showed that neurexin interacted with CD2AP, a cytoplasmic molecule binding to nephrin [21]. Ephrin-B1 is a protein of Eph-ephrin family, which has many biological functions in several types of tissues [43]. Ephrin-B1 interacts with nephrin via their extracellular portions. The podocyte selective ephrin-B1 KO mice displayed mild but significant proteinuria [44], indicating that ephrin-B1 participates in maintaining the barrier function of the slit diaphragm. However, the molecular components forming a sieve structure of the extracellular domain of the slit diaphragm are not well clarified yet.
Multiple scaffold proteins participate in the link of slit diaphragm to the cytoskeleton
Multiple cytoplasmic proteins linking the membrane proteins at the slit diaphragm to the cytoskeleton were identified. Schematic interaction of the scaffold proteins and transmembrane proteins at the slit diaphragm was shown in Fig. 1. ZO-1 (zonula occludens-1) was reported to be localized at the cytoplasm just below the cell surface at the insertion point of slit diaphragm [45]. ZO-1 is the first molecule identified as a component of the slit diaphragm. ZO-1 was originally identified as a component of tight junction and belongs to a family of the membrane–associated guanylate kinase (MAGUK) molecules with PDZ domains [46]. ZO-1 directly interacts with NEPH1 by binding of the first PDZ domain of ZO-1 to the C-terminus PDZ binding motif of NEPH1 [47]. The interaction between NEPH1 and ZO-1 was lost in response to the ischemic injury leading to podocyte foot process effacement and proteinuria [48]. The recent study intending novel drug development showed that isodesmosine, a naturally occurring compound, can protect podocyte from injury by stabilizing the ZO-1-NEPH1 complex [49]. These reports suggested that the interaction of ZO-1 and NEPH1 is essential for maintaining the slit diaphragm structure, and the interaction could be a target for a drug.

Schematic diagram of the slit diaphragm. Nephrin, NEPH1, ephrin-B1, FAT, and neurexin form the extracellular region of the slit diaphragm. The extracellular components of the slit diaphragm interact with the scaffold proteins, podocin, CD2AP, ZO-1, Nck, MAGI, and Par-complex molecules (Par-3, Par-6, aPKC)
Podocin is identified as a product of a gene mutated in familial steroid-resistant nephrotic syndrome [50]. Since podocin has a single transmembrane region and both N- and C-terminus of podocin are localized at cytoplasm, podocin is considered to have a hairpin-like structure [51]. Podocin interacts with nephrin, NEPH1 and CD2AP [52,53,54]. The expression and the localization of podocin are altered in several types of acquired glomerular diseases such as FSGS [55], lupus nephritis [56] and IgA nephropathy [57], and in experimental nephrotic models [58]. Podocin is a mammalian homolog of the C. elegans stomatin family protein Mec-2. Mec-2 is recruited to the putative mechanosensory complex in C. elegans touch sensory neurons [59]. Podocin interacts with TRPC6, one of the key regulators of slit diaphragm function. Knockdown of podocin markedly increased stretch-evoked activation of TRPC6. It is also reported that podocin deficiency results in Ca2+ overload in foot processes [60]. Podocin regulates the barrier function of the slit diaphragm by acting as a switch to determine the preferred mode of TRPC6 activation.
CD2AP, an 80 kDa protein, has been shown to interact with nephrin [61]. CD2AP was identified as an SH3-containing protein that binds to the cytoplasmic domain of CD2, a membrane protein on T cell and natural killer cell. CD2AP anchors nephrin to the cytoskeleton, since CD2AP has an actin-binding site at the N-terminus. Mice lacking CD2AP exhibit morphological alterations such as loss of foot process, severe proteinuria [62]. Kim et al. reported that two human patients with focal segmental sclerosis had a mutation predicted to ablate the expression of one CD2AP allele [63]. It has been shown that lack of CD2AP leads to the increased expression of TGF-β and promotes the TGF-β-induced apoptosis [64]. The study indicated that CD2AP regulates the survival of podocyte by regulating the expression of TGF-β. It is also reported that dendrin binds CD2AP and nephrin at the slit diaphragm [16]. The report showed that dendrin relocates to the nucleus of injured podocytes and that nuclear dendrin modulates TGF-β-induced apoptosis. Following the report, Yaddanapudi et al. reported that loss or downregulation of CD2AP allowed for an increase in TGF-β signaling and the translocation of dendrin from the slit diaphragm into the nucleus. Dendrin is a transcription factor specifically promoting the expression of cytosolic CatL. Cytosolic CatL, in turn, drove the reorganization of the actin cytoskeleton. Then, it was concluded that CD2AP functions as the gatekeeper of the podocyte TGF-β response through its regulation of cytosolic CatL expression [65]. Very recently, Tossidou et al. reported that CD2AP is a phosphorylation target of receptor tyrosine kinases stimulated by VEGF-A [66]. They demonstrated that phosphorylation of tyrosine at position Y10 of the SH3-1 domain of CD2AP could change the affinity of CD2AP to nephrin and is indispensable for CD2AP function.
MAGI proteins (MAGI-1, MAGI-2, MAGI-3) belong to the MAGUK family function as molecular scaffolds, coordinating signaling complexes by linking cell surface receptors to the cytoskeleton. MAGI-1 interacts with junctional adhesion molecule 4 (JAM4), and both MAGI-1 and JAM-4 are expressed in podocytes [67]. Immunoelectron microscopy shows that the localization of MAGI-1 is restricted to the slit diaphragm, whereas JAM4 is distributed at the slit diaphragm and on apical membranes. The in vitro interaction assay showed that MAGI-1 binds nephrin via the middle PDZ domains of MAGI-1 and the carboxyl terminus nephrin [68, 69]. It is understood that MAGI-1 forms a tripartite complex with nephrin and JAM4 at the slit diaphragm [68]. The studies with nephrotic models showed that MAGI-1 and JAM 4 are downregulated in the proteinuric states [68, 70]. MAGI-2 is also expressed in podocyte and directly binds the carboxyl terminus of nephrin. Shirata et al. reported that podocyte-specific conditional MAGI-2-knockout (MAGI-2-CKO) mice exhibited slit diaphragm disruption, morphologic abnormalities of foot processes, and podocyte apoptosis leading to podocyte loss [71]. MAGI-2 interacts with dendrin and plays a role in retaining it at the slit diaphragm. In MAGI-2 CKO mice, dendrin is translocated from the slit diaphragm to the nucleus, and podocyte apoptosis is promoted. Thereby the lack of MAGI-2 in podocyte results in FSGS.
The partitioning-defective (Par)-complex (Par-3/Par-6/aPKC) is understood to be a central player in regulating cell polarity in several cell types. Hartleben et al. reported that Par-3 and aPKC are expressed at podocyte slit diaphragm and NEPH1-nephrin complex binds to the Par-complex [72]. Recently, Takamura et al. demonstrated that Par-3 binds to nephrin, and Par-6 binds to ephrin-B1, another transmembrane protein at slit diaphragm [73]. The mice administered with a dominant-negative aPKC construct showed significant proteinuria, and the loss of foot process architecture was detected in the isolated glomeruli treated with an inhibitor of aPKC. These observations clearly showed that the NEPH1–Nephrin–Par complex is essential for the maintenance of the barrier function of the slit diaphragm. The selective depletion of aPKCλ/ι in mouse podocyte results in slit diaphragm displacement, foot process effacement, proteinuria, and renal failure [74, 75]. It is known that aPKC has two isoforms: aPKCλ/ι and aPKCζ. The double aPKCλ/ι and aPKCζ knockout in podocyte results in severe proteinuria and perinatal death [76]. The developmental study with neonatal mice by Huber et al. showed that Par polarity complex translocated from apical to basal during glomerulogenesis and that the translocation proceeds slit diaphragm formation [76, 77]. These findings suggested that aPKC signaling is a fundamental mechanism for the development and maintenance of podocyte slit diaphragm.
Synaptopodin plays a critical role in maintaining podocyte function and is reported to be a target of cyclosporine, an immunosuppresive agent which is used for patients with nephrotic syndrome [78]. Synaptopodin was originally identified as an actin-associated protein of podocytes [13]. Synaptopodin interacts with α-actinin-4 and regulates its actin-bundling activity at foot processes [79]. Synaptopodin interacts with CD2AP at the slit diaphragm [80, 81]. Yu et al. reported that synaptopodin affects the localization and function of TRPC6 [82]. Synaptopodin participates in the regulation of the slit diaphragm function through regulating the TRPC6 function.
Slit diaphragm functions as a signaling platform
The slit diaphragm functions not only as a barrier but also as a signaling platform transfer the signal to the inside of the cell. Nephrin, a key transmembrane protein of the extracellular domain of the slit diaphragm, has several tyrosine residues. The tyrosine residues of nephrin can be phosphorylated by Src family kinases, including Src, Fyn, Lyn, and Yes [83,84,85]. The tyrosine residues inducing signaling pathways were divided into two groups. The first group is Y1114 (YEES), Y1138/9 (YYRS) (human numbering system). Phosphorylation of the first group tyrosines induces the binding to p85/PI3K [86, 87]. The second group tyrosine is YDxV motif, which includes Y1176 (YDEV), Y1193 (YDEV), and Y1217 (YDQV). Phosphorylation of tyrosines of the second group promotes the recruitment of NCK, an SH2/SH3 containing adaptor protein [88, 89]. It is also reported that nephrin phosphorylated at the tyrosine residue of this group linked to another SH2/SH3 containing protein PLB-γ1 [90, 91].
There is still a lack of consensus on whether phosphorylation of tyrosines of nephrin is associated with promotion or protection of podocyte injury, and it is understood that there are site-specific differences in phosphorylation in baseline, injury, and recovery. Tyrosine phosphorylation of nephrin induces two forms of the structure of actin in culture. One form is lamellipodia, in which two-dimensional actin mesh is observed. Lamellipodia is sometimes associated with foot process effacement of podocyte seen in the pathogenic state in vivo and is mediated by the phosphorylation of tyrosines of the first group [85, 86]. Another form is growth of actin polymer (production of actin tail at nephrin), which is considered to be associated with the stabilization of the foot process and the maintenance of the slit diaphragm structure. The production of the actin tail is basically mediated by the phosphorylation of the second group tyrosines [89]. The reduced phosphorylation level of the second group tyrosines was detected in human glomerular diseases such as minimal change disease [92], membranous nephropathy [93]. The recent study shows that the knock-in mice of which three tyrosine residues of Y1191, Y1208, and Y1232 of mouse numbering system, which corresponds to the second group tyrosine residues in human, were converted to phenylalanine, developed progressive proteinuria accompanied by structural changes [94]. The results indicated that phosphorylation of these tyrosines is required for stabilization of podocyte morphology. Phosphorylation of tyrosines of nephrin is negatively regulated by several tyrosine phosphatases. Alterations in levels of tyrosine phosphatases are also detected in several pathogenic states. Protein tyrosine phosphatase 1B, which can directly dephosphorylate tyrosines of the second group, is upregulated in rat puromycin aminonucleoside nephropathy [95]. SH2 domain-containing phosphatase 1, which de-phosphorylates tyrosines of the second group, is increased in models of diabetic nephropathy [96]. C1-Ten, which is recently identified as nephrin tyrosine phosphatase targeting tyrosines of the first group (Y1114 and Y1138) is also upregulated in diabetic nephropathy [97]. These recent findings showed that a reduction of nephrin phosphorylation level is involved in the development of podocyte injury in diabetic nephropathy. It is also reported that the phosphorylation status of nephrin regulated its own endocytosis. The studies on the endocytosis mechanism of nephrin will be described in the next section. A recent study demonstrated that nephrin signaling results in integrin β1 activation. The finding implied that nephrin-mediated signal regulates podocyte attachment to glomerular basement membrane [98].
NEPH1 also can transduce outside-in signals. Four tyrosine residues (Y637, Y638, Y716, Y719) of NEPH1 were identified that became phosphorylated. NEPH1 phosphorylation results in the recruitment of Grb2. Fyn is necessary for NEPH1-Grb2 interaction. The interaction results in actin polymerization [99]. Phosphorylated NEPH1 and nephrin coordinate distinct signaling pathway through binding to different SH2 domain proteins Grb2 and NCK. NEPH1 is phosphorylated in several injured podocyte models [48, 100, 101]. Since inhibiting NEPH1 phosphorylation protects podocyte from injury, it is assumed that inhibiting NEPH1 signaling is therapeutically significant in preventing podocyte damage [48].
Ephrin-B1 at slit diaphragm also functions as a signaling molecule. The nephrin-binding ephrin-B1 at slit diaphragm transfers the signals nephrin detected to the inside of the cell via different ways from nephrin-mediated signaling [44]. If ephrin-B1 is phosphorylated, nephrin and Par-6 were dissociated from ephrin-B1 [44, 73]. Phosphorylation of ephrin-B1 promoted mobility of podocyte through activation of JNK. It is estimated that the promoted mobility participates in podocyte injury. Regulation of phospho-status of ephrin-B1 could be a therapeutic target. Roles of phosphorylation of tyrosine residues of nephrin, NEPH1, and ephrin-B1 are summarized in Fig. 2.

Roles of tyrosine phosphorylation of nephrin, NEPH1, and ephrin-B1. Phosphorylation (P) of tyrosine residues (Y) of nephrin and NEPH1 regulates actin polymerization and reorganization. Ephrin-B1 phosphorylation regulates JNK signaling
Mechanisms regulating the slit diaphragm function
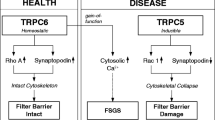
The dynamics of podocyte morphology and the function of the slit diaphragm are regulated by calcium signaling. TRPC5 and TRPC6 channels have been identified in podocyte, and they play a central role in regulating calcium influx and affect the reorganization of the actin cytoskeleton of podocyte [102]. TRPC6 couples with RhoA, while TRPC5 couples with Rac. Although TRPC5-mediated signals have been considered to cause podocyte injury leading to cytoskeletal collapse [103], the function of TRPC5 in podocytes is not clearly understood. Winn et al. reported that a mutation of TRPC6, proline to glutamine at position 112 (P112Q), was detected in patients of familiar FSGS [104]. Reiser et al. reported that other TRPC6 mutations R895C and E897K were detected in the patients [105]. They showed that these mutations lead to increased calcium influx. It is understood that the gain-of-function alteration causes podocyte injury leading to FSGS. On the contrary, recently a loss of function type of TRPC6 mutation was found to be associated with human FSGS [106]. The report suggests that unbalanced calcium entry dysregulated by TRPC6 causes podocyte damage. TRPC6 affects not only FSGS but also other diseases with podocyte injury. It is observed that the expression of TRPC6 was increased in diabetic nephropathy [107,108,109]. TRPC6 interacts with podocin, which suggests that the regulation of calcium signaling mediated by TRPC6 is highly associated with the maintenance of slit diaphragm function.
Inhibiting angiotensin II (AngII) action has been known to be beneficial in many types of kidney diseases. Several studies have suggested that ACEI and AngII type 1 receptor blockade (ARB) have a protective role for slit diaphragm by promoting the expression of nephrin, podocin, and NEPH1 [110,111,112,113]. Although the pharmacological mechanism of these drugs is not fully explained yet, it is reported that AngII action affects the slit diaphragm by regulating the TRPC6 function. The study analyzing the mechanism of AngII-induced apoptosis showed that the protein level of TRPC6 was increased markedly in response to Ang II and that the intracellular Ca2+ concentration was elevated [114]. The study also showed that if TRPC6 was knocked down with siRNA, Ang II-induced podocyte apoptosis and the transient Ca2+ influx were inhibited. The role of TRPC6 in AngII-induced podocyte injury was also demonstrated by in vivo study [115]. The study revealed that TRPC6-deficient mice had significantly less albuminuria. These observations showed that AngII action causes podocyte injury by enhancing the TRPC6 function. Nijenhuis et al. proposed an attractive hypothesis explaining the mechanism of AngII-induced podocyte injury [116]. AngII type 1 receptor stimulation by AngII results in Ca2+ influx mediated by TRPC6. Ca2+-dependent calcineurin activation leads to activation and nuclear translocation of NFAT, which enhances transcription of NFAT-responsive genes such as TRPC6. A consequent increase in TRPC6 expression at the cell membrane could result in a positive feedback regulatory circuit. The positive feedback mechanism can result in persistent calcineurin activation and promotes the podocyte injury. It is also proposed that calcineurin inhibitor such as cyclosporine ameliorates podocyte injury by blocking the positive circuit. The breaking down of the positive circuit can be a plausible strategy of a novel therapy for protecting podocyte.
Since AT1R is a member of Gq-coupled receptors, the beneficial effect of agents inhibiting the activation of AT1R may be mediated at least in part by inhibition of Gq signaling. Wang et al. reported that constitutive active Gq-alpha subunit promoted podocyte injury by stimulating calcineurin activity, resulting in calcineurin-dependent upregulation of TRPC6 [117]. It is also reported that other G protein-coupled receptors (GPCRs) contribute to kidney injury by activating TRPC6. Roshanravan et al. demonstrated that ATP evoked activation of TRPC6 through G protein-coupled pathway from P2Y receptors [118]. A recent study by Wang et al. demonstrated that activation of group I mGluRs induced TRPC6-dependent Ca2+ influx [119]. An alternative treatment strategy might be to target the signaling pathway from GPCRs. Especially, agents inhibiting Gq activation and therapy targeting downstream signaling cascade linked to Gq activation might be useful novel therapy for podocyte injury.
Recently, Verheijden et al. [120] demonstrated that the stimulation of TRPC6-dependent calcium influx increased calpain-1 and calcineurin activity and reduced the expression of a calpain target Talin-1. The study also showed that in kidneys of patients with FSGS, calpain, and calcineurin activity, as well as TRPC6 expression were increased, and the expression of Talin-1 was clearly reduced. Talin-1 links the actin cytoskeleton to integrins, and is critical for podocyte cytoskeletal stability. Very recently, it was reported that for activation of calpain, the physical interaction between TRPC6 and calpain is important and TRPC6 channel activity is independent [121]. It is conceivable that calpain-1 inhibition could be future therapeutic options to treat patients with FSGS.
A slit diaphragm is understood to be a highly dynamic unit. Regular replacement of the slit diaphragm components is necessary for maintaining the integrity of slit diaphragm. However, little is known about the mechanism regulating endocytosis and recycling back to the plasma membrane of the transmembrane proteins of the slit diaphragm. In podocytes two endocytic pathways, clathrin-dependent and clathrin-independent endocytosis, have been identified. Quack et al. reported that β-arrestin mediated clathrin-dependent endocytosis of nephrin [122]. The study demonstrated that β -arrestin interacts with nephrin dephosphorylated at Y1193. Phosphorylation of nephrin Y1193 by Fyn enhances the interaction of nephrin with podocin, and prevents the interaction with β -arrestin, and attenuates the β -arrestin-mediated nephrin endocytosis. It is also reported that Fyn-mediated phosphorylation of nephrin promoted nephrin endocytosis via the clathrin-independent pathway [84, 123]. It is plausible that the phosphorylated state shifts nephrin to the different endocytic pathways. Recent report showed that ShcA, a SH2-containing protein, binds to the nephrin phosphorylated at Y1193 and promotes nephrin endocytosis [124]. These observations showed that the phosphorylation status of nephrin Y1193 determines the slit diaphragm integrity. It is proposed that the regulation of the phosphorylation status of nephrin Y1193 could be a strategy for a novel therapy for proteinuria. The study by Soda et al. showed that dynamin participates in the mechanism of nephrin endocytosis [20]. It is reported that activation of Notch signaling induces nephrin internalization via a β -arrestin/dynamin-dependent route [125]. Activation of Notch signaling was detected in podocyte injury with proteinuria of several types of human glomerular disease [126, 127]. Notch activation is considered to function as a molecular switch that triggers irreversible podocyte injury and promotes disease progression in proteinuric glomerulopathy. The therapeutic approach targeting Notch may promote the repair of the glomerular filtration barrier and prevent terminal podocyte injury.
Future lines of study: a strategy for the establishment of a novel therapy for proteinuria
In this review, we overview the podocyte functional molecules that play a critical role in the maintenance of the barrier function of the slit diaphragm. The molecular structure of the slit diaphragm and the mechanism regulating the function of the slit diaphragm are much more complicated than we thought in the 1990s. Although a lot of studies were piled up, the nature of the slit diaphragm is not fully clarified yet, and the therapy targeting the slit diaphragm molecules is not established yet.
The transmembrane proteins forming a molecular sieve of the slit diaphragm such as nephrin, NEPH1, and ephrin-B1 must be prior targets for a novel therapy for proteinuria. Of course, nephrin should be the most important target; however, some recent reports suggested that the stabilization of NEPH1 is an essential strategy to protect podocyte from serious damages [49]. The authors have been analyzing the expression of the slit diaphragm molecules in several nephrotic models and observed that nephrotic model in which NEPH1 is downregulated from the early phase shows persistent proteinuria and progress to irreversible podocyte injury [32]. However, our knowledge on the mechanism of turnover of NEPH1 is very limited. The transcriptional mechanism and the endocytosis mechanism of NEPH1 should be clarified.
The modifications of signaling pathways from the slit diaphragm should be one of the important strategies for novel therapy for the podocyte injuries. Although the pathological significance of the signaling pathway from nephrin is not precisely understood, several studies showed that the phosphorylation level of each tyrosine residues of nephrin is strictly regulated. It was reported that the phosphorylation level of nephrin is regulated by the activity of the phosphatase such as protein tyrosine phosphatase 1B and C1-Ten [95,96,97]. These phosphatases may be targets for novel therapy. The study with podocyte-specific conditional knock out mice showed that ephrin-B1 plays a central role in regulating the JNK signaling pathway of podocyte [44]. JNK is a stress-activated kinase and is considered to regulate cell motility of podocyte. It is conceivable that the regulation of the JNK pathway mediated by ephrin-B1 is a rational strategy for protecting podocyte from irreversible injury.
Slit diaphragm is considered to be a variant of tight junction [8, 9, 12, 45], and the transition of slit diaphragm to tight junction appears in several nephrotic conditions [128, 129]. The Par complex is essential for the establishment of cell polarity and the formation of the tight junction. It is reported that the tight link between the Par complex to the slit diaphragm components is critical for maintaining the structure of the slit diaphragm [72, 73]. Reinforcement of the linking between the transmembrane proteins and the Par complex molecules is also a rational strategy.
In this article, we reviewed that the slit diaphragm has common characteristics with synapse. Several synapse-associated molecules are highly expressed in podocytes, and they play a critical role in maintaining the slit diaphragm function. Synaptic vesicle-associated proteins are involved in the maintenance of the slit diaphragm [17,18,19,20]. Dendrin, which is originally identified in dendrite of rat neuron, plays a critical role in stabilizing the scaffold proteins of the slit diaphragm [16]. These neuron-associated molecules could be targets for therapy for protecting podocyte. It is expected that some of the drugs and chemical agents developed for neuronal diseases have an effect on podocyte. We established the in vitro system to analyze the expression of these neuronal molecules with cultured podocytes. However, to select the effective agent more sensitive screening system should be established. Recently, the culture condition to induce the cultured podocytes with long arborized processes was reported [130]. The method may help for establishing a better screening system.
Reducing the level of proteinuria is critical for preventing the progression to end-stage kidney. Although proteinuria is accepted to be one of the most important risk factors of stroke and cardiovascular diseases, the causal relationship between proteinuria and these diseases remains to be unclear. We suppose that proteinuria may work as “canary in a coal mine” to predict other disease, because the glomerular capillary wall preventing proteinuria seems to be a very delicate and fragile unit. Therapeutic intervention of proteinuria from the early phase will help to prevent stroke, cardiovascular disease, and some other diseases which have a common pathogenic mechanism with proteinuria. A novel effective therapy for proteinuria targeting slit diaphragm should be established as soon as possible.
References
Madison JR, Spies C, Schatz IJ, Masaki K, Chen R, Yano K, et al. Proteinuria and risk for stroke and coronary heart disease during 27 years of follow-up: the Honolulu Heart Program. Arch Intern Med. 2006;166(8):884–9.
Tryggvason K, Pettersson E. Causes and consequences of proteinuria: the kidney filtration barrier and progressive renal failure. J Intern Med. 2003;254(3):216–24.
Menzel S, Moeller MJ. Role of the podocyte in proteinuria. Pediatr Nephrol. 2011;26(10):1775–800.
Kawachi H, Shimizu F. Molecular composition and function of the slit diaphragm: nephrin, the molecule responsible for proteinuria. Clin Exp Nephrol. 2000;4(3):161–72.
Kawachi H, Suzuki K, Miyauchi N, Hashimoto T, Otaki Y, Shimizu F. Slit diaphragm dysfunction in proteinuric states: identification of novel therapeutic targets for nephrotic syndrome. Clin Exp Nephrol. 2009;13(4):275–80.
Rodewald R, Karnovsky MJ. Porous substructure of the glomerular slit diaphragm in the rat and mouse. J Cell Biol. 1974;60(2):423–33.
Arakawa M. A scanning electron microscope study of the human glomerulus. Am J Pathol. 1971;64(2):457–66.
Schnabel E, Dekan G, Miettinen A, Farquhar MG. Biogenesis of podocalyxin—the major glomerular sialoglycoprotein—in the newborn rat kidney. Eur J Cell Biol. 1989;48(2):313–26.
Kawachi H, Abrahamson DR, St John PL, Goldstein DJ, Shia MA, Matsui K, et al. Developmental expression of the nephritogenic antigen of monoclonal antibody 5–1–6. Am J Pathol. 1995;147(3):823–33.
Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11(1):1–8.
Inoue T, Yaoita E, Kurihara H, Shimizu F, Sakai T, Kobayashi T, et al. FAT is a component of glomerular slit diaphragms. Kidney Int. 2001;59(3):1003–122.
Fukasawa H, Bornheimer S, Kudlicka K, Farquhar MG. Slit diaphragms contain tight junction proteins. J Am Soc Nephrol. 2009;20(7):1491–503.
Mundel P, Heid HW, Mundel TM, Kruger M, Reiser J, Kriz W. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139(1):193–204.
Ahola H, Heikkila E, Astrom E, Inagaki M, Izawa I, Pavenstadt H, et al. A novel protein, densin, expressed by glomerular podocytes. J Am Soc Nephrol. 2003;14(7):1731–7.
Peitsch WK, Hofmann I, Endlich N, Pratzel S, Kuhn C, Spring H, et al. Cell biological and biochemical characterization of drebrin complexes in mesangial cells and podocytes of renal glomeruli. J Am Soc Nephrol. 2003;14(6):1452–63.
Asanuma K, Campbell KN, Kim K, Faul C, Mundel P. Nuclear relocation of the nephrin and CD2AP-binding protein dendrin promotes apoptosis of podocytes. Proc Natl Acad Sci USA. 2007;104(24):10134–9.
Rastaldi MP, Armelloni S, Berra S, Li M, Pesaresi M, Poczewski H, et al. Glomerular podocytes possess the synaptic vesicle molecule Rab3A and its specific effector rabphilin-3a. Am J Pathol. 2003;163(3):889–99.
Miyauchi N, Saito A, Karasawa T, Harita Y, Suzuki K, Koike H, et al. Synaptic vesicle protein 2B is expressed in podocyte, and its expression is altered in proteinuric glomeruli. J Am Soc Nephrol. 2006;17(10):2748–59.
Fukusumi Y, Wakamatsu A, Takashima N, Hasegawa E, Miyauchi N, Tomita M, et al. SV2B is essential for the integrity of the glomerular filtration barrier. Lab Invest. 2015;95(5):534–45.
Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, Giovedi S, et al. Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J Clin Invest. 2012;122(12):4401–11.
Saito A, Miyauchi N, Hashimoto T, Karasawa T, Han GD, Kayaba M, et al. Neurexin-1, a presynaptic adhesion molecule, localizes at the slit diaphragm of the glomerular podocytes in kidneys. Am J Physiol Regul Integr Comp Physiol. 2011;300(2):R340–R348348.
Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1(4):575–82.
Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestila M, Jalanko H, et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci USA. 1999;96(14):7962–7.
Kiselyov VV, Berezin V, Maar TE, Soroka V, Edvardsen K, Schousboe A, et al. The first immunoglobulin-like neural cell adhesion molecule (NCAM) domain is involved in double-reciprocal interaction with the second immunoglobulin-like NCAM domain and in heparin binding. J Biol Chem. 1997;272(15):10125–34.
Obrink B. CEA adhesion molecules: multifunctional proteins with signal-regulatory properties. Curr Opin Cell Biol. 1997;9(5):616–26.
Sonderegger P, Rathjen FG. Regulation of axonal growth in the vertebrate nervous system by interactions between glycoproteins belonging to two subgroups of the immunoglobulin superfamily. J Cell Biol. 1992;119(6):1387–94.
Orikasa M, Matsui K, Oite T, Shimizu F. Massive proteinuria induced in rats by a single intravenous injection of a monoclonal antibody. J Immunol. 1988;141(3):807–14.
Kawachi H, Koike H, Kurihara H, Yaoita E, Orikasa M, Shia MA, et al. Cloning of rat nephrin: expression in developing glomeruli and in proteinuric states. Kidney Int. 2000;57(5):1949–61.
Topham PS, Kawachi H, Haydar SA, Chugh S, Addona TA, Charron KB, et al. Nephritogenic mAb 5–1–6 is directed at the extracellular domain of rat nephrin. J Clin Invest. 1999;104(11):1559–666.
Holthofer H, Ahola H, Solin ML, Wang S, Palmen T, Luimula P, et al. Nephrin localizes at the podocyte filtration slit area and is characteristically spliced in the human kidney. Am J Pathol. 1999;155(5):1681–7.
Nakatsue T, Koike H, Han GD, Suzuki K, Miyauchi N, Yuan H, et al. Nephrin and podocin dissociate at the onset of proteinuria in experimental membranous nephropathy. Kidney Int. 2005;67(6):2239–53.
Otaki Y, Miyauchi N, Higa M, Takada A, Kuroda T, Gejyo F, et al. Dissociation of NEPH1 from nephrin is involved in development of a rat model of focal segmental glomerulosclerosis. Am J Physiol Renal Physiol. 2008;295(5):F1376–F13871387.
Furness PN, Hall LL, Shaw JA, Pringle JH. Glomerular expression of nephrin is decreased in acquired human nephrotic syndrome. Nephrol Dial Transplant. 1999;14(5):1234–7.
Huh W, Kim DJ, Kim MK, Kim YG, Oh HY, Ruotsalainen V, et al. Expression of nephrin in acquired human glomerular disease. Nephrol Dial Transplant. 2002;17(3):478–84.
Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, et al. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52(4):1023–30.
Langham RG, Kelly DJ, Cox AJ, Thomson NM, Holthofer H, Zaoui P, et al. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: effects of angiotensin converting enzyme inhibition. Diabetologia. 2002;45(11):1572–6.
Garovic VD, Wagner SJ, Petrovic LM, Gray CE, Hall P, Sugimoto H, et al. Glomerular expression of nephrin and synaptopodin, but not podocin, is decreased in kidney sections from women with preeclampsia. Nephrol Dial Transplant. 2007;22(4):1136–43.
Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol. 2001;21(14):4829–36.
Barletta GM, Kovari IA, Verma RK, Kerjaschki D, Holzman LB. Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J Biol Chem. 2003;278(21):19266–71.
Grahammer F, Wigge C, Schell C, Kretz O, Patrakka J, Schneider S, et al. A flexible, multilayered protein scaffold maintains the slit in between glomerular podocytes. JCI Insight. 2016;1(9):e86177.
Ciani L, Patel A, Allen ND, Ffrench-Constant C. Mice lacking the giant protocadherin mFAT1 exhibit renal slit junction abnormalities and a partially penetrant cyclopia and anophthalmia phenotype. Mol Cell Biol. 2003;23(10):3575–82.
Hashimoto T, Karasawa T, Saito A, Miyauchi N, Han GD, Hayasaka K, et al. Ephrin-B1 localizes at the slit diaphragm of the glomerular podocyte. Kidney Int. 2007;72(8):954–64.
Miao H, Wang B. Eph/ephrin signaling in epithelial development and homeostasis. Int J Biochem Cell Biol. 2009;41(4):762–70.
Fukusumi Y, Zhang Y, Yamagishi R, Oda K, Watanabe T, Matsui K, et al. Nephrin-binding ephrin-B1 at the slit diaphragm controls podocyte function through the JNK pathway. J Am Soc Nephrol. 2018;29(5):1462–74.
Schnabel E, Anderson JM, Farquhar MG. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J Cell Biol. 1990;111(3):1255–63.
Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103(3):755–66.
Huber TB, Schmidts M, Gerke P, Schermer B, Zahn A, Hartleben B, et al. The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J Biol Chem. 2003;278(15):13417–21.
Wagner MC, Rhodes G, Wang E, Pruthi V, Arif E, Saleem MA, et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J Biol Chem. 2008;283(51):35579–89.
Sagar A, Arif E, Solanki AK, Srivastava P, Janech MG, Kim SH, et al. Targeting Neph1 and ZO-1 protein-protein interaction in podocytes prevents podocyte injury and preserves glomerular filtration function. Sci Rep. 2017;7(1):12047.
Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24(4):349–54.
Roselli S, Gribouval O, Boute N, Sich M, Benessy F, Attie T, et al. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol. 2002;160(1):131–9.
Huber TB, Kottgen M, Schilling B, Walz G, Benzing T. Interaction with podocin facilitates nephrin signaling. J Biol Chem. 2001;276(45):41543–6.
Sellin L, Huber TB, Gerke P, Quack I, Pavenstadt H, Walz G. NEPH1 defines a novel family of podocin interacting proteins. FASEB J. 2003;17(1):115–7.
Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108(11):1621–9.
Koop K, Eikmans M, Baelde HJ, Kawachi H, De Heer E, Paul LC, et al. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol. 2003;14(8):2063–71.
Perysinaki GS, Moysiadis DK, Bertsias G, Giannopoulou I, Kyriacou K, Nakopoulou L, et al. Podocyte main slit diaphragm proteins, nephrin and podocin, are affected at early stages of lupus nephritis and correlate with disease histology. Lupus. 2011;20(8):781–91.
Fukuda H, Hidaka T, Takagi-Akiba M, Ichimura K, Oliva Trejo JA, Sasaki Y, et al. Podocin is translocated to cytoplasm in puromycin aminonucleoside nephrosis rats and in poor-prognosis patients with IgA nephropathy. Cell Tissue Res. 2015;360(2):391–400.
Kawachi H, Koike H, Kurihara H, Sakai T, Shimizu F. Cloning of rat homologue of podocin: expression in proteinuric states and in developing glomeruli. J Am Soc Nephrol. 2003;14(1):46–56.
Huber TB, Schermer B, Muller RU, Hohne M, Bartram M, Calixto A, et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci USA. 2006;103(46):17079–866.
Anderson M, Kim EY, Hagmann H, Benzing T, Dryer SE. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am J Physiol Cell Physiol. 2013;305(3):C276–C289289.
Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science. 1999;286(5438):312–5.
Shih NY, Li J, Cotran R, Mundel P, Miner JH, Shaw AS. CD2AP localizes to the slit diaphragm and binds to nephrin via a novel C-terminal domain. Am J Pathol. 2001;159(6):2303–8.
Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300(5623):1298–300.
Schiffer M, Mundel P, Shaw AS, Bottinger EP. A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem. 2004;279(35):37004–12.
Yaddanapudi S, Altintas MM, Kistler AD, Fernandez I, Moller CC, Wei C, et al. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J Clin Invest. 2011;121(10):3965–80.
Tossidou I, Teng B, Worthmann K, Müller-Deile J, Jobst-Schwan T, Kardinal C, et al. Tyrosine phosphorylation of CD2AP affects stability of the slit diaphragm complex. J Am Soc Nephrol. 2019;30(7):1220–377.
Hirabayashi S, Tajima M, Yao I, Nishimura W, Mori H, Hata Y. JAM4, a junctional cell adhesion molecule interacting with a tight junction protein, MAGI-1. Mol Cell Biol. 2003;23(12):4267–82.
Hirabayashi S, Mori H, Kansaku A, Kurihara H, Sakai T, Shimizu F, et al. MAGI-1 is a component of the glomerular slit diaphragm that is tightly associated with nephrin. Lab Invest. 2005;85(12):1528–43.
Ni J, Bao S, Johnson RI, Zhu B, Li J, Vadaparampil J, et al. MAGI-1 Interacts with nephrin to maintain slit diaphragm structure through enhanced Rap1 activation in podocytes. J Biol Chem. 2016;291(47):24406–17.
Harita Y, Miyauchi N, Karasawa T, Suzuki K, Han GD, Koike H, et al. Altered expression of junctional adhesion molecule 4 in injured podocytes. Am J Physiol Renal Physiol. 2006;290(2):F335–F34444.
Shirata N, Ihara KI, Yamamoto-Nonaka K, Seki T, Makino SI, Oliva Trejo JA, et al. Glomerulosclerosis induced by deficiency of membrane-associated guanylate kinase inverted 2 in kidney podocytes. J Am Soc Nephrol. 2017;28(9):2654–69.
Hartleben B, Schweizer H, Lubben P, Bartram MP, Moller CC, Herr R, et al. Neph-Nephrin proteins bind the Par3-Par6-atypical protein kinase C (aPKC) complex to regulate podocyte cell polarity. J Biol Chem. 2008;283(34):23033–8.
Takamura S, Fukusumi Y, Zhang Y, Narita I, Kawachi H. Par-6–ephrin-B1 interaction is regulated by nephrin-mediated signal and is crucial in maintaining slit diaphragm of podocyte. Am J Pathol. 2020;190(2):333–46.
Huber TB, Hartleben B, Winkelmann K, Schneider L, Becker JU, Leitges M, et al. Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J Am Soc Nephrol. 2009;20(4):798–806.
Hirose T, Satoh D, Kurihara H, Kusaka C, Hirose H, Akimoto K, et al. An essential role of the universal polarity protein, aPKClambda, on the maintenance of podocyte slit diaphragms. PLoS ONE. 2009;4(1):e4194.
Hartleben B, Widmeier E, Suhm M, Worthmann K, Schell C, Helmstadter M, et al. aPKClambda/iota and aPKCzeta contribute to podocyte differentiation and glomerular maturation. J Am Soc Nephrol. 2013;24(2):253–67.
Hartleben B, Widmeier E, Wanner N, Schmidts M, Kim ST, Schneider L, et al. Role of the polarity protein Scribble for podocyte differentiation and maintenance. PLoS ONE. 2012;7(5):e36705.
Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14(9):931–8.
Patrie KM, Drescher AJ, Welihinda A, Mundel P, Margolis B. Interaction of two actin-binding proteins, synaptopodin and alpha-actinin-4, with the tight junction protein MAGI-1. J Biol Chem. 2002;277(33):30183–90.
Huber TB, Kwoh C, Wu H, Asanuma K, Godel M, Hartleben B, et al. Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn, and synaptopodin. J Clin Invest. 2006;116:1337–455.
Yanagida-Asanuma E, Asanuma K, Kim K, Donnelly M, Young Choi H, Hyung Chang J, et al. Synaptopodin protects against proteinuria by disrupting Cdc 42:IRSp53: Mena signaling complexes in kidney podocytes. Am J Pathol. 2007;171(2):415–27.
Yu H, Kistler A, Faridi MH, Meyer JO, Tryniszewska B, Mehta D, et al. Synaptopodin limits TRPC6 podocyte surface expression and attenuates proteinuria. J Am Soc Nephrol. 2016;27(11):3308–19.
Verma R, Wharram B, Kovari I, Kunkel R, Nihalani D, Wary KK, et al. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem. 2003;278(23):20716–23.
Li H, Lemay S, Aoudjit L, Kawachi H, Takano T. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J Am Soc Nephrol. 2004;15(12):3006–155.
Lahdenpera J, Kilpelainen P, Liu XL, Pikkarainen T, Reponen P, Ruotsalainen V, et al. Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int. 2003;64(2):404–13.
Zhu J, Sun N, Aoudjit L, Li H, Kawachi H, Lemay S, et al. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008;73(5):556–66.
Garg P, Verma R, Cook L, Soofi A, Venkatareddy M, George B, et al. Actin-depolymerizing factor cofilin-1 is necessary in maintaining mature podocyte architecture. J Biol Chem. 2010;285(29):22676–88.
Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440(7085):818–23.
Blasutig IM, New LA, Thanabalasuriar A, Dayarathna TK, Goudreault M, Quaggin SE, et al. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol. 2008;28(6):2035–46.
Harita Y, Kurihara H, Kosako H, Tezuka T, Sekine T, Igarashi T, et al. Phosphorylation of nephrin triggers Ca2+ signaling by recruitment and activation of phospholipase C-{gamma}1. J Biol Chem. 2009;284(13):8951–62.
Kanda S, Harita Y, Shibagaki Y, Sekine T, Igarashi T, Inoue T, et al. Tyrosine phosphorylation-dependent activation of TRPC6 regulated by PLC-gamma1 and nephrin: effect of mutations associated with focal segmental glomerulosclerosis. Mol Biol Cell. 2011;22(11):1824–35.
Uchida K, Suzuki K, Iwamoto M, Kawachi H, Ohno M, Horita S, et al. Decreased tyrosine phosphorylation of nephrin in rat and human nephrosis. Kidney Int. 2008;73(8):926–32.
Ohashi T, Uchida K, Asamiya Y, Tsuruta Y, Ohno M, Horita S, et al. Phosphorylation status of nephrin in human membranous nephropathy. Clin Exp Nephrol. 2010;14(1):51–5.
New LA, Martin CE, Scott RP, Platt MJ, Keyvani Chahi A, Stringer CD, et al. Nephrin tyrosine phosphorylation is required to stabilize and restore podocyte foot process architecture. J Am Soc Nephrol. 2016;27(8):2422–35.
Aoudjit L, Jiang R, Lee TH, New LA, Jones N, Takano T. Podocyte protein, nephrin, is a substrate of protein tyrosine phosphatase 1B. J Signal Transduct. 2011;2011:376543.
Denhez B, Lizotte F, Guimond MO, Jones N, Takano T, Geraldes P. Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes. J Biol Chem. 2015;290(1):350–8.
Lee J, Koh A, Jeong H, Kim E, Ha TS, Saleem MA, et al. C1-Ten is a PTPase of nephrin, regulating podocyte hypertrophy through mTORC1 activation. Sci Rep. 2017;7(1):12346.
Dlugos CP, Picciotto C, Lepa C, Krakow M, Stöber A, Eddy ML, et al. Nephrin signaling results in integrin β1 activation. J Am Soc Nephrol. 2019;30(6):1006–199.
Garg P, Verma R, Nihalani D, Johnstone DB, Holzman LB. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol Cell Biol. 2007;27(24):8698–712.
Harita Y, Kurihara H, Kosako H, Tezuka T, Sekine T, Igarashi T, et al. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J Biol Chem. 2008;283(14):9177–86.
Arif E, Rathore YS, Kumari B, Ashish F, Wong HN, Holzman LB, et al. Slit diaphragm protein Neph1 and its signaling: a novel therapeutic target for protection of podocytes against glomerular injury. J Biol Chem. 2014;289(14):9502–18.
Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, et al. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal. 2010;3(145):ra77.
Wieder N, Greka A. Calcium, TRPC channels, and regulation of the actin cytoskeleton in podocytes: towards a future of targeted therapies. Pediatr Nephrol. 2016;31(7):1047–54.
Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308(5729):1801–4.
Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37(7):739–44.
Riehle M, Buscher AK, Gohlke BO, Kassmann M, Kolatsi-Joannou M, Brasen JH, et al. TRPC6 G757D loss-of-function mutation associates with FSGS. J Am Soc Nephrol. 2016;27(9):2771–833.
Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, Staruschenko A. Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci Rep. 2015;5:17637.
Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, et al. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am J Pathol. 2014;184(6):1715–26.
Staruschenko A. TRPC6 in diabetic kidney disease: good guy or bad guy? Kidney Int. 2019;95(2):256–8.
Suzuki K, Han GD, Miyauchi N, Hashimoto T, Nakatsue T, Fujioka Y, et al. Angiotensin II type 1 and type 2 receptors play opposite roles in regulating the barrier function of kidney glomerular capillary wall. Am J Pathol. 2007;170(6):1841–53.
Takahashi A, Fukusumi Y, Yamazaki M, Kayaba M, Kitazawa Y, Tomita M, et al. Angiotensin II type 1 receptor blockade ameliorates proteinuria in puromycin aminonucleoside nephropathy by inhibiting the reduction of NEPH1 and nephrin. J Nephrol. 2014;27(6):627–34.
Zoja C, Corna D, Gagliardini E, Conti S, Arnaboldi L, Benigni A, et al. Adding a statin to a combination of ACE inhibitor and ARB normalizes proteinuria in experimental diabetes, which translates into full renoprotection. Am J Physiol Renal Physiol. 2010;299(5):F1203–F12111211.
Fukuda A, Fujimoto S, Iwatsubo S, Kawachi H, Kitamura K. Effects of mineralocorticoid and angiotensin II receptor blockers on proteinuria and glomerular podocyte protein expression in a model of minimal change nephrotic syndrome. Nephrology (Carlton). 2010;15(3):321–6.
Zhang H, Ding J, Fan Q, Liu S. TRPC6 up-regulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-kappaB translocation. Exp Biol Med (Maywood). 2009;234(9):1029–36.
Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, et al. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol. 2011;22(3):526–35.
Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179(4):1719–32.
Wang L, Jirka G, Rosenberg PB, Buckley AF, Gomez JA, Fields TA, et al. Gq signaling causes glomerular injury by activating TRPC6. J Clin Invest. 2015;125(5):1913–26.
Roshanravan H, Dryer SE. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: role of podocin and reactive oxygen species. Am J Physiol Renal Physiol. 2014;306(9):F1088–F10971097.
Wang Q, Wang D, Shibata S, Ji T, Zhang L, Zhang R, et al. Group I metabotropic glutamate receptor activation induces TRPC6-dependent calcium influx and RhoA activation in cultured human kidney podocytes. Biochem Biophys Res Commun. 2019;511(2):374–80.
Verheijden KAT, Sonneveld R, Bakker-van Bebber M, Wetzels JFM, van der Vlag J, Nijenhuis T. The calcium-dependent protease calpain-1 links TRPC6 activity to podocyte injury. J Am Soc Nephrol. 2018;29(8):2099–109.
Farmer LK, Rollason R, Whitcomb DJ, Ni L, Goodliff A, Lay AC, et al. TRPC6 binds to and activates calpain, independent of its channel activity, and regulates podocyte cytoskeleton, cell adhesion, and motility. J Am Soc Nephrol. 2019;30(10):1910–24.
Quack I, Rump LC, Gerke P, Walther I, Vinke T, Vonend O, et al. beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity. Proc Natl Acad Sci USA. 2006;103(38):14110–5.
Qin XS, Tsukaguchi H, Shono A, Yamamoto A, Kurihara H, Doi T. Phosphorylation of nephrin triggers its internalization by raft-mediated endocytosis. J Am Soc Nephrol. 2009;20(12):2534–45.
Martin CE, Petersen KA, Aoudjit L, Tilak M, Eremina V, Hardy WR, et al. ShcA adaptor protein promotes nephrin endocytosis and is upregulated in proteinuric nephropathies. J Am Soc Nephrol. 2018;29(1):92–103.
Waters AM, Wu MY, Huang YW, Liu GY, Holmyard D, Onay T, et al. Notch promotes dynamin-dependent endocytosis of nephrin. J Am Soc Nephrol. 2012;23(1):27–35.
Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, et al. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14(3):290–8.
Sharma M, Callen S, Zhang D, Singhal PC, Vanden Heuvel GB, Buch S. Activation of Notch signaling pathway in HIV-associated nephropathy. AIDS. 2010;24(14):2161–70.
Gong Y, Sunq A, Roth RA, Hou J. Inducible expression of claudin-1 in glomerular podocytes generates aberrant tight junctions and proteinuria through slit diaphragm destabilization. J Am Soc Nephrol. 2017;28(1):106–17.
Succar L, Boadle RA, Harris DC, Rangan GK. Formation of tight junctions between neighboring podocytes is an early ultrastructural feature in experimental crescentic glomerulonephritis. Int J Nephrol Renovasc Dis. 2016;24(9):297–312.
Yaoita E, Yoshida Y, Nameta M, Takimoto H, Fujinaka H. Induction of interdigitating cell processes in podocyte culture. Kidney Int. 2018;93(2):519–24.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared no competing interest.
Research involving human participants or animals
This article does not contain any original studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kawachi, H., Fukusumi, Y. New insight into podocyte slit diaphragm, a therapeutic target of proteinuria. Clin Exp Nephrol 24, 193–204 (2020). https://doi.org/10.1007/s10157-020-01854-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-020-01854-3