
Abstract
To initiate breeding programs for kelp grouper (Epinephelus bruneus), the establishment of genetic linkage maps becomes essential accompanied by the search for quantitative trait loci that may be utilized in selection programs. We constructed a high-resolution genetic linkage map using 1055 simple sequence repeat (SSR) markers in an F1 family. Genome-wide and chromosome-wide significances of growth-related quantitative trait loci (QTLs) (body weight (BW) and total length (TL)) were detected using non-parametric mapping, Kruskal-Wallis (K-W) analysis, simple interval mapping (IM) and a permutation test (PT). Two stages and two families of fish were used to confirm the QTL regions. Ultimately, 714 SSR markers were matched that evenly covered the 24 linkage groups. In total, 509 and 512 markers were localized to the female and male maps, respectively. The genome lengths were approximately 1475.95 and 1370.39 cM and covered 84.68 and 83.21 % of the genome, with an average interval of 4.1 and 4.0 cM, in females and males, respectively. One major QTL affecting BW and TL was found on linkage group EBR 17F that identified for 1 % of the genome-wide significance and accounted for 14.6–18.9 and 14.7–18.5 % of the phenotypic variance, and several putative QTL with 5 % chromosome-wide significance were detected on eight linkage groups. Furthermore, the confirmed results of the regions harboring the major and putative QTLs showed consistent significant experiment-wide values of 1 and 5 % as well as a chromosome-wide value of 5 %. We identified growth-related QTLs that could be applied to find candidate genes for growth traits in further studies, and potentially useful in MAS breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The kelp grouper (Epinephelus bruneus) is a commercially important marine fish in East Asia. This species belongs to the subfamily Ephinephelinae, family Serranidae, and order Perciformes. Groupers, or Serranidae in general, are protogynous, which means they first start life as a female fish and then later switch into being males once they pass a certain size threshold or due to social cues (Lee et al. 2002; Tsuchihashi et al. 2003; Yeh et al. 2003) and matures at more than 6 years of age (Liu et al. 2013). The kelp grouper is a carnivorous fish that feeds on small fish and crustaceans. Generally, juvenile kelp groupers are found in shallow water estuaries (Heemstra and Randall 1995) and coastal areas, while the adult fish inhabit waters ranging 20–200 m of depth (An et al. 2011) around coral reefs, rocky reefs, and mud bottom areas. The maximum size of a kelp grouper is reported to be about 128 cm in length and 33 kg in body weight (Tupper and Sheriff 2008). At least 16 species of grouper, including the kelp grouper, have been used successfully in aquaculture in many countries in East Asia (Tupper and Sheriff 2008).
In Japan, the kelp grouper has a high value because of its high market demand and low quantity of catch in natural waters (Mitcheson et al. 2003). Recently, this species was listed as a vulnerable species by the International Union for Conservation of Nature and Natural Resources (the IUCN Red List of Threatened Species) because of the rapid decrease in the natural population (Thierry et al. 2008). The kelp grouper is a target species for aquaculture in Japan (Fui et al. 2014); however, during artificial larval rearing, high mortality is frequent in the early life stages (Sawada et al. 1999). In addition, the kelp grouper grows slowly in farms and a prolonged farming period is required to reach a marketable size. To date, domestication of broodstock and a selective breeding program on a commercial scale for the kelp grouper in Japan have not yet been fully developed. Marker-assisted selection (MAS) based on quantitative trait loci (QTLs) is an effective method to improve quantitative traits (Max and Anatoly 2007) such as slow growth and high mortality in the larval stage of groupers.
In the recent decades, several genetic linkage maps of fin fish have been constructed using genetic markers, such as those for rainbow trout (Oncorhynchus mykiss), using simple sequence repeats (SSRs); Atlantic salmon (Salmo salar) using amplified fragment length polymorphisms (AFLPs) and SSRs; brown trout (Salmo trutta, AFLPs and SSRs); Nile tilapia (Oreochromis niloticus, AFLPs and SSRs); channel catfish (Ictalurus punctatus, AFLPs and SSRs), Japanese flounder (Paralichthys olivaceus, AFLPs and SSRs); ayu (Plecoglossus altivelis, AFLPs and SSRs; and yellowail (Seriola quinqueradiata, SSRs) (Danzmann and Ghabi 2007). A genetic linkage map of the kelp grouper was produced based on microsatellite markers (Liu et al. 2013). Several studies on growth-related quantitative traits (QTLs) have been carried out recently on fishes such as the rainbow trout, Nile tilapia, Arctic char (Salvelinus alpinus) (Danzmann and Ghabi 2007), Atlantic salmon (Baranski et al. 2010), barramundi (Lates calcarifer) (Wang et al. 2008), and turbot (Scophthalmus maximus) (Molano et al. 2011).
SSR markers are highly polymorphic and show high inheritance and codominance of inheritance, making them suitable to identify homozygotes and heterozygotes. They are usually evenly distributed throughout the genome, and their results are simple to interpret, highly reproducible, and easily automated (Liu 2007). Thus, they are useful to construct a genetic linkage map. Nevertheless, the detection of a reasonable proportion of QTLs segregating in a population requires a large number of markers to increase the accuracy of QTL detection.
Economic traits in aquaculture fish, especially growth-related quantitative traits, are the main goals for improvement in a genetic breeding program. Growth-related traits have been measured and reported in several economically important marine fishes (Yue 2013). To study growth-related traits in fish and other species by molecular tools is complex, because growth-related traits are influenced not only by genetics, but also by the environment (Abraham et al. 2007; Molano et al. 2011).
In 2013, the first-generation genetic linkage map for the kelp grouper was constructed using 222 microsatellite markers, covering 23 and 25 linkage groups in the male and female maps, with marker intervals of 5.0 and 6.7 cM, respectively (Liu et al. 2013). In the present study, a high-resolution genetic linkage map and a genome scan for QTLs affecting growth-related traits (BW and TL) in F1 progeny of kelp groupers were conducted. These results could be used to investigate candidate genes that will accelerate genetic improvement using MAS breeding programs in the kelp grouper.
Materials and Methods
Reference Family and DNA Extraction
Paternal half-sib F1 progeny from two families (families A and B) produced from two females and a single male were used. The fish were taken from recently derived wild broodstock of the kelp grouper maintained at the Ehime Fisheries Research Center, Japan. Fish were measured at two timepoints. Stage I at 5 months post-hatching (average total length of 150 mm), and stage II at 11 months post-hatching. At stage I, individual fish were tracked using an embedded passive integrated transponder (PIT) tag. This facilitated comparisons of fast growth phases that occur in the fish at this point in their development. All fish were measured for body weight (BW) and total length (TL). In total, 360 and 163 progeny in stages I and II of family A; and 112 and 45 progeny in stages I and II of family B were measured for BW and TL. Fin clip samples were collected and kept in absolute ethanol (99.9 % ethanol solution). DNA extraction was carried out from these samples using the Agincourt DNAdvance Genomic DNA Isolation Kit (Beckman Coulter, USA), following the manufacturer’s recommended protocol. The quality and quantity of the extracted DNA was quantified using a spectrophotometer (Untrospec 2100 pro, GE USA) and the DNA was diluted to 10 ng/μL for PCR.
The high-resolution genetic linkage maps to find candidate growth-related QTL regions were constructed by using the parents and 90 F1 progeny in stage II of family A. After that, all progeny in both stages of families A and B were used to confirm the candidate QTL regions.
SSR Markers and Genotyping
A total of 2348 microsatellite-enriched segments from the kelp grouper were developed using next-generation sequencing (NGS) by the GS FLX system (Roche, Switzerland) (denoted as the EBR series) (Kubota et al. 2014) and 889 simple tandem repeats (STR) markers were obtained from the NCBI database of a cross section of species in the subfamily Epinephelinae (denoted as the STR series) (Chapman et al. 1999; Dong et al. 2008; Liu et al. 2008; Lo and Yue 2007; Mokhtar et al. 2011; Ramirez et al. 2006; Renshaw et al. 2010; Rivera et al. 2003; Zeng et al. 2008; Zhao et al. 2009a, b; Zhu et al. 2005). In total, 1867 SSR markers (1466 EBR markers and 401 STR markers) were designed using the TROLL program at http://wsmartins.net/websat/ (Martins et al. 2009) under the default settings and considering a product size of 100–250 bp. For the SSR markers, the forward primers were labeled with tetrachloro-6-carboxy-fluorescein (TET) fluorescent dye at the 5′-end. Polymerase chain reactions (PCR) were performed in 11 μl volumes containing 50 ng of genomic DNA, 1× Ex Taq buffer (Mg2+ free), 2.0 mM MgCl2, 0.2 mM dNTP, 1 % BSA, 0.025 U of Taq polymerase (Takara: Ex-Taq™ (Mg2+ free buffer)), 0.5 pmol/μL of the reverse primer, and 0.05 pmol/μL of the forward primer. Cycle amplification was performed on an MJ PTC-100 (Bio-Rad, USA), with the program conditions of 95 °C for 5 min for initial denaturation; followed by 36 cycles of 30 s at 95 °C, 1 min at the annealing temperature 56 °C, and 1 min at 72 °C, and a final extension at 72 °C for 10 min. The amplified products were mixed with an equal volume of loading buffer (98 % formaldehyde, 10 mM EDTA, and 0.05 % bromophenol blue), heated for 10 min at 95 °C and then immediately cooled on ice. The samples were separated on a 6 % polyacrylamide gel containing 7 M urea and 0.5× Trizma base/Boric Acid/EDTA-2Na (TBE) buffer and 40 % Page-plus (Amresco, USA) with a 500-bp DNA ladder (GeneScan™-500 TAMRA™). Electrophoresis was performed using 0.5× TBE buffer at a constant voltage of 1800 V for 1.5 h. After electrophoresis, the gel was scanned and imaged using an FMBIO III Multi-View fluorescence image analyzer (Hitachi-soft, Japan).
Linkage Analysis
Linkage analysis was performed using LINKMFEX version 2.3 (Danzmann 2006). This application can separate alleles that originated from males or females. To avoid errors during genotyping, the accuracy of genotypes in their progeny was checked from parental male and female alleles. Genotype data were converted to a backcross format even though the grandparent genotype was unknown. Pairwise analysis was performed, and markers were sorted into linkage groups at a logarithm of odds (LOD) threshold of 4.0. Linkage phases were determined retrospectively by examining the assortment of alleles among linked markers. The goodness of fit of the chi-square analysis ( χ 2) was used to test for Mendelian segregation distortion of the locus. Therefore, the distance of the marker was estimated on each linkage group, assuming the Kosambi mapping function. Double recombination was checked using the application in Map Manager QTX (Manly et al. 2001). Graphical representation of the linkage groups was performed using MAPCHART version 2.1 (Voorrips 2002). In addition, a consensus linkage map was constructed using JoinMap version 4 (Ooijen 2006) and the module of the combined group for map integration was used to integrate the sex-specific linkage maps.
Estimation of Genome Size and Coverage
A sex-specific map of genome length was estimated by two different calculation methods. First, genome estimation size 1 (G e1) was calculated by adding 2s, where s is the average framework marker spacing that was calculated by dividing the summed length of all the genetic linkage groups by the number of intervals (number of markers minus the number of genetic linkage groups) to the length of each genetic linkage group, accounting for chromosome ends beyond the terminal markers coverage. Second, genome estimation size 2 (G e2) was calculated by multiplying the length of each genetic linkage group by a factor (m + 1)/(m − 1). Where m is the number of framework markers for each genetic linkage group (Chakravarti et al. 1990). The estimated genome length (G e) for each sex was used as an average of the two estimates (Fishman et al. 2001; Sanchez et al. 2010). The genome coverage for each sex was calculated as the observed genome length (G oa) divided by the estimated genome length (G e) (Song et al. 2013) while the observed genome length (G oa) was taken as the combination of total length in all linkage group.
QTL Analysis
First, the normality of the phenotypes (BW and TL) was tested using the Kolmogorov-Smirnov test (N > 50) and Shapiro-Wilk test (N < 50), implemented in SPSS 16.0 package. The data were converted to Z scores before analysis using MapQTL software.
QTL analysis was carried out using MapQTL 5 software (Ooijen 2004). Ninety F1 progeny from stage II of family A were used to find candidate QTLs. A non-parametric Kruskal-Wallis analysis was used to determine the significance level of all marker loci associated with the growth-related traits (BW and TL). Meanwhile, simple interval mapping was used to detect significant associations with growth-related traits and marker loci in the data sets under the significant threshold of genome-wide (P value < 0.01 and P value < 0.05) and chromosome-wide (P value < 0.05) analyses. A minimum LOD threshold of 4.0 was used for determining a significant QTL and the percentage of phenotypic variance of each QTL. Permutation tests were performed (1000 replicates) to determine the LOD threshold by type one error. The significant thresholds derived from the permutation tests was estimated by dividing the nominal P value by the total number of chromosomes (Churchill and Doerge 1994; Ozaki et al. 2013). A graphical representation of the significant QTLs was constructed using MAPCHART version 2.1 and MapQTL 5. The results of the growth-related QTL regions of stage II family A were confirmed to be reproducible in the other stage and family.
Results
Correlation of Phenotypes and Growth-Related Traits in Families A and B
The correlation of phenotypes was tested using Pearson’s correlation coefficient. The results showed a high correlation between BW and TL in both stages of the two families (Table 1). The normal distribution of the phenotype was tested by a Kolmogorov-Smirnov test or Shapiro-Wilk test depending on the number of samples (Table 2, Additional file 1). The high correlation between BW and TL and normal distribution of phenotypes in stage II of family A led us to select family A to construct the high genetic linkage map and to screen candidate QTL regions.
High-Resolution Genetic Linkage Map and Genome Coverage
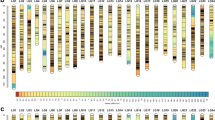
A total of 1867 SSR markers were designed. Of them, approximately 1050 SSR markers were polymorphic (56.2 %), and composed 905 EBR and 145 STR SSR markers. Ultimately, 714 SSR markers were used to construct a linkage map with reference species. The list of SSR markers used for mapping is given in additional file 2. Twenty-four genetic linkage groups (LG1–LG24) were identified. The female linkage map contained 509 markers distributed in 24 linkage groups (EBR 1F–EBR 24F) (Fig. 1). The total genome size of the female map was estimated as 1249.8 cM. The number of markers per linkage group varied from 5 to 29, with an average of 21; the longest linkage group of the female map extended to 65.4 cM (EBR 7F). Meanwhile, 512 markers were distributed in 24 linkage groups of the male map (EBR 1M–EBR 24M) (Fig. 1). The total genome was estimated at 1140.3 cM. The longest linkage group of the male extended to 58.0 cM (EBR 1), while the average number of markers per linkage group was 21, and varying from 9 to 31. The framework interval in each group was estimated based on the distance between clusters or markers, because some markers located on the same cluster. The female and male linkage maps comprised 305 and 285 framework, respectively, and the average interval between markers was 4.1 and 4.0 cM, respectively (Tables 3 and 4).




Kelp grouper female (left) and male (right) maps, linkage groups EBR 1–EBR 24. a EBR 1. b EBR 2. c EBR 3. d EBR 4. e EBR 5. f EBR 6. g EBR 7. h EBR 8. i EBR 9. j EBR 10. k EBR 11. l EBR 12. m EBR 13. n EBR 14. o EBR 15. p EBR 16. q EBR 17. r EBR 18. s EBR 19. t EBR 20. u EBR 21. v EBR 22. w EBR 23. x EBR 24. Total lengths of linkage groups are expressed in Kosambi cM. SSR markers are coded as “EBR” and “STR”. Bold letters indicate co-segregating microsatellite loci between the female and male maps
Recombination rate between the sex-specific genetic linkages were estimated by co-segregation markers. At least two SSR markers shared loci in the female and male maps and could be used to calculate the recombination rate among adjacently paired markers. The total length of genetic distance obtained from 24 genetic linkage groups (LGs) were 1249.8 and 1140.3 cM in female and male maps, respectively. The relative recombination ratio between females and males in these pairs was 1.12:1, which indicated that female LGs had a higher recombination rate than male except for LGs 1, 8, 11, 12, 14, and 19 (Table 5).
Genome length (G e) was estimated as approximately 1475.95 and 1370.39 cM in the female and male maps, respectively. The female map was 1.07 times longer than the male map. Only nine LGs (1, 6, 11, 12, 14, 19, 21, 23, and 24) on the male map were longer than the female map. The genome coverages of the female and male maps were estimated at 84.68 and 83.21 %, respectively (Table 4).
Screening Candidate QTL Regions
Screening for candidate QTL of BW using the Kruskal-Wallis analysis of stage II family A (90 progeny) identified 5, 23, and 6 of the 34 total markers were significant (P < 0.01) on three linkage groups corresponding to chromosomes EBR 13F, EBR 17F, and EBR 18M (data not shown). The results of simple interval mapping and the permutation test showed a significant major QTL (qBW17f) at the 1 and 5 % genome-wide level on the linkage group EBR 17F (Fig. 2). The LOD score of qBW17f (LOD = 4.09) was higher than the genome-wide LOD significance threshold of 3.7. This candidate major QTL region was detected close to the SSR markers Ebr00153FRA, Ebr00702FRA, Ebr00314FRA, and EguSTR119DB, and accounted for 14.6–18.9 % of the phenotypic variance with 1.00–1.13 of the additive effect (Table 6). While seven putative QTLs (qBW5f, qBW10m, qBW13f, qBW15m, qBW18m, qBW19f, and qBW21f) were significant at a 5 % chromosome-wide level on linkage groups EBR 5F, EBR 13F, EBR 19F, and EBR 21F of the female map, and linkage groups EBR 10M, EBR 15M, and EBR 18M of the male map (Fig. 3) and could explain 7.5–12 % of the phenotypic variance with 0.70–0.92 of the additive effect (Table 6).

Localization of a significant marker for body weight traits in linkage group EBR 17F of family A. EBR (linkage group) F; marker distance on the female map. qBW17f: QTL for body weight on EBR 17F. Map positions and LOD scores were based on simple interval mapping. QTL analysis was performed using the software MapQTL 5. LOD limit of detection (significance threshold), P g genome-wide significance threshold

Localization of putative QTLs for body weight traits in the female map of family A. EBR (linkage group) F marker distance on the female map; EBR (linkage group) M marker distance on the male map. a qBW5f: QTL for body weight on EBR5F. b qBW13f: QTL for body weight on EBR13F. c qBW19f: QTL for body weight on EBR19F. d qBW21f: QTL for body weight on EBR 21F. e qBW10m: QTL for body weight on EBR 10M. f qBW15m: QTL for body weight on EBR 15M. g qBW18m: QTL for body weight on EBR 18M. Map positions and LOD scores were based on simple interval mapping, QTL analysis was performed using the software MapQTL 5. LOD limit of detection (significance threshold), P c chromosome-wide significance threshold
Due to a high significance level (P < 0.01) in the non-parametric K-W analysis and the LOD score of a candidate major and two putative QTLs exceeded the high significance thresholds (P < 0.01 and P < 0.05) of genome- and chromosome-wide after simple interval mapping and permutation analysis. Then, 35 representative microsatellite markers (Additional file 3) related with these QTLs from three linkage groups (EBR 13F , EBR 17F, and EBR 18M) were used to confirm the candidate QTL regions in all progeny in the two stages of families A and B.
Confirmation of the Candidate QTL Regions
Thirty-five marker loci from three candidate QTL regions of three linkage groups affecting BW in stage II family A were used to confirm the QTL region in the other stage of the same family and in the other family by collecting genotype data in both stages of the two families. In the case of the stage II family analysis, the number of progeny analyzed for the trait analysis increased from 90 to 163 progeny. For family A, the K-W test results showed that eight markers from linkage groups EBR 13F and EBR 17F of the female map showed consistently significant results (P < 0.001) in stage II. Of them, three markers (Ebr00254FRA, Ebr00314FRA, and EguSTR119DB) showed the highest consistently significant results (P < 0.0005), while only two markers (ElaSTR366DB and Ebr00443FRA) showed consistently significant results (P < 0.005) in the male map (Table 7). Simple interval mapping on a chromosome-wide basis was then performed in each stage. The results showed only three QTLs (qBW13f, qBW17f, and qBW18m) in stage II were still significant. However, the results of interval mapping in stage II showed decreasing LOD scores (4.09 to 3.17) from the genome-wide analysis, with an LOD experimental-wide significance threshold of 2.0 (Fig. 4a) with the LOD maximum locus (qBW17f) could explain phenotypic variance ranging 5.9–8.6 % with 0.49–0.59 of the additive effect of the BW traits. In contrast, for two candidate QTLs (qBW13f, qBW18m) on linkage groups EBR 13F and EBR 18M, their LOD scores increased from 2.5 to 3.38, and from 2.47 to 2.9, respectively, under the experiment-wide analysis. LOD significant threshold of 2.0 and 2.0 (Fig. 4b, c) with the region of LOD maximum locus (qBW13f and qBW18m) could explain phenotypic variance ranging 4.2–9.1 and 5–7.9 % with 0.42–0.62 and 0.44–0.56 of the additive effect of BW traits (Table 8). Nevertheless, we could not find any consistently significant results in stage I of family A.

Localization of major and putative QTLs for the body weight trait in the female and male maps, based on confirmed QTL regions of family A. EBR (linkage group) F marker distance on the female map; EBR (linkage group) M marker distance on the male map. a qBW17f: QTL for body weight on EBR17F. b qBW13f: QTL for body weight on EBR 13F. c qBW18m: QTL for body weight on EBR 18M. Map positions and LOD scores were based on a simple interval mapping. QTL analysis was performed using the software MapQTL 5. LOD limit of detection (significance threshold), P e experiment-wide significance threshold, P c chromosome-wide significance threshold
In family B, the results showed only one marker (Ebr00702FRA) on linkage group EBR 17M in stage I, which presented consistent highly significant results (P < 0.001), was a putative QTL (qBW17m-1). It had a LOD score of 2.65, which was higher than the chromosome-wide LOD significance threshold of 2.0, with a range of 10.3 % of the phenotypic variance with 0.64 of the additive effect (Table 8, Fig. 5). Meanwhile, other significant regions in all linkages were rejected as QTLs in stage II of family B.

Localization of a suggested QTL for body weight traits in the male map of family B. EBR (linkage group) M marker distance on the male map. qBW17m-1: QTL number 1 for body weight on EBR 17M; Map positions and LOD score based on simple interval mapping. QTL analysis was performed using the software MapQTL 5. LOD limit of detection (significance threshold), P e experiment-wide significance threshold, P c ; chromosome-wide significance threshold
Association of Growth-Related Trait QTL Regions and TL
In this study, we also measured another phenotype, TL, which was highly correlated with BW of fish (Pearson correlation coefficient test P < 0.01), particularly in stage II of both families. For stage II of family A, the results of the K-W analysis and simple interval mapping showed significant loci in eight linkage groups (EBR 5F, EBR 7F, EBR 8F, EBR 13F, EBR 10M, EBR 17F, EBR 18M, and EBR 22M). The LOD score of a major QTL (qTL17f) effected to TL in linkage group EBR 17F was 4.0. with genome-wide significance (P < 0.01). This QTL region could explain 14.7–18.5 % of the phenotypic variance and 0.99–1.12 of the additive effect of TL trait. Meanwhile, another region with an LOD maximum locus in the other linkage group had a value that exceeded the chromosome-wide value and could explain 7.0–11.3 % of the phenotypic variance and 0.69–0.89 of the additive effect of the TL trait (Table 9). Moreover, we confirmed all the candidate QTL regions that affected TL using 35 markers in both stages of families A and B, just as we did for the BW trait. The K-W analysis results revealed eight markers from linkage groups EBR 13F and EBR 17F of the female map that showed consistently significant results in stage II. Of them, three markers (Ebr00254FRA, Ebr00314FRA, and EguSTR119DB) showed the highest consistently significant results (P < 0.0005). While only two markers (ElaSTR366DB and Ebr00443FRA) in linkage group EBR 18M of the male map showed consistently significant results (P < 0.005) (Table 10). LOD analysis showed a decreasing LOD score from 4.00 to 3.25 at an LOD experimental-wide significance threshold of 2.0, in the candidate major QTL (qBW17f) on the linkage group EBR 17F. By contrast, the confirmation of two candidate putative QTL regions (qTL13f and qTL18m) demonstrated LOD scores that increased from 2.34 to 3.24 and 2.32 to 2.46, respectively, on the experiment-wide scale. LOD significant threshold of 2.0 and 2.0. The region of the LOD maximum locus (qTL13f and qTL18m) could explain phenotypic variance ranging from 3.9–8 .to 4.8–6.7 % of the phenotypic variance and 0.40–0.61 and 0.43–0.52 of the additive effect of the TL trait. As with the results for BW, we could not find any consistently significant values for stage I of family A or for both stages of family B (Table 8).
Discussion
The high-resolution genetic linkage maps of the kelp grouper produced in this study greatly enhanced the previous genetic linkage map for the kelp grouper which was developed by using 222 microsatellite markers. The previous female and male map consisted of 25 and 23 linkage groups with 67.2 and 67.8 % of genome coverage and 1.5:1 of average recombination ratio (Liu et al. 2013). In the new genetic linkage map, 714 SSR markers were mapped in the 24 linkage groups, which is consistent with the diploid chromosome number of the kelp grouper (2N = 48) (Lan 2009). About 509 and 512 markers were identified and evenly covered the 24 linkage groups of the female and male maps, respectively. Only 10 of 714 markers remained as single markers. All of the microsatellite markers used in the previous genetic linkage map were also included and were consistently assigned in the same order and linkage groups in the present study, except for six markers. Of these, three markers (EguStr125DB, MiniSTR267DB, and Ebr00025FRA) and three other markers (MiniSTR266DB, Ebr00270FRA, and Ebr00253FRA) in linkage group EBR 24 and EBR 25 of the female map were moved to linkage groups EBR 23 and EBR 5 in the new female map, respectively. In addition, the genome coverage and average ratio of recombination between female and male maps were about 84.68, 83.21, and 1.12:1, respectively. This result revealed a large number of markers in the F1 progeny that filled several gaps of the new linkage map, which led to a reduction in the average mapping interval and an increase of the genome coverage. Considering the average interval and the genome coverage, we conclude that the high-resolution genetic linkage map of the kelp grouper of this study offers a sufficient marker density to permit a preliminary genome-wide scan for QTLs for growth-related traits (Massault et al. 2008). In addition, markers from other grouper species could speed up the construction and completion of a genetic linkage map of the kelp grouper in the near future.
The recombination rate of a gene located on a chromosome (autosomal) is different between females and males because of the number of crossing-over events that occur during meiosis I. Differences in recombination rates between sexes have been identified in many species; for example, humans (Dib et al. 1996), dogs (Wong et al. 2010), crocodiles (Miles et al. 2009), and fish. In fish, recombination rates have generally been reported to be higher in females compared to males ranging from 3.25:1 in rainbow trout (Sakamoto et al. 2000), 7.4:1 in the Japanese flounder (Coimbra et al. 2003), 1.37:1 in Atlantic salmon (Lien et al. 2011), 2.2:1 in the silver carp (Guo et al. 2013), 2:1 in the Atlantic halibut (Reid et al. 2007), 1.5:1 in the kelp grouper (Liu et al. 2013), 1.03:1 in the orange-spotted grouper (You et al. 2013), and 1.19:1 in the white grouper (Dor et al. 2014). In this study, the recombination rate ratio between females and males was 1.12:1, which was lower than previous reports. This may reflect the increased number of markers linked to the male map rather than the female map, which would affect not only the density of the markers but also the recombination rate in all linkage groups. In the present study, we found that markers in the female and male maps were irregularly distributed and showed high clustering of markers in all linkage groups. These markers tended to be compressed in the telomeric and centromeric regions of the female and male maps. A higher rate of recombination in the female and male maps probably occurred near the centromeric and the telomeric regions (You et al. 2013). This could be explained by the higher frequency of recombination in females near the centromeric regions during oogenesis. Similarly, more frequent recombination in males was also found near the telomeres during meiosis (Strachan and Read 2011; You et al. 2013). For indicating the centromeric or telomeric region in female and male maps, these two regions were observed by the map distance between markers. In the case of high recombination, the maps will present high distance between markers or clusters. The distances between markers in the centrometric region were assessed to be larger than other sites (telemetric). Similar to the male map, the markers or clusters in telemetric regions were estimated to have a larger distance than the centrometric region. The difference in sex recombination is an important factor in the implementation of marker-assisted selection using QTL-associated mapping.
The growth-related quantitative trait QTLs in this study were identified using F1 progeny of the kelp grouper. This was different from other studies that performed QTL mapping using F2 generation from F1 crosses in a genetically different line or F2 back-cross (Hayashi and Awata 2004), such as the Pacific white leg shrimp (Andriantahina et al. 2013). Kelp groupers are protogynous hermaphrodites and it would take a long time to produce an F2 generation. This type of reproductive system takes a longer time for the sex reversal from male to female when they exceed a certain age or body size. In the kelp grouper, it takes more than 6 years of culture for the fish to reach maturity (before the first maturation and spawning). This is too long to create an F2 generation. This explains our choice of producing F1 progeny for the QTL study. In the past decade, the analysis of QTLs using F1 progeny was developed and successfully applied to Asian seabass (Wang et al. 2006). Under the criteria of heritability of traits of interest, the power of QTL detection depends on the heritability of the traits, the effect of alleles involved, the recombination distance of the associated marker, and the sample size (Mackay 1996). We found a major QTL affecting BW in the kelp grouper that was located on linkage group EBR 17F of the female map under genome-wide linkage analysis. We also found putative QTLs affecting BW that were located in seven linkage groups under a chromosome-wide analysis. The phenotypic variance of the major QTL was 14.6–18.9 and was 7.5–12 % for the putative QTLs. Similar results were obtained for the total length trait. One major QTL was detected in the same linkage group of BW that explained 14.7–18.5 % of the phenotypic variance. The putative QTLs accounted for 7–11.3 % of the phenotypic variance. These results indicated that several QTL region-associated BW and TL traits are determined by multiple genes. Our result also revealed that the growth-related traits of the kelp grouper might be controlled by a few QTLs with large effects.
The candidate QTLs were confirmed in two developmental stages in families A and B, with 35 representative markers. The results showed a highly significant level for major QTL in stage II of family A after adding the number of progeny, which were rejected in stage I of family A and both stages of family B. For the putative QTL regions in stage II of family A on linkage groups EBR 13F and EBR 18M, the results were rejected for stage I family A and stage II of family B. However, they were accepted for stage II family A and stage I family B with same regions on linkage group EBR 17M of the male map (qBW17m-1). From these results, we considered that the explanation lay in the parental fish, the distribution of the phenotype, and the number of progeny. In addition, we noticed the significance of the LOD score of the candidate major QTL decreased after confirmation of the significant QTL region, while the LOD of the putative QTL region increased. This was particularly true for the putative QTL affecting BW and TL on linkage group EBR 13F after we increased the number of progeny. It is possible that given a sufficiently large number of progeny, more major QTL regions could be detected and confirmed.
Herein, the most important finding was a single peak of QTL associated with BW and TL within the proximal region of linkage group EBR 17F. Both QTL (qBW17f and qTL17f) peaks were located at position 22.4 cM, with 99 % confidence interval mapping within 4.4 cM of the most proximal markers from Ebr00702FRA to Ebr00092FRA by simple interval mapping. The narrowness of the interval marker of the candidate QTL region should be considered as a fine approximation, given the large QTL effect and high recombination rate found in kelp grouper females. These results could be used to investigate candidate genes in a future study of growth-related traits of the kelp grouper.
Conclusions
This study constructed the first high-resolution genetic linkage map of the kelp grouper. The map provided an increased SSR marker density from 222 microsatellite markers on the first-generation genetic linkage map (Liu et al. 2013) to 716 SSR markers. Twenty-four linkage groups were identified, consistent with the 24 haploid chromosome number of the kelp grouper (2N = 48). The female and male maps accounted for 84.68 and 83.21 % coverage and produced average mapping intervals of 4.1 and 4.0, respectively. Considering the average mapping interval and genome covered, these linkage maps would be sufficient for genome-wide linkage analysis and could increase the power of statistics to detect growth-related QTL traits.
Three significant QTLs affecting both phenotypes (BW and TL) were detected and confirmed. One major QTL was significant (1 and 5 % at the experiment-wide significance level) in linkage group EBR 17F of the female map, which showed 6–8.6 and 6.1–8.8 % of the phenotypic variance. Two putative QTLs affecting both phenotypes (BW and TL) (5 % chromosome-wide significance level) were located on linkage groups EBR 13F and EBR 18M of the female and male maps, explaining 1.8–9.1 and 1.2–8.7 % of the phenotypic variance. These results suggested that the growth-related quantitative traits are controlled by multiple genes.
We anticipate that the high resolution of genetic linkage map and growth-related QTLs found in this study could be applied to find candidate genes, will be powerful tools for a future MAS breeding program and may provide further insights into the genetic control of growth traits in the kelp grouper.
References
Abraham K, Andrey S, Avner C, Eric MH (2007) Detection and analysis of quantitative trait loci (QTL) for economic traits in aquatic species. In: Liu ZJ (ed) Aquaculture genome technology, 1st edn. Blackwell Publishing, Iowa, pp 169–196
An HS, Kim JW, Lee JW, Kim SK, Lee BI, Kim DJ, Kim YC (2011) Development and characterization of microsatellite markers for an endangered species, Epinephelus bruneus, to establish a conservation program. Anim Cells Syst 16:50–56
Andriantahina F, Liu X, Huang H (2013) Genetic map construction and quantitative trait locus (QTL) detection of growth-related traits in Litopenaeus vannamei for selective breeding applications. PLoS One 8:e75206
Baranski M, Moen T, Vege DI (2010) Mapping of quantitative trait loci for flesh colour and growth traits in Atlantic salmon (Salmo salar). Genet Sel Evol 42:17
Chakravarti A, Lasher KL, Reefer JE (1990) A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 128:175–182
Chapman RW, Sedberry GR, Koenig CC, Eleby BM (1999) Stock identification of gag, Mycteroperca microlepis, along the southeast coast of the United States. Mar Biotechnol 1:137–146
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Coimbra MRM, Kobayashi K, Koretsugu S, Hasegawa O, Ohara E, Ozaki A, Sakamoto T, Naruse K, Okamoto N (2003) A genetic linkage map of the Japanese flounder, Paralichthys olivaceus. Aquaculture 220:203–218
Danzmann RG (2006) Linkage analysis package for outcrossed families with male or female exchange of the mapping parent, version 2.3. University of Guelph, Ontario
Danzmann RG, Ghabi K (2007) Linkage mapping in aquaculture species. In: Liu ZJ (ed) Aquaculture genome technology, 1st edn. Blackwell Publishing, Iowa, pp 39–167
Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Karzan J, Seboune E, Lathrop M, Gyapay G, Morissette J, Weissenbach J (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380:152–154
Dong Y, Han J, Cai H (2008) Cross-species amplification and characterization of polymorphic microsatellite loci in Hong Kong grouper, Epinephelus akaara. J Beijing Normal Univ (Nat Sci) 44:511–514
Dor L, Shirak A, Gorshkov S, Band RM, Korol A, Ronin Y, Curzon A, Hulata G, Seroussi E, Ron M (2014) Construction of a microsatellites-based linkage map for the white grouper (Epinephelus aeneus). G3 (Bethesda) 4:1455–1464
Fishman L, Kelly AJ, Morgan E, Willis JH (2001) A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics 159:1701–1716
Fui CF, Miura A, Nakagawa Y, Kato K, Senoo S, Sakamoto W, Takii K, Miyashita S (2014) Flow field control via aeration adjustment for the enhancement of larval survival of the kelp grouper Epinephelus bruneus (Perciformes: Serranidae). Aquac Res 45:874–881
Guo W, Tong J, Yu X, Zhu C, Feng X, Fu B, He S, Zeng F, Wang X, Liu H, Liu L (2013) A second generation genetic linkage map for silver carp (Hypophthalmichthys molitrix) using microsatellite markers. Aquaculture 412–413:97–106
Hayashi T, Awata T (2004) Efficient method for analysis of QTL using F1 progenies in an outcrossing species. Genetica 122:173–183
Heemstra PC, Randall JE (1995) Groupers of the world. FAO species catalogue, vol 16. FAO, Rome
Kubota S, Liu Q, Kessuwan K, Okamoto N, Sakamoto T, Nakamura Y, Shigenobu Y, Sugaya T, Sano M, Uji S, Nomura K, Ozaki A (2014) High-throughput simple sequence repeat (SSR) markers development for the kelp grouper and cross-species amplifications for Epinephelinae species. Adv Biosci Biotechnol 5:117–130
Lan GM (2009) Comparative Studies of Epinephelus moara and Epinephelus bruneus. PhD. dissertation, Xiamen University
Lee YD, Park SH, Takemura A, Takano K (2002) Histological observations of seasonal reproductive and lunar-related spawning cycle in the female honeycomb grouper Epinephelus merra in Okinawa waters. Fish Sci 68:872–877
Lien S, Gidskehaug L, Moen T, Hayes BJ, Berg PR, Davidson WS, Omholt SW, Kent MP (2011) A dense SNP-based linkage map for Atlantic salmon (Salmo salar) reveals extended chromosome homeologies and striking differences in sex-specific recombination patterns. BMC Genomics 12:615
Liu ZJ (2007) Microsatellite markers and assessment of marker utility. In: Liu ZJ (ed) Aquaculture genome technology, 1st edn. Blackwell Publishing, Iowa, pp 43–57
Liu L, Liu CW, Guo YS, Dong QF, Xu TJ (2008) Isolation and population genetic diversity analysis of microsatellite DNA markers in yellow grouper (Epinephelus awoara). J Fish Sci China 15:22–29
Liu Q, Sakamoto T, Satoshi K, Okamoto N, Yamashita H, Takagi M, Shigenobu Y, Sugaya T, Nakamura Y, Sano M, Wuthisuthimethavee S, Ozaki A (2013) A genetic linkage map of kelp grouper (Epinephelus bruneus) based on microsatellite markers. Aquaculture 414–415:63–81
Lo LC, Yue GH (2007) Microsatellites for broodstock management of the tiger grouper, Epinephelus fuscoguttatus. Anim Genet 39:90–91
Mackay TFC (1996) The nature of quantitative genetic variation revisited: lessons from Drosophila bristles. BioEssays 18:113–121
Manly KF, Cudmore RH Jr, Meer JM (2001) Manager QTX, cross-platform software for genetic mapping. Mamm Genome 12:930–932
Martins WS, Lucas DCS, Neves KFS, Bertioli DJ (2009) WebSat—a web software for microsatellite marker development. Bioinformation 3:282–283
Massault C, Bovenhuis H, Haley C, Koning DJ (2008) QTL mapping designs for aquaculture. Aquaculture 285:23–29
Max FR, Anatoly R (2007) Marker-assisted selection for aquaculture species. In: Liu ZJ (ed) Aquaculture genome technology, 1st edn. Blackwell Publishing, Iowa, pp 199–213
Miles LG, Isberg SR, Glenn TC, Lance SL, Dalzell P, Thomson PC, Moran C (2009) A genetic linkage map for the saltwater crocodile (Crocodylus porosus). BMC Genomics 10:339
Mitcheson YS, Craig MT, Bertoncini AA, Carpenter KE, Chaung WWL, Choat JH, Cornish AS, Fennessy ST, Ferreira BP, Heemstra PC, Liu M, Myers RF, Pollard DA, Rhodes KL, Rocha LA, Russell BC, Samoilys MA, Sanciangco J (2003) Fishing groupers towards extinction: a global assessment of threat and extinction risks in a billion dollar fishery. Fish Fish 14:119–136
Mokhtar MA, Normah MN, Kumar SV, Baharum SN (2011) Characterization of 10 novel microsatellite loci for the brown marbled grouper, Epinephelus fuscoguttatus (Serranidae). Genet Mol Res 10:885–888
Molano ES, Cerna EA, Toro MA, Bouza C, Hemida M, Pardo BG, Cabaleiro S, Fernandez J, Martinez P (2011) Detection of growth-related QTL in turbot (Scophthalmus maximus). BMC Genomics 12:473
Ooijen JWV (2004) MapQTL 5.0 Software for the mapping of quantitative trait loci in experimental population. Plant Research International, Wageningen
Ooijen JWV (2006) JoinMap 4.0 Software for the calculation of genetic linkage maps in experimental populations. Plant Research International, Wageningen
Ozaki A, Yoshida K, Fuji K, Kubota S, Kai W, Aoki J, Kawabata Y, Suzuki J, Akita K, Koyama T, Nakagawa M, Hotta T, Tsuzaki T, Okamoto N, Araki K, Sakamoto T (2013) Quantitative trait loci (QTL) associated with resistance to a monogenean parasite (Benedenia seriolae) in yellowtail (Seriola quinqueradiata) through genome wide analysis. PLoS One 8:e64987
Ramirez MA, Patricia AJ, Planas S, Carlin JL, Funk SM, McMillan WO (2006) New microsatellite resources for groupers (Serranidae). Mol Ecol Notes 6:813–817
Reid DP, Smith CA, Rommens M, Blanchard B, Martin RD, Reith M (2007) A genetic linkage map of Atlantic halibut (Hippoglossus hippoglossus L.). Genetics 177:1193–1205
Renshaw MA, Portnoy DS, Gold JR (2010) PCR primers for nuclear-encoded microsatellites of the groupers Cephalopholis fulva (coney) and Epinephelus guttatus (red hind). Conserv Genet 11:1197–1202
Rivera MAJ, Graham GC, Roderick GK (2003) Isolation and characterization of nine microsatellite loci from the Hawaiian grouper Epinephelus quernus (Serranidae) for population genetic analyses. Mar Biotechnol 5:126–129
Sakamoto T, Danzmann RG, Gharbi K, Howard P, Ozaki A, Khoo SK, Woram RA, Okamoto N, Ferguson MM, Holm LE, Guyomard R, Hoyheim B (2000) A microsatellite linkage map of rainbow trout (Oncorhynchus mykiss) characterized by large sex-specific differences in recombination rates. Genetics 155:1331–1345
Sanchez C, Fuji CK, Ozaki A, Hasegawa O, Sakamoto T, Morishima K, Nakayama I, Fujiwara A, Masaoka T, Okamoto H, Hayashida K, Tagami M, Kawai J, Hayashizaki Y, Okamoto N (2010) A second generation genetic linkage map of Japanese flounder (Paralichthys olivaceus). BMC Genomics 11:554
Sawada, Kate YK, Okada T, Kurata M, Mukai Y, Miyashita S, Murata O, Kumai H (1999) Growth and morphological development of larval and juvenile Epinephelus bruneus (Perciformes: Serranidae). Ichthyol Res 46:245–257
Song W, Miao G, Zhao Y, Niu Y, Pang R, Liao X, Shao C, Chen S (2013) Construction of a microsatellite-based genetic linkage map for half-smooth tongue sole Cynoglossus semilaevis. Curr Zool 59:99–108
Strachan T, Read A (2011) Genetic mapping of Mendelian characters. In: Strachan T (ed) Human molecular genetics, 4th edn. Garland Science, UK, pp 442–465
Thierry C, Sadovy Y, To AWL (2008) Epinephelus bruneus. The IUCN red list of threatened species. Version 2014.3. www.iucnredlist.org. Accessed 1 June 2014
Tsuchihashi Y, Tanaka H, Kuromiya Y, Kashiwagi M, Yoshioka M (2003) Control of induction of sex reversal in the sevenband grouper, Epinephelus septemfasciatus. Suisanzoshoku 51:189–196
Tupper M, Sheriff N (2008) Capture-based aquaculture of groupers. In: Lovatelli A, Holthus PF (eds) Capture-based aquaculture. Global overview, vol 508. FAO, Rome, pp 217–229
Voorrips RE (2002) MapChart software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang CM, Lo LC, Zhu ZY, Yue GH (2006) A genome scan for quantitative trait loci affecting growth-related traits in an F1 family of Asian seabass (Lates calcarifer). BMC Genomics 7:274
Wang CM, Lo LC, Feng Z, Zhu YGH (2008) Identification and verification of QTL associated with growth traits in two genetic backgrounds of barramundi (Lates calcarifer). Anim Genet 39:34–39
Wong AK, Ruhe AL, Dumont BL, Robertson KR, Guerrero G, Shull SM, Ziegle JS, Millon LV, Broman KW, Payseur BA, Neff MW (2010) A comprehensive linkage map of the dog genome. Genetics 184:595–605
Yeh SL, Kuo CM, Ting YY, Chang CF (2003) The effects of exogenous androgens on ovarian development and sex change in female orange-spotted protogynous grouper, Epinephelus coioides. Aquaculture 218:729–739
You X, Shu L, Li S, Chen J, Luo J, Lu J, Mu Q, Bai J, Xia Q, Chen Q, Cai Y, Zhang H, Chen G, Lin H, Zhang Y, Shi Q (2013) Construction of high-density genetic linkage maps for orange-spotted grouper Epinephelus coioides using multiplexed shotgun genotyping. BMC Genet 14:113
Yue GH (2013) Recent advances of genome mapping and marker-assisted selection in aquaculture. Fish Fish 15:376–396
Zeng HS, Ding SX, Wang J, Su YQ (2008) Characterization of eight polymorphic microsatellite loci for the giant grouper (Epinephelus lanceolatus Bloch). Mol Ecol Resour 8:805–807
Zhao L, Shao C, Liao X, Chen S (2009a) Isolation and characterization of polymorphic microsatellite loci from a dinucleotide-enriched genomic library of seven-band grouper (Epinephelus septemfasciatus) and cross-species amplification. Conserv Genet 10:627–629
Zhao L, Shao C, Liao X, Ma H, Zhu X, Chen S (2009b) Twelve novel polymorphic microsatellite loci for the yellow grouper (Epinephelus awoara) and cross-species amplifications. Conserv Genet 10:743–745
Zhu ZY, Lo LC, Lin G, Xu YX, Yue GH (2005) Isolation and characterization of polymorphic microsatellites from red coral grouper (Plectropomus maculatus). Mol Ecol Resour 5:579–581
Acknowledgments
This research was supported by SATREPS (Science and Technology Research Partnership for Sustainable Development, JST/JICA, Japan). We thank Ms. Kishiko Kubo and Ms. Naomi Yamasaki for their assistance with the genotyping and their contributions throughout the study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional file 1
Normal distribution and Pearson correlation coefficients of total length and body weight in stage I and II of families A and B. a Distribution of total length and body weight and Pearson correlation coefficients between the total length and body weight in stage I of family A (360 progeny). b Distribution of total length and body weight and Pearson correlation coefficients between the total length and body weight in stage II of family A (163 progeny). c Distribution of total length and body weight and Pearson correlation coefficients between the total length and body weight in stage I of family B (112 progeny). d Distribution of total length and body weight and Pearson correlation coefficients between the total length and body weight in stage I of family B (45 progeny). (JPEG 921 bytes)
Additional file 2
List of SSR markers used to construct a high-resolution genetic linkage map of the kelp grouper. EBR markers were developed using microsatellite-enriched segments from next-generation sequencing (NGS) using the GS FLX system (Roche, Switzerland); STR markers were developed using simple sequence repeats (SSRs) of a cross species of grouper from the NCBI database. (XLSX 204 kb)
Additional file 3
List of SSR markers used to confirm the QTL regions of the kelp grouper. EBR markers were developed using microsatellite-enriched segments from next generation sequencing (NGS) using the GS FLX system (Roche, Switzerland); STR markers were developed using simple sequence repeats (SSRs) of a cross species of grouper from the NCBI database. (XLSX 23 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kessuwan, K., Kubota, S., Liu, Q. et al. Detection of Growth-Related Quantitative Trait Loci and High-Resolution Genetic Linkage Maps Using Simple Sequence Repeat Markers in the Kelp Grouper (Epinephelus bruneus). Mar Biotechnol 18, 57–84 (2016). https://doi.org/10.1007/s10126-015-9673-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-015-9673-5