
Abstract
When bound to a pair of F atoms and a phenyl ring, a pyramidal pnicogen (Z) atom can form a pnicogen bond wherein an NH3 base lies opposite one F atom. In addition to this σ-hole complex, the ZF2C6H5 molecule can distort in such a way that the NH3 approaches on the opposite side to the lone pair on Z, where there is a so-called π-hole. The interaction energies of these π-hole dimers are roughly 30 kcal/mol, much larger than the equivalent quantities for the σ-hole complexes, which are only 4–13 kcal/mol. On the other hand, this large interaction energy is countered by the considerable deformation energy required for the Lewis acid to adopt the geometry necessary to form the π-hole complex. The overall energetics of the complexation reaction are thus more exothermic for the σ-hole dimers than for the π-hole dimers.
Similar content being viewed by others


Introduction
Recent years have witnessed a dramatic growth in interest in noncovalent interactions. New insights have been gleaned from a palette of computational tools based on quantum chemistry. While any single noncovalent interaction is weak in comparison to a covalent bond, such interactions nonetheless exert a powerful impact on numerous processes linked to biochemistry or crystal engineering, nanoparticle self-assembly, drug binding, and biomolecular folding processes [1,2,3,4,5,6,7,8,9,10,11,12,13,14]. After decades of scientific attention devoted to the hydrogen bond [4, 8, 15,16,17,18,19,20], a number of different but related noncovalent interactions have enjoyed the limelight more recently. Many of these bonds derive from the σ-hole concept, wherein—even though it does not carry a partial positive charge—an electronegative atom can nonetheless attract a nucleophile via an anisotropic electronic distribution which provides a positive electrostatic potential in a constricted region lying opposite a covalent bond [21,22,23,24,25,26,27,28]. The σ-hole area may be treated as an acidic binding site that attracts an incoming nucleophile, which may take the form of a lone pair, an anion, or even a π-electron system. In addition to the electrostatic attraction, noncovalent bonds benefit from electron transfer from a Lewis base to a σ* antibonding orbital of the acid [29,30,31,32,33], which also contributes to the directionality of the σ-hole bond [34,35,36].
In addition to σ-holes, which appear directly opposite covalent bonds, certain molecules can also develop π-holes, which lie above the plane of the system [37,38,39,40,41,42,43] and give rise to a π-hole-bonded complex [44]. These π-holes have been identified in numerous molecules, such as carbonyls, trisubstituted centers, and nitro- and acyl-carbon-containing entities [40, 45, 46]. The ensuing π-hole interactions share many of the same features with their σ-hole cousins [47] and can be responsible for even stronger bonds [48]. Although the study of π-hole interactions is accelerating, direct comparisons of σ- and π-bonds for the same pair of subunits [44, 48,49,50,51,52,53,54,55,56,57,58,59,60] remain limited, leaving many questions unanswered.
The pnicogen bond is a case in point. It has been reasonably widely examined [34, 61,62,63,64,65,66,67,68,69,70,71,72,73,74], but little has been determined in terms of competing configurations of a given pair of molecules. For example, the complexes PH2R···BrCl (R = H, F, OH, OCH3, CH3) were studied at the MP2 level [74], focusing on two possible σ-hole arrangements: halogen-bonded vs pnicogen-bonded. The halogen bonds were calculated to be stronger and driven primarily by electrostatics, while the weaker pnicogen bonds relied mostly on dispersion. With respect to π-electron donors, Zhu and coworkers observed a reversal in that pnicogen bonds were stronger than halogen bonds in complexes between PH2Cl and substituted benzene [68]. The strongest σ-hole bonding in PH2R complexes with formaldehyde [75] was found to occur when R = NO2, followed by R = F and R = Cl. Pnicogen bonds exhibit cooperativity with H-bonds, as noted in complexes of π-electron systems such as benzene with PCl3 [65]. There have been some studies of π-hole pnicogen bonds, but those studies focused on the hypervalent PO2Cl molecule [76], PO2R [77], or the (H2C=PH2)+ ion [78], none of which can contain a σ-hole. π-Hole pnicogen bonds have been identified in unconventional bonding situations such as NNO [79], but these are quite weak and there is no competing σ-hole interaction.
Although there has been little examination of σ-hole versus π-hole bonding in pnicogen bonds, there has been some study of this question for the related tetrel bond wherein TF4 (T = Si, Ge, Sn) molecules were paired with pyridine derivatives [80]. The approach of the Lewis base prompted large-scale geometry distortion in TF4, changing its shape from tetrahedral to trigonal bipyramidal. This transformation led to the formation of two types of dimers with σ-hole and π-hole characteristics. While the latter were more stable with regard to interaction energies (surpassing 50 kcal/mol), the accompanying deformation of the monomer geometry was so high in these configurations that the overall energetics favored the σ-hole complexes. The ability of tetrel atoms to engage in both σ-hole and π-hole interactions was recently examined [81], and it was learned that π-complexes can be stronger than their σ analogues, although different molecules were used for each class of interaction. Aerogen bonds are capable of forming both σ-hole and π-hole interactions, and recent calculations [82] suggest that it is the latter that are the stronger. In another somewhat related study, σ-hole pnicogen bonds were compared with a π-hole tetrel bond [83].
The ability of TF4 molecules to undergo [80] a geometrical transformation so as to form either a σ-hole or π-hole tetrel bond inspired us to wonder if something of this sort is also possible for pnicogen atoms. If such is the case, then there are a number of obvious and important questions. Would σ-hole or π-hole complexes be more stable, and how much deformation energy might be required for each to form? It would be interesting to determine the underlying sources of the stability of each to see what differences there might be. Do both sorts of bonds require the same proportions of electrostatic, polarization, and dispersion energy contributions? How do the quantitative aspects of the σ- and π-holes of the properly distorted monomer differ from each other, and do their magnitudes correlate with the strength of the interaction with a base? It would be interesting to determine whether both sorts of bonds undergo the same systematic trends as the pnicogen atom grows larger, i.e., P → As → Sb → Bi. The present communication details the results of calculations intended to answer these questions.
Systems and computational methods
The proper selection of electron donors and acceptor is crucial to deriving a systematic understanding of the nature of the bonding. In the Lewis acid, sufficiently potent electron-withdrawing substituents must be attached to the pnicogen atom to ensure the presence of regions of positive potential that can attract a base. The entire molecule must be flexible enough that geometrical deformations to accommodate both sorts of bonding are feasible. For the Lewis acid, then, the set of molecules ZF2C6H5 (Z = P, As, Sb, Bi) was selected. The F atoms act as electron-withdrawing agents, and the phenyl ring can rotate as needed. NH3 was chosen as the Lewis base due to its small size, which minimizes complicating secondary interactions, as well as its easily available lone pair of electrons.
The geometries of dimeric complexes between ZF2C6H5 (Z = P, As, Sb, Bi) and NH3 were optimized at the MP2 level in conjunction with the aug-cc-pVDZ basis set [84, 85]. Optimization was also carried out at the BLYP-D3/Def2TZVPP level of theory. Energies were additionally computed at the CCSD(T)/aug-cc-pVDZ level (using MP2-generated minima) for the purposes of comparison and validation [86,87,88,89,90,91,92]. For accurate electronic descriptions of the heavy Sb and Bi atoms, the aug-cc-pVDZ-PP basis set with pseudopotentials taken from the EMSL library was employed [93]. Structures were verified as true minima by checking that all vibrational frequencies were positive. Computations were performed via the Gaussian 09 software package [94]. Molecular electrostatic potential (MEP) analysis was applied to identify and quantify all MEP extrema using the WFA-SAS and MultiWFN programs [95,96,97]. Interaction energies were calculated as the difference in energy between the complex and the sum of monomers (with the same geometries as they adopt within the complex). Binding energies were computed relative to the monomers in their isolated optimized structures. Both quantities were corrected for basis set superposition error (BSSE) using the counterpoise protocol [98]. The electron density topology was analyzed using AIMAll software [99]. Energy decomposition analysis (EDA) was performed at the BLYP-D3/ZORA/TZ2P level using DFT-optimized geometries with the aid of ADF software [100,101,102]. In order to analyze interorbital connections and charge flow between the monomers, the natural bond orbital (NBO) procedure (using GenNBO 6.0) was utilized using the wavefunction generated at the DFT level [103]. The CSD (Cambridge Structural Database) [104] was searched for pertinent experimental crystal structures similar to those described in this manuscript.
Results
Monomer characteristics
The isolated ZF2C6H5 (Z = P, As, Sb, Bi) monomers were fully optimized at the MP2/aug-cc-pVDZ and BLYP-D3/Def2TZVPP levels. The equilibrium geometries of these monomers calculated at the MP2 level are similar; they all have a highly pyramidal Z atom close to the Cs point group. This general structure is presented as conformer A in Fig. 1. Despite appearing to be in close proximity, AIM analysis does not find any bond critical point between the F and ring H atoms when Z = P, As, or Bi. In the case of BiF2C6H5, however, one of the F atoms is nearly coplanar with the aromatic ring, with a dihedral angle φ(F–Bi–C–C) = 11.3° (see Table S1 in the “Electronic supplementary material,” ESM), and AIM finds a BCP between this F and the nearby ring H atom. On the other hand, this interaction is rather weak, with an electron density at the BCP of only 0.0095 au.

Structures of the A and B conformers of the isolated ZF2C6H5 monomers
Unlike MP2, the DFT functional identified two separate stable conformers of ZF2C6H5. In addition to the A geometry discussed above, the B conformer rotates the ZF2 group such that both F atoms lie below the phenyl ring, and the Z lone pair points above this ring, belonging to the Cs point group. Structure B is slightly less stable than A, by 1.04, 0.70, and 0.58 kcal/mol for Z = P, As, and Sb, respectively; these differences are even smaller in terms of ∆G. DFT, like MP2, does not identify a B geometry for BiF2C6H5.
The molecular electrostatic potential surrounding the A geometry of PF2C6H5 is displayed in Fig. 2; similar diagrams are obtained for the other ZF2C6H5 systems. The most intense σ-holes lie opposite the F atoms, along extensions of the P–F bonds. The value of Vs,max on this surface increases with the size of the Z atom from 19.4 kcal/mol for Z = P to 52.6 kcal/mol for Z = Bi, as may be seen in the first column of Table 1. There is a second, but weaker, σ-hole opposite the C–P bond, with smaller Vs,max values listed in the last column of Table 1. The calculated Vs,min for the isolated ammonia molecule that acts as a Lewis base is −37.7 kcal/mol.

MEPs of the isolated PF2C6H5 σ-hole donor molecule, computed on the 0.001 au isodensity surface at the MP2/aug-cc-pVDZ level. Color ranges, in kcal/mol, are: red greater than 15, yellow between 8 and 15, green between 0 and 8, blue below 0 kcal/mol
σ-Hole and π-hole dimer interactions
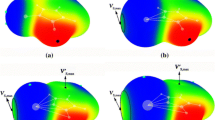
When each ZF2C6H5 molecule was paired with NH3, two types of dimer geometries were identified. As illustrated in the top half of Fig. 3, in the first case the NH3 approaches Z along the direction of one of the two Z–F σ-holes. Another dimer structure is related to the B monomer geometry, wherein the NH3 lies roughly perpendicular to the C–Z covalent bond, in between the two Z–F bonds. These two structures are designated σ-hole and π-hole complexes, respectively.

MP2/aug-cc-pVDZ-optimized structure (top and side views) of complexes of NH3 with ZF2C6H5
The geometric details of these two types of dimers are listed in Table 2. F1 refers to the F atom that lies directly opposite the NH3 N atom in the σ-hole structure, and F2 refers to the other F atom. The angle θ(F1–Z···N) in the σ-hole geometries ranges between 155° and 166°, and is more linear for the smaller Z atoms. The intermolecular distances R(Z···N) are surprisingly insensitive to the nature of the Z atom, and are longest for P and shortest for As. In contrast, this same distance is highly dependent upon Z for the π-hole complexes, elongating from 1.932 Å for P to 2.343 Å for Bi. This pattern fits the simple idea of a progressively larger Z atom along this series. Note also that the N atom lies on the same side of the phenyl plane as the two F atoms, with the angles θ(F–Z–N) all less than 80°. The bond lengths r(Z–F1) for the F lying opposite the N in the σ-hole dimers are consistently longer than r(Z–F2) by about 0.01 Å, consistent with the idea of charge transfer into the σ*(Z–F1) antibonding orbital (see below). The Z–F bond lengths are considerably longer in the π-hole complexes.
The substituents around the central Z atom constitute a pyramidal structure. Its deviation from planarity can be quantified as the sum of the three angles θ(X–Z–Y), where X and Y refer to the substituent atoms (C or F). This sum would be equal to 360° for a fully planar structure, so deviations from this sum can be considered as a measure of the nonplanarity, i.e., the “pyramidality.” As shown in the last column of Table 2, this angle sum is quite small for the σ-hole complexes, less than 290°. The deviation from planarity gets larger as the Z atom grows in size. On the other hand, the uncomplexed monomers are also quite nonplanar, as may be seen in the last column of Table S1. The increase in nonplanarity caused by complexation, i.e., the difference in ∑θ(Z) between the monomer and complex, is between 4° and 7°. The angle sum is much closer to 360° for the π-hole complexes. This change from the monomer amounts to a 40–50° loss of pyramidal character, i.e., a flattening of the pyramid at the central Z atom. This alteration has important repercussions for the energetics of this process, as elaborated below.
The interaction energies of the various heterodimers obtained at three different levels of theory are presented in Table 3. The most striking trend in these values is that the interactions involved in the π-hole complexes are much stronger than those for the σ-hole complexes. Eint lies in the range 24–34 kcal/mol for these structures—much larger than the 4–13 kcal/mol range for the σ-hole geometries. Taking the CCSD(T)/aug-cc-pVDZ values as a benchmark, the MP2 quantities for the π-hole dimers are slightly inflated, whereas those calculated at the BLYP-D3/Def2TZVPP level are significantly underestimated. The three levels provide much more uniform data for the σ-hole geometries. With regard to the former structures, Eint tends to drop slowly as the Z atom becomes larger; the opposite pattern emerges for the σ-hole dimers, where NH3 is bound more than three times more strongly for Z = Bi than for P.
AIM analysis of these complexes helps us to understand these energetic trends. The relevant molecular graphs are displayed in Fig. S1 of the ESM, where all structures are shown to contain a bond critical point between the pnicogen and the N atom of the base. The relevant characteristics of this critical point are listed in Table 4, along with any secondary critical points between the two molecules. All of the data support the stronger binding of the π-hole dimers as compared to the σ-hole dimers. For example, ρ is between 2 and 5 times larger for the former than the latter, and ∇2ρ is much larger as well. H is much more negative for the π-hole dimers than for the σ-hole dimers, again consistent with the stronger binding in the former. Less consistent are the values within each series. ρ only grows slowly with increasing Z size for the σ-hole set, while the interaction energy grows much more quickly. While the π-hole Eint is insensitive to the identity of Z, there is a clear tendency for ρ to decrease as the size of Z increases. It can also be observed that additional NH···F H-bonds are indicated by AIM, which add to the larger interaction energies for the π-hole dimers. Even though the corresponding bond path is not observed in each of the dimers investigated, the CH···F or NH···F interactions may contribute to the overall interaction energy [105, 106].
NBO evaluation of the charge transfer from orbitals of the base to those of the Lewis acid provides an alternative perspective on the nature of the bonding. The second and sixth columns of Table 5 present the total charge transferred from the N lone pair to any of the orbitals of the Lewis base for σ-hole-bonded and π-hole-bonded complexes, respectively. In other columns of the table, this total charge transferred is split into that transferred to LV orbitals of the Z atom, defined by NBO as one-center unfilled nonbonded valence orbitals of Z, and that transferred to the σ*(Z–C) antibonding orbital involving the phenyl C, respectively. Another two columns of the table refer to CT the total charge transferred, i.e., the total natural charge on all atoms of each subunit. The NBO data tend to mimic the interaction energies in Table 3 to some extent. As an illustration of this, the relationship between CT and interaction energy is presented in Fig. S2 of the ESM. The much stronger binding of the π-hole complexes is reflected in the interorbital and total charge transfer. Also reproduced is the diminishing π-hole bond strength with larger Z. With the exception of Z = Bi, the σ-hole data show a trend for increasing energy as Z gets larger.
Decomposition of the interaction energy into its constituent parts provides further insights into the nature of these dimers. The data in Table 6 illustrate that roughly 60% of the interactions in both σ-hole and π-hole complexes are based on electrostatic attraction. This percentage increases a little as the Z atom grows heavier. Orbital interactions account for a larger share of the interaction energy in the π-hole dimers (35–46%) as compared to their σ-hole analogues (only 30–32%). This distinction is consistent with the larger NBO charge transfer values. The σ-hole geometries make up the difference with a larger fraction of dispersion energy, which makes virtually no contribution to the π-hole structure. However, there is quite a distinction between the σ-hole and π-hole dimers in that the overall quantities are much larger for the latter structures. A large part of this increase is due to their much shorter intermolecular distances, as detailed in Table 2.
Monomer deformations
The above analyses considered the interactions between the two subunits after each has altered its internal geometry to that which it adopts within the context of the dimer. This atomic rearrangement requires a certain amount of energy. The deformation energies required for this transformation are reported in Table 7. These values show that it is the Lewis acid ZF2C6H5 which undergoes the major transformation, as the NH3 requires less than 1 kcal/mol. The deformation energy of the Lewis acid is also quite small for the σ-hole dimers—on the order of 1 kcal/mol or less. However, the deformation energies of the π-hole structures are dramatically different; they are all greater than 15 kcal/mol. This quantity is smallest for BiF2C6H5, but it climbs rapidly as Z shrinks, reaching over 43 kcal/mol for Z = P.
As described earlier, a major aspect of the internal rearrangement of each Lewis acid is a change in the pyramidization of the Z atom. This deformation results in an intensification of the positive region of the MEP surrounding the Z atom. The Vs,max in each properly deformed Lewis acid is displayed in the last column of Table 7, along with the increase relative to the undistorted monomer in parentheses. One can see that this increase is relatively modest for the σ-hole geometries in the upper part of the table—only about 10% or less. However, the much weaker pyramidization of the Z atom in the π-hole dimers is accompanied by substantial enhancement of Vs,max. This enhancement is as much as nearly 50% in the case of Z = P. The much more intense holes in the π-hole cases are largely responsible for their much larger interaction energies as compared to the σ-hole cases.
However, an apparent anomaly is also present, but only for π-hole-bonded complexes. The π-holes in the deformed Lewis acids become progressively more intense as the Z atom grows larger, but at the same time there is no corresponding increase in the interaction energy. In fact, the latter quantity steadily decreases as Z grows larger. The electrostatic component of the interaction energy from Table 6 also diminishes in the sequence P → Bi, in contrast to the rising pattern of Vs,max seen in Table 7. This contrary behavior is an indication that the value of the MEP of a particular point in the vicinity of a molecule does not always correlate with the actual electrostatic element of an intermolecular interaction. It should also be emphasized that this lack of correlation can be explained as the consequence of the large contribution of the orbital interaction component.
When considering the overall complexation reaction, all species (monomers and dimer alike) are typically considered to be in their optimized geometries. This reaction, or binding, energy must therefore incorporate any deformation energies of each species. The binding energies Eb obtained when the deformation energies are appropriately combined with the interaction energies are presented in Table 8. Comparison with Table 3 shows that the energetics of the σ-hole complexes barely change, since their deformation energies are small. The large deformation energies of the π-hole dimers, on the other hand, lead to dramatic changes. Eb is quite a bit less exothermic than Eint, even to the point of becoming endothermic for the smaller Z atoms. Another notable reversal is that it is the σ-hole rather than the π-hole complexes that are more stable. This preference is quite large, 14 kcal/mol for Z = P, but it then diminishes for larger Z atoms, dropping to only 1 or 2 kcal/mol for Bi. The trend in the binding energies also changes to a trend noted earlier for the interaction energies. Whereas the latter quantity is insensitive to the nature of the Z atom for the π-hole complexes, the binding energies display strongly increasing exothermicity as Z transitions from P to Bi.
Vibrational analysis
Certain features of the vibrational spectrum can shed light on the fundamental nature of molecular interactions. Selected vibrational frequencies and intensities of the monomers and their complexes are displayed in Table 9. The first two rows of this table document progressively redshifted symmetric and antisymmetric Z–F stretching frequencies as Z grows larger, along with slowly reducing intensities. With respect to the σ-hole complexes, the N···Z stretching frequency shifts to the blue as Z becomes larger, consistent with the strengthening interaction energy. The interaction becomes slightly weaker in the π-hole dimers, which is also consistent with the redshifting Z···N stretching frequency. The much higher interaction energies of the π-hole complexes are reflected in the considerably greater Z···N stretching frequencies.
In fact, there are very strong correlations between these intermolecular stretching frequencies and other aspects of the binding. The correlation with the interaction energy exhibited in Fig. 4a for both the σ-hole and π-hole complexes suggests a linear relationship. This correlation is especially good for the σ-hole structures, with R2 = 0.946. The correlation is even better with the intensities of the σ-holes and π-holes, with R2 reaching 0.999 for the σ-holes. However, it is perhaps surprising to note a negative slope for the π-holes in Fig. 4b, which links a more intense π-hole to a reduced ν(N···Z). This opposite behavior was also noted above for the relationship of Vs,max to the full electrostatic component.

Linear relationships between ν(N···Z) and a Eint and b Vs,max
The formation of σ-hole complexes is expected to result in the transfer of electron density into the σ*(Z–F1) antibonding orbital. This shift will lengthen this bond, as already noted above, and will weaken the bond and thereby shift the stretching frequency to the red. A comparison of the frequency νa(ZF2) of each monomer with ν(Z–F1) in the corresponding σ-dimer confirms this expectation, with a redshift of about 40 cm−1. This change in frequency is accompanied by band intensification, with I increasing by a factor of 1.5 for all Z except Bi. For the π-geometries, we can compare the νa(ZF2) values for the dimer and monomer. This frequency shifts heavily to the red by 126–227 cm−1, with the greatest shifts occurring for the smallest Z atoms.
Discussion and conclusions
The Z atom in ZF2C6H5 is not the only site that can attract a base such as NH3. There are positive regions of the MEP that surround the aryl H atoms as well, as may be seen in Fig. 2. Table S2 in the ESM shows that Vs,max lies in the vicinity of +20 kcal/mol for these H atoms. It is no surprise, then, that there are a number of secondary minima on the potential energy surfaces of these heterodimers that contain a CH···N H-bond. These structures are depicted in Table S3 of the ESM, along with selected geometric parameters and interaction energies. The latter are all between 2 and 3 kcal/mol, very much smaller than those of the primary σ-hole and π-hole complexes that are the focus of this work. Even the complexes which combine a CH···N H-bond with NH···F interactions in the last row of Table S3 of the ESM are still much more weakly bound than the primary structures.
In any computational elucidation of bimolecular complexes, there is always the issue of experimental confirmation. A survey of the CSD (Cambridge Structural Database) [104] provides experimental evidence of the existence of both σ- and π-types of tetracoordinate pnicogen complexes. The first two examples collected in Table S4 of the ESM [107,108,109,110] clearly indicate the ability of a pnicogen atom, in this case As, to bind a Lewis base through a σ-hole that lies directly opposite a F atom. Three examples of π-systems are also provided, including systems incorporating As and Bi.
The large deformation energies noted here for the π-hole complexes are not without precedent. A recent study of hypervalent pnicogen and other bonds [111] noted a rearrangement from a trigonal bipyramidal ZF5 monomer to a square-pyramidal complex, which was associated with a large deformation energy. This sort of deformation occurs not only in ZF5 but also in TF4 [112] (T = tetrel atom), which would not be considered hypervalent. These distortion energies in tetrel bonds can control the preferred equilibrium geometry [80] and tend to lessen as the size of the central tetrel atom increases [113], consonant with the findings here for pnicogen bonds. Similar distortions were observed [81] in both TR4(σ) and TR2=CH2(π) tetrel-bonding molecules.
In conclusion, ZF2C6H5 molecules containing a pair of F atoms and a phenyl ring surrounding a pnicogen atom Z form a fairly strong complex with an NH3 molecule. For each Z, there are two possible geometric arrangements, depending upon the positions of the two F atoms. When one F is located above the aromatic ring plane and the other below it, the base positions itself directly opposite one of the two F atoms in a σ-hole arrangement. If both F atoms lie on the same side of the ring and almost in the same plane, the NH3 is located directly above the Z atom, perpendicular to the ring plane, in a π-hole orientation. The interaction energies in the latter system are considerably larger than those in the former, on the order of 30 kcal/mol. However, at the same time, the π-hole dimers require a good deal more deformation of the ZF2C6H5 monomer, meaning that the overall dimerization process is more exothermic for the σ-hole structures. This overall preference for the σ-hole is most substantial for the smaller Z atoms, nearly disappearing for Z = Bi.
References
Bulfield D, Huber SM (2016) Halogen bonding in organic synthesis and organocatalysis. Chem Eur J 22(41):14434–14450. https://doi.org/10.1002/chem.201601844
Cordier P, Tournilhac F, Soulié-Ziakovic C, Leibler L (2008) Self-healing and thermoreversible rubber from supramolecular assembly. Nature 451:977. https://doi.org/10.1038/nature06669. https://www.nature.com/articles/nature06669#supplementary-information
Hobza P, Muller-Dethlefs K (2009) Non-covalent interactions: theory and experiment. Royal Society of Chemistry, London
Kendall K, Roberts AD (2015) van der Waals forces influencing adhesion of cells. Philos Trans R Soc Lond Ser B Biol Sci 370(1661):20140078. https://doi.org/10.1098/rstb.2014.0078
Lacour J, Moraleda D (2009) Chiral anion-mediated asymmetric ion pairing chemistry. Chem Commun 46:7073–7089. https://doi.org/10.1039/B912530B
Lehn J-M (2002) Toward self-organization and complex matter. Science 295(5564):2400. https://doi.org/10.1126/science.1071063
Lim JYC, Beer PD (2018) Sigma-hole interactions in anion recognition. Chem 4(4):731–783. https://doi.org/10.1016/j.chempr.2018.02.022
Müller-Dethlefs K, Hobza P (2000) Noncovalent interactions: a challenge for experiment and theory. Chem Rev 100(1):143–168. https://doi.org/10.1021/cr9900331
Strekowski L, Wilson B (2007) Noncovalent interactions with DNA: an overview. Mutat Res Fundam Mol Mech Mutagen 623(1):3–13. https://doi.org/10.1016/j.mrfmmm.2007.03.008
Stupp SI, LeBonheur V, Walker K, Li LS, Huggins KE, Keser M, Amstutz A (1997) Supramolecular materials: self-organized nanostructures. Science 276(5311):384. https://doi.org/10.1126/science.276.5311.384
Wagner JP, Schreiner PR (2015) London dispersion in molecular chemistry—reconsidering steric effects. Angew Chem Int Ed 54(42):12274–12296. https://doi.org/10.1002/anie.201503476
Whitesides GM, Mathias JP, Seto CT (1991) Molecular self-assembly and nanochemistry: a chemical strategy for the synthesis of nanostructures. Science 254(5036):1312. https://doi.org/10.1126/science.1962191
Zhao Y, Cotelle Y, Sakai N, Matile S (2016) Unorthodox interactions at work. J Am Chem Soc 138(13):4270–4277. https://doi.org/10.1021/jacs.5b13006
Zhou P, Huang J, Tian F (2012) Specific noncovalent interactions at protein-ligand Interface: implications for rational drug design. Curr Med Chem 19(2):226–238. https://doi.org/10.2174/092986712803414150
Grabowski SJ (2006) Hydrogen bonding—new insights, vol 3. Springer, Berlin
Desiraju GR, Steiner T (2006) The weak hydrogen bond in structural chemistry and biology. Oxford University Press, Oxford
Gilli G, Gilli P (2009) The strength of the H-bond: definitions and thermodynamics. In: The nature of the hydrogen bond: outline of a comprehensive hydrogen bond theory. Oxford University Press, Oxford
Zundel G, Sandorfy C, Schuster P (1976) The hydrogen bond: recent developments in theory and experiments, vol 2. North-Holland, Amsterdam
Gilli G, Gilli P (2009) The nature of the hydrogen bond: outline of a comprehensive hydrogen bond theory. Oxford University Press, Oxford, p 23
Scheiner S (1997) Hydrogen bonding: a theoretical perspective. Oxford University Press, New York
Clark T, Hennemann M, Murray JS, Politzer P (2007) Halogen bonding: the sigma-hole. J Mol Model 13(2):291–296. https://doi.org/10.1007/s00894-006-0130-2
Murray JS, Lane P, Clark T, Politzer P (2007) Sigma-hole bonding: molecules containing group VI atoms. J Mol Model 13(10):1033–1038. https://doi.org/10.1007/s00894-007-0225-4
Murray JS, Lane P, Politzer P (2007) A predicted new type of directional noncovalent interaction. Int J Quantum Chem 107(12):2286–2292. https://doi.org/10.1002/qua.21352
Politzer P, Lane P, Concha MC, Ma YG, Murray JS (2007) An overview of halogen bonding. J Mol Model 13(2):305–311. https://doi.org/10.1007/s00894-006-0154-7
Politzer P, Murray JS, Lane P (2007) Sigma-hole bonding and hydrogen bonding: competitive interactions. Int J Quantum Chem 107(15):3046–3052. https://doi.org/10.1002/qua.21419
Politzer P, Murray JS, Concha MC (2008) Sigma-hole bonding between like atoms; a fallacy of atomic charges. J Mol Model 14(8):659–665. https://doi.org/10.1007/s00894-008-0280-5
Murray JS, Riley KE, Politzer P, Clark T (2010) Directional weak intermolecular interactions: sigma-hole bonding. Aust J Chem 63(12):1598–1607. https://doi.org/10.1071/Ch10259
Politzer P, Murray JS (2013) Halogen bonding: an interim discussion. ChemPhysChem 14(2):278–294. https://doi.org/10.1002/cphc.201200799
Isaacs ED, Shukla A, Platzman PM, Hamann DR, Barbiellini B, Tulk CA (1999) Covalency of the hydrogen bond in ice: a direct X-ray measurement. Phys Rev Lett 82(3):600–603. https://doi.org/10.1103/PhysRevLett.82.600
Ghanty TK, Staroverov VN, Koren PR, Davidson ER (2000) Is the hydrogen bond in water dimer and ice covalent? J Am Chem Soc 122(6):1210–1214. https://doi.org/10.1021/ja9937019
Grabowski SJ (2011) What is the covalency of hydrogen bonding? Chem Rev 111(4):2597–2625. https://doi.org/10.1021/cr800346f
Bauzá A, Frontera A (2015) Aerogen bonding interaction: a new supramolecular force? Angew Chem Int Ed 54(25):7340–7343. https://doi.org/10.1002/anie.201502571
Gao M, Cheng J, Li W, Xiao B, Li Q (2016) The aerogen—π bonds involving π systems. Chem Phys Lett 651:50–55. https://doi.org/10.1016/j.cplett.2016.03.021
Guan LY, Mo YR (2014) Electron transfer in pnicogen bonds. J Phys Chem A 118(39):8911–8921. https://doi.org/10.1021/jp500775m
Politzer P, Murray JS, Clark T (2010) Halogen bonding: an electrostatically-driven highly directional noncovalent interaction. Phys Chem Chem Phys 12(28):7748–7757. https://doi.org/10.1039/c004189k
Stone AJ (2013) Are halogen bonded structures electrostatically driven? J Am Chem Soc 135(18):7005–7009. https://doi.org/10.1021/ja401420w
Esrafili MD, Mohammadian-Sabet F, Baneshi MM (2015) Cooperative and substitution effects in enhancing the strength of fluorine bonds by anion-pi interactions. Can J Chem 93(11):1169–1175. https://doi.org/10.1139/cjc-2015-0154
McDowell SAC, Joseph JA (2015) A comparative study of model halogen-bonded, pi-hole-bonded and cationic complexes involving NCX and H2O (X = F, Cl, Br). Mol Phys 113(1):16–21. https://doi.org/10.1080/00268976.2014.939116
Bauza A, Frontera A, Mooibroek TJ (2016) Pi-hole interactions involving nitro compounds: directionality of nitrate esters. Cryst Growth Des 16(9):5520–5524. https://doi.org/10.1021/acs.cgd.6b00989
Echeverria J (2017) Alkyl groups as electron density donors in pi-hole bonding. CrystEngComm 19(42):6289–6296. https://doi.org/10.1039/c7ce01259d
Zhang JR, Li WZ, Cheng JB, Liu ZB, Li QZ (2018) Cooperative effects between pi-hole triel and pi-hole chalcogen bonds. RSC Adv 8(47):26580–26588. https://doi.org/10.1039/c8ra04106g
Bauza A, Mooibroek TJ, Frontera A (2015) Directionality of pi-holes in nitro compounds. Chem Commun 51(8):1491–1493. https://doi.org/10.1039/c4cc09132a
Murray JS, Lane P, Clark T, Riley KE, Politzer P (2012) Sigma-holes, pi-holes and electrostatically-driven interactions. J Mol Model 18(2):541–548. https://doi.org/10.1007/s00894-011-1089-1
Wang H, Wang W, Jin WJ (2016) σ-Hole bond vs π-hole bond: a comparison based on halogen bond. Chem Rev 116(9):5072–5104. https://doi.org/10.1021/acs.chemrev.5b00527
Jabłoński M (2019) In search for a hydride-carbene bond. J Phys Org Chem e3949. https://doi.org/10.1002/poc.3949
Jabłoński M (2018) Hydride-triel bonds. J Comput Chem 39(19):1177–1191. https://doi.org/10.1002/jcc.25178
Bauza A, Mooibroek TJ, Frontera A (2015) The bright future of unconventional σ/π-hole interactions. ChemPhysChem 16(12):2496–2517. https://doi.org/10.1002/cphc.201500314
Gao L, Zeng Y, Zhang X, Meng L (2016) Comparative studies on group III σ-hole and π-hole interactions. J Comput Chem 37(14):1321–1327. https://doi.org/10.1002/jcc.24347
Solimannejad M, Ramezani V, Trujillo C, Alkorta I, Sanchez-Sanz G, Elguero J (2012) Competition and interplay between sigma-hole and pi-hole interactions: a computational study of 1:1 and 1:2 complexes of nitryl halides (O2NX) with ammonia. J Phys Chem A 116(21):5199–5206. https://doi.org/10.1021/jp300540z
Zhao XR, Wang H, Jin WJ (2013) The competition of C–X⋯O=P halogen bond and π-hole⋯O=P bond between halopentafluorobenzenes C6F5X (X=F, Cl, Br, I) and triethylphosphine oxide. J Mol Model 19(11):5007–5014. https://doi.org/10.1007/s00894-013-2007-5
Esrafili MD, Vakili M (2014) Halogen bonds enhanced by σ-hole and π-hole interactions: a comparative study on cooperativity and competition effects between X⋯N and S⋯N interactions in H3N⋯XCN⋯SF2 and H3N⋯XCN⋯SO2 complexes (X = F, Cl, Br and I). J Mol Model 20(6):2291. https://doi.org/10.1007/s00894-014-2291-8
Esrafili MD, Vakili M, Solimannejad M (2014) Cooperative interaction between π-hole and single-electron σ-hole interactions in O2S⋯NCX⋯CH3 and O2Se⋯NCX⋯CH3 complexes (X = F, Cl, Br and I). Mol Phys 112(16):2078–2084. https://doi.org/10.1080/00268976.2014.884730
Gao L, Zeng Y, Zhang X, Meng L (2016) Comparative studies on group III sigma-hole and pi-hole interactions. J Comput Chem 37(14):1321–1327. https://doi.org/10.1002/jcc.24347
Nziko VDN, Scheiner S (2016) Comparison of pi-hole tetrel bonding with sigma-hole halogen bonds in complexes of XCN (X = F, Cl, Br, I) and NH3. Phys Chem Chem Phys 18(5):3581–3590. https://doi.org/10.1039/c5cp07545a
Zhou PP, Yang X, Ye WC, Zhang LW, Yang F, Zhou DG, Liu SB (2016) Competition and cooperativity of sigma-hole and pi-hole intermolecular interactions between carbon monoxide and bromopentafluorobenzene. New J Chem 40(11):9139–9147. https://doi.org/10.1039/c6nj01904h
Fanfrlik J, Svec P, Ruzickova Z, Hnyk D, Ruzicka A, Hobza P (2017) The interplay between various sigma- and pi-hole interactions of trigonal boron and trigonal pyramidal arsenic triiodides. Crystals 7(7):225. https://doi.org/10.3390/cryst7070225
Saha S, Desiraju GR (2017) Sigma-hole and pi-hole synthon mimicry in third-generation crystal engineering: design of elastic crystals. Chem Eur J 23(20):4936–4943. https://doi.org/10.1002/chem.201700813
Naseer MM, Bauza A, Alnasr H, Jurkschat K, Frontera A (2018) Lone pair-pi vs. sigma-hole-pi interactions in bromine head-containing oxacalix[2]arene[2]triazines. CrystEngComm 20(23):3251–3257. https://doi.org/10.1039/c8ce00666k
Wei YX, Li QZ (2018) Comparison for sigma-hole and pi-hole tetrel-bonded complexes involving cyanoacetaldehyde. Mol Phys 116(2):222–230. https://doi.org/10.1080/00268976.2017.1377851
Politzer P, Murray JS (2018) Sigma-holes and -holes: similarities and differences. J Comput Chem 39(9):464–471. https://doi.org/10.1002/jcc.24891
Alkorta I, Elguero J, Del Bene JE (2013) Pnicogen bonded complexes of PO2X (X = F, Cl) with nitrogen bases. J Phys Chem A 117(40):10497–10503. https://doi.org/10.1021/jp407097e
Del Bene JE, Alkorta I, Elguero J (2013) Properties of complexes H2C=(X)P:PXH2, for X = F, Cl, OH, CN, NC, CCH, H, CH3, and BH2: P⋯P pnicogen bonding at sigma-holes and pi-holes. J Phys Chem A 117(45):11592–11604. https://doi.org/10.1021/jp409016q
Scheiner S (2013) Detailed comparison of the pnicogen bond with chalcogen, halogen, and hydrogen bonds. Int J Quantum Chem 113(11):1609–1620. https://doi.org/10.1002/qua.24357
Xu HY, Wang W, Zou JW (2013) Theoretical study of pnicogen bonding interactions between PH2X and five-membered heterocycles. Acta Chim Sin 71(8):1175–1182. https://doi.org/10.6023/A13030332
Xu HY, Wang W, Zou JW, Xu XL (2014) Theoretical calculations of pi-type pnicogen bonds in the triad intermolecular complexes. J Theor Comput Chem 13(8):1450068. https://doi.org/10.1142/S0219633614500680
Lo R, Svec P, Ruzickova Z, Ruzicka A, Hobza P (2016) On the nature of the stabilisation of the E⋯π pnicogen bond in the SbCl3⋯toluene complex. Chem Commun 52(17):3500–3503. https://doi.org/10.1039/c5cc10363k
Esrafili MD, Sadr-Mousavi A (2017) Modulating of the pnicogen-bonding by a H⋯π interaction: an ab initio study. J Mol Graph Model 75:165–173. https://doi.org/10.1016/j.jmgm.2017.04.017
Zhu JQ, Cao SW, Wang W, Xu XL, Xu HY (2017) The substituent effects on π-type pnicogen bond interaction. Iop C Ser Earth Env 63(1):012027. https://doi.org/10.1088/1755-1315/63/1/012027
Benz S, Poblador-Bahamonde AI, Low-Ders N, Matile S (2018) Catalysis with pnictogen, chalcogen, and halogen bonds. Angew Chem Int Ed 57(19):5408–5412. https://doi.org/10.1002/anie.201801452
Lee J, Lee LM, Arnott Z, Jenkins H, Britten JF, Vargas-Baca I (2018) Sigma-hole interactions in the molecular and crystal structures of N-boryl benzo-2,1,3-selenadiazoles. New J Chem 42(13):10555–10562. https://doi.org/10.1039/c8nj00553b
Li Y, Xu ZF (2018) Competition between tetrel bond and pnicogen bond in complexes of TX3-ZX2 and NH3. J Mol Model 24(9):247. https://doi.org/10.1007/s00894-018-3732-6
McDowell SAC, Buckingham AD (2018) A computational study of chalcogen-containing H2X...YF and (CH3)2X...YF (X = O, S, Se; Y = F, Cl, H) and pnicogen-containing H3X′...YF and (CH3)3X′...YF (X′ = N, P, As) complexes. ChemPhysChem 19(14):1756–1765. https://doi.org/10.1002/cphc.201800179
Esrafili MD, Mousavian P, Mohammadian-Sabet F (2019) Tuning of pnicogen and chalcogen bonds by an aerogen-bonding interaction: a comparative ab initio study. Mol Phys 117(1):58–66. https://doi.org/10.1080/00268976.2018.1492746
Wu JY, Yan H, Zhong AG, Chen H, Jin YX, Dai GL (2019) Theoretical and conceptual DFT study of pnicogen- and halogen-bonded complexes of PH2X⋯BrCl. J Mol Model 25(1):28. https://doi.org/10.1007/s00894-018-3905-3
Liu Y-Z, Yuan K, Yuan Z, Zhu Y-C, Zhao X (2015) Theoretical exploration of pnicogen bond noncovalent interactions in HCHO⋯PH2X (X = CH3, H, C6H5, F, Cl, Br, and NO2) complexes. J Chem Sci 127(10):1729–1738. https://doi.org/10.1007/s12039-015-0933-8
Wang YH, Zeng YL, Li XY, Meng LP, Zhang XY (2016) The mutual influence between pi-hole pnicogen bonds and sigma-hole halogen bonds in complexes of PO2Cl and XCN/C6H6 (X = F, Cl, Br). Struct Chem 27(5):1427–1437. https://doi.org/10.1007/s11224-016-0762-5
Esrafili MD, Mohammadian-Sabet F (2015) Pnicogen-pnicogen interactions in O2XP:PH2Y complexes (X = H, F, CN; Y = H, OH, OCH3, CH3, NH2). Chem Phys Lett 638:122–127. https://doi.org/10.1016/j.cplett.2015.08.045
Del Bene JE, Alkorta I, Elguero J (2015) Exploring the (H2C=PH2)(+):N-base potential surfaces: complexes stabilized by pnicogen, hydrogen, and tetrel bonds. J Phys Chem A 119(48):11701–11710. https://doi.org/10.1021/acs.jpca.5b06828
Alkorta I, Legon AC (2018) An ab initio investigation of the geometries and binding strengths of tetrel-, pnictogen-, and chalcogen-bonded complexes of CO2, N2O, and CS2 with simple Lewis bases: some generalizations. Molecules 23(9):2250. https://doi.org/10.3390/molecules23092250
Zierkiewicz W, Michalczyk M, Wysokiński R, Scheiner S (2019) Dual geometry schemes in tetrel bonds: complexes between TF4 (T = Si, Ge, Sn) and pyridine derivatives. Molecules 24(2):376
Zierkiewicz W, Michalczyk M, Scheiner S (2018) Comparison between tetrel bonded complexes stabilized by sigma and pi hole interactions. Molecules 23(6):1416. https://doi.org/10.3390/molecules23061416
Zierkiewicz W, Michalczyk M, Scheiner S (2018) Aerogen bonds formed between AeOF2 (Ae = Kr, Xe) and diazines: comparisons between sigma-hole and pi-hole complexes. Phys Chem Chem Phys 20(7):4676–4687. https://doi.org/10.1039/c7cp08048d
Dong WB, Wang Y, Cheng JB, Yang X, Li QZ (2019) Competition between sigma-hole pnicogen bond and pi-hole tetrel bond in complexes of CF2=CFZH2 (Z = P, As, and Sb). Mol Phys 117(3):251–259. https://doi.org/10.1080/00268976.2018.1508782
Moller C, Plesset MS (1934) Note on an approximation treatment for many-electron systems. Phys Rev 46(7):0618–0622. https://doi.org/10.1103/PhysRev.46.618
Dunning TH (1989) Gaussian-basis sets for use in correlated molecular calculations .1. The atoms boron through neon and hydrogen. J Chem Phys 90(2):1007–1023. https://doi.org/10.1063/1.456153
Purvis GD, Bartlett RJ (1982) A full coupled-cluster singles and doubles model: the inclusion of disconnected triples. J Chem Phys 76(4):1910–1918. https://doi.org/10.1063/1.443164
Pople JA, Head-Gordon M, Raghavachari K (1987) Quadratic configuration interaction. A general technique for determining electron correlation energies. J Chem Phys 87(10):5968–5975. https://doi.org/10.1063/1.453520
Lee CT, Yang WT, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron-density. Phys Rev B 37(2):785–789. https://doi.org/10.1103/PhysRevB.37.785
Raghavachari K, Trucks GW, Pople JA, Headgordon M (1989) A 5th-order perturbation comparison of electron correlation theories. Chem Phys Lett 157(6):479–483. https://doi.org/10.1016/S0009-2614(89)87395-6
Becke AD (1993) Density-functional thermochemistry. 3. The role of exact exchange. J Chem Phys 98(7):5648–5652. https://doi.org/10.1063/1.464913
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7(18):3297–3305. https://doi.org/10.1039/b508541a
Weigend F (2006) Accurate coulomb-fitting basis sets for H to Rn. Phys Chem Chem Phys 8(9):1057–1065. https://doi.org/10.1039/b515623h
Peterson KA (2003) Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J Chem Phys 119(21):11099–11112. https://doi.org/10.1063/1.1622923
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, vol E.01. Gaussian, Inc., Wallingford
Bulat FA, Toro-Labbe A, Brinck T, Murray JS, Politzer P (2010) Quantitative analysis of molecular surfaces: areas, volumes, electrostatic potentials and average local ionization energies. J Mol Model 16(11):1679–1691. https://doi.org/10.1007/s00894-010-0692-x
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592. https://doi.org/10.1002/jcc.22885
Bijina PV, Suresh CH (2016) Molecular electrostatic potential analysis of non-covalent complexes. J Chem Sci 128(10):1677–1686. https://doi.org/10.1007/s12039-016-1162-5
Boys SF, Bernardi F (1970) Calculation of small molecular interactions by differences of separate total energies—some procedures with reduced errors. Mol Phys 19(4):553. https://doi.org/10.1080/00268977000101561
Keith AT (2014) AIMAll (version 14.11.23). TK Gristmill Software, Overland Park
Guerra CF, Snijders JG, te Velde G, Baerends EJ (1998) Towards an order-N DFT method. Theor Chem Accounts 99(6):391–403. https://doi.org/10.1007/s002140050021
te Velde G, Bickelhaupt FM, Baerends EJ, Guerra CF, Van Gisbergen SJA, Snijders JG, Ziegler T (2001) Chemistry with ADF. J Comput Chem 22(9):931–967. https://doi.org/10.1002/jcc.1056
Scientific Computing & Modelling NV (2014) ADF2014. Vrije Universiteit, Amsterdam
Glendening ED, Landis CR, Weinhold F (2013) NBO 6.0: natural bond orbital analysis program. J Comput Chem 34(16):1429–1437. https://doi.org/10.1002/jcc.23266
Groom CR, Bruno IJ, Lightfoot MP, Ward SC (2016) The Cambridge Structural Database. Acta Crystallogr B 72:171–179. https://doi.org/10.1107/S2052520616003954
Shahbazian S (2018) Why bond critical points are not “bond” critical points. Chem Eur J 24(21):5401. https://doi.org/10.1002/chem.201705163
Jabłoński M (2018) Bond paths between distant atoms do not necessarily indicate dominant interactions. J Comput Chem 39(26):2183–2195. https://doi.org/10.1002/jcc.25532
Vrana J, Jambor R, Ruzicka A, Lycka A, De Proft F, Dostal L (2013) N→As intramolecularly coordinated organoarsenic(III) chalcogenides: isolation of terminal As-S and As-Se bonds. J Organomet Chem 723:10–14. https://doi.org/10.1016/j.jorganchem.2012.10.029
Carmalt CJ, Cowley AH, Culp RD, Jones RA, Kamepalli S, Norman NC (1997) Synthesis and structures of intramolecularly base-coordinated group 15 aryl halides. Inorg Chem 36(13):2770–2776. https://doi.org/10.1021/ic9701165
James SC, Norman NC, Orpen AG (1999) Pyridine adducts of arylbismuth(III) halides. J Chem Soc Dalton Trans 2837–2843. https://doi.org/10.1039/a900823c
Dostal L, Jambor R, Ruzicka A, Jirasko R, Holecek J, De Proft F (2011) OCO and NCO chelated derivatives of heavier group 15 elements. Study on possibility of cyclization reaction via intramolecular ether bond cleavage. Dalton Trans 40(35):8922–8934. https://doi.org/10.1039/c1dt10234f
Scheiner S, Lu J (2018) Halogen, chalcogen, and pnicogen bonding involving hypervalent atoms. Chemistry 24(32):8167–8177. https://doi.org/10.1002/chem.201800511
Zierkiewicz W, Michalczyk M, Scheiner S (2018) Implications of monomer deformation for tetrel and pnicogen bonds. Phys Chem Chem Phys 20(13):8832–8841. https://doi.org/10.1039/c8cp00430g
Scheiner S (2018) Steric crowding in tetrel bonds. J Phys Chem A 122(9):2550–2562. https://doi.org/10.1021/acs.jpca.7b12357
Acknowledgements
This work was financed in part by a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Science and Technology. The generous computer time allocated by the Wroclaw Supercomputer and Networking Center is acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts of interest to declare.
Additional information
This paper is dedicated to Zdzisław Latajka, honoring his many contributions to chemistry, on the occasion of his retirement. This paper belongs to Topical Collection Z. Latajka Festschrift.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to the Topical Collection Zdzislaw Latajka 70th Birthday Festschrift
Electronic supplementary material
ESM 1
(PDF 613 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zierkiewicz, W., Michalczyk, M., Wysokiński, R. et al. On the ability of pnicogen atoms to engage in both σ and π-hole complexes. Heterodimers of ZF2C6H5 (Z = P, As, Sb, Bi) and NH3. J Mol Model 25, 152 (2019). https://doi.org/10.1007/s00894-019-4031-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4031-6