
Abstract
A purine nucleoside phosphorylase from the alkaliphile Bacillus halodurans Alk36 was cloned and overexpressed in Escherichia coli. The enzyme was purified fivefold by membrane filtration and ion exchange. The purified enzyme had a V max of 2.03 × 10−9 s −1 and a K m of 206 μM on guanosine. The optimal pH range was between 5.7 and 8.4 with a maximum at pH 7.0. The optimal temperature for activity was 70°C and the enzyme had a half life at 60°C of 20.8 h.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
5-Methyluridine is a non-natural nucleoside that can be used as an intermediate in the synthesis of thymidine, and in the synthesis of nucleoside analogues AZT and stavudine, both of which are used in highly active antiretroviral treatment of HIV/AIDS patients. As the compound needs to be formed as a single isomer, 5-methyluridine can be synthesised through the transglycosylation of d-ribose-1-phosphate, using guanosine as a donor, and thymine as receptor (Ge et al. 2009; Medici et al. 2008; Rocchietti et al. 2004). Enzymes provide regio- and stereoselectivity, and hence are an ideal option for nucleoside transglycosylation (Prasad et al. 1999; Utagawa 1999). The hydrolysis reaction for the ribose decoupling reaction can be achieved using purine nucleoside phosphorylase (PNPase; EC 2.4.2.1) (Fig. 1).

Phosphorolysis of guanosine by purine nucleoside phosphorylase
However, the reagents guanosine and thymine are relatively insoluble, and are particulate substrates with poor reaction kinetics. The most effective method of solubilising these materials is in hot aqueous solutions. Therefore, it would be preferable to utilise thermostable enzymes to catalyse these reactions. Enzymes that can be used in this transglycosylation reaction include PNPase, thymidine phosphorylase (TPase; EC 2.4.2.4) and uridine phosphorylase (UPase; EC 2.4.2.3) (Bzowska et al. 2000; Pugmire and Ealick 2002). TPase and UPase are functionally both pyrimidine nucleoside phosphorylases (PyNPase; EC 2.4.2.2.), although UPase is closer in sequence identity to PNPase than PyNPase (Lewkowicz and Iribarren 2006). PNPases are divided into two different classes depending on their tertiary and quaternary structures (Bzowska et al. 2000). Type I PNPases tend to be bacterial in origin and, based on sequence and structural information, appear to have a hexameric structure. Type II PNPases tend to be found in eukaryotes and, based on the sequence and structural information, are trimeric in structure (Bzowska et al. 2000; Pugmire and Ealick 2002). Bzowska et al. (2000) refer to type I PNPases as high molecular mass PNPases, and the type II PNPases as low molecular mass PNPases on the basis of their quaternary structures (Bzowska et al. 2000). In general, prokaryotic PNPases are more amenable to these transglycosylation reactions as they have broader specificity than their mammalian counterparts (Tonon et al. 2004). In addition, thermophiles have been shown to harbour enzymes that exhibit a much greater thermostability. For example, the thermophile Geobacillus stearothermophilus (previously Bacillus stearothermophilus) has two purine nucleoside phosphorylases which have been characterised (Hori et al. 1989a, b; Saunders et al. 1969) and applied in the synthesis of 5-methyluridine (Hori et al. 1989c, 1991). However, although these enzymes are thermostable, they have low levels of expression in the wild type. The genes have both been subsequently successfully expressed in E. coli at high levels (Hamamoto et al. 1996, 1997a, b).
The present work investigated the nucleoside phosphorylases present in the moderately thermophilic and alkaliphilic organism Bacillus halodurans Alk36 (Louw et al. 1993; Crampton et al. 2007). This organism was chosen due to its moderately thermophilic nature. It describes the cloning, heterologous expression in E. coli, purification, and evaluation of BHPNP1, a purine nucleoside phosphorylase from B. halodurans Alk36.
Methods
Materials
All restriction enzymes and the T4-DNA ligase were purchased from Fermentas (Lithuania). The Roche high-fidelity PCR mix was used for all polymerase chain reactions. SDS-PAGE markers were purchased from Fermentas.
Bioinformatics
Owing to the high level of sequence identity between the genomes of B. halodurans Alk36 and B. halodurans C-125 the genome sequence of B. halodurans C-125 (Takami et al. 2000) (NC_002570), as published in the DNA Data Bank of Japan (http://gib.genes.nig.ac.jp), was searched for novel nucleoside phosphorylase gene sequences using the genomic BLAST (basic local alignment search tool; Altschul et al. 1990) located at the NCBI to confirm that no other PNPases or PyNPases were present apart from the annotated ones. Two PNPase and one PyNPase genes were identified. The two PNPases are BH1531 and BH1532, and the PyNPase was designated BH1533. Primers for the amplification of the PNPase gene BH1531 were designed based on the genome sequence. Isolation and characterisation of the gene corresponding to BH1531 from B. halodurans strain Alk36 was subsequently performed. This gene was termed BHPNP1.
Genomic DNA isolation
Bacillus halodurans Alk36 was grown overnight at 42°C in Luria Broth (LB) pH 8.5 (10 g/L NaCl; 10 g/L tryptone, 5 g/L yeast extract). Genomic DNA was isolated from B. halodurans according to the method of Lovett and Keggins (1979).
Cloning of the B. halodurans Alk36 PNPase gene
The PNPase gene designated BHPNP1 was amplified using the following primers: BH1531F 5′—GGACATATGCTTAACGTAACTCAATTG (NdeI site, underlined) and BH1531R 5′—GGTAAGCTTTTACATGTCTTTAACGATTGC (HindIII site, underlined). PCR was performed using the high-fidelity polymerase from Roche (Germany). The PCR amplification protocol employed was as follows: a single 10-min hold at 95°C was followed by 25 cycles of 1 min at 95°C, 1 min at 55°C and 1 min at 72°C. A final 10-min incubation at 72°C was followed by a 4°C hold. The size of the amplified product was confirmed on a 0.8% agarose gel and isolated using the Geneclean kit™ (Qbiogene, USA). The PCR product was ligated into pGEM-T Easy (Promega, USA). Restriction digests were performed with NdeI and HindIII to release the BHPNP1 insert.
The BHPNP1 gene was subsequently ligated using T4-DNA ligase (Fermentas) into the pMS470Δ8 expression vector (Balzer et al. 1992) restricted with NdeI and HindIII. This gave plasmid pMSPNP. This plasmid was transformed into E. coli JM109 (DE3) for expression analysis.
DNA sequencing
The insert was sequenced at Inqaba Biotechnology (Pretoria, South Africa) using the PCR primers described above. The sequence was compared with the known nucleotide and amino acid sequence of the BH1531 gene from B. halodurans C-125 (protein sequence—BAB05250) and has been submitted to GenBank under the accession number GQ390428.
Homology modelling
Multiple sequence alignments were performed using ClustalW (Larkin et al. 2007). Homology modelling was performed using Accelrys Discovery Studio 2.0. A trimeric model was based on the bovine structure 1LVU (Bzowska et al. 2004). A second model was based on the monomeric structure 1VFN (Koellner et al. 1997). Bovine PNPase and BHPNP1 have 49% sequence identity and 61% sequence similarity (Table 1).
Growth and induction
Recombinant E. coli strains were grown in 50 ml LB medium with 100 μg/ml ampicillin, at 37°C with shaking at 200 rpm. Cultures were induced with 0.25 mM IPTG when they had reached an OD600 between 0.05 and 0.1. Cultures were subsequently grown at 30°C with shaking at 150 rpm overnight for enzyme expression.
Batch fermentations
A 1.5 l InFors HT batch fermentor (Labfors, Switzerland) containing 1 l of GMO 20 medium was inoculated with a 50 ml inoculum (overnight culture of E. coli JM109 [pMSPNP] in LB medium). The composition of the GMO 20 medium was as follows: 14.6 g/l K2HPO4, 2 g/l (NH4)2SO4, 3.6 g/l Na2HPO4, 2.5 g/l citric acid, 1.2 g/l MgSO4, 5 g/l NH4NO3, and 20 g/l yeast extract. Glucose (17.5 g/l) and trace element solution (5 ml/l) was sterilized separately and added to the fermenters before inoculation. Ampicillin (100 μg/ml) was aseptically added to the flasks containing the glucose and trace element solution. The trace element solution consisted of the following: 0.4 g/l CaCl2.2H2O, 16.7 g/l FeCl3.6H2O, 0.15 g/l MnCl2.4H2O, 0.18 g/l ZnSO4.7H2O, 0.125 g/l CuCl2.2H2O. 0.18 g/l CoCl2.6H2O, and 20.1 g/l Na2EDTA. The pH of the fermentations was controlled at pH 7.2 with 33% m/v NH4OH or 20% m/v H2SO4. The temperature was controlled at 37°C and the aeration set to 1 v/v/min. The starting agitation was set at 300 rpm and ramped up manually to control the pO2 above 30% saturation. PNPase expression was induced at mid-log phase by adding IPTG to a final broth concentration of 0.5 mM. Fermentations were run for a further 4 h after induction.
Preparation of crude extract
After induction, the bacteria were harvested by centrifugation (15,000g, 20 min). The pellet was re-suspended in minimal sterile deionised water, and subjected to a freeze–thaw cycle alternating between +20 and −20°C. Liberated protein was separated by centrifugation (14,000g, 10 min) and stored. The pellet was re-suspended in 1 l sterile deionised water and further disrupted using a cell disruptor (2 Plus, Constant Systems, UK) with 1 pass at 40 kpsi. Cellular debris was removed by centrifugation (15,000g, 10 min). The resultant protein solution (supernatants from freeze–thaw and cell disruption processes) were concentrated and washed once with sterile deionised water by ultrafiltration using an Amicon (Millipore, USA) stirred cell ultrafiltration unit (30 kDa cut-off polyethersulfone membrane). The final preparation was lyophilised in the presence of 1% maltose and 1% PEG 8000 (Vertis Genesis 25 l). A portion of the lyophilised product, equivalent to 200 ml original fermentation broth, was re-suspended in 20 mM Tris–HCl buffer (pH 7.5) for further purification.
Column chromatography
Anion exchange chromatography of each sample was performed on an AKTA Prime (Amersham Biosciences, UK) using Toyopearl SuperQ650m anion exchange resin (Tosoh BioSep, USA). Protein was first eluted from the column using a salt gradient of between 50 and 500 mM NaCl in 20 mM Tris–HCl pH 7.2, over 400 ml at a flow rate of 4 ml/min. PNPase activity was assayed in all fractions (5 ml fractions collected) and those containing activity were separately pooled and concentrated by ultrafiltration (30 kDa membrane, Millipore USA). This sample was then re-applied to the anion exchange column and eluted over a salt gradient of 150–400 mM NaCl. Active fractions were pooled and concentrated by ultrafiltration as above.
Tertiary conformation
Denatured (5 min, 95°C) and non-denatured preparations of the enzyme were analysed on a 12% SDS-PAGE gel. The gel was overlaid with a 0.5% agarose solution to determine the position of active subunits. The agarose solution contained 10 mM inosine, 0.2 U/ml xanthine oxidase and 10 mM INT (iodonitrotetrazolium violet) in 20 mM sodium phosphate buffer, pH 7.5. Active PNPase is indicated by a red/pink band on the gel due to the cascade action of the PNPase and xanthine oxidase leading to the reduction of INT to its tetrazolium salt. Positions of active and non-active units were then confirmed by staining the gel with Coomassie brilliant blue G-250.
PNPase assay
A volume (10 μl) of suitably diluted sample was added to 190 μl of 50 mM sodium phosphate buffer containing 0.5 mM inosine and 0.2 U/ml of xanthine oxidase in UV compatible microtitre plates (Thermomix) (Erion et al. 1997). The change in absorbance at 293 nm due to the liberation of uric acid was measured on a Powerwave HT microplate spectrophotometer (Biotek, USA). One unit of PNPase is defined as the amount of enzyme required to liberate 1 μmol of uric acid from inosine, in the presence of an excess of xanthine oxidase, in 1 min. The extinction coefficient (ε) under these conditions was determined to be 7,454 cm2/mol.
Physical characteristics
A pH profile was performed using reaction mixtures (1 ml) containing 1 mM guanosine in 50 mM universal buffer (50 mM Tris, 50 mM boric acid, 33 mM citric acid 50 mM Na2PO4), adjusted to pH values between 3 and 11 with either HCl or NaOH. PNPase (0.025 U) was added to initiate the reaction. After 10 min of incubation at 40°C, the reaction was stopped by the addition of 0.5 ml of a 5 M NaOH solution. Guanosine conversion and guanine formation were analysed by HPLC on a Waters 2690 HPLC (interfaced with Waters Millennium Software) equipped with Waters 996 Photodiode Array Detector at 260 nm and a Phenomenex Synergi 4u Max-RP 80A, 150 × 4.60 mm column at 22°C. The mobile phase was 25 mM ammonium acetate (pH 4.0), at a flow rate of 1.0 ml/min.
The temperature optimum was determined with reaction mixtures (1 ml) containing 1 mM guanosine in 50 mM sodium phosphate buffer, pH 8.0. PNPase (0.025 U) was added to initiate the reaction. After 10 min of incubation at temperatures between 30°C and 90°C, the reaction was stopped by the addition of 0.5 ml of a 5 M NaOH solution. Guanosine conversion and guanine formation were analysed by HPLC as above. For temperature stability, enzyme solutions were incubated at temperatures between 40 and 70°C. Samples were removed and analysed every 30 min for the first 2 h, followed by less frequent sampling for a further 18 h.
Kinetic parameters
The kinetic parameters for PNPase were determined for both inosine (standard assay) and guanosine (assay as described for temperature optimum study) as starting substrates. Initial substrate concentrations were varied between 0.05 and 1.0 mM. The reaction was stopped at 1, 2, 3, 4, 6 and 10 min to ensure measurements remained in the linear range. Michaelis–Menten plots and the linear transformations (Lineweaver–Burk, Hanes–Woolf and Eadie–Hofstee) were used to determine kinetic parameters.
Results and discussion
Sequence analysis and homology modelling
Bacillus halodurans is unusual amongst the Bacilli that have been completely sequenced so far in that it contains two type II PNPases as opposed to the types I and II PNPases present in other Bacillus species (unpublished data). The gene sequence of BHPNP1 was identical to that of BH1531 from B. halodurans C-125 except for a silent substitution at nucleotide 519 (C–T), and hence the protein sequence was identical to that expressed by B. halodurans C-125. BLAST analysis indicated that the closest related structure deposited in the protein data base (PDB) was that of the bovine PNP (Table 1; Fig. 2) which is 47% identical to BHPNP1. On the basis of sequence identity, BH1531 is a member of the type II PNPases.

Multiple sequence alignment comparing BHPNP1 to other type II PNPases BHPNP1 was aligned with G. stearothermophilus PNP1 (P77834) (Hamamoto et al. 1997a), bovine PNP (P55859) (Bzowska et al. 1995), human PNP (P00491) (Williams et al. 1984), B. subtilis PNP (P46354) (Schuch et al. 1999), the second B. halodurans PNP (BH1532, BAB05251) (Takami et al. 2000) and E. coli XapA (NP_416902) (Dandanell et al. 2005). The alignment was generated using ClustalW (Larkin et al. 2007; http://www.ebi.ac.uk/clustalw). Residues shown to be important in the binding site of bovine and human PNPases are underlined (Mao et al. 1997, 1998; Montgomery et al. 1993; Narayana et al. 1997). Dots (. and :) indicate partial similarity and asterisks (*) indicate a 100% match
BHPNP1 has low levels of identity to type I PNPases, such as E. coli PNP and G. stearothermophilus PNP2 (17 and 18.3% identity, respectively). It has higher levels of identity to type II PNPases. Selected type II PNPases were aligned using ClustalW (Larkin et al. 2007). Amino acids known to be involved in the activity of the bovine and human enzymes (Lewkowicz and Iribarren 2006; Ealick et al. 1990) were generally conserved, with the exception of Tyr192, Ser234, Met236 and Ala237 in BHPNP1 and Cys115, Tyr193, Met194, Ile210, Asp236, Met237 and Ala238 in BH1532. These were all conservative substitutions with the exception of Met236 (BHPNP1), Cys115, Met194 and Met237 (BH1532). The position and orientation of these residues are shown in Fig. 3. Hence, it is likely that the active site, and overall tertiary structure, of BHPNP1 should resemble the bovine and human PNPases and is, therefore, likely to be a homotrimer. The structure of BHPNP1 was, hence, modelled based on the structure of the bovine PNPase (Fig. 3). This structure shows the modelled monomer, including a putative substrate, namely hypoxanthine. Residues that are known to be important in structure and function of the mammalian proteins and are conserved in BHPNP1 are indicated in yellow. Residues that are known to be important in structure and function of the mammalian proteins and are not conserved in BHPNP1 are indicated in red.

Homology modelled three-dimensional structure of BHPNP1. Homology modelling of the BHPNP1 structure was performed using the bovine structure 1VFN (Koellner et al. 1997) as a template. The monomer was modelled along with the substrate, hypoxanthine
Tertiary conformation
BHPNP1 and G. stearothermophilus PNP1 are 74.9% identical. These proteins are likely to have similar structural characteristics. As already mentioned by Hamamoto et al. (1997b), the high level of sequence identity between the G. stearothermophilus PNP1 and the eukaryotic PNPases strongly suggest structural similarity. This would also apply to the BHPNP1 protein. However, gel filtration analysis of the G. stearothermophilus PNP1 gave an apparent molecular weight of 68,000, potentially indicating a dimeric protein (Hori et al. 1989b).
To confirm whether BHPNP1 is a trimer, which the sequence data suggests, or a dimer based on information about G. stearothermophilus PNP1 an overlay experiment was performed. The stained gel and overlay are shown in Fig. 4. The experiment indictated that the predominant tertiary confirmation was a dimer. The expected monomeric subunit was visible in both the heat-treated and non-heat-treated samples at approximately 27 kDa. Another dominant band at approximately 50 kDa on the Coomassie stained gel (lane 2, Fig. 4a) was related to the active band on the overlay (lane 2, Fig. 4b). Hence, in contrast to the mammalian system, this enzyme appears to be dimeric.

12% SDS PAGE gel (a) and corresponding activity gel overlay (b). M marker, lane 1 heat-treated PNPase preparation (95°C, 5 min); lane 2 non-heat-treated PNPase preparation. Arrows indicate the position of the monomer and the dimer
Enzyme expression and purification
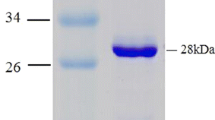
The productivity of B. halodurans Alk36 BHPNP1 heterologously expressed in E. coli JM109 (DE3) was 700 U/l/h in shake flasks, but increased to 3,007 U/l/h under the controlled fermentation conditions. PNPase was purified to 42% purity (by density analysis in SDS-PAGE, Fig. 5) and a specific activity of 30.2 U/mg total protein with a fold purification of 5.0 from the culture broth (Table 2).

Denaturing SDS-PAGE gel (12%) showing successive purification steps. Various fractions from a purification of BHPNP were resolved using 12% SDS-PAGE. Lane 1 protein marker sizes in kilodaltons, lane 2 crude extract, lane 3 concentrated sample after anion exchange column 1, lane 4 final sample after anion exchange column 2
Physical characteristics
PNPase showed a pH optimum of 7.0, retaining 60% activity between pH 5.7 and 7.4 (Fig. 6). PNPase had optimum activity at 70°C and a broad activity range, retaining 60% activity between 30 and 74°C (Fig. 7). Although the optimum temperature of PNPase was shown to be 70°C, only 7.2% activity remained after 30 min incubation at this temperature (t 1/2 − 15.2 min). PNPase did, however, show good stability at 60°C (t 1/2 − 20.8 h) and excellent stability at 40°C, with no change in activity over the time period (19 h).

pH optimum profiles of BHPNP1. A pH profile was performed using reaction mixtures (1 ml) containing 1 mM guanosine in 50 mM universal buffer (50 mM Tris, 50 mM boric acid, 33 mM citric acid; 50 mM Na2PO4), adjusted to pH values between 3 and 11 with either HCl or NaOH. PNPase (0.025 U) was added to initiate the reaction. After 10 min of incubation at 40°C, the reaction was stopped by the addition of 0.5 ml of a 5 M NaOH solution. Guanosine conversion and guanine formation were analysed by HPLC at 260 nm. The mobile phase was 25 mM ammonium acetate (pH 4.0), at a flow rate of 1.0 ml/min

Temperature optimum profile of BHPNP1. The temperature optimum of BHPNP1 was determined with reaction mixtures containing 1 mM guanosine in 50 mM sodium phosphate buffer, pH 8.0. PNPase (0.025 U) was added to initiate the reaction. After 10 min of incubation at temperatures between 30 and 90°C, the reaction was stopped by the addition of 0.5 ml of a 5 M NaOH solution. Guanosine conversion and guanine formation were analysed by HPLC
Kinetic characterisation
Linear transformation of velocity data obtained for varying initial substrate concentrations showed good linear regression fit where inosine was used (R 2 > 99% for all plots) and adequate fit for guanosine experiments (R 2 > 94%). From the plots (Lineweaver–Burk, Eadie–Hofstee and Hanes–Woolf), K m and V max were determined with <7% deviation in the values calculated from the three plots. Subsequently, the turnover number (k cat ) and the specificity constant were calculated. These values are summarised in Table 3.
The PNP1 from the thermophile G. stearothermophilus has been previously expressed and characterised (Hori et al. 1989a; Hamamoto et al. 1997a). The G. stearothermophilus enzyme has a pI of 4.7, with an optimal pH range between 7.5 and 11, in contrast to the optimal pH range of BHPNP1 of between 5.7 and 8.4 and a predicted pI of 5.1. The G. stearothermophilus PNPase is more thermostable then BHPNP1 as it is stable at 70°C for greater then 30 h. In contrast, the half life of PNPase BHPNP1 at 60°C is 20 h. G. stearothermophilus PNPase K m for inosine was similar at 0.22 mM, but had a greater affinity for guanosine at K m of 0.14 mM.
Conclusions
The thermostable type II nucleoside phosphorylase from the bacterium B. halodurans Alk36 was expressed heterologously in E. coli, purified, and functionally characterised. The enzyme was capable of phosphorolysis of guanosine to yield guanine and ribose-1-phosphate, the latter of which may be used in the enzymatic glycosylation of nucleosides.
Abbreviations
- BHPNP1:
-
Bacillus halodurans purine nucleoside phosphorylase 1
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Balzer D, Ziegelin G, Pansegrau W, Kruft V, Lanka E (1992) KorB protein of promiscuous plasmid RP4 recognizes inverted sequence repetitions in regions essential for conjugative plasmid transfer. Nucl Acids Res 20:1851–1858
Bzowska A, Luić M, Schröder W, Shugar D, Saenger W, Koellner G (1995) Calf spleen purine nucleoside phosphorylase: purification, sequence and crystal structure of its complex with an N(7)-acycloguanosine inhibitor. FEBS Lett 367:214–218
Bzowska A, Kulikowska E, Shugar D (2000) Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacol Therapeut 88:349–425
Bzowska A, Koellner G, Wielgus-Kutrowska B, Stroh A, Raszewski G, Holý A, Steiner T, Frank J (2004) Crystal structure of calf spleen purine nucleoside phosphorylase with two full trimers in the asymmetric unit: important implications for the mechanism of catalysis. J Mol Biol 342:1015–1032
Crampton M, Berger E, Reid S, Louw M (2007) The development of a flagellin surface display expression system in a moderate thermophile, Bacillus halodurans Alk36. Appl Microbiol Biotechnol 75:599–607
Dandanell G, Szczepanowski RH, Kierdaszuk B, Shugar D, Bochtler M (2005) Escherichia coli purine nucleoside phosphorylase II, the product of the xapA gene. J Mol Biol 348:113–125
Ealick SE, Rule SA, Carter DC, Greenhough TJ, Babu YS, Cook WJ, Habash J, Helliwell JR, Stoeckler JD, Parks RE Jr (1990) Three-dimensional structure of human erythrocytic purine nucleoside phosphorylase at 3.2 A resolution. J Biol Chem 265:1812–1820
Erion MD, Takabayashi K, Smith HB, Kessi J, Wagner S, Hönger S, Shames SL, Ealick SE (1997) Purine nucleoside phosphorylase. 1. Structure–function studies. Biochemistry 36:11725–11734
Ge C, Ouyang L, Ding Q, Ou L (2009) Co-expression of recombinant nucleoside phosphorylase from Escherichia coli and its application. Appl Biochem Biotechnol 159:168–177
Hamamoto T, Noguchi T, Midorikawa Y (1996) Purification and characterization of purine nucleoside phosphorylase and pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus TH 6-2. Biosci Biotechnol Biochem 60:1179–1180
Hamamoto T, Okuyama K, Noguchi T, Midorikawa Y (1997a) Cloning and expression of purine nucleoside phosphorylase I gene from Bacillus stearothermophilus TH 6-2. Biosci Biotechnol Biochem 61:272–275
Hamamoto T, Noguchi T, Midorikawa Y (1997b) Cloning of purine nucleoside phosphorylase II gene from Bacillus stearothermophilus TH 6-2 and characterization of its gene product. Biosci Biotechnol Biochem 61:276–280
Hori N, Watanabe M, Yamazaki Y, Mikami Y (1989a) Purification and characterisation of thermostable purine nucleoside phosphorylase of Bacillus stearothermophilus JTS 859. Agric Biol Chem 53:2205–2210
Hori N, Watanabe M, Yamazaki Y, Mikami Y (1989b) Purification and characterization of second thermostable purine nucleoside phosphorylase in Bacillus stearothermophilus JTS 859. Agric Biol Chem 53:3219–3224
Hori N, Watanabe M, Yamazaki Y, Mikami Y (1989c) Synthesis of 5-methyluridine by a thermophile, Bacillus stearothermophilus JTS 859. Agric Biol Chem 53:197–202
Hori N, Watanabe M, Sunagawa K, Uehara K, Mikami Y (1991) Production of 5-methyluridine by immobilized thermostable purine nucleoside phosphorylase and pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus JTS 859. J Biotechnol 17:121–131
Koellner G, Luić M, Shugar D, Saenger W, Bzowska A (1997) Crystal structure of calf spleen purine nucleoside phosphorylase in a complex with hypoxanthine at 2.15 Å resolution. J Mol Biol 265:202–216
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lewkowicz E, Iribarren A (2006) Nucleoside phosphorylases. Curr Org Chem 10:1197–1215
Louw ME, Reid SJ, Watson TG (1993) Characterization, cloning and sequencing of a thermostable endo-(1, 3-1, 4) beta-glucanase-encoding gene from an alkalophilic Bacillus brevis. Appl Microbiol Biotechnol 38:507–513
Lovett PS, Keggins KM (1979) Bacillus subtilis as a host for molecular cloning. Method Enzymol 68:342–357
Mao C, Cook WJ, Zhou M, Koszalka GW, Krenitsky TA, Ealick SE (1997) The crystal structure of Escherichia coli purine nucleoside phosphorylase: a comparison with the human enzyme reveals a conserved topology. Structure 5:1373–1383
Mao C, Cook WJ, Zhou M, Federov AA, Almo SC, Ealick SE (1998) Calf spleen purine nucleoside phosphorylase complexed with substrates and substrate analogues. Biochemistry 37:7135–7146
Medici R, Porro MT, Lewkowicz E, Montserrat J, Iribarren AM (2008) Coupled biocatalysts applied to the synthesis of nucleosides. Nucleic Acids Symp Ser 2008:541–542
Montgomery JA, Niwas S, Rose JD, Secrist JA, Babu YS, Bugg CE, Erion MD, Guida WC, Ealick SE (1993) Structure-based design of inhibitors of purine nucleoside phosphorylase. 1. 9-(arylmethyl) derivatives of 9-deazaguanine. J Med Chem 36:55–69
Narayana SV, Bugg CE, Ealick SE (1997) Refined structure of purine nucleoside phosphorylase at 2.75 Å resolution. Acta Crystallogr D 53:131–142
Prasad A, Trikha S, Parmar V (1999) Nucleoside synthesis mediated by glycosyl transferring enzymes. Bioorg Chem 27:135–154
Pugmire MJ, Ealick SE (2002) Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem J 361:1–25
Rocchietti S, Ubiali D, Terreni M, Albertini AM, Fernández-Lafuente R, Guisán JM, Pregnolato M (2004) Immobilization and stabilization of recombinant multimeric uridine and purine nucleoside phosphorylases from Bacillus subtilis. Biomacromolecules 5:2195–2200
Saunders PP, Wilson BA, Saunders GF (1969) Purification and comparative properties of a pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus. J Biol Chem 244:3691–3697
Schuch R, Garibian A, Saxild HH, Piggot PJ, Nygaard P (1999) Nucleosides as a carbon source in Bacillus subtilis: characterization of the drm-pupG operon. Microbiology 145:2957–2966
Takami H, Nakasone K, Takaki Y, Maeno G, Sasaki R, Masui N, Fuji F, Hirama C, Nakamura Y, Ogasawara N, Kuhara S, Horikoshi K (2000) Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res 28:4317–4331
Tonon G, Capra E, Orsini G, Zuffi G (2004) Novel immobilized biocatalysts usable for the production of natural nucleosides and modified analogues by enzymatic transglycosylation reactions. US Patent Number 20040142438
Utagawa T (1999) Enzymatic preparation of nucleoside antibiotics. J Mol Catal B Enzym 6:215–222
Williams SR, Goddard JM, Martin DW (1984) Human purine nucleoside phosphorylase cDNA sequence and genomic clone characterization. Nucleic Acids Res 12:5779–5787
Acknowledgments
We would like to thank Dr D. R. Walwyn (CEO of Arvir Technologies) for support for this project, as well as Zimkhitha Sotenjwa, Dr Anu Idicula and Dr Neeresh Rohitlall who helped lay the foundations for this research. We gratefully acknowledge the financial support, and resources from CSIR Biosciences, LIFElab and Arvir Technologies in funding this project, without which this work would not have been possible.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Visser, D.F., Hennessy, F., Rashamuse, K. et al. Cloning, purification and characterisation of a recombinant purine nucleoside phosphorylase from Bacillus halodurans Alk36. Extremophiles 14, 185–192 (2010). https://doi.org/10.1007/s00792-009-0297-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-009-0297-4