
Abstract
Quantitative nuclear magnetic resonance (qNMR) in combination with metrological weighing is optimised to demonstrate the power of the qNMR measurement method. It is shown that with 1H-qNMR it is possible to certify the purity of organic reference materials (expressed as mass fraction) with relative expanded uncertainties of <0.1 % for a 95 % confidence interval (k = 2). Following well-defined selection criteria, a set of twelve different chemical compounds is evaluated and certified to serve as internal references for 1H-qNMR measurements. A series of comparison measurements is made amongst a subset of the selected compounds. The purity of maleic acid is determined by six different 1H-qNMR measurement series, and all results show full consistency. All the six mean values are covered within the range of ±0.05 %. In two more measurement series, four different nuclei are analysed within the same sample against one calibrator. Even with non-optimised signal intensity ratios and varying signal pattern, a high consistency was obtained. Therefore, the validity and robustness of 1H-qNMR measurement results are demonstrated. 1H-qNMR measurement results are directly traceable to a variety of internationally accepted primary reference materials, and therefore, traceability to SI units is obtained. All experiments are performed under ISO/IEC 17025 and ISO Guide 34 accreditation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since first described in 1946 [1, 2], nuclear magnetic resonance (NMR) spectroscopy has become an essential analytical technique for chemical structure elucidation as well as purity and impurity control. Nowadays, it is used in a variety of different applications in industry as well as academic research. Over the last two decades, the importance of quantification using NMR, especially by 1H-NMR, has been significantly increased [3–6]. In 1998 and 2005, Holzgrabe et al. [7] published reviews which cover different applications of NMR spectroscopy in pharmacy, in particular the application of quantitative NMR (qNMR) [8]. Furthermore several international pharmacopoeias describe as well methods which are used to determine the impurity profile of drugs by qNMR.
Quantitative NMR has several outstanding advantages compared to other analytical techniques with regard to purity determination of organic CRMs. One of the most important pros is the insensitivity against impurities. As long as only one sample signal is pure, that is, not affected by impurities, the measurement result is not affected by any impurities in the sample. This also includes non-detectable impurities such as salts and residual water. Jancke et al. proposed NMR spectroscopy as a relative primary analytical method because it can be described completely by mathematical equations from which a full uncertainty budget may be derived, and so it can be employed at the highest metrological level. He also clarified that 1H-NMR spectroscopy is appropriate for quantitative analysis because of the high sensitivity of the proton nuclei combined with relative short relaxation times and nearly 100 % natural abundance. The intensity of the NMR signal is directly proportional to the number of protons that are giving rise to the signal [9]. So, the quantification is done by measuring the sample peak area of interest with respect to a signal which comes from an arbitrary internal standard, that is, an internationally accepted reference material. In case, a primary reference material is used this leads to traceability to an SI unit. As a consequence for NMR quantification, it is not necessary to have a reference standard available which is of the same chemical structure as the sample. For many organic samples, a direct traceability to a small set of internationally accepted reference standards can be achieved [10, 11].
In 2005, Malz and Jancke [12] presented an approach for the validation of qNMR. They developed a protocol for the application of qNMR experiments which has been tested furthermore in a round robin test. Following this protocol, they obtained an expanded measurement uncertainty of 1.5 % with a coverage factor of k = 2 [13, 14]. In 2009, Ihara et al. from the National Metrology Institute of Japan (AIST) published their results for the certification of pesticides by 1H-qNMR and their optimised 1H-qNMR methods lead to expanded measurement uncertainty values between 0.3 % and 1.2 % [15].
Experimental
Chemicals
Potassium hydrogen phthalate and benzoic acid were used from NIST. These materials are named NIST-KHP (SRM 84k, acidimetric standards: 99.9911 % ± 0.0054 % and SRM 84l, acidimetric standard: 99.9934 % ± 0.0076 %), NIST-BA1 (SRM 350b, acidimetric standard: 99.9978 % ± 0.0044 %), and NIST-BA2 (SRM 39j, calorimetric standard with indicated purity of (0.999996 ± 0.000072) mol/mol) in this article. Other certified reference materials were used from Sigma-Aldrich: maleic acid, MA (Fluka no.: 92816), 3,5-dinitrobenzoic acid, DNB (Fluka no.: 15639), benzoic acid, BA (Fluka no.: 06185), dimethyl sulfone, DMSO2 (Fluka no.: 41867), ethyl 4-(dimethylamino)-benzoate, EDAB (Fluka no.: 42582), and caffeine (Fluka no.: 56396). All deuterated solvents were purchased from Sigma-Aldrich: D2O (Aldrich no.: 151882, 99.9 atom% D), NaOD (Aldrich no.: 372072; 0.40 g/g in D2O, 99.5 atom% D), DMSO-D6 (Aldrich no.: 151874, 99.9 atom% D), CDCl3 (Aldrich no.: 151823, 99.8 atom% D) and Acetic acid-D4 (Aldrich no.: 151785, 99.5 atom% D).
Weighing and sample preparation
All weighing steps were performed on a Mettler Toledo UMT 5 ultra-microbalance. Air buoyancy correction was considered for the final mass determination. The ratio of the masses was calculated to ensure approximately 1:1 ratios for the signals of the calibrant and the sample. In most cases, between 20 and 100 mg of substance was weighed. In all experiments, ten different samples were prepared by accurately weighing internal standard and sample together into a HPLC vial. After adding the suitable deuterated solvent, the samples were thoroughly sonicated to completely dissolve both components and then the solution was transferred to a 5-mm NMR tube.
Pretests
A series of pretests were carried out prior to any qNMR quantification experiments. First, the chemical compatibility between sample and internal standard has been checked by acquiring a proton NMR spectrum of the mixture right after preparation and again after 24 h. Several tests are applied to ensure that no impurity lies underneath the peaks of interest. This is done with chromatographic methods in combination with 2D NMR experiments where impurities of <0.05 % signal intensity portion can be detected. The T 1 relaxation time was evaluated with an inversion recovery experiment, since the relaxation time can vary depending on the mixture and the solvent. Checking the hygroscopy or volatility of the candidate substance is also very important. Only non-volatile and non-hygroscopic sample candidates were chosen to obtain best weighing results. A sample was defined to be non-volatile or non-hygroscopic when no change in weighing value of greater than 0.02 mg was obtained over a time period of 10 min.
NMR experiments
All steps leading to a content determination by the use of qNMR and generation of certified reference materials were performed under ISO-accredited procedures (ISO/IEC 17025 and ISO Guide 34). This includes stability tests at different temperatures and homogeneity studies, both an ISO Guide 34 requirement.
All NMR experiments were carried out on a Bruker AVANCE III 600 MHz spectrometer operating at 600.2 MHz, equipped with a BBO (broadband observe) probe head with z gradient. Temperature stability was controlled by a BVT 3200 unit (Bruker variable temperature) at 298.2 K.
Quantitative NMR experiments were carried out with ten different samples for each qNMR series. A number of 16 transients with 65,536 data points each were collected to ensure a signal to noise ratio of >300 for the relevant peaks, with a standard single pulse experiment without decoupling. All experiments were carried out under non-spinning conditions with regard to the high magnetic field and to avoid spinning side bands. To receive fully relaxed NMR spectra with maximum signal intensity, a 90° pulse was applied. Based on previous T 1 inversion recovery experiments, the T 1 relaxation delay was checked for each mixture and set accordingly. In most cases, a relaxation delay of 60 s was chosen in view of 5–7 times T 1. Prior to Fourier transformation, a window function was applied, and the spectra were processed with a line broadening of 0.1 Hz, zero filling was done once. After carefully manual phasing and automatic baseline correction, the integration of the signals was done manually.
The integration of the calibrant signal and the sample signal is always done in the very same way, that is, both signals integrated with or without 13C satellites. The transmitter frequency offset (O 1) is always set in a way so that neither the calibrant signal nor the sample signal is affected.
Calculation of CRM content
The purity of a sample is expressed as a mass fraction and is calculated according to Eq. (1).
- b S :
-
Air buoyancy correction factor for the sample
- b Ref :
-
Air buoyancy correction factor for the reference
- I S :
-
Integral area of the sample signal
- I Ref :
-
Integral area of the reference signal
- M S :
-
Molecular mass of the sample, g/mol
- M Ref :
-
Molecular mass of the reference, g/mol
- N S :
-
Number of protons generating the sample signal
- N Ref :
-
Number of protons generating the reference signal
- P S :
-
Purity of the sample as mass fraction, g/g
- P Ref :
-
Purity of the reference as mass fraction, g/g
- W S :
-
Weighing value of the sample, g
- W Ref :
-
Weighing value of the reference, g
Results and discussion
Metrological traceability scheme for qNMR
According to De Bièvre et al. [16], a metrological traceability scheme as shown in Fig. 1 can be drawn. An internationally accepted primary reference material serves as the primary calibrator and ensures SI traceability. A set of well-selected substances serves as 1H-qNMR references whereby the certified mass fraction content can be assigned to each of these calibrators. This set of 1H-qNMR calibrators is then used to assign certified values to a wide range of organic substances (samples), that is, pesticides, pollutants, drugs, metabolites, and many others. Sometimes the sample purity (expressed as a mass fraction) can also be assigned by direct measurement against a primary calibrator. In these cases, no (secondary) calibrator is used. In Fig. 1, an example is shown where caffeine as the sample is certified using 1H-qNMR with respect to maleic acid as the (secondary) calibrator, and maleic acid is certified by 1H-qNMR against the primary calibrator KHP from NIST. Therefore, the traceability to the SI unit ampere is obtained by coulometry as the primary measurement technique. The assigned relative expanded standard uncertainties for (secondary) calibrators—in our case the set of twelve 1H-qNMR references—are in the range of 0.08–0.17 %. These secondary calibrators are then used as a reference for the certification of a variety of organic samples. Due to uncertainty propagation, this leads to typical values of the relative expanded standard uncertainty in the range of 0.15–0.3 % for certified samples.

Example of a traceability chain of an organic CRM: caffeine is certified by qNMR through comparison to maleic acid (secondary calibrator) and KHP (primary calibrator) and finally to the SI unit ampere
Calculation of the uncertainty budget
All uncertainty calculations are based on well-established guidelines [13, 14] and are visualised in Fig. 2. For the purity determination, the combined standard uncertainty u c(P s) can be calculated by Eq. (2):

Simplified cause-effect diagram of the uncertainty contributions for a qNMR measurement

The combined relative standard uncertainty u c(P S) is determined by statistical (type A) as well as non-statistical (type B) contributions whereby the statistical contribution u(I Rep) arise from the repeatability of weighing and signal integration.
On the other hand, there are various systematic contributions, for example, the air buoyancy correction, balance parameters, molecular masses, and the purity of the reference (expressed as a mass fraction).
For all standard reference materials from NIST, the uncertainty contributions were increased in cases when the samples were smaller than the recommended minimum sample size. Mass determination uncertainty (weighing and air buoyancy correction) is calculated according to common literature. Details are described in [17], and no further details are given in this article. The cutting (integration) of the signals is done manually, and small differences have been observed depending on the operator. This individual or person-to-person influence is considered in the overall budget as “Individual integration contribution” I Ind. The uncertainty of I Ind is calculated based on a series of experiments where different operators analysed various sets of analysis data at different points in time. As indicated in Fig. 3, this contribution is the major contribution to the overall uncertainty budget, and it contributes with roughly 0.03 % to the standard uncertainty budget. The repeatability of the ten individual replicate measurements (ten different sample preparations by the same operator) represents another significant uncertainty contribution. It is noteworthy that no reduction by square root of ten (number of replicate measurements) has been applied in any case, that is, the replicate measurements have not been supposed to be independent. This approach was chosen to ensure a conservative uncertainty budget evaluation.

Overview on typical contributions to the relative standard uncertainty (absolute contributions given as single standard deviations) for the quantification of maleic acid using KHP from NIST (SRM 84k) as the primary calibrator
Suitable substances as qNMR reference materials
As basis for the 1H-qNMR-certification of organic substances, a set of selected references is needed to serve as calibrators. These substances must fulfil a series of criteria to be considered as an adequate 1H-qNMR reference candidate:
-
availability in very high purity
-
non-hygroscopic and non-volatile
-
low chemical reactivity and toxicity
-
simple signal pattern (few signals only)
-
chemical shift of signals covering different areas
-
low ratio of isochronic protons to molecular mass
-
solubility in multiple deuterated solvents
Based on these criteria, only few substances are suitable to serve as a good 1H-qNMR reference. Following the procedures described in this article, twelve substances were selected and certified by 1H-qNMR for purity under double accreditation ISO/IEC 17025 and ISO Guide 34 (Fig. 4) [18]. All these CRMs are certified with relative expanded standard uncertainty values between 0.08 % and 0.17 %. With this set of 1H-qNMR reference standards, a wide range of different chemical shifts and also different solubilities are covered.

Set of 1H-NMR spectra of twelve CRM to serve as qNMR standards covering a wide range of chemical shifts and different solubilities. They are all chemically highly stable, non-hygroscopic, and non-volatile
Validation of qNMR measurement results
With regard to the obtained very low expanded measurement uncertainties of <0.1 % relative, the consistency of the data was extensively proofed by a series of cross-checking experiments. The experimental design of this study is shown in Fig. 5.

Validation of qNMR measurement results for the certification of maleic acid purity through six different traceability chains. Upper values refer to the assigned purity expressed as mass fraction in %, lower values refer to the corresponding expanded absolute uncertainty of the mass fraction value in % (k = 2). The six different values assigning a purity value for MA are given in bold. The comparability of these six measurement results is further illustrated in Fig. 6. Full chemical names of the used abbreviations are given in the “Experimental” section
First, maleic acid (MA) was chosen as a (secondary) calibrator and its purity (expressed as a mass fraction) was quantified by 1H-qNMR with respect to the three different (primary) reference materials NIST-BA1, NIST-BA2, and NIST-KHP. The corresponding experiments are described below as Exp.A to Exp.C. Three independent different values for the mass fraction of MA were obtained, whereby “value” always describes the assigned purity expressed as mass fraction including its expanded uncertainty at the 95 % confidence level (k = 2):
-
Exp.A: A value was assigned to MA using NIST-BA1 as primary calibrator (w MA,BA1 = 99.751 %, U(w MA) = 0.085 %)
-
Exp.B: A value was assigned to MA using NIST-BA2 as primary calibrator (w MA,BA2 = 99.806 %, U(w MA) = 0.079 %)
-
Exp.C: A value was assigned to MA using NIST-KHP as primary calibrator (w MA,KHP = 99.818 %, U(w MA) = 0.088 %)
Based on the assigned mass fraction values of MA through these three different measurement series, the MA was then taken as the calibrator for the following measurement series Exp.D to Exp.F:
-
Exp.D: A value was assigned to DNB based on MA as calibrator (w DNB,MA = 99.420 %, U(w DNB,MA) = 0.109 %). Then, DNB was taken as the calibrator, and a value was assigned to BA (w BA,DNB = 99.958 %, U(w BA,DNB) = 0.131 %). Finally, BA was taken as the calibrator, and a value was assigned to MA (w MA,BA = 99.732 %, U(w MA,BA) = 0.155 %).
-
Exp.E: DNB was taken as the calibrator, and a value was assigned to DMSO2 (\( w_{{{\text{DMSO}}_{ 2} ,{\text{DNB}}}} \) = 99.642 %, \( U(w_{{{\text{DMSO}}_{ 2} ,{\text{DNB}}}} ) \) = 0.123 %). Then, DMSO2 was taken as the calibrator to assign a value for MA (\( w_{{{\text{MA}},{\text{DMSO}}_{ 2} }} \) = 99.733 %, \( U(w_{{{\text{MA}},{\text{DMSO}}_{ 2} }} ) \) = 0.146 %).
-
Exp.F: BA was taken as the calibrator to assign a value to DMSO2 (\( w_{{{\text{DMSO}}_{ 2} ,{\text{BA}}}} \) = 99.643 %, \( U(w_{{{\text{DMSO}}_{ 2} ,{\text{BA}}}} ) \) = 0.151 %). Then, DMSO2 was taken as the calibrator to assign a value for MA (\( w_{{{\text{MA}},{\text{DMSO}}_{ 2} ,{\text{BA}}}} \) = 99.734 %, \( U(w_{{{\text{MA}},{\text{DMSO}}_{ 2} ,{\text{BA}}}} ) \) = 0.165 %).
Six different mass fraction values for the same MA were observed in this study, whereby three of the results came from a direct comparison measurement to a primary calibrator (Exp.A to Exp.C), two of the results were generated by a three-step comparison measurement (Exp.D and Exp.E), and one result was observed from a four-step comparison measurement (Exp.F). All the six results show full consistency in terms of overlapping of the uncertainty budgets with all the other mean values (Fig. 6). The relative standard deviation of the six mean values from Exp.A to Exp.F is 0.04 %, and all the six measurement results are covered within a range of ±0.05 %. Of course, the measurement uncertainties of experiments A to F are different, whereby the direct measurements (Exp.A to Exp.C) show the lowest uncertainties (all below 0.09 % relative), and due to uncertainty propagation, the uncertainty values are higher for Exp.D to Exp.F, whereby Exp.F comprises four-comparison measurements and therefore shows the highest uncertainty. Nevertheless, Exp.F shows the enormous potential of the 1H-qNMR method. The content of MA was confirmed within an expanded relative standard uncertainty of <0.2 % although this value was generated over a series of four comparison measurements using three intermediate references.

Comparison of six different purity results (expressed as mass fraction, %) of maleic acid. These data refer to the experiments described in Fig. 5 for measuring the purity of MA by different traceability chains
A second study was performed similar to the one presented in this article whereby this time DMSO2 was defined as the secondary calibrator instead of MA. A mass fraction of DMSO2 was assigned against the primary calibrator NIST-BA1. Then, MA, BA, and DNB were taken as the cross-checking samples, and following the above-described design of experiment, four different mass fraction values for DMSO2 were assigned through the different traceability chains. Again, all the four results did show full consistency in terms of overlapping of all uncertainties with all mean values.
Influence of signal intensity and pattern
As mentioned in this article, all experiments were set-up so that the integrated signals of calibrant and sample were optimised to a 1:1 ratio. Nevertheless, for most organic samples, more than one signal can be selected for the purity determination. Also in terms of the signal pattern in most cases, there are multiple choices, that is, singlet signals versus multiplets. In the following two experiments, it was evaluated if these two effects are of significant relevance or not. To do so, EDAB was chosen as a sample with five different signals with different signal intensity and pattern.
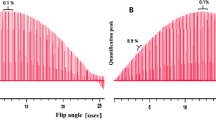
Two different measurement series were applied, both with MA as the internal calibrator: In the first experiment (Exp.1), the mass ratio of sample and calibrator was optimised to obtain a 1:1 peak ratio of MA and the six methyl protons of the amino group of EDAB. In the second experiment (Exp.2), the mass ratio of sample and calibrator was optimised for a 1:1 peak ratio of MA and the ethyl group of EDAB (2 isochronic nuclei). In both experiments, all EDAB signals were then analysed against the MA calibrant reference signal. One signal generated by one pair of acrylic CH was not analysed due to overlapping signals from trace impurities. The data are summarised in Fig. 7 and Table 1.

Using one calibrator (maleic acid as reference), the purity of EDAB was assigned four times evaluating four different protons. Two experiments were realised with different optimised ratios of signal intensities between EDAB and calibrator. The optimal ratio of 1:1 signal intensity for each experiment is indicated by a black dot
There are several interpretations which can be made, even when it would need an in-depth study to fully understand all the details of these effects. First, comparability of experiments with optimised signal intensity ratio is extremely high. This is indicated by almost identical values of the three results shown with black dots. Nevertheless, all eight values show a good consistency in the range of ±0.07 % relative standard deviation of the mean values. Second, signal pattern (singlet versus multiplet) seems not to be of highest relevance in terms of accuracy, again indicated by the three black dots. Nevertheless, the result where a singlet was analysed (N-(CH3)2) is the one having the smallest uncertainty.
Limitations
As described in this article, 1H-qNMR has the potential to generate highly accurate measurement results with expanded uncertainties significantly below the 0.1 % level. But there are certain preconditions which have to be fulfilled and also some limitations for the generation of excellent results with 1H-qNMR. First of all, good results require excellent weighing capabilities including the right metrological weighing equipment and also a good understanding of the most relevant effects. Of course, air buoyancy correction must be applied since this effect can influence the accuracy of the mass determination in the range of 0.1 %. In addition, adequate NMR settings must be defined, whereby the relaxation delay is probably most crucial. Some molecules show relaxation times of more than 10 s. So, if the relaxation delay is set too short, this will lead to molecule specific loss of intensity and therefore to a significant bias.
There are also restrictions with regard to the nature and chemistry of the sample. Highly accurate 1H-qNMR measurement results can only be obtained with highly pure substances. When corrections for impurities have to be made, this normally increases the overall uncertainty. Mixtures can also be measured by 1H-qNMR but only when a limited number of components are present in the mixture. Complex mixtures of complex molecules normally can not be measured. This is even more true for matrix samples. Also chemically highly reactive substances cannot be measured since they will react with either the solvent or the calibrator. Volatile and hygroscopic samples can be measured under special handling conditions, but this also leads to higher measurement uncertainties.
Conclusions
Many articles about the potential of 1H-qNMR have been published over the last few years. But until now, it could not be demonstrated that 1H-qNMR does have the potential to compete against established (primary) metrological techniques. In this article, it is demonstrated that 1H-qNMR combined with metrological weighing can be optimised to obtain results with <0.1 % expanded uncertainty. For this reason, the authors decided to call this approach high-performance qNMR (HP-qNMR®). Several experimental series showed the validity and accuracy of the 1H-qNMR results. Only a few restrictions are limiting the fields of application of 1H-qNMR such as impure samples or complex mixtures.
Compared to chromatographic techniques 1H-qNMR does not need a reference material of the same chemical constitution as the sample. With 1H-qNMR, only a limited number of properly selected substances are needed to serve as calibrators. In this article, the development of twelve available 1H-qNMR reference calibrators is described. Having these 1H-qNMR references available, it is possible to certify thousands of organic substances at the 0.1–0.4 % expanded uncertainty level. In addition, the certification of organic CRM with 1H-qNMR leads to a direct traceability to an SI unit. Until now more than 200 organic CRM have been certified and made available to the analytical community. Amongst them are pesticides, organic pollutants (PAHs, PCBs), fatty acids and esters, amino acids, drugs and APIs, impurities, phytochemicals, metabolites, and many more.
References
Bloch F, Hansen W, Packard ME (1946) Phys Rev 69:127
Purcell EM, Torrey HC, Pound RV (1946) Phys Rev 69:37–38
Fujiwara S, Wainai T (1961) Anal Chem 33:1085–1087
Jungnickel JL, Forbes JW (1963) Anal Chem 35:938–942
Hollis DP (1963) Anal Chem 35:1682–1684
Wells RJ, Cheung J, Hook JM (2004) Acc Qual Assur 9:450–456
Holzgrabe U, Diehl BWK, Wawer I (1998) J Pharm Biomed Anal 17:557–616
Holzgrabe U, Deubner R, Schollmayer C, Waibel B (2005) J Pharm Biomed Anal 38:806–812
Jancke H (1998) CCQM/98 98:1–12
Al-Deen TS, Hibbert DB, Hook JM, Wells RJ (2004) Accred Qual Assur 9:55–63
Saito T, Ihara T, Koike M, Kinugasa S, Fujimine Y, Nose K, Hirai T (2009) Accred Qual Assur 14:79–86
Malz F, Jancke H (2005) J Pharm Biomed Anal 38:813–823
BIPM (1995) Guide to the expression of uncertainty measurement, 2nd edn. International Organization for Standardization, Switzerland
EURACHEM/CITAG guide (2000) Quantifying uncertainty in analytical measurements, 2nd edn. ISBN 0 948926 15 5
Ihara T, Saito T, Sugimoto N (2009) Synthesiol Engl Ed 2:12–22
DeBièvre P, Dybkaer R, Fajgelj A, Hibbert B (2011) Pure Appl Chem 83(10):1873–1935
Reichmuth A, Wunderli S, Weber M, Meier VR (2004) Microchim Acta 148:133–141
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Weber, M., Hellriegel, C., Rück, A. et al. Using high-performance quantitative NMR (HP-qNMR®) for certifying traceable and highly accurate purity values of organic reference materials with uncertainties <0.1 %. Accred Qual Assur 18, 91–98 (2013). https://doi.org/10.1007/s00769-012-0944-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00769-012-0944-9