
Abstract
Amino acids not only participate in intermediary metabolism but also stimulate insulin-mechanistic target of rapamycin (MTOR)-mediated signal transduction which controls the major metabolic pathways. Among these is the pathway of autophagy which takes care of the degradation of long-lived proteins and of the elimination of damaged or functionally redundant organelles. Proper functioning of this process is essential for cell survival. Dysregulation of autophagy has been implicated in the etiology of several pathologies. The history of the studies on the interrelationship between amino acids, MTOR signaling and autophagy is the subject of this review. The mechanisms responsible for the stimulation of MTOR-mediated signaling, and the inhibition of autophagy, by amino acids have been studied intensively in the past but are still not completely clarified. Recent developments in this field are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For maintenance of cellular homeostasis, it is not only essential that cell components are synthesized and assembled when required but also that these components are removed and degraded when they are aberrantly synthesized, become damaged or when they are functionally redundant. When these processes are not carried out properly, the cell may either die or turn into a tumor cell in which cell growth proceeds unrestrained.
The major protein degradation systems include the ubiquitin–proteasome pathway (responsible for the quality control of newly synthesized proteins and the degradation of short-lived proteins) (Ciechanover 2012), macroautophagy (responsible for the degradation of long-lived proteins, protein aggregates and entire organelles) (Klionsky and Codogno 2013; Choi et al. 2013; Shen and Mizushima 2014) and chaperone-mediated autophagy (taking care of the removal of specific cytosolic proteins carrying a lysosomal target motif) (Cuervo and Wong 2014). Cross talk between these systems is also possible (Wang et al. 2013).
The process of macroautophagy (hereafter referred to as “autophagy”) has gained tremendous scientific interest in recent years. This is not only because of the partial unraveling of the protein and lipid machinery participating in this complicated cell biological process (Ohsumi 2014; Feng et al. 2014) but also because of the control of autophagic flux by growth factor- and amino acid-dependent signal transduction (Meijer and Codogno 2009; Russell et al. 2014). Above all, however, autophagy gained general interest because dysregulation of the process is implicated in many pathologies. These include, for example, cancer, neurodegeneration, obesity, type 2 diabetes, aging, heart and liver disease, lysosomal storage disorders, bacterial/viral infection and immunity diseases (Rubinsztein et al. 2012; Lavallard et al. 2012; Lieberman et al. 2012; Lorin et al. 2013a; Jiang and Mizushima 2014). In addition, autophagic activity in neurons of the hypothalamus appears to play an essential role in the control of body energy expenditure, appetite and body weight (Kaushik et al. 2011; Lavallard et al. 2012; Quan and Lee 2013). After a brief description of the process of autophagy as we know it today, the focus in this review will be on the regulation of autophagy by amino acids. The history of this fascinating topic, the discovery of amino acid-dependent signaling and possible mechanisms contributing to the inhibition of autophagy by amino acids, with recent developments in this field, will be discussed.
Autophagy
According to current opinion, the primary function of autophagy is to allow the cell to survive under stress conditions rather than to function as a cell death mechanism (Kroemer and Levine 2008). In the course of autophagy, macromolecules are degraded to small molecule precursors in order to support essential metabolic pathways under these conditions. A classical example at the whole body level is the autophagic production of amino acids, in the liver or elsewhere in the body, for hepatic gluconeogenesis during starvation (Schworer and Mortimore 1979; Ueno et al. 2012), glucose being essential as energy source for brain and erythrocytes under all circumstances.
During autophagy, which occurs in all eukaryotic cells, part of the cytoplasm is surrounded by a double membrane to form an autophagosome that acquires hydrolytic enzymes by fusion with endocytic compartments and lysosomes to form an autophagolysosome. In this process, the outer autophagosomal membrane fuses with the lysosomal membrane, and the inner autophagosomal membrane vesicle is released in the lysosomal lumen ((Meijer and Codogno 2009), for literature) upon which this vesicle, including its sequestered material, becomes degraded. The degradation products (e.g., amino acids) are transported to the cytosol via specific permeases (Mizushima and Klionsky 2007). The rate-limiting step in the entire autophagic pathway is the formation of the autophagosome. This formation starts with the expansion of a membrane core, the isolation membrane, or so-called phagophore (Seglen and Bohley 1992; Klionsky and Seglen 2010). Although progress concerning the origin and the biogenesis of the isolation membrane has been made, many questions still remain to be answered (Lamb et al. 2013; Shibutani and Yoshimori 2014). Very recent data suggest that the phagophore may be built up from the ER-mitochondria contact site (Hamasaki et al. 2013). However, other compartments such as the endoplasmic reticulum-Golgi intermediate compartment (Ge et al. 2013), endosomes and the plasma membrane also contribute to the formation of autophagosomes (Puri et al. 2013). Many different proteins are involved in autophagosome formation. More than 30 proteins have been identified in yeast (Ohsumi 2014; Feng et al. 2014) which are referred to as ATG (Autophagy related) proteins. Seventeen of these proteins are engaged in the biogenesis of the autophagosome, most of which are conserved in mammalian cells (including human cells) but carry different names in order to distinguish them from their yeast counterparts (Stanley et al. 2014). Among these is LC3-I (equivalent to Atg8 in yeast) which, when lipidated with phosphatidylethanolamine (and then named LC3-II), is widely used as a marker of autophagosomes (Klionsky et al. 2012).
Autophagosome formation and degradation can be extremely rapid. For example, in rat hepatocytes in vivo, the autophagic sequestration rate may vary from 0.2 % in the fed state to 1–1.5 % of the cell volume/h in the fasted state (Schworer and Mortimore 1979; Kovacs et al. 1982). In vitro, with isolated hepatocytes or in the perfused liver, in the absence of amino acids, this rate can be as high as 4 %/h (Blommaart et al. 1997a).
In the liver, autophagosomes are synthesized and degraded with a half-life of 8 min (Pfeifer 1977, 1978; Schworer and Mortimore 1979), and such short half-lives also apply to other cell types (Hailey and Lippincott-Schwartz 2009; Shibutani and Yoshimori 2014). Because of this high turnover rate, the steady-state volume of autophagosomes in the cell is low. It is important to stress that the measurement of the steady-state level of autophagosomes (a situation to be compared with the concentration of an intermediate in a metabolic pathway) does not give any information on the magnitude of the autophagic flux as is often assumed, and this notion not only applies to hepatocytes but to other cells as well (Meijer 2009; Klionsky et al. 2012). In order to estimate autophagic flux, at least for in vitro studies with perfused organs or cultured cells, several methods have been recommended. Among these, the most popular are the rate of 3-methyladenine (an inhibitor of autophagosome formation, see below)-sensitive degradation of long-lived proteins and the rate of accumulation of LC3-II (not the level of LC3-II at one time point), which is assumed to represent the rate of autophagosome formation, when lysosomal function and/or fusion is compromised by specific inhibitors (e.g., by chloroquine or bafilomycin, respectively) (Klionsky et al. 2012). The latter method may be complicated by possible feedback interactions caused by the accumulation of autophagosomes (Ktistakis et al. 2011) so that the accumulation of LC3-II may not always be linear with time. Another approach to quantify autophagic flux measures the rate of disappearance of p62/SQSTM1, an adapter protein that serves to carry protein cargo to be degraded to the phagophore (Bjorkoy et al. 2009). However, caution must be taken because in some cell lines amino acids upregulate the transcription of SQSTM1 (Sahani et al. 2014). Other methods, including sophisticated fluorescence techniques, are also available. For a complete description of these techniques, the reader is referred to a recent paper (Klionsky et al. 2012).
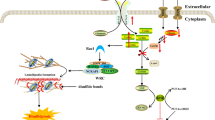
Autophagy is initiated by activation of a protein complex containing the protein kinases ULK1 and 2 (the mammalian counterpart of yeast Atg1), the proteins ATG13, FIP200 (yeast Atg17), ATG101 and many other components (Wong et al. 2013; Russell et al. 2014). Phosphorylation of ATG13 by ULK1/2 promotes the association of these proteins and is essential for initiation of autophagy (Fig. 1). ULK1 binds to membranes through its C-terminal domain (Chan et al. 2009). Downstream of the ULK1/2 complex is a lipid kinase, PIK3C3 (yeast Vps34), which produces PI(3)P and which is part of another protein complex also containing the regulatory protein PIK3R4 (yeast Vps15), the proteins Beclin1 (yeast Atg6/Vps30), ATG14 (also known as BARKOR). In the Beclin1-PIK3C3 core complex, Beclin1 can interact with several proteins such as AMBRA1, UVRAG and VMP1 to control autophagosome formation and/or maturation (Wirth et al. 2013) (cf. also Fig. 2). ULK1 phosphorylates both AMBRA1 and Beclin1 to initiate autophagosome formation (Russell et al. 2013; Lorin et al. 2013a). This results in activation of PIK3C3. Production of PI(3)P recruits PI(3)P-binding proteins involved in the initial formation of the autophagosome, WIPI1/2 (yeast Atg18), ZFYVE1/DFCP1 and two ubiquitin-like conjugation systems ATG12–ATG5-ATG16L and LC3-phosphatidylethanolamine (LC3-II) (Polson et al. 2010; Weidberg et al. 2011; McAlpine et al. 2013). PI(3)P is also required to join the ends of the autophagosomal membrane in statu nascendi (termed the “omegasome” because of its cup-formed shape) (Axe et al. 2008). The final fusion of the newly formed autophagosome with the lysosome requires small Rab GTPases, such as Rab7, Rab8 and Rab24, and the transmembrane protein LAMP2 (Simonsen and Tooze 2009; Ao et al. 2014).

Overview of the autophagic machinery. (Macro)autophagy starts with the nucleation of an isolation membrane, named the phagophore, which surrounds a fraction of the cytoplasm destined for degradation. Upon induction of autophagy, e.g., in starvation, the ULK1 complex localizes to a specialized domain of the ER called the omegasome. This privileged site for the biogenesis of the phagophore forms a cradle where the autophagosomal membrane elongates and acts as a template for the spherical form of the autophagosome. Downstream the ULK1 complex is the PIK3C3 complex, which produces phosphatidylinositol 3-phosphate (PI(3)P) to allow the recruitment of the PI(3)P-binding proteins WIPI1/2 and ZFYVE1/DFCP1. Both contribute to the expansion and the closure of the autophagosome together with the ATG12–ATG5-ATG16L complex and the LC3-phosphatidylethanolamine (LC3-PE) conjugate. Whereas ATG12–ATG5-ATG16L only transiently associates with the autophagosomal membrane, LC3–PE constitutes a specific marker of the autophagosome as it remains associated with the autophagosomal inner membrane. The newly formed autophagosome receives input from the endocytic pathway and ultimately fuses with a lysosome, allowing the degradation of autophagic substrates by lysosomal hydrolases. Fusion of the autophagosome with the lysosome requires the small Rab GTPases (such as Rab7, 8B and 24) and the transmembrane lysosomal protein LAMP2. The products of autophagic degradation, such as amino acids, are recycled to the cytosol where they exert a negative feedback on autophagy initiation. In addition to amino acids, autophagy is also controlled by upstream signaling pathways governed by insulin/growth factors, reactive oxygen species (ROS) and the energy status (through AMPK). Most of these factors regulate the two initiation complexes, ULK1 and PIK3C3. As a master regulator of autophagy, MTORC1 integrates multiple of these upstream signals and controls the activity of the ULK1 complex

Regulation of autophagy by amino acids. Autophagosome formation is regulated by two major modulators of autophagy, the MTORC1 and PIK3C3 complexes, which integrate amino acid signaling. Under fed conditions, when MTORC1 is fully active, MTORC1 downregulates autophagy by phosphorylating ULK1 and ATG13, which inhibits the ULK1 complex. MTORC1 also inhibits the synthesis of ATG proteins and the synthesis of proteins involved in the biogenesis of lysosomes at the transcription level, by preventing the translocation of TFEB to the nucleus. MTORC1 is activated in two ways: first, by insulin/growth factor signaling which involves PIK3C1, PDK1, PKB and TSC1/TSC2 as signaling components, and second, by amino acids through the Rag GTPases. To be active, MTORC1 has to localize at the lysosomal membrane, where its co-activator RhebGTP resides. In response to amino acids, Rag promotes the translocation of MTORC1 to the lysosomal membrane and its consecutive activation. Rag proteins are heterodimers of two subunits: RagA/B and RagC/D in which RagA/B is linked to GTP and RagC/D to GDP in the most active form of the dimer. The Rag GTPases are regulated by the v-ATPase, Ragulator and leucyl-tRNA synthetase (LRS). The nucleotide status of RagA/B and of RagC/D is regulated by GATOR and folliculin, respectively (not shown in the figure, for the purpose of clarity; see main text). In response to a rise in the intralysosomal pool of amino acids, the v-ATPase, present in the lysosomal membrane, changes its conformation and recruits Ragulator which displays a guanine nucleotide exchange factor (GEF) activity toward RagA/B. This results in the formation of RagA/BGTP and the activation of MTORC1. In this mechanism, the transporter PAT1, responsible for the efflux of amino acids from the lysosome, controls the concentration of amino acids in the lysosomal lumen and thus the extent of MTORC1 activation. In the presence of cytosolic leucine, binding of leucine to LRS reveals its GTPase-activating protein (GAP) activity toward RagC/D, resulting in the formation of RagC/DGDP and activation of MTORC1. The activity of Rag is also promoted by glutamate dehydrogenase (GDH). This mitochondrial enzyme, which plays a central role in amino acid catabolism, is allosterically activated by leucine. The production of 2-oxoglutarate by GDH may stimulate the loading of RagB with GTP. GDH may also activate MTORC1, and inhibit autophagy, through other distinct mechanisms. (1) The production of NAD(P)H by GDH may lead to the reduction of ROS, a potent activator of autophagy which acts through MTORC1-dependent and MTORC1-independent pathways (i.e., by inhibiting MTORC1 and PKB and by activating AMPK and ATG4). In addition to NAD(P)H, 2-oxoglutarate can also act as a scavenger of ROS, which oxidizes 2-oxoglutarate to succinate non-enzymatically. (2) The production of 2-oxoglutarate by GDH replenishes the citric acid cycle intermediates, increases the rate of ATP production and inhibits AMPK. The fall in AMPK activity may inhibit autophagy by MTORC1-dependent and MTORC1-independent mechanisms (i.e., by inhibition of TSC1/TSC2, stimulation of MTORC1 and inhibition of the ULK1 complex and of Beclin1). Probably acting in parallel with the Rag GTPase pathway, MAP4K3 and IPMK are other proteins involved in the regulation of MTORC1 by amino acids. The extracellular pool of amino acids may be sensed by the plasma membrane amino acid receptor T1R1/T1R3, which regulates MTORC1 and autophagy. The other major protein complexes controlling autophagy contain the Beclin1 protein. The core proteins of these two complexes are Beclin1, PIK3C3 and PIK3R4. When associated with ATG14 and AMBRA1, Beclin1 stimulates the early steps of autophagosome formation, downstream of the ULK1 complex. When associated with UVRAG, Beclin1 is mainly involved in the formation and maturation of autophagosomes. In response to amino acids, the protein kinase JNK1 becomes inhibited, leading to the formation of a stable complex between Beclin1 and Bcl-2 which sequesters Beclin1 and results in inhibition of autophagy. Beclin1 is also inhibited by PKB-dependent phosphorylation, which likewise inhibits autophagy. Long-term regulation of autophagy by PKB occurs by phosphorylation of FoxO3, another transcription factor (in addition to TFEB) responsible for the synthesis of ATG proteins. For further details, see main text. For reasons of clarity, the control of the inhibitory acetylation of ATG proteins by mitochondrial amino acid catabolism, which increases the concentration of acetyl CoA in the cytosol, is not indicated in the figure
Association of Beclin1 with PIK3C3 is an essential step in autophagosome formation. However, Beclin1 is a BH3 (Bcl-2 homology domain) protein and can also associate with the anti-apoptotic proteins Bcl-2/Bcl-xL which contain a BH3-binding groove. This means that association of Beclin1 with these anti-apoptotic proteins is inhibitory for autophagy and their dissociation is essential to allow Beclin1 to bind to PIK3C3 and to initiate autophagy. The dissociation of the inhibitory Beclin1–Bcl-2 complex is promoted by either JNK-1-mediated phosphorylation of Bcl-2, by phosphorylation of Beclin1 mediated by the tumor suppressor DAPK (a Ca2+-calmodulin-activated protein kinase), or by displacement of Beclin1 from its complex with Bcl-2 by other BH3-containing proteins such as the proapoptotic protein BAD and the proautophagic protein BNIP3 ((Meijer and Codogno 2009; Mariño et al. 2014a), for literature) (Fig. 2).
The insulin-amino acid-MTOR signaling pathway
The initiation of autophagosome formation is controlled by the insulin/growth factor-PI3KC1-PKB-TSC-MTOR-mediated signal-transduction pathway, which integrates hormonal, nutritional, cellular energy and oxidative stress inputs and which controls all major metabolic pathways (Meijer and Codogno 2009; Avruch et al. 2009; Kim and Guan 2011; Laplante and Sabatini 2012; Shanware et al. 2013; Cornu et al. 2013; Kim et al. 2013b).
The initial part of this signaling pathway, upstream of MTOR, an evolutionary conserved serine/threonine protein kinase which was first described by (Heitman et al. 1991), involves the insulin receptor, IRS1 and IRS2, PIK3C1, producing PI(3,4,5)P3 and PI(3,4)P2, PDK1 and PKB (Fig. 2). For activation of MTOR, the presence of insulin alone is not sufficient: The presence of amino acids is indispensable (Blommaart et al. 1995; Hara et al. 1998; Meijer and Codogno 2009; Barazzoni et al. 2012) (see below).
The second part of the insulin-signaling pathway, downstream of MTOR, may involve components such as S6K, 4E-BP1, eIF2α kinase and eEF2 kinase. Phosphorylation of these proteins by MTOR promotes protein synthesis.
MTOR itself is part of two multiprotein complexes. MTORC1 contains MTOR, RAPTOR, mLST8, and the inhibitory proteins DEPTOR and PRAS40. MTORC2 (not indicated in Fig. 2) contains MTOR, RICTOR, mLST8, mSin1, PROTOR and DEPTOR. Activation of MTORC1 simultaneously inhibits autophagy and stimulates protein synthesis (Meijer and Codogno 2009; Laplante and Sabatini 2012). Activation of MTORC2 does not have these effects but rather phosphorylates, and activates, PKB. In liver, for example, this results in stimulation of glycolysis and lipid biosynthesis (Hagiwara et al. 2012). MTORC1 and MTORC2 can be distinguished on the basis of their sensitivity toward inhibition by rapamycin which only inhibits MTORC1 (Sarbassov et al. 2006). Although the kinase activity of MTOR in MTORC2 is insensitive to acute rapamycin treatment, chronic exposure to the drug can disrupt its structure and results in insulin resistance (Sarbassov et al. 2006; Ye et al. 2012).
The activity of MTOR in MTORC1 is inhibited by the heterodimer TSC1/TSC2: it acts as a GTPase-activating protein complex for the small G-protein Rheb. RhebGTP, not RhebGDP, binds and activates MTOR. PKB phosphorylates TSC2, which inactivates the TSC1/TSC2 complex and stimulates MTORC1 (Fig. 2). In addition, MTORC1 is stimulated by PKB-dependent phosphorylation of PRAS40 (Manning and Cantley 2007).
Interaction of the insulin-amino acid-MTOR signaling pathway with autophagy
The insulin/growth factor-PIK3C1-PKB-MTOR signaling pathway inhibits autophagosome formation at several levels.
MTOR phosphorylates ULK1/2 and ATG13 which results in their dissociation and in inactivation of ULK (Ganley et al. 2009; Jung et al. 2009; Mizushima and Komatsu 2011; Lorin et al. 2013a). MTOR also inactivates PIK3C3, albeit indirectly, i.e., when the lipid kinase is associated with ATG14, by phosphorylating ATG14 (Yuan et al. 2013). Long-term regulation of autophagy by MTOR occurs by phosphorylation of the transcription factor EB (TFEB), a master regulator of the synthesis of ATG proteins and of the biogenesis of lysosomes. This phosphorylation prevents translocation of TFEB to the nucleus (Settembre et al. 2012). In addition, the activity of TFEB is inhibited by nutrient- and growth factor-dependent extracellular signal-regulated kinase 2 in an MTOR-independent fashion (Settembre et al. 2011) (Fig. 2). The same protein kinase can also inhibit autophagy in the short term by phosphorylation of TSC1/TSC2 (Kim et al. 2013b).
The complexity of the regulation of autophagy by MTOR is illustrated by the fact that MTOR not only inhibits ULK1 but that, conversely, ULK1 also inhibits MTOR by phosphorylation. This inhibition of MTORC1 by ULK1 may serve to amplify and stabilize initially small changes in nutrient signaling (Chang et al. 2009; Jung et al. 2011).
Autophagy is also controlled by PKB. Short-term regulation occurs by PKB-dependent phosphorylation of Beclin1 (Wang et al. 2012) (Fig. 2). Long-term regulation by PKB occurs by phosphorylation of FoxO3, another transcription factor responsible for the synthesis of ATG proteins (Mammucari et al. 2007). This set of ATG proteins differs from that controlled by TFEB (Settembre et al. 2011).
In addition to these various phosphorylation mechanisms, PI(3,4,5)P3 and PI(3,4)P2, the products of PIK3C1, are inhibitors of autophagy, possibly because these lipids compete with PI(3)P (Petiot et al. 2000; Meijer and Codogno 2009; Farrell et al. 2013).
Regulation of autophagy by amino acids and hormones: some history
Initial studies on the regulation of autophagy were primarily carried out with liver, both in vitro with the perfused liver and isolated hepatocytes, and in vivo. The process was known to be product inhibited by amino acids (Schworer and Mortimore 1979; Seglen et al. 1980), stimulated by glucagon (Ashford and Porter 1962; Deter et al. 1967) and inhibited by insulin (Mortimore and Mondon 1970), but the mechanisms underlying these effects were unknown at the time. Interestingly, in the perfused liver insulin and glucagon did only affect autophagy at intermediate amino acid concentrations but had no effect in the absence of amino acids when autophagic flux was maximal, nor did they have an effect in the presence of high concentrations of amino acids that inhibited autophagy maximally (Schworer and Mortimore 1979; Mortimore et al. 1987). It was also clear that regulation of autophagy by these factors was primarily exerted at the level of autophagosome formation (the autophagic sequestration step), although effects at later stages in the autophagic pathway could not be excluded (Hoyvik et al. 1991; Luiken et al. 1996). One confounding factor in experiments in vitro has been the accumulation of ammonia, derived from amino acid degradation which was found to increase the intralysosomal pH because of its acidotropic properties (Seglen 1977; Kadowaki and Kanazawa 2003). Even today, this complication is not always appreciated and unawareness of it may lead to erroneous conclusions (Meijer 2009).
Our own interest in the regulation of autophagy by amino acids was triggered by experiments performed with freshly isolated, perifused, rat hepatocytes under true steady-state conditions. Intriguingly, we found that a simple combination of leucine and alanine (which was selected on the basis of existing literature) at near-physiological concentrations could mimic the inhibitory effect on autophagic proteolysis of a complete mixture of all amino acids; either alanine or leucine alone had no effect (Leverve et al. 1987). Similar observations were made by Mortimore and colleagues for the perfused liver (Mortimore et al. 1988). Combinations of low concentrations of leucine and either proline, glutamine, or asparagine were also effective as inhibitors of autophagy (Caro et al. 1989). With leucine present, the inhibition by alanine, but not by proline, glutamine, or asparagine, was sensitive to inhibition by amino-oxyacetate, a transaminase inhibitor (Leverve et al. 1987; Caro et al. 1989). This indicated that metabolism of alanine, but not that of leucine, was required for its inhibitory effect on autophagy. As conversion of proline and glutamine to glutamate and of asparagine to aspartate does not require transamination reactions, we suspected that a combination of leucine with intracellular glutamate or aspartate (which does not readily leave hepatocytes) was sufficient to inhibit autophagy maximally. Indeed, in the presence of leucine and either alanine, proline, glutamine, or asparagine, there was an inverse relationship between the rate of autophagic proteolysis and the intracellular concentration of glutamate plus aspartate (Caro et al. 1989). However, the underlying mechanism for the inhibition of autophagy was entirely unknown. Interestingly, the same amino acids that inhibited hepatic autophagic proteolysis were also known to inhibit proteolysis in other tissues, including skeletal muscle, heart and kidney, with leucine being most effective (Blommaart et al. 1997a).
A new factor in the control of autophagic protein degradation was introduced with the observation, initially reported for liver but later extended to other cell types, that an increase in cell volume was able to mimic several of the anabolic properties of insulin, including inhibition of glycogenolysis (Lang et al. 1989), inhibition of proteolysis (Hallbrucker et al. 1991), stimulation of protein synthesis (Stoll et al. 1992) and lipogenesis (Baquet et al. 1991). Cell swelling was induced by either hypo-osmotic conditions or by Na+-dependent concentrative transport of amino acids across the plasma membrane. We suspected that intracellular accumulation of impermeant metabolites such as glutamate and aspartate, derived from amino acid degradation, also helped to increase the intracellular osmolarity and in this way contributed to cell swelling. In freshly isolated hepatocytes, cell swelling was found to mimic the effect of insulin in that it potentiated the ability of a complete mixture of amino acids, or of a combination of leucine with tyrosine and phenylalanine, to inhibit autophagic proteolysis (Meijer et al. 1993; Luiken et al. 1994).
Around the same time, our group in Amsterdam, in collaboration with the group of Hue in Brussels, studied the mechanism of the stimulation by amino acids of glycogen synthesis from glucose. This stimulation, first reported by Katz and colleagues (Katz et al. 1976), had been investigated by many laboratories but its mechanism was never elucidated. We discovered that the synthesis of glycogen, too, was due to an amino acid-induced increase in cell volume (Baquet et al. 1990). Leucine, which did not increase cell volume, had no effect, indicating that the mechanism of the stimulation of glycogen synthesis by amino acids differed from that of their inhibition of autophagy. It was known at the time that, in response to an increase in cell volume, cells undergo “regulatory volume decrease” in which they try to restore, at least in part, the original volume by releasing KCl (Hoffmann and Simonsen 1989; Häussinger 1996). We then found that the enzyme glycogen synthase phosphatase was inhibited by intracellular chloride and that the fall in chloride in response to cell swelling was sufficient to activate this enzyme and thus the synthesis of glycogen (Meijer et al. 1992). With regard to autophagy, the mechanism of inhibition by cell swelling was, and still is, not clear. Perhaps chloride ions are involved because they accompany protons, driven by the ATP-dependent lysosomal proton pump, to the lysosomal interior (Ishida et al. 2013). Very recent evidence suggests that this may, indeed, be the case (Hosogi et al. 2014). An effect of cell swelling on the cytoskeleton, mediated by integrins, is another, or additional, possibility (Häussinger et al. 2006).
In summary, in the control of autophagy, leucine appeared to be the most inhibitory amino acid. In addition, its anti-proteolytic effect was potentiated by insulin and by cell swelling.
Discovery of amino acid signaling and its relationship with autophagy
Early studies by Seglen and coworkers with protein kinase and protein phosphatase inhibitors indicated that protein phosphorylation was involved in the regulation of autophagy in hepatocytes, but the connection with amino acids was not made (Holen et al. 1992, 1993).
A breakthrough in our understanding of the mechanism by which amino acids inhibit autophagy (and stimulate protein synthesis) was obtained by the Amsterdam group in a study carried out with [32P]Pi-labeled rat hepatocytes. It was discovered that the same amino acid mixtures that inhibited autophagy greatly stimulated (up to fivefold) the phosphorylation of a protein that was identified as ribosomal protein S6, with rapid kinetics (t 1/2, 10 min). The stimulation of S6 phosphorylation by amino acids was not due to the inhibition of a protein phosphatase acting on S6 (Luiken et al. 1994; Blommaart et al. 1995). Dephosphorylation of S6 followed similar rapid kinetics upon withdrawal of amino acids or by addition of rapamycin. We observed synergy between either insulin or cell swelling and low concentrations of a complete mixture of amino acids, not only with regard to their inhibition of autophagic proteolysis but also with regard to their ability to promote S6 phosphorylation. Addition of insulin alone, in the absence of added amino acids, did not stimulate S6 phosphorylation. In the presence of high concentrations of amino acids, S6 phosphorylation was maximal and not further increased by insulin. Glucagon, on the other hand, stimulated autophagy and inhibited S6 phosphorylation at low, but not at high concentrations of amino acids or in their absence. Among various amino acids, leucine, not valine, appeared to be very effective. Under several incubation conditions, with different amino acid mixtures, in the absence or presence of insulin and glucagon, there appeared to be a linear relationship between the percentage of inhibition of autophagic proteolysis (measured in the presence of a low concentration cycloheximide sufficient to inhibit simultaneous protein synthesis) and the degree of phosphorylation of S6 (Blommaart et al. 1995). Amino acid-induced S6 phosphorylation was completely prevented by rapamycin, indicating that MTOR and S6K were components of the signaling pathway. Of great significance was the fact that rapamycin could partly (albeit not completely, this will be explained later) reverse the inhibition of autophagy by amino acids. It should be stressed, however, that in the absence of cycloheximide, rapamycin also did not completely inhibit protein synthesis. Because at that time MTOR signaling was known to be involved in the regulation of protein synthesis, it was concluded that (global) protein synthesis and (autophagic) protein degradation were under the opposite control by the same signaling pathway, which was considered to be metabolically efficient (Blommaart et al. 1995).
These studies were the first to demonstrate that amino acids were essential for MTOR-mediated signaling and that this signaling was connected to the regulation of autophagy. They were also the first demonstration that rapamycin was able to stimulate autophagy.
The stimulation of autophagy by rapamycin was confirmed several years later for other cell types (Mordier et al. 2000; Eskelinen et al. 2002; Moazed and Desautels 2002), including yeast (Noda and Ohsumi 1998), and rapamycin is nowadays widely used as an activator of autophagy (Rubinsztein et al. 2012). Torin1, a newly developed ATP-competitive inhibitor of MTOR, acts similarly (Thoreen et al. 2009).
The ability of amino acids to stimulate signaling even in the absence of insulin, and the synergy of amino acids with insulin, was confirmed for hepatocytes (Krause et al. 1996) and for other insulin-sensitive cell types, including muscle cells, adipocytes, hepatoma cells, CHO cells and pancreatic β-cells (Hara et al. 1998; Wang et al. 1998; Fox et al. 1998; Patti et al. 1998; Xu et al. 1998; Iiboshi et al. 1999). In these studies, it was shown that, in addition to S6, other downstream targets of MTOR such as S6K, 4E-BP1, eIF2α kinase (the equivalent of Gcn2 in yeast) and eEF2 kinase were found to be phosphorylated in response to amino acids. However, these studies also indicated that amino acids did not affect PKB activity and PIK3C1, signaling proteins upstream of MTOR that were known to be stimulated by insulin. It was also demonstrated that in case insulin alone, in the absence of added amino acids, stimulated MTOR signaling, this was dependent on the presence of amino acids that were generated by autophagic proteolysis (Shigemitsu et al. 1999a; Beugnet et al. 2003; Duran et al. 2011). Noteworthy is that the kinetics of the stimulation by amino acids of MTOR-mediated phosphorylations in these various cell types were similar to those observed by Blommaart et al. (1995) with regard to S6 phosphorylation in hepatocytes. They were also similar to the rapid phosphorylations reported very recently in an extensive study on the early temporal dynamics of the phosphoproteome in breast cancer cells upon initiation of autophagy by either withdrawal of amino acids or by addition of rapamycin (Rigbolt et al. 2014).
Most studies agreed that leucine (but not the other branched-chain amino acids), independent of the cell type, was the most effective amino acid in stimulating signaling but that, in addition, some other amino acids were required (Hara et al. 1998; Wang et al. 1998; Fox et al. 1998; Patti et al. 1998; Xu et al. 1998; Kimball et al. 1999; Shigemitsu et al. 1999b; van Sluijters et al. 2000; Lynch et al. 2000; Atherton et al. 2010). In analogy with the inhibition of autophagy and the stimulation of S6 phosphorylation by amino acids (see above), it was proposed that leucine, in combination with amino acid-induced cell swelling, would be sufficient to stimulate signaling (van Sluijters et al. 2000). In line with this, Krause et al. (2002b) showed synergy between glutamine, a potent amino acid in promoting cell swelling, and leucine with regard to S6K phosphorylation in hepatocytes. In intestinal cells, with their rapid growth, in addition to glutamine and leucine, arginine has also been mentioned as an activator of MTOR signaling (Nakajo et al. 2005; Marc and Wu 2009). In CHO cells, arginine also stimulated signaling albeit less effective than leucine (Hara et al. 1998). Although never considered, it is possible that the effect of arginine may be attributed, at least in part, to glutamate produced from arginine by the combined actions of arginase, ornithine aminotransferase and pyrroline 5-carboxylate dehydrogenase. Involvement of NO production from arginine can also not be excluded (Angcajas et al. 2014).
In order to account for the synergy between amino acids and insulin with respect to the activation of MTOR, it had to be assumed that MTOR received two parallel input signals, one from the insulin-PIK3C1-PKB signaling branch and one from amino acids (cf. (van Sluijters et al. 2000; Shah et al. 2000), for reviews). We now know that this is, indeed, the case (cf. Fig. 2): Insulin activates MTOR through PKB-dependent phosphorylation of TSC2, which results in activation of Rheb, and of PRAS40 as discussed earlier, while amino acids separately activate MTOR by mechanisms to be discussed below. However, the relative contribution of insulin and amino acids in the regulation of MTORC1 and autophagy may be tissue dependent. Thus, in mice in vivo, insulin appeared to be more potent in stimulating MTORC1 and inhibiting autophagy in muscle than in liver. By contrast, amino acids were more potent in the control of MTORC1 and autophagy in the liver than in muscle (Naito et al. 2013). This difference between the two tissues was tentatively ascribed to the regulation of MTORC1 and of autophagy by glucagon in the liver, but not in muscle. Another possibility is that there are differences in the intramuscular and intrahepatic amino acid concentrations.
S6K was also shown to phosphorylate, and inhibit, IRS1 (Tremblay and Marette 2001). Although this feedback effect of S6K has been proposed to participate in the etiology of insulin resistance (Um et al. 2004; Efeyan et al. 2014), it may also act as a mechanism to prevent overactivation of the MTOR pathway in order to allow some autophagy to continue even under nutrient-rich conditions (Meijer and Codogno 2009).
The inhibition of MTOR signaling by glucagon was later shown to proceed through a mechanism dependent on protein kinase A (Kimball et al. 2004), which stimulates autophagy (Mavrakis et al. 2006). It must be pointed out that, in contrast to mammalian cells, activation of protein kinase A in yeast cells inhibits autophagy (Stephan et al. 2009). In this context, it is important to stress that the function of cAMP in yeast, in contrast to that in mammalian cells, is to stimulate cell growth (Thevelein and de Winde 1999).
The requirement of PI(3)P for autophagy
Although in the presence of amino acids, rapamycin was able to stimulate autophagy, interruption of signaling by the PIK3C1 inhibitors wortmannin and LY294002, unexpectedly, did not. Even more surprising was the finding that in the absence of amino acids, with maximal autophagic flux, these two compounds strongly inhibited autophagy (Blommaart et al. 1997b). At the time, mammalian cells were known to contain both PIK3C1 and PIK3C3 while yeast only contained the homolog of PIK3C3, Vps34 (and its adapter protein Vps15). Vps34 was known to be involved in processes requiring membrane flow (Schu et al. 1993), but its relationship with autophagy was not yet studied in this organism. Because the phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 were not specific in that they could not distinguish between PIK3C1 and PIK3C3, it was postulated that perhaps PIK3C3, and thus its product PI(3)P, might be essential for autophagy (Blommaart et al. 1997a, b). This hypothesis was tested, and confirmed, in transfection experiments using HT-29 cells, a human colon cancer cell line, which showed that PI(3)P was, indeed, required for autophagy. Moreover, the products of PIK3C1, PI(3,4)P2 and PI(3,4,5)P3 acted as inhibitors of autophagy (Petiot et al. 2000). In further support of this, it was found that overexpression of PTEN, which removes the phosphate from the 3-position of PI(3,4)P2 and PI(3,4,5)P3, increased autophagy (Arico et al. 2001).
These observations were the first to show that PI(3)P participated in the process of autophagy. They have been amply confirmed for both mammalian cells and yeast (Kihara et al. 2001; Suzuki et al. 2001; Jaber et al. 2012; Schink et al. 2013; Cheng et al. 2014).
3-Methyladenine
An important spin-off from these studies was our finding that 3-methyladenine, a specific inhibitor of autophagy described by Gordon and Seglen in as early as 1982 (Seglen and Gordon 1982), and which has proven to be extremely useful in studies on autophagy (to this day), turned out to be an inhibitor of PIK3C3. This provided a satisfactory explanation for its mechanism of action (Blommaart et al. 1997b; Petiot et al. 2000).
Autophagy regulation and energy: the role of AMPK
Apart from being activated by insulin and amino acids, MTOR was also found to be affected by the cellular energy state. Initial studies with HEK293 cells indicated that MTOR may also act as a sensor of the intracellular ATP concentration (Dennis et al. 2001). It was noted that among various protein kinases the K m of MTOR for ATP in vitro was exceptionally high and within the physiological (mM) range of ATP concentrations. Because intracellular AMP is much more sensitive than ATP as an indicator of the cellular energy state, being connected to ATP in the reversible adenylate kinase reaction, AMPK was later considered as another candidate controlling MTOR activity. Indeed, studies simultaneously reported by several laboratories, including our own, demonstrated that, rather than the decreased ATP concentration, it was the activation of AMPK that was responsible for the inhibition of MTOR when energy falls short (Fig. 1) (Dubbelhuis and Meijer 2002; Bolster et al. 2002; Larsen et al. 2002; Krause et al. 2002a; Kimura et al. 2003). In agreement with the notion that inhibition of MTOR results in stimulation of autophagy (as discussed earlier), and also in line with its function to stimulate catabolism (Hardie 2007), it was reported, first by us (Meley et al. 2006) and later by others (Hoyer-Hansen et al. 2007; Liang et al. 2007), that AMPK is essential for autophagy. The activation of AMPK also underlied the stimulation of autophagy in cerebral (Adhami et al. 2006) and cardiac ischemia (Matsui et al. 2007).
The mechanism by which AMPK inhibited MTOR was twofold. The first mechanism proceeded via AMPK-mediated phosphorylation, and activation, of TSC2 (Inoki et al. 2003; Corradetti et al. 2004) which catalyzes the conversion of RhebGTP to RhebGDP. The second mechanism proceeded via phosphorylation, and inactivation, of RAPTOR (Gwinn et al. 2008) (Fig. 2).
The stimulation of autophagy by AMPK, however, was not only through inhibition of MTOR but also occurred by AMPK-mediated phosphorylation, and activation, of ULK1 (Egan et al. 2011; Kim et al. 2011a). Interestingly, ULK1 is thus phosphorylated by both MTOR and AMPK, but at different sites, with opposing effects on ULK1 activity (Meijer and Codogno 2011). In addition, it was recently shown that in response to glucose starvation AMPK phosphorylates, and activates, Beclin1 provided it is associated with PIK3C3 and ATG14 (Kim et al. 2013a). Thus, as ULK1, Beclin1 is phosphorylated at different sites, with opposing effects: in this case by AMPK (activation) and, as discussed earlier, by PKB (inhibitory) (Fig. 2).
Inhibition of AMPK activity by amino acids (cf. next paragraph), and thus inhibition of Beclin1, would account for an earlier observation showing that amino acids decreased Beclin1-associated PIK3C3 activity (Tassa et al. 2003). An additional explanation is that amino acids promote the association between Beclin1 and Bcl-2, and thus Beclin1 sequestration (Pattingre et al. 2005), through inhibition of JNK1-mediated phosphorylation of Bcl-2 (Wei et al. 2008).
Yet another mechanism by which AMPK can promote autophagy may proceed through activation of Sirtuin1, an NAD-dependent protein deacetylase (Canto et al. 2009; Ruderman et al. 2010). Sirtuin1, which increases in starvation, is known to stimulate autophagy by increasing the deacetylation of several ATG proteins (Lee et al. 2008). In addition, Sirtuin1 stimulates the deacetylation of FoxO3 which also results in activation (Canto et al. 2009). Conversely, acetylation of ATG proteins, which inhibits their activity, is brought about by the acetyltransferase P300 (also known as EP300) (Lee and Finkel 2009).
In principle, the inhibition of AMPK by amino acids would be an attractive mechanism to account for their ability to inhibit autophagy and to stimulate MTOR activity. Such an effect of amino acids on AMPK could be mediated by glutamate dehydrogenase, which plays a central role in amino acid catabolism and which is known to be specifically stimulated by leucine, not by the other branched-chain amino acids (Sener and Malaisse 1980; Fahien et al. 1990). This enzyme produces 2-oxoglutarate, replenishes citric acid cycle intermediates and thus helps to increase the rate of ATP production. Indeed, in many studies, inhibition of AMPK by amino acids has been observed (Xiao et al. 2011; Ghislat et al. 2012; Li et al. 2013). This was not always the case, however (Krause et al. 2002a; Kim et al. 2011a; Wauson et al. 2012; Duran et al. 2013; Rahman et al. 2014).
Because metabolism differs among cell types, e.g., with differences in the use of oxidizable substrates, it is understandable that the effect of amino acids on AMPK may vary. Although it is possible that AMPK is involved in amino acid regulation of autophagy and of MTOR signaling in some cell types, other amino acid sensing mechanisms may also exist. These will be discussed below.
Mechanisms of amino acid signaling
The mechanism by which amino acids stimulate MTOR activity, and inhibit autophagy, has remained, and still is to some extent, an enigma for a long time. Amino acids do not directly stimulate signaling upstream of MTOR, as discussed above, nor do they stimulate MTOR directly (van Sluijters et al. 2000; Kim and Guan 2011; Laplante and Sabatini 2012). However, as discussed in the previous paragraph, they can do so indirectly by inhibition of AMPK, depending on the cell type. As also mentioned earlier, in most cell types, among the various amino acids, leucine, but not the other branched-chain amino acids valine and isoleucine, is most potent in inhibiting autophagy and stimulating MTOR, and non-metabolizable analogs of leucine could mimic its effect. Catabolism of leucine does not seem to be required (Lynch et al. 2003). Indeed, fibroblasts from patients with defects in leucine catabolism displayed enhanced MTOR activity (Schriever et al. 2013). However, a role for leucine catabolites in MTOR signaling cannot entirely be ruled out. Thus, in experiments in man, administration of the leucine metabolite β-hydroxy-β-methylbutyrate, a compound that in clinical settings is used to treat disease-related muscle wasting, has been shown to stimulate skeletal muscle growth through the MTOR pathway (Wilkinson et al. 2013). It cannot be excluded that this compound, by serving as an oxidizable substrate, decreases AMPK activity and in this way stimulates MTOR signaling. A link between β-hydroxy-β-methylbutyrate-stimulated MTOR activity and the control of autophagy was not established.
It is concluded that any mechanism of amino acid sensing must account for the high leucine specificity.
Several factors involved in amino acid-MTOR signaling have been described in the past and have been reviewed in detail elsewhere (Dann and Thomas 2006; Avruch et al. 2009; Meijer and Codogno 2009; Kim and Guan 2011; Laplante and Sabatini 2012) (Fig. 2). These include, a.o., MAP4K3, PIK3C3, the G-protein Rheb, proton-assisted amino acid transporters in the lysosomal membrane, the Rag GTPases, leucyl-tRNA synthetase, the adapter protein p62, phospholipase D and inositol polyphosphate multikinase. The evidence in support of these various factors relied on the observation that elimination of any one of them strongly interfered with the ability of amino acids to stimulate MTOR signaling. In none of these studies, however, the nature of the primary amino acid sensor was identified. As will become clear, some of these factors are part of one mechanism responsible for the activation of MTOR activity by amino acids, whereas others participate in mechanisms that are independent of, but act in parallel to, each other.
Rheb
A potential mechanism of amino acid sensing was suggested by the observation that amino acids, leucine in particular, promote the association of Rheb with MTOR (Long et al. 2005). Although the effect was said not to be due to increased loading of Rheb with GTP (Long et al. 2005), other data indicate that amino acids did in fact promote the loading of Rheb with GTP (Smith et al. 2005; Roccio et al. 2006; Tzatsos and Kandror 2006; Sun and Chen 2008), but the reason for this increase in GTP loading was not explained. It is important to stress, however, that in TSC knockdown cells in which Rheb was fully charged with GTP, irrespective of the presence of amino acids, activation of MTOR remained amino acid dependent (Smith et al. 2005; Roccio et al. 2006). This indicated at least one other mechanism for amino acid activation of MTOR activity.
Rag
A big step forward in the search for a mechanism of amino acid sensing was the demonstration that the Rag GTPases are essential for the activation of MTOR by amino acids (Kim et al. 2008; Sancak et al. 2008) and that active MTORC1 appeared to be localized at the lysosomal membrane (Sancak et al. 2010). The Rag proteins form heterodimers between RagA/B and RagC/D, and in its most active form, RagA/B is in the GTP form and RagC/D in the GDP form (Tsun et al. 2013). Interestingly, amino acids increased the charging of RagA/B with GTP (Sancak et al. 2008). It was proposed that the v-ATPase in the lysosomal membrane, in addition to its role in proton pumping, acts as the amino acid sensor in MTOR signaling (Zoncu et al. 2011). The v-ATPase responds to an increase in the intralysosomal, rather than the cytosolic, amino acid concentration with a conformational change. This causes increased binding of Ragulator, a scaffolding protein complex consisting of 5 different proteins with guanine nucleotide exchange activity toward RagA and RagB, which anchors the Rag proteins to the lysosomal surface (Bar-Peled et al. 2012). At the extralysosomal surface MTORC1 interacts with Rheb and becomes activated.
Recruitment of the MTOR–Rag protein complex to the lysosome requires binding to the tumor suppressor protein folliculin. Folliculin is a GTPase-activating protein that specifically regulates the nucleotide status of RagC/D, not of RagA/B. Folliculin binding to the lysosome further requires association with the folliculin interacting proteins-1 and 2 (Petit et al. 2013; Tsun et al. 2013). A protein complex, called GATOR, composed of two subcomplexes, GATOR-1 and -2, also interacts with the Rags. As folliculin, GATOR-1 acts as a GTPase-activating protein but in this case toward RagA and RagB, not RagC and D, and thereby suppresses MTORC1 activity (Bar-Peled et al. 2013). Inactivating mutations of its components, which may occur in cancer, makes MTORC1 signaling resistant to amino acid deprivation. GATOR-2, in turn, negatively regulates GATOR-1 so that inhibition of GATOR-2 suppresses MTORC1 signaling.
The v-ATPase-mediated mechanism of amino acid sensing by MTORC1 at the lysosomal surface requires the proton gradient across the lysosomal membrane (Settembre et al. 2012). The activity of the lysosomal proton-assisted amino acid transporter PAT1, responsible for the efflux of amino acids from the lysosomes, may control the concentration of amino acids within the lysosomal lumen and thus the extent of MTORC1 activation (Zoncu et al. 2011), but the participation of other amino acid transporters in the lysosomal membrane cannot be ruled out (Efeyan et al. 2012). If the mechanism is correct, it must be speculated that among the various amino acids leucine is specially active in inducing the conformation change of the v-ATPase. In vitro experiments with isolated lysosomes in which the conditions for binding to the lysosomal membrane, and activation, of MTORC1 were analyzed have suggested that this may, indeed, be the case (Zoncu et al. 2011).
The fact that MTOR activity in MTORC1 is determined by the size of the intralysosomal pool of amino acids implies that the use of compounds such as the v-ATPase inhibitor bafilomycin or the acidotropic agent chloroquine cannot be recommended to estimate autophagic flux by monitoring the accumulation of the autophagosomal marker LC3-II. This is because inhibition of proteolysis within the lysosomes will directly affect the intralysosomal pool of amino acids, and thus leads to underestimation of MTOR activity and overestimation of autophagic flux (Juhasz 2012; Klionsky et al. 2012).
Puzzling is a recent report showing that <10 % of total intracellular Rag colocalized with the lysosomes, although loss of Rag from the lysosomal fraction to the cytosol during cell disruption and fractionation by differential centrifugation on sucrose density gradients could not be excluded (Oshiro et al. 2013).
In addition to the localization of active MTOR, as part of MTORC1, at the lysosomal membrane, MTOR present in MTORC2 is localized at the mitochondria (Schieke et al. 2006; Ramanathan and Schreiber 2009; Betz et al. 2013), presumably at the mitochondrial-endoplasmic reticulum contact site where it controls mitochondrial function (Betz et al. 2013).
Leucyl-tRNA synthetase
An early hypothesis proposed that free, uncharged, tRNA participates in amino acid sensing (Hara et al. 1998; Iiboshi et al. 1999). This hypothesis was based on observations with yeast in which, on amino acid starvation, free tRNA binds with high affinity to the protein kinase GCN2 (the equivalent of eIF2α-kinase) because the active center of GCN2 strongly resembles that of aminoacyl-tRNA synthetases (Hinnebusch 1997; Dong et al. 2000). GCN2 activation results in phosphorylation of eIF2α which then results in the derepression of GCN4 mRNA translation. GCN4 (equivalent to mammalian ATF4) is a transcriptional activator that promotes the transcription of many genes involved in nitrogen metabolism, not only genes involved in amino acid biosynthesis but also in autophagy (Natarajan et al. 2001; Tallóczy et al. 2002; B’chir et al. 2013). The particular potency of leucine in activating MTOR was proposed to be related to the frequency of utilization of this amino acid in protein synthesis and by the existence of multiple leucyl-tRNA synthetases arising from the sixfold codon degeneracy (Hara et al. 1998). Because uncharged tRNA did not affect the in vitro activity of immunoprecipitated MTOR, it was postulated that free tRNA interacted with an as yet unknown, signal-transduction component regulating MTOR (Iiboshi et al. 1999). Incompatible with the tRNA hypothesis, however, were kinetic considerations (the K m of leucine for leu-tRNA synthetase being orders of magnitude lower than prevailing intracellular leucine concentrations) and because specific aminoalcohols, inhibiting leu-tRNA synthetase, did not affect MTOR signaling (Lynch et al. 2000) (contrast (Iiboshi et al. 1999)). Finally, at least in HEK-293 cells, free tRNA levels did appear to change by amino acid starvation (Dennis et al. 2001).
Interest in the tRNA mechanism revived with the finding that leucyl-tRNA synthetase directly binds to Rag GTPase in a leucine-dependent manner and functions as a GTPase-activating protein (GAP) for Rag GTPase to activate MTORC1 (Han et al. 2012). In this mechanism, leucylation of the tRNA is not required. It is sufficient that leucine binds to the leucine-binding domain of the leucyl-tRNA synthetase and activates the enzyme, as measured by ATP-[32P]PPi exchange activity (Han et al. 2012). In this context, it is noteworthy that diadenosine tetraphosphate (Ap4A), a byproduct of the aminoacyl-tRNA synthetase reaction, was previously proposed by us as a factor involved in MTOR stimulation by amino acids because Ap4A is a strong inhibitor of AMPK (Meijer 2008).
It is important to stress that the two mechanisms of amino acid sensing, discussed in the previous paragraphs, detect different pools of amino acids: The v-ATPase senses the intralysosomal pool of amino acids, while the leucyl-tRNA synthetase senses cytosolic leucine. It is perfectly possible that these two mechanisms coexist in amino acid sensing (Yoon et al. 2011; Duran and Hall 2012a).
Glutamate dehydrogenase
Some years ago, on the basis of existing literature, we hypothesized that glutamate dehydrogenase (GDH), in addition to its role in amino acid catabolism, is involved in amino acid sensing and in controlling autophagy (Meijer 2008; Meijer and Codogno 2008, 2009). The arguments were as follows. As discussed earlier, this mitochondrial enzyme is specifically activated by leucine. In pancreatic β-cells, the ability of leucine (but not of valine or isoleucine) to stimulate production of insulin and to stimulate rapamycin-sensitive S6K phosphorylation was ascribed to stimulation of GDH (Xu et al. 2001). Moreover, a mutation in GDH, which results in overactivation of the enzyme, underlies the hyperinsulinism/hyperammonia (HHS) syndrome (Li et al. 2012b). A combination of glutamine (a glutamate donor) and leucine, which maximizes the flux through GDH, is most effective in stimulating MTOR and in inhibiting autophagic flux in several cell types, including a.o., β-cells and hepatocytes ((Meijer and Codogno 2009), for literature), as discussed earlier (cf. section “Discovery of amino acid signaling and its relationship with autophagy”). Recent studies by Duran et al. (2012) using both genetic and pharmacological methods have now provided strong experimental evidence that in the course of glutamine metabolism, GDH does, indeed, play a crucial role in the activation of MTOR. It was also demonstrated that it is the production of 2-oxoglutarate by GDH which stimulates loading of RagB with GTP. The link between 2-oxoglutarate and MTORC1 was proposed to be prolylhydroxylase which, in a HIF-1α-independent manner, somehow results in increased RagBGTP (Duran et al. 2013). In these studies, the possibility of increased production of GTP by succinyl CoA synthetase in the course of 2-oxoglutarate oxidation in the mitochondria, suggested by us in the past (Meijer and Codogno 2009), was not explored.
By contrast, other data have indicated that the charging of RagA/B with GTP does not play a role in the mechanism of amino acid sensing. Thus, Rag heterodimers extracted from [32P]Pi-labeled whole cells or from the pool associated with the lysosomal membrane exhibited constitutive [32P]GTP charging that was unaltered by amino acid withdrawal. In addition, in cells with mutant Rag which was unable to bind GTP, activation of MTOR by amino acids still occurred (Oshiro et al. 2013). Another concern is that dimethyl-2-oxoglutarate, a cell-permeable analog of 2-oxoglutarate, has been shown to inhibit, rather than stimulate, MTORC1 activity (Tan and Hagen 2013). The reason for these differences in results is not clear.
The importance of GDH in the activation of MTOR was also indicated, albeit indirectly, by studies of van der Vos et al. (2012) showing that overexpression of glutamine synthetase inhibited MTOR activity, inhibited the translocation of MTOR to the lysosomes and activated autophagy. Surprisingly, it was concluded that glutamine itself functions as an inhibitor of MTOR, and thus as an activator of autophagy, a conclusion which is in contrast to existing literature (see above). However, the fact that increased flux through glutamine synthetase results in increased flux through GDH, in this case in the direction of amination, i.e., from 2-oxoglutarate to glutamate, because of the use of glutamate for glutamine synthesis, was overlooked (Duran and Hall 2012b). In agreement with this interpretation is the finding that in glutamine-depleted cells pharmacological inhibition of glutamine synthetase greatly stimulated MTOR activity (Tardito et al. 2012).
Apart from a role of GDH in the production of 2-oxoglutarate for the activation of MTOR, it is also possible that NADPH, another product of the deamination reaction, activates MTOR, and inhibits autophagy, by eliminating reactive oxygen species (ROS) which are predominantly produced in the mitochondria (Meijer and Codogno 2009). This may occur by, e.g., the glutathione–glutathione reductase system, and/or through direct prevention by NADPH of ROS production at the FMN-a site of complex I of the mitochondrial respiratory chain (Albracht et al. 2011). In this context, it is worthwhile to note that glutaminase-2 (liver-type), in contrast to glutaminase-1 (kidney-type), is not sensitive to product inhibition (Kovacevic and McGivan 1983; Mates et al. 2013). This allows a high flux through glutaminase and GDH at a relatively high steady-state intramitochondrial glutamate concentration. This is not only of importance for production of NADPH via GDH with its high K m for glutamate but it is also favorable for the synthesis of glutathione. Interestingly, not only does a high flux through GDH stimulate MTORC1 but, conversely, does MTORC1 activate GDH through transcriptional repression of SIRT4, the mitochondrial localized sirtuin that inhibits GDH (Csibi et al. 2013). This is very efficient from the point of view of metabolic regulation.
In addition to NADPH, 2-oxoglutarate, the other product of the GDH reaction, can also act as a scavenger of ROS, which oxidizes 2-oxoglutarate to succinate non-enzymically (Mailloux et al. 2007).
When the production of ROS exceeds its degradation, excessive ROS levels induce oxidative stress and damage of cellular components including DNA, proteins and lipids. A rise in ROS levels (e.g., in starvation) activates autophagy as a protective mechanism, and this occurs in a manner that is sensitive to antioxidants (Scherz-Shouval et al. 2007; Li et al. 2012a; Morales et al. 2014; Rahman et al. 2014) which, simultaneously, stimulate MTOR activity (Li et al. 2013). The importance of ROS in the initiation of autophagy was also indicated by the observation that knockdown of the antioxidant transcription factor Nrf2 in breast cancer cells substantially increased autophagy in response to oxidative stress (Rao et al. 2010). Possible targets of ROS contributing to the stimulation of autophagy by ROS are ATG4 (Scherz-Shouval and Elazar 2011), PKB (Rahman et al. 2014), Beclin1 (Bolisetty and Jaimes 2013) and AMPK (Toyoda et al. 2004).
Experiments very recently carried out in our laboratories (Lorin et al. 2013b) showed that the production of ROS by starved HeLa cells, a cervix cancer cell line, was suppressed by either a complete mixture of all amino acids or by a combination of glutamine plus leucine alone. Knockdown of GDH prevented these effects. At the same time, knockdown of GDH stimulated autophagy and inhibited MTOR signaling in the presence of amino acids. By what mechanism oxidative stress inhibits MTOR is unknown. Possibilities are increased AMPK and decreased PKB activities (cf. the previous paragraph). It is also possible that one, or more, of the components involved in the amino acid sensing mechanisms discussed above is redox sensitive. A very recent development is the finding that TSC1/TSC2 is a target for ROS, in this case of ROS produced by peroxisomes. Interestingly, both TSC1/TSC2 and Rheb were found to be bound to peroxisomes and TSC1/TSC2 became activated by ROS produced by the peroxisomes. This resulted in the conversion of peroxisome-bound RhebGTP to RhebGDP and in inhibition of lysosome-bound MTORC1 (Zhang et al. 2013). This was accompanied by activation of autophagy. How peroxisomal activation of TSC1/TSC2 and inhibition of Rheb caused inactivation of lysosomal MTORC1 was not clear because MTORC1 did not colocalize with the peroxisomes ((Benjamin and Hall 2013; Betz and Hall 2013), for discussion). An attractive mechanism would be that MTORC1, TSC1/TSC2 and/or Rheb shuttle between various sites in the cell where ROS is produced locally to bring the signal to the lysosomes which is considered to be the site responsible for amino acid activation of MTORC1. In line with this is, the very recent finding that deprivation of insulin and of amino acids recruits the TSC complex to the lysosomes where it acts to inactivate Rheb (Menon et al. 2014; Demetriades et al. 2014). It will be of interest to see whether a system similar to that found for peroxisomal ROS is also present for ROS produced by the mitochondria. Indeed, association of Rheb and MTORC1 with mitochondria cannot be excluded (Schieke et al. 2006; Groenewoud and Zwartkruis 2013).
In this context, it is important to note that glutamine deprivation in pancreatic cancer cells also resulted in increased ROS production (Son et al. 2013). Suppression of ROS by glutamine was ascribed to production of oxaloacetate from glutamine-derived aspartate, followed by its reduction to malate and subsequent production of NADPH (and pyruvate) by malic enzyme in the cytosol. Elimination of cytosolic aspartate aminotransferase, indeed, greatly increased production of ROS, which could be counteracted, at least in part, by addition of oxaloacetate (Son et al. 2013). However, net production of oxaloacetate from glutamine requires net production 2-oxoglutarate, and this cannot proceed without the participation of GDH or of glutamate pyruvate transaminase (Yang et al. 2009). The finding that addition of oxaloacetate in the absence of aspartate aminotransferase could suppress ROS accumulation is not surprising because α-oxoacids react non-enzymically with ROS (Holleman 1904; Andrae et al. 1985; Mailloux et al. 2007).
In retrospect, the participation of glutamate dehydrogenase in the control of autophagy is in agreement with the observation that autophagy was stimulated pharmacologically by the green tea component epigallocatechin gallate (Li et al. 2011; Zhou et al. 2014). This compound is a powerful inhibitor of glutamate dehydrogenase (Li et al. 2006), but the link between these phenomena was not made.
Involvement of GDH in amino acid sensing has serious consequences for the use of chloroquine in the measurement of autophagic flux. This compound not only increases the intralysosomal pH and in this way affects the intralysosomal amino acid pool, as discussed above (cf. section “Rag”) but, in addition, is also a potent inhibitor of GDH (Jarzyna et al. 1997). This, too, leads to overestimation of autophagic flux. Along the same line, stimulation of autophagy by ammonia (Eng et al. 2010; Harder et al. 2014) may be ascribed to the fact that this metabolite drives the GDH reaction in the direction of glutamate synthesis. An alternative, or perhaps additional, mechanism is activation of AMPK by ammonia (Harder et al. 2014). It must be pointed out that in the latter experiments, ammonia was used at mM concentrations. Although the data indicated that ammonia increased the rate of autophagosome formation, it is highly likely that under these conditions the intralysosomal pH was also affected. Autophagic flux under turnover conditions, as measured by 3-methyladenine-sensitive proteolysis, was not analyzed, however.
PIK3C3 and p62/SQSTM1
Some years ago it was proposed that PIK3C3 is required for amino acid signaling, an effect that is mediated by an amino acid-induced rise in cytosolic Ca++, which results in increased binding of Ca++/calmodulin to, and activation of, PIK3C3 (Nobukuni et al. 2005; Gulati et al. 2008). Apart from the fact that a rise in Ca++/calmodulin stimulates autophagy through activation of AMPK by calmodulin-dependent kinase–kinase-β (Hoyer-Hansen et al. 2007; Meijer and Codogno 2009; Pfisterer et al. 2011; Ghislat et al. 2012) and that it is more likely that cytosolic Ca++ decreases rather than increases in the presence of amino acids (Meijer and Codogno 2009; Ghislat et al. 2012), although this issue has been controversial (Wauson et al. 2012), these observations were puzzling because PIK3C3 also participates in the formation of autophagosomes.
A similar problem relates to the proposal that p62/SQSTM1 participates in amino acid signaling (Duran et al. 2011), because this protein does not inhibit autophagy but rather is required for this process.
In order to solve the problem with PIK3C3, it was proposed that the enzyme is part of different protein complexes with different functions (Kim and Guan 2011; Ktistakis et al. 2012). Recent experiments indicate that this is, indeed, the case and that different PIK3C3 complexes exist which are differentially regulated by amino acids (Yuan et al. 2013; Kim et al. 2013a).
It has been proposed that amino acids, by locally activating the formation of PI(3)P, actually stimulate the production of PI(3,5)P2 by phosphatidylinositol-3-phosphate-5-kinase. PI(3,5)P2 is able to bind to RAPTOR where it serves to recruit MTOR downstream targets and/or to bring MTORC1 to the proper location in the cell (Bridges et al. 2012; Jin et al. 2014).
p62/SQSTM1 may also be compartmented because only a small part of the total cellular p62 is bound to the MTORC1 complex through its association with the Rag GTPases (Duran et al. 2011). As an alternative explanation, however, we propose that autophagy itself produces amino acids which then stimulate MTOR activity (Shigemitsu et al. 1999a; Beugnet et al. 2003; Yu et al. 2010; Inoki et al. 2012). Thus, overexpression of PIK3C3 or p62 initially activates autophagy, resulting in increased production of amino acids from proteins within the lysosomes. This increases the size of the intralysosomal pool of amino acids which is sensed by the v-ATPase in the lysosomal membrane and activates MTOR according to the mechanism discussed above. Conversely, if PIK3C3 or p62 becomes inhibited, autophagic flux declines, the intralysosomal amino acid pool decreases and MTOR becomes inhibited. The observation that in skeletal muscle of mice deficient of myotubularin, the lipid phosphatase responsible for the degradation of PI(3)P, autophagy is defective and MTOR overactivated (Fetalvero et al. 2013) may be explained similarly. Perhaps initially, autophagy is overactivated because of the rise in PI(3)P. This results in increased autophagic proteolysis, a rise in the concentration of lysosomal amino acids, upon which MTOR becomes activated and autophagy inhibited again.
Phospholipase D
The phosphatidic acid-producing enzyme phospholipase D (PLD) (Fang et al. 2001) has also been implicated as one of the components taking part in the mechanism by which amino acids activate MTORC1.
It has been reported that amino acids activate PLD1 in a PIK3C3-dependent manner and that PLD1 is indispensable for translocation of MTORC1 to the lysosomes and its activation by amino acids (Yoon et al. 2011; Xu et al. 2011). In the absence of PIK3C3, addition of phosphatidic acid only activated MTORC1 when amino acids were also present. Addition of exogenous PI(3)P stimulated PLD1 activity in the absence of amino acids but did not activate MTORC1 (Yoon et al. 2011). These and other observations led to the suggestion that PLD1 may be part of the protein complex anchoring MTORC1 to the lysosomal membrane and that the PIK3C3-PLD1 pathway acts in parallel to the Rag pathway in regulating amino acid activation of MTORC1 (Yoon et al. 2011; Wiczer and Thomas 2012). Unexpectedly, the effect of PLD1 on autophagy has been controversial: both stimulation (Dall’Armi et al. 2010) and inhibition (Jang et al. 2014) of autophagy have been reported. It was suggested that this dual effect of PLD on autophagy, as with PIK3C3, is dependent on the subcellular localization of PLD (Jang et al. 2014). However, as discussed in the previous section, it cannot be excluded that the PIK3C3-phosphatidic acid pathway is actually required for autophagy but that MTORC1 becomes activated after the intralysosomal pool of amino acids has sufficiently expanded by autophagic proteolysis.
Inositol polyphosphate multikinase
Inositol polyphosphate multikinase is another component implicated in the mechanism responsible for stimulation of MTOR activity by amino acids (Kim et al. 2011b). Independent of its catalytic activity the enzyme appeared to stabilize the binding between MTOR and RAPTOR in the MTORC1 complex through its aminoterminal amino acid sequence which forms a unique mammalian MTOR binding site (Kim et al. 2011b).
MAP4K3
MAP4K3 is another protein that acts upstream (Findlay et al. 2007; Yan et al. 2010) of MTOR which becomes activated by amino acids. Its role in amino acid sensing is not entirely clear. It is unlikely that this kinase participates in either the v-ATPase or the leucyl-tRNA synthetase mechanism of amino acid sensing. Presumably, MAP4K3 is part of another pathway leading to amino acid-induced MTOR activation (Kim and Guan 2011).
Plasma membrane amino acid receptor
Early evidence indicated that the amino acid receptor was intracellular rather than extracellular (Martin and Sutherland 2001; Christie et al. 2002; Beugnet et al. 2003). However, this may not entirely be true. There are indications that the plasma membrane can also contribute to amino acid sensing. Thus, it has been suggested that the plasma membrane of hepatocytes contains a leucine-specific receptor protein (which does not transport leucine) which controls autophagy independently of MTOR (Kanazawa et al. 2004).
In analogy with yeast, plasma membrane amino acid transporters have also been implicated in the sensing of (extracellular) amino acid availability by mammalian cells (Hundal and Taylor 2009; Kim and Guan 2011). Such a role was attributed to a transport protein that mediates the exchange between extracellular leucine and intracellular glutamine, which allows leucine to be transported against a concentration gradient (Nicklin et al. 2009). It must be pointed out, however, that an intracellular localization of the amino acid sensor implies that any process affecting the intracellular concentration of leucine, whether it is its transport across the plasma membrane or the rate of the intracellular metabolism of leucine, will affect the ability of leucine to stimulate MTOR activity.
Very recently, it was reported that the G-protein-coupled taste receptor complex T1R1/T1R3, an amino acid receptor in the plasma membrane, originally discovered in gustatory neurons as a detector of the umami flavor and present in many tissues, is an early sensor of extracellular amino acid availability (Wauson et al. 2012). Reduced expression of T1R1/T1R3 impaired the activation of MTOR by amino acids, caused mislocalization of MTORC1, and accelerated autophagy under nutrient-rich conditions. Interestingly, the intracellular concentration of amino acids, leucine included, was not affected by knockdown of the taste receptor even though the expression of several plasma membrane amino acid transporters greatly increased under these conditions (Wauson et al. 2012). The question of how inactivation of MTOR and activation of autophagy in T1R1/T1R3 receptor knockdown cells could occur in the absence of changes in intracellular amino acid concentrations remained unanswered. An obvious explanation could be that GDH was downregulated after knockdown of the taste receptor. But this is unlikely because this enzyme plays a central role in amino acid catabolism and its downregulation would have resulted in increased intracellular amino acid concentrations under these conditions, which was not observed. Another, plausible, possibility is that AMPK was activated. However, AMPK was inhibited, instead (Wauson et al. 2012). A third possibility, i.e., that leucyl-tRNA synthetase was affected by T1R1/T1R3 receptor knockdown, was not explored.
Amino acid sensing and the concentration of cytosolic acetyl CoA
A very recent development has been the finding that the acetyltransferase EP300, responsible for the inhibitory acetylation of several ATG proteins, because of its low affinity (high K m) for acetyl CoA, acts as a sensor of cytosolic acetyl CoA which translates increases in cytosolic acetyl CoA into inhibition of autophagy (Mariño et al. 2014b). Thus, several experimental manipulations designed to alter cytosolic acetyl CoA, both in cultured cells and in vivo in mice, resulted in predicted changes in autophagic flux. Because the level of cytosolic acetyl CoA also strongly correlated with the activity of MTORC1 the possibility that the acetylation-dependent control of autophagy was indirect and mediated by MTORC1 could not be excluded (Mariño et al. 2014b).
In view of these fascinating observations, it is possible that amino acid catabolism may actually result in increased cytosolic acetyl CoA and in this way inhibits autophagy and stimulates MTORC1 signaling. This, then, would yet be another mechanism by which amino acids affect these pathways. Although this remains to be demonstrated, glutamate dehydrogenase, by stimulating synthesis of citrate followed by exit of citrate from the mitochondria and its cleavage by ATP-citrate lyase in the cytosol, can be expected to play an important role here, too.
Amino acid signaling and autophagy: summary
In summary, on the basis of current literature, it is clear that there is not one unique mechanism of amino acid sensing. Rather, several mechanisms of amino acid sensing, leading to activation of MTOR, can operate. Whether these mechanisms are context- and/or cell type-dependent, or act in parallel, is currently not known.
Although amino acids can inhibit autophagy by activation of MTOR, they can also do so in an MTOR-independent manner. Thus, as we have seen, amino acids can decrease Beclin1-associated PIK3C3 activity because they promote the association between Beclin 1 and Bcl-2 through inhibition of JNK1-mediated phosphorylation of Bcl-2. In addition, amino acids may decrease AMPK activity which results in dephosphorylation, and inhibition, of Beclin1 and ULK1. This may explain why the inhibition of autophagy by amino acids is not always fully reversed by rapamycin. The inhibition of autophagy by insulin through protein kinase B-induced phosphorylation of Beclin1 and the transcription factor FoxO3 and through the direct inhibitory effects of PI(3,4)P2 and PI(3,4,5)P3 is also MTORC1 independent, and thus rapamycin insensitive.
Conclusions
Our original discoveries of amino acid-stimulated, MTOR-mediated, signaling and its role in the control of autophagy, the regulation of these two pathways by the energy sensor AMPK and the role of phosphatidylinositol 3-phosphate in autophagy have opened new perspectives in the understanding of the regulation of cell metabolism. Given the enormous impact of autophagy on cell function, and its regulation by amino acid signaling, it is not surprising that research in these fields has expanded exponentially over the last two decades (Klionsky 2007; Ohsumi 2014). Proper in vivo manipulation of autophagy, either pharmacologically or by dietary restriction, under many pathological conditions may be used to the benefit of patients (Hermans et al. 2013). The safest way is dietary restriction. Although it is known for a very long time to be beneficial for health, e.g., in obesity, type 2 diabetes and aging, the underlying mechanisms are still not clear. A simple explanation is that dietary restriction acts as a two-sided sword. On the one hand, it reduces the redox pressure on the mitochondrial respiratory chain, and thus diminishes overproduction of potentially harmful ROS. On the other hand, dietary restriction also stimulates autophagy and thereby contributes to the improvement of cell function (Cavallini et al. 2008; Blagosklonny 2010; Gelino and Hansen 2012; Eisenberg et al. 2014).
Abbreviations
- 4E-BP1:
-
Eukaryotic translation initiation factor 4E binding protein 1
- AA:
-
Amino acids
- AMBRA1:
-
Activating molecule in Beclin1-regulated autophagy protein 1
- AMPK:
-
AMP-activated protein kinase
- ATF4:
-
Activating transcription factor 4
- ATG:
-
Autophagy related
- BAD:
-
Bcl-2-associated death promoter
- BARKOR:
-
Beclin1-associated autophagy-related key regulator
- Bcl-2:
-
B cell lymphoma 2
- Bcl-xL:
-
B cell lymphoma extra large
- Beclin1:
-
Bcl-2-interacting coiled-coil protein 1
- BH3:
-
Bcl-2 homology domain
- BNIP3:
-
Bcl-2/adenovirus E1B 19 kDa interacting protein 3
- CHO:
-
Chinese hamster ovary
- DAPK:
-
Death-associated protein kinase
- DEPTOR:
-
DEP domain-containing MTOR-interacting protein
- DFCP1:
-
Double FYVE-domain-containing protein 1
- eEF2 kinase:
-
Eukaryotic translation elongation factor 2 kinase
- eIF2α kinase:
-
Eukaryotic initiation factor 2α kinase
- EP300:
-
E1A binding protein p300
- E1A:
-
Adenovirus early region 1A
- FIP200:
-
Focal adhesion kinase family-interacting protein of 200 kDa
- FoxO3:
-
Forkhead box O3
- GAP:
-
GTPase-activating protein
- GATOR:
-
GTPase-activating protein toward Rags
- Gcn2:
-
General control non-depressible 2
- Gcn4:
-
General control non-depressible 4
- GDH:
-
Glutamate dehydrogenase
- GEF:
-
Guanine nucleotide exchange factor
- HEK293:
-
Human embryonic kidney 293
- HIF-1α:
-
Hypoxia-inducible factor 1α
- IPMK:
-
Inositol polyphosphate multikinase
- IRS:
-
Insulin receptor substrate
- Lamp2:
-
Lysosomal-associated membrane protein 2
- LC3-I:
-
Microtubule-associated protein 1A/1B-light chain 3
- LC3-II:
-
Microtubule-associated protein 1A/1B-light chain 3 conjugated with phosphatidylethanolamine
- LRS:
-
Leucyl-tRNA synthetase
- MAP4K3:
-
Mitogen-activated protein kinase kinase kinase kinase 3
- mLST8:
-
Mammalian lethal with sec-13 protein 8 (also GβL)
- mSin1:
-
Mammalian stress-activated MAP kinase-interacting protein 1
- MTOR:
-
Mechanistic target of rapamycin (formerly mTOR, mammalian target of rapamycin)
- MTORC:
-
Mechanistic target of rapamycin complex
- Nrf2:
-
NF-E2-related factor 2
- PDK1:
-
Phosphoinositide-dependent kinase 1
- PE:
-
Phosphatidylethanolamine
- Pi :
-
Inorganic phosphate
- PI(3)P:
-
Phosphatidylinositol 3-phosphate
- PI(3,4)P2:
-
Phosphatidylinositol 3,4-biphosphate
- PI(3,4,5)P3:
-
Phosphatidylinositol 3,4,5-triphosphate
- PIK3C1:
-
Phosphatidylinositol 3-kinase class I
- PIK3C3:
-
Phosphatidylinositol 3-kinase class III
- PIK3R4:
-
Phosphatidylinositol 3-kinase regulatory subunit 4; also hVps15, formerly called p150
- PKB:
-
Protein kinase B (formerly AKT)
- PLD:
-
Phospholipase D
- PRAS40:
-
Proline-rich AKT substrate 40
- PROTOR:
-
Protein observed with RICTOR
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome 10
- Rag:
-
Ras-related GTP-binding protein
- RAPTOR:
-
Regulatory-associated protein of MTOR
- Rheb:
-
Ras homolog enriched in brain
- RICTOR:
-
Rapamycin-insensitive companion of TOR
- ROS:
-
Reactive oxygen species
- S6:
-
Ribosomal protein S6
- S6K:
-
70 kDa S6 kinase (formerly p70S6K)
- SQSTM1:
-
Sequestosome 1
- T1R1:
-
Taste receptor type 1 member 1
- T1R3:
-
Taste receptor type 1 member 3
- TFEB:
-
Transcription factor EB
- TSC:
-
Tuberous sclerosis
- ULK1/2:
-
Unc-51-like kinase1/2
- UVRAG:
-
UV irradiation resistance-associated gene
- v-ATPase:
-
Vacuolar ATPase
- VMP1:
-
Vacuole membrane protein 1
- Vps15:
-
Vacuolar protein sorting 15
- Vps34:
-
Vacuolar protein sorting 34
- WIPI-1/2:
-
WD repeat domain phosphoinositide-interacting protein 1/2
References
Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY (2006) Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol 169:566–583
Albracht SP, Meijer AJ, Rydstrom J (2011) Mammalian NADH:ubiquinone oxidoreductase (Complex I) and nicotinamide nucleotide transhydrogenase (Nnt) together regulate the mitochondrial production of H(2)O(2)–implications for their role in disease, especially cancer. J Bioenerg Biomembr 43:541–564
Andrae U, Singh J, Ziegler-Skylakakis K (1985) Pyruvate and related alpha-ketoacids protect mammalian cells in culture against hydrogen peroxide-induced cytotoxicity. Toxicol Lett 28:93–98
Angcajas AB, Hirai N, Kaneshiro K, Karim MR, Horii Y, Kubota M, Fujimura S, Kadowaki M (2014) Diversity of amino acid signaling pathways on autophagy regulation: a novel pathway for arginine. Biochem Biophys Res Commun 446:8–14
Ao X, Zou L, Wu Y (2014) Regulation of autophagy by the Rab GTPase network. Cell Death Differ 21:348–358
Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E (2001) The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem 276:35243–35246
Ashford TP, Porter KR (1962) Cytoplasmic components in hepatic cell lysosomes. J Cell Biol 12:198–202
Atherton PJ, Smith K, Etheridge T, Rankin D, Rennie MJ (2010) Distinct anabolic signalling responses to amino acids in C2C12 skeletal muscle cells. Amino Acids 38:1533–1539
Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N (2009) Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab 296:E592–E602
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182:685–701
Baquet A, Hue L, Meijer AJ, van Woerkom GM, Plomp PJ (1990) Swelling of rat hepatocytes stimulates glycogen synthesis. J Biol Chem 265:955–959
Baquet A, Maisin L, Hue L (1991) Swelling of rat hepatocytes activates acetyl-CoA carboxylase in parallel to glycogen synthase. Biochem J 278:887–890
Barazzoni R, Short KR, Asmann Y, Coenen-Schimke JM, Robinson MM, Nair KS (2012) Insulin fails to enhance mTOR phosphorylation, mitochondrial protein synthesis, and ATP production in human skeletal muscle without amino acid replacement. Am J Physiol Endocrinol Metab 303:E1117–E1125
Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150:1196–1208
Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM (2013) A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340:1100–1106
B’chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A (2013) The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41:7683–7699
Benjamin D, Hall MN (2013) TSC on the peroxisome controls mTORC1. Nat Cell Biol 15:1135–1136
Betz C, Hall MN (2013) Where is mTOR and what is it doing there? J Cell Biol 203:563–574
Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN (2013) Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 110:12526–12534
Beugnet A, Tee AR, Taylor PM, Proud CG (2003) Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem J 372:555–566
Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T (2009) Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452:181–197
Blagosklonny MV (2010) Linking calorie restriction to longevity through sirtuins and autophagy: any role for TOR. Cell Death Dis 1:e12
Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ (1995) Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 270:2320–2326
Blommaart EF, Luiken JJ, Meijer AJ (1997a) Autophagic proteolysis: control and specificity. Histochem J 29:365–385
Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ (1997b) The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 243:240–246
Bolisetty S, Jaimes EA (2013) Mitochondria and reactive oxygen species: physiology and pathophysiology. Int J Mol Sci 14:6306–6344
Bolster DR, Crozier SJ, Kimball SR, Jefferson LS (2002) AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem 277:23977–23980
Bridges D, Ma JT, Park S, Inoki K, Weisman LS, Saltiel AR (2012) Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1. Mol Biol Cell 23:2955–2962
Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458:1056–1060
Caro LH, Plomp PJ, Leverve XM, Meijer AJ (1989) A combination of intracellular leucine with either glutamate or aspartate inhibits autophagic proteolysis in isolated rat hepatocytes. Eur J Biochem 181:717–720
Cavallini G, Donati A, Gori Z, Bergamini E (2008) Towards an understanding of the anti-aging mechanism of caloric restriction. Curr Aging Sci 1:4–9
Chan EY, Longatti A, McKnight NC, Tooze SA (2009) Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol 29:157–171
Chang YY, Juhasz G, Goraksha-Hicks P, Arsham AM, Mallin DR, Muller LK, Neufeld TP (2009) Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem Soc Trans 37:232–236
Cheng J, Fujita A, Yamamoto H, Tatematsu T, Kakuta S, Obara K, Ohsumi Y, Fujimoto T (2014) Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphate asymmetries. Nat Commun 5:3207
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368:651–662
Christie GR, Hajduch E, Hundal HS, Proud CG, Taylor PM (2002) Intracellular sensing of amino acids in Xenopus laevis oocytes stimulates p70 S6 kinase in a target of rapamycin-dependent manner. J Biol Chem 277:9952–9957
Ciechanover A (2012) Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Neurodegener Dis 10:7–22
Cornu M, Albert V, Hall MN (2013) mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev 23:53–62
Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL (2004) Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 18:1533–1538
Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, Haigis MC, Cantley LC, Stephanopoulos G, Yu J, Blenis J (2013) The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153:840–854
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24:92–104
Dall’Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, Duff K, Frohman MA, Wenk MR, Yamamoto A, Di Paolo G (2010) The phospholipase D1 pathway modulates macroautophagy. Nat Commun 1:142
Dann SG, Thomas G (2006) The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett 580:2821–2829
Demetriades C, Doumpas N, Teleman AA (2014) Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156:786–799
Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G (2001) Mammalian TOR: a homeostatic ATP sensor. Science 294:1102–1105
Deter RL, Baudhuin P, De DC (1967) Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol 35:C11–C16
Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG (2000) Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell 6:269–279
Dubbelhuis PF, Meijer AJ (2002) Hepatic amino acid-dependent signaling is under the control of AMP-dependent protein kinase. FEBS Lett 521:39–42
Duran RV, Hall MN (2012a) Leucyl-tRNA synthetase: double duty in amino acid sensing. Cell Res 22:1207–1209
Duran RV, Hall MN (2012b) Glutaminolysis feeds mTORC1. Cell Cycle 11:4107–4108
Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT (2011) p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 44:134–146
Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN (2012) Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 47:349–358
Duran RV, MacKenzie ED, Boulahbel H, Frezza C, Heiserich L, Tardito S, Bussolati O, Rocha S, Hall MN, Gottlieb E (2013) HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 32:4549–4556
Efeyan A, Zoncu R, Sabatini DM (2012) Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 18:524–533
Efeyan A, Schweitzer LD, Bilate AM, Chang S, Kirak O, Lamming DW, Sabatini DM (2014) RagA, but Not RagB, Is Essential for Embryonic Development and Adult Mice. Dev Cell 29:321–329
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–461
Eisenberg T, Schroeder S, Andryushkova A, Pendl T, Küttner V, Bhukel A, Mariño G, Pietrocola F, Harger A, Zimmermann A, Moustafa T, Sprenger A, Jany E, Büttner S, Carmona-Gutierrez D, Ruckenstuhl C, Ring J, Reichelt W, Schimmel K, Leeb T, Moser C, Schatz S, Kamolz LP, Magnes C, Sinner F, Sedej S, Fröhlich KU, Juhasz G, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, Madeo F (2014) Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab 19:431–444
Eng CH, Yu K, Lucas J, White E, Abraham RT (2010) Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal 3:ra31
Eskelinen EL, Prescott AR, Cooper J, Brachmann SM, Wang L, Tang X, Backer JM, Lucocq JM (2002) Inhibition of autophagy in mitotic animal cells. Traffic 3:878–893
Fahien LA, Teller JK, Macdonald MJ, Fahien CM (1990) Regulation of glutamate dehydrogenase by Mg2+ and magnification of leucine activation by Mg2+. Mol Pharmacol 37:943–949
Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294:1942–1945
Farrell O, Rusten TE, Stenmark H (2013) Phosphoinositide 3-kinases as accelerators and brakes of autophagy. FEBS J 280:6322–6337
Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24:24–41
Fetalvero KM, Yu Y, Goetschkes M, Liang G, Valdez RA, Gould T, Triantafellow E, Bergling S, Loureiro J, Eash J, Lin V, Porter JA, Finan PM, Walsh K, Yang Y, Mao X, Murphy LO (2013) Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol Cell Biol 33:98–110
Findlay GM, Yan L, Procter J, Mieulet V, Lamb RF (2007) A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem J 403:13–20
Fox HL, Kimball SR, Jefferson LS, Lynch CJ (1998) Amino acids stimulate phosphorylation of p70S6k and organization of rat adipocytes into multicellular clusters. Am J Physiol 274:C206–C213
Ganley IG, Lam dH, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284:12297–12305
Ge L, Melville D, Zhang M, Schekman R (2013) The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2:e00947
Gelino S, Hansen M (2012) Autophagy - an emerging anti-aging mechanism. J Clin Exp Pathol Suppl 4:006
Ghislat G, Patron M, Rizzuto R, Knecht E (2012) Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-beta (CaMKK-beta). J Biol Chem 287:38625–38636
Groenewoud MJ, Zwartkruis FJ (2013) Rheb and mammalian target of rapamycin in mitochondrial homoeostasis. Open Biol 3:130185
Gulati P, Gaspers LD, Dann SG, Joaquin M, Nobukuni T, Natt F, Kozma SC, Thomas AP, Thomas G (2008) Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab 7:456–465
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226
Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Ruegg MA, Hall MN (2012) Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 15:725–738
Hailey DW, Lippincott-Schwartz J (2009) Using photoactivatable proteins to monitor autophagosome lifetime. Methods Enzymol 452:25–45
Hallbrucker C, vom Dahl S, Lang F, Häussinger D (1991) Control of hepatic proteolysis by amino acids. The role of cell volume. Eur J Biochem 197:717–724
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495:389–393
Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, Ha SH, Ryu SH, Kim S (2012) Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149:410–424
Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J (1998) Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 273:14484–14494
Harder LM, Bunkenborg J, Andersen JS (2014) Inducing autophagy: a comparative phosphoproteomic study of the cellular response to ammonia and rapamycin. Autophagy 10:339–355
Hardie DG (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8:774–785
Häussinger D (1996) The role of cellular hydration in the regulation of cell function. Biochem J 313:697–710
Häussinger D, Reinehr R, Schliess F (2006) The hepatocyte integrin system and cell volume sensing. Acta Physiol (Oxf) 187:249–255
Heitman J, Movva NR, Hall MN (1991) Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253:905–909
Hermans G, Casaer MP, Clerckx B, Güiza F, Vanhullebusch T, Derde S, Meersseman P, Derese I, Mesotten D, Wouters PJ, Van Cromphaut S, Debaveye Y, Gosselink R, Gunst J, Wilmer A, Van den Berghe G, Vanhorebeek I (2013) Effect of tolerating macronutrient deficit on the development of intensive-care unit acquired weakness: a subanalysis of the EPaNIC trial. Lancet Respir Med 1:621–629
Hinnebusch AG (1997) Translational regulation of yeast GCN4. A window on factors that control initiator-trna binding to the ribosome. J Biol Chem 272:21661–21664
Hoffmann EK, Simonsen LO (1989) Membrane mechanisms in volume and pH regulation in vertebrate cells. Physiol Rev 69:315–382
Holen I, Gordon PB, Seglen PO (1992) Protein kinase-dependent effects of okadaic acid on hepatocytic autophagy and cytoskeletal integrity. Biochem J 284:633–636
Holen I, Gordon PB, Seglen PO (1993) Inhibition of hepatocytic autophagy by okadaic acid and other protein phosphatase inhibitors. Eur J Biochem 215:113–122
Holleman MAF (1904) Notice sur l’action de l’eau oxygenee sur les acides acetoniques et sur les dicetones 1,2. Receuil des Travaux Chimiques des Pays-Bas et de la Belgique 23:169–172
Hosogi S, Kusuzaki K, Inui T, Wang X, Marunaka Y (2014) Cytosolic chloride ion is a key factor in lysosomal acidification and function of autophagy in human gastric cancer cell. J Cell Mol Med. doi:10.1111/jcmm.12257
Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M (2007) Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 25:193–205
Hoyvik H, Gordon PB, Berg TO, Stromhaug PE, Seglen PO (1991) Inhibition of autophagic-lysosomal delivery and autophagic lactolysis by asparagine. J Cell Biol 113:1305–1312
Hundal HS, Taylor PM (2009) Amino acid transceptors: gate keepers of nutrient exchange and regulators of nutrient signaling. Am J Physiol Endocrinol Metab 296:E603–E613
Iiboshi Y, Papst PJ, Kawasome H, Hosoi H, Abraham RT, Houghton PJ, Terada N (1999) Amino acid-dependent control of p70(s6k). Involvement of tRNA aminoacylation in the regulation. J Biol Chem 274:1092–1099
Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590
Inoki K, Kim J, Guan KL (2012) AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol 52:381–400
Ishida Y, Nayak S, Mindell JA, Grabe M (2013) A model of lysosomal pH regulation. J Gen Physiol 141:705–720
Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, Zong WX (2012) Class III PI3 K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci USA 109:2003–2008
Jang YH, Choi KY, Min DS (2014) Phospholipase D-mediated autophagic regulation is a potential target for cancer therapy. Cell Death Differ 21:533–546
Jarzyna R, Lenarcik E, Bryla J (1997) Chloroquine is a potent inhibitor of glutamate dehydrogenase in liver and kidney-cortex of rabbit. Pharmacol Res 35:79–84
Jiang P, Mizushima N (2014) Autophagy and human diseases. Cell Res 24:69–79
Jin N, Mao K, Jin Y, Tevzadze G, Kauffman EJ, Park S, Bridges D, Loewith R, Saltiel AR, Klionsky DJ, Weisman LS (2014) Roles for PI(3,5)P2 in nutrient sensing through TORC1. Mol Biol Cell 25:1171–1185
Juhasz G (2012) Interpretation of bafilomycin, pH neutralizing or protease inhibitor treatments in autophagic flux experiments: novel considerations. Autophagy 8:1875–1876
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20:1992–2003
Jung CH, Seo M, Otto NM, Kim DH (2011) ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 7:1212–1221
Kadowaki M, Kanazawa T (2003) Amino acids as regulators of proteolysis. J Nutr 133:2052S–2056S
Kanazawa T, Taneike I, Akaishi R, Yoshizawa F, Furuya N, Fujimura S, Kadowaki M (2004) Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem 279:8452–8459
Katz J, Golden S, Wals PA (1976) Stimulation of hepatic glycogen synthesis by amino acids. Proc Natl Acad Sci USA 73:3433–3437
Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, Cuervo AM, Singh R (2011) Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab 14:173–183
Kihara A, Noda T, Ishihara N, Ohsumi Y (2001) Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 152:519–530
Kim J, Guan KL (2011) Amino acid signaling in TOR activation. Annu Rev Biochem 80:1001–1032
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL (2008) Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10:935–945
Kim J, Kundu M, Viollet B, Guan KL (2011a) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141
Kim S, Kim SF, Maag D, Maxwell MJ, Resnick AC, Juluri KR, Chakraborty A, Koldobskiy MA, Cha SH, Barrow R, Snowman AM, Snyder SH (2011b) Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab 13:215–221
Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013a) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152:290–303
Kim SG, Buel GR, Blenis J (2013b) Nutrient regulation of the mTOR complex 1 signaling pathway. Mol Cells 35:463–473
Kimball SR, Shantz LM, Horetsky RL, Jefferson LS (1999) Leucine regulates translation of specific mRNAs in L6 myoblasts through mTOR-mediated changes in availability of eIF4E and phosphorylation of ribosomal protein S6. J Biol Chem 274:11647–11652
Kimball SR, Siegfried BA, Jefferson LS (2004) Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem 279:54103–54109
Kimura N, Tokunaga C, Dalal S, Richardson C, Yoshino K, Hara K, Kemp BE, Witters LA, Mimura O, Yonezawa K (2003) A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells 8:65–79
Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937
Klionsky DJ, Codogno P (2013) The mechanism and physiological function of macroautophagy. J Innate Immun 5:427–433
Klionsky DJ, Seglen PO (2010) The Norse god of autophagy. Interviewed by Daniel J Klionsky. Autophagy 6:1017–1031
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Bánhegyi G, Bartholomew CR, Bassham DC, Bast RC Jr, Batoko H, Bay BH, Beau I, Béchet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin JP, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahová M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LA, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae HJ, Chai CY, Chan DC, Chan EY, Chang RC, Che CM, Chen CC, Chen GC, Chen GQ, Chen M, Chen Q, Chen SS, Chen W, Chen X, Chen X, Chen X, Chen YG, Chen Y, Chen Y, Chen YJ, Chen Z, Cheng A, Cheng CH, Cheng Y, Cheong H, Cheong JH, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang HL, Chiarelli R, Chiba T, Chin LS, Chiou SH, Chisari FV, Cho CH, Cho DH, Choi AM, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang TH, Chueh SH, Chun T, Chwae YJ, Chye ML, Ciarcia R, Ciriolo MR, Clague MJ, Clark RS, Clarke PG, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coombs GH, Coppens I, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronjé MJ, Cuervo AM, Cullen JJ, Czaja MJ, D’Amelio M, Darfeuille-Michaud A, Davids LM, Davies FE, De Felici M, de Groot JF, de Haan CA, De Martino L, De Milito A, De Tata V, Debnath J, Degterev A, Dehay B, Delbridge LM, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di Gioacchino M, Di Paolo G, Di Pietro C, Díaz-Araya G, Díaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding WX, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA, Duff K, Dugail I, Durbeej M, Duszenko M, Edelstein CL, Edinger AL, Egea G, Eichinger L, Eissa NT, Ekmekcioglu S, El-Deiry WS, Elazar Z, Elgendy M, Ellerby LM, Eng KE, Engelbrecht AM, Engelender S, Erenpreisa J, Escalante R, Esclatine A, Eskelinen EL, Espert L, Espina V, Fan H, Fan J, Fan QW, Fan Z, Fang S, Fang Y, Fanto M, Fanzani A, Farkas T, Farré JC, Faure M, Fechheimer M, Feng CG, Feng J, Feng Q, Feng Y, Fésüs L, Feuer R, Figueiredo-Pereira ME, Fimia GM, Fingar DC, Finkbeiner S, Finkel T, Finley KD, Fiorito F, Fisher EA, Fisher PB, Flajolet M, Florez-McClure ML, Florio S, Fon EA, Fornai F, Fortunato F, Fotedar R, Fowler DH, Fox HS, Franco R, Frankel LB, Fransen M, Fuentes JM, Fueyo J, Fujii J, Fujisaki K, Fujita E, Fukuda M, Furukawa RH, Gaestel M, Gailly P, Gajewska M, Galliot B, Galy V, Ganesh S, Ganetzky B, Ganley IG, Gao FB, Gao GF, Gao J, Garcia L, Garcia-Manero G, Garcia-Marcos M, Garmyn M, Gartel AL, Gatti E, Gautel M, Gawriluk TR, Gegg ME, Geng J, Germain M, Gestwicki JE, Gewirtz DA, Ghavami S, Ghosh P, Giammarioli AM, Giatromanolaki AN, Gibson SB, Gilkerson RW, Ginger ML, Ginsberg HN, Golab J, Goligorsky MS, Golstein P, Gomez-Manzano C, Goncu E, Gongora C, Gonzalez CD, Gonzalez R, González-Estévez C, González-Polo RA, Gonzalez-Rey E, Gorbunov NV, Gorski S, Goruppi S, Gottlieb RA, Gozuacik D, Granato GE, Grant GD, Green KN, Gregorc A, Gros F, Grose C, Grunt TW, Gual P, Guan JL, Guan KL, Guichard SM, Gukovskaya AS, Gukovsky I, Gunst J, Gustafsson AB, Halayko AJ, Hale AN, Halonen SK, Hamasaki M, Han F, Han T, Hancock MK, Hansen M, Harada H, Harada M, Hardt SE, Harper JW, Harris AL, Harris J, Harris SD, Hashimoto M, Haspel JA, Hayashi S, Hazelhurst LA, He C, He YW, Hébert MJ, Heidenreich KA, Helfrich MH, Helgason GV, Henske EP, Herman B, Herman PK, Hetz C, Hilfiker S, Hill JA, Hocking LJ, Hofman P, Hofmann TG, Höhfeld J, Holyoake TL, Hong MH, Hood DA, Hotamisligil GS, Houwerzijl EJ, Høyer-Hansen M, Hu B, Hu CA, Hu HM, Hua Y, Huang C, Huang J, Huang S, Huang WP, Huber TB, Huh WK, Hung TH, Hupp TR, Hur GM, Hurley JB, Hussain SN, Hussey PJ, Hwang JJ, Hwang S, Ichihara A, Ilkhanizadeh S, Inoki K, Into T, Iovane V, Iovanna JL, Ip NY, Isaka Y, Ishida H, Isidoro C, Isobe K, Iwasaki A, Izquierdo M, Izumi Y, Jaakkola PM, Jäättelä M, Jackson GR, Jackson WT, Janji B, Jendrach M, Jeon JH, Jeung EB, Jiang H, Jiang H, Jiang JX, Jiang M, Jiang Q, Jiang X, Jiang X, Jiménez A, Jin M, Jin S, Joe CO, Johansen T, Johnson DE, Johnson GV, Jones NL, Joseph B, Joseph SK, Joubert AM, Juhász G, Juillerat-Jeanneret L, Jung CH, Jung YK, Kaarniranta K, Kaasik A, Kabuta T, Kadowaki M, Kagedal K, Kamada Y, Kaminskyy VO, Kampinga HH, Kanamori H, Kang C, Kang KB, Kang KI, Kang R, Kang YA, Kanki T, Kanneganti TD, Kanno H, Kanthasamy AG, Kanthasamy A, Karantza V, Kaushal GP, Kaushik S, Kawazoe Y, Ke PY, Kehrl JH, Kelekar A, Kerkhoff C, Kessel DH, Khalil H, Kiel JA, Kiger AA, Kihara A, Kim DR, Kim DH, Kim DH, Kim EK, Kim HR, Kim JS, Kim JH, Kim JC, Kim JK, Kim PK, Kim SW, Kim YS, Kim Y, Kimchi A, Kimmelman AC, King JS, Kinsella TJ, Kirkin V, Kirshenbaum LA, Kitamoto K, Kitazato K, Klein L, Klimecki WT, Klucken J, Knecht E, Ko BC, Koch JC, Koga H, Koh JY, Koh YH, Koike M, Komatsu M, Kominami E, Kong HJ, Kong WJ, Korolchuk VI, Kotake Y, Koukourakis MI, Kouri Flores JB, Kovács AL, Kraft C, Krainc D, Krämer H, Kretz-Remy C, Krichevsky AM, Kroemer G, Krüger R, Krut O, Ktistakis NT, Kuan CY, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung HJ, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, László L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee AJ, Lee BW, Lee GM, Lee J, Lee JH, Lee M, Lee MS, Lee SH, Leeuwenburgh C, Legembre P, Legouis R, Lehmann M, Lei HY, Lei QY, Leib DA, Leiro J, Lemasters JJ, Lemoine A, Lesniak MS, Lev D, Levenson VV, Levine B, Levy E, Li F, Li JL, Li L, Li S, Li W, Li XJ, Li YB, Li YP, Liang C, Liang Q, Liao YF, Liberski PP, Lieberman A, Lim HJ, Lim KL, Lim K, Lin CF, Lin FC, Lin J, Lin JD, Lin K, Lin WW, Lin WC, Lin YL, Linden R, Lingor P, Lippincott-Schwartz J, Lisanti MP, Liton PB, Liu B, Liu CF, Liu K, Liu L, Liu QA, Liu W, Liu YC, Liu Y, Lockshin RA, Lok CN, Lonial S, Loos B, Lopez-Berestein G, López-Otín C, Lossi L, Lotze MT, Lőw P, Lu B, Lu B, Lu B, Lu Z, Luciano F, Lukacs NW, Lund AH, Lynch-Day MA, Ma Y, Macian F, MacKeigan JP, Macleod KF, Madeo F, Maiuri L, Maiuri MC, Malagoli D, Malicdan MC, Malorni W, Man N, Mandelkow EM, Manon S, Manov I, Mao K, Mao X, Mao Z, Marambaud P, Marazziti D, Marcel YL, Marchbank K, Marchetti P, Marciniak SJ, Marcondes M, Mardi M, Marfe G, Mariño G, Markaki M, Marten MR, Martin SJ, Martinand-Mari C, Martinet W, Martinez-Vicente M, Masini M, Matarrese P, Matsuo S, Matteoni R, Mayer A, Mazure NM, McConkey DJ, McConnell MJ, McDermott C, McDonald C, McInerney GM, McKenna SL, McLaughlin B, McLean PJ, McMaster CR, McQuibban GA, Meijer AJ, Meisler MH, Meléndez A, Melia TJ, Melino G, Mena MA, Menendez JA, Menna-Barreto RF, Menon MB, Menzies FM, Mercer CA, Merighi A, Merry DE, Meschini S, Meyer CG, Meyer TF, Miao CY, Miao JY, Michels PA, Michiels C, Mijaljica D, Milojkovic A, Minucci S, Miracco C, Miranti CK, Mitroulis I, Miyazawa K, Mizushima N, Mograbi B, Mohseni S, Molero X, Mollereau B, Mollinedo F, Momoi T, Monastyrska I, Monick MM, Monteiro MJ, Moore MN, Mora R, Moreau K, Moreira PI, Moriyasu Y, Moscat J, Mostowy S, Mottram JC, Motyl T, Moussa CE, Müller S, Muller S, Münger K, Münz C, Murphy LO, Murphy ME, Musarò A, Mysorekar I, Nagata E, Nagata K, Nahimana A, Nair U, Nakagawa T, Nakahira K, Nakano H, Nakatogawa H, Nanjundan M, Naqvi NI, Narendra DP, Narita M, Navarro M, Nawrocki ST, Nazarko TY, Nemchenko A, Netea MG, Neufeld TP, Ney PA, Nezis IP, Nguyen HP, Nie D, Nishino I, Nislow C, Nixon RA, Noda T, Noegel AA, Nogalska A, Noguchi S, Notterpek L, Novak I, Nozaki T, Nukina N, Nürnberger T, Nyfeler B, Obara K, Oberley TD, Oddo S, Ogawa M, Ohashi T, Okamoto K, Oleinick NL, Oliver FJ, Olsen LJ, Olsson S, Opota O, Osborne TF, Ostrander GK, Otsu K, Ou JH, Ouimet M, Overholtzer M, Ozpolat B, Paganetti P, Pagnini U, Pallet N, Palmer GE, Palumbo C, Pan T, Panaretakis T, Pandey UB, Papackova Z, Papassideri I, Paris I, Park J, Park OK, Parys JB, Parzych KR, Patschan S, Patterson C, Pattingre S, Pawelek JM, Peng J, Perlmutter DH, Perrotta I, Perry G, Pervaiz S, Peter M, Peters GJ, Petersen M, Petrovski G, Phang JM, Piacentini M, Pierre P, Pierrefite-Carle V, Pierron G, Pinkas-Kramarski R, Piras A, Piri N, Platanias LC, Pöggeler S, Poirot M, Poletti A, Poüs C, Pozuelo-Rubio M, Prætorius-Ibba M, Prasad A, Prescott M, Priault M, Produit-Zengaffinen N, Progulske-Fox A, Proikas-Cezanne T, Przedborski S, Przyklenk K, Puertollano R, Puyal J, Qian SB, Qin L, Qin ZH, Quaggin SE, Raben N, Rabinowich H, Rabkin SW, Rahman I, Rami A, Ramm G, Randall G, Randow F, Rao VA, Rathmell JC, Ravikumar B, Ray SK, Reed BH, Reed JC, Reggiori F, Régnier-Vigouroux A, Reichert AS, Reiners JJ Jr, Reiter RJ, Ren J, Revuelta JL, Rhodes CJ, Ritis K, Rizzo E, Robbins J, Roberge M, Roca H, Roccheri MC, Rocchi S, Rodemann HP, Rodríguez de Córdoba S, Rohrer B, Roninson IB, Rosen K, Rost-Roszkowska MM, Rouis M, Rouschop KM, Rovetta F, Rubin BP, Rubinsztein DC, Ruckdeschel K, Rucker EB 3rd, Rudich A, Rudolf E, Ruiz-Opazo N, Russo R, Rusten TE, Ryan KM, Ryter SW, Sabatini DM, Sadoshima J, Saha T, Saitoh T, Sakagami H, Sakai Y, Salekdeh GH, Salomoni P, Salvaterra PM, Salvesen G, Salvioli R, Sanchez AM, Sánchez-Alcázar JA, Sánchez-Prieto R, Sandri M, Sankar U, Sansanwal P, Santambrogio L, Saran S, Sarkar S, Sarwal M, Sasakawa C, Sasnauskiene A, Sass M, Sato K, Sato M, Schapira AH, Scharl M, Schätzl HM, Scheper W, Schiaffino S, Schneider C, Schneider ME, Schneider-Stock R, Schoenlein PV, Schorderet DF, Schüller C, Schwartz GK, Scorrano L, Sealy L, Seglen PO, Segura-Aguilar J, Seiliez I, Seleverstov O, Sell C, Seo JB, Separovic D, Setaluri V, Setoguchi T, Settembre C, Shacka JJ, Shanmugam M, Shapiro IM, Shaulian E, Shaw RJ, Shelhamer JH, Shen HM, Shen WC, Sheng ZH, Shi Y, Shibuya K, Shidoji Y, Shieh JJ, Shih CM, Shimada Y, Shimizu S, Shintani T, Shirihai OS, Shore GC, Sibirny AA, Sidhu SB, Sikorska B, Silva-Zacarin EC, Simmons A, Simon AK, Simon HU, Simone C, Simonsen A, Sinclair DA, Singh R, Sinha D, Sinicrope FA, Sirko A, Siu PM, Sivridis E, Skop V, Skulachev VP, Slack RS, Smaili SS, Smith DR, Soengas MS, Soldati T, Song X, Sood AK, Soong TW, Sotgia F, Spector SA, Spies CD, Springer W, Srinivasula SM, Stefanis L, Steffan JS, Stendel R, Stenmark H, Stephanou A, Stern ST, Sternberg C, Stork B, Strålfors P, Subauste CS, Sui X, Sulzer D, Sun J, Sun SY, Sun ZJ, Sung JJ, Suzuki K, Suzuki T, Swanson MS, Swanton C, Sweeney ST, Sy LK, Szabadkai G, Tabas I, Taegtmeyer H, Tafani M, Takács-Vellai K, Takano Y, Takegawa K, Takemura G, Takeshita F, Talbot NJ, Tan KS, Tanaka K, Tanaka K, Tang D, Tang D, Tanida I, Tannous BA, Tavernarakis N, Taylor GS, Taylor GA, Taylor JP, Terada LS, Terman A, Tettamanti G, Thevissen K, Thompson CB, Thorburn A, Thumm M, Tian F, Tian Y, Tocchini-Valentini G, Tolkovsky AM, Tomino Y, Tönges L, Tooze SA, Tournier C, Tower J, Towns R, Trajkovic V, Travassos LH, Tsai TF, Tschan MP, Tsubata T, Tsung A, Turk B, Turner LS, Tyagi SC, Uchiyama Y, Ueno T, Umekawa M, Umemiya-Shirafuji R, Unni VK, Vaccaro MI, Valente EM, Van den Berghe G, van der Klei IJ, van Doorn W, van Dyk LF, van Egmond M, van Grunsven LA, Vandenabeele P, Vandenberghe WP, Vanhorebeek I, Vaquero EC, Velasco G, Vellai T, Vicencio JM, Vierstra RD, Vila M, Vindis C, Viola G, Viscomi MT, Voitsekhovskaja OV, von Haefen C, Votruba M, Wada K, Wade-Martins R, Walker CL, Walsh CM, Walter J, Wan XB, Wang A, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H, Wang HG, Wang HD, Wang J, Wang K, Wang M, Wang RC, Wang X, Wang X, Wang YJ, Wang Y, Wang Z, Wang ZC, Wang Z, Wansink DG, Ward DM, Watada H, Waters SL, Webster P, Wei L, Weihl CC, Weiss WA, Welford SM, Wen LP, Whitehouse CA, Whitton JL, Whitworth AJ, Wileman T, Wiley JW, Wilkinson S, Willbold D, Williams RL, Williamson PR, Wouters BG, Wu C, Wu DC, Wu WK, Wyttenbach A, Xavier RJ, Xi Z, Xia P, Xiao G, Xie Z, Xie Z, Xu DZ, Xu J, Xu L, Xu X, Yamamoto A, Yamamoto A, Yamashina S, Yamashita M, Yan X, Yanagida M, Yang DS, Yang E, Yang JM, Yang SY, Yang W, Yang WY, Yang Z, Yao MC, Yao TP, Yeganeh B, Yen WL, Yin JJ, Yin XM, Yoo OJ, Yoon G, Yoon SY, Yorimitsu T, Yoshikawa Y, Yoshimori T, Yoshimoto K, You HJ, Youle RJ, Younes A, Yu L, Yu L, Yu SW, Yu WH, Yuan ZM, Yue Z, Yun CH, Yuzaki M, Zabirnyk O, Silva-Zacarin E, Zacks D, Zacksenhaus E, Zaffaroni N, Zakeri Z, Zeh HJ 3rd, Zeitlin SO, Zhang H, Zhang HL, Zhang J, Zhang JP, Zhang L, Zhang L, Zhang MY, Zhang XD, Zhao M, Zhao YF, Zhao Y, Zhao ZJ, Zheng X, Zhivotovsky B, Zhong Q, Zhou CZ, Zhu C, Zhu WG, Zhu XF, Zhu X, Zhu Y, Zoladek T, Zong WX, Zorzano A, Zschocke J, Zuckerbraun B (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544
Kovacevic Z, McGivan JD (1983) Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev 63:547–605
Kovacs AL, Reith A, Seglen PO (1982) Accumulation of autophagosomes after inhibition of hepatocytic protein degradation by vinblastine, leupeptin or a lysosomotropic amine. Exp Cell Res 137:191–201
Krause U, Rider MH, Hue L (1996) Protein kinase signaling pathway triggered by cell swelling and involved in the activation of glycogen synthase and acetyl-CoA carboxylase in isolated rat hepatocytes. J Biol Chem 271:16668–16673
Krause U, Bertrand L, Hue L (2002a) Control of p70 ribosomal protein S6 kinase and acetyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in isolated hepatocytes. Eur J Biochem 269:3751–3759
Krause U, Bertrand L, Maisin L, Rosa M, Hue L (2002b) Signalling pathways and combinatory effects of insulin and amino acids in isolated rat hepatocytes. Eur J Biochem 269:3742–3750
Kroemer G, Levine B (2008) Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9:1004–1010
Ktistakis NT, Andrews S, Long J (2011) What is the advantage of a transient precursor in autophagosome biogenesis? Autophagy 7:118–122
Ktistakis NT, Manifava M, Schoenfelder P, Rotondo S (2012) How phosphoinositide 3-phosphate controls growth downstream of amino acids and autophagy downstream of amino acid withdrawal. Biochem Soc Trans 40:37–43
Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14:759–774
Lang F, Stehle T, Häussinger D (1989) Water, K+, H+, lactate and glucose fluxes during cell volume regulation in perfused rat liver. Pflugers Arch 413:209–216
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293
Larsen AK, Moller MT, Blankson H, Samari HR, Holden L, Seglen PO (2002) Naringin-sensitive phosphorylation of plectin, a cytoskeletal cross-linking protein, in isolated rat hepatocytes. J Biol Chem 277:34826–34835
Lavallard VJ, Meijer AJ, Codogno P, Gual P (2012) Autophagy, signaling and obesity. Pharmacol Res 66:513–525
Lee IH, Finkel T (2009) Regulation of autophagy by the p300 acetyltransferase. J Biol Chem 284:6322–6328
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA 105:3374–3379
Leverve XM, Caro LH, Plomp PJ, Meijer AJ (1987) Control of proteolysis in perifused rat hepatocytes. FEBS Lett 219:455–458
Li C, Allen A, Kwagh J, Doliba NM, Qin W, Najafi H, Collins HW, Matschinsky FM, Stanley CA, Smith TJ (2006) Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. J Biol Chem 281:10214–10221
Li W, Zhu S, Li J, Assa A, Jundoria A, Xu J, Fan S, Eissa NT, Tracey KJ, Sama AE, Wang H (2011) EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem Pharmacol 81:1152–1163
Li L, Ishdorj G, Gibson SB (2012a) Reactive oxygen species regulation of autophagy in cancer: implications for cancer treatment. Free Radic Biol Med 53:1399–1410
Li M, Li C, Allen A, Stanley CA, Smith TJ (2012b) The structure and allosteric regulation of mammalian glutamate dehydrogenase. Arch Biochem Biophys 519:69–80
Li L, Chen Y, Gibson SB (2013) Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal 25:50–65
Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB (2007) The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 9:218–224
Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A (2012) Autophagy in lysosomal storage disorders. Autophagy 8:719–730
Long X, Ortiz-Vega S, Lin Y, Avruch J (2005) Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem 280:23433–23436
Lorin S, Hamai A, Mehrpour M, Codogno P (2013a) Autophagy regulation and its role in cancer. Semin Cancer Biol 23:361–379
Lorin S, Tol MJ, Bauvy C, Strijland A, Pous C, Verhoeven AJ, Codogno P, Meijer AJ (2013b) Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy 9:850–860
Luiken JJ, Blommaart EF, Boon L, van Woerkom GM, Meijer AJ (1994) Cell swelling and the control of autophagic proteolysis in hepatocytes: involvement of phosphorylation of ribosomal protein S6? Biochem Soc Trans 22:508–511
Luiken JJ, Aerts JM, Meijer AJ (1996) The role of the intralysosomal pH in the control of autophagic proteolytic flux in rat hepatocytes. Eur J Biochem 235:564–573
Lynch CJ, Fox HL, Vary TC, Jefferson LS, Kimball SR (2000) Regulation of amino acid-sensitive TOR signaling by leucine analogues in adipocytes. J Cell Biochem 77:234–251
Lynch CJ, Halle B, Fujii H, Vary TC, Wallin R, Damuni Z, Hutson SM (2003) Potential role of leucine metabolism in the leucine-signaling pathway involving mTOR. Am J Physiol Endocrinol Metab 285:E854–E863
Mailloux RJ, Beriault R, Lemire J, Singh R, Chenier DR, Hamel RD, Appanna VD (2007) The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS ONE 2:e690
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6:458–471
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129:1261–1274
Marc RJ, Wu G (2009) Glutamine, arginine, and leucine signaling in the intestine. Amino Acids 37:111–122
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G (2014a) Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15:81–94
Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, Scoazec M, Durand S, Enot DP, Fernández ÁF, Martins I, Kepp O, Senovilla L, Bauvy C, Morselli E, Vacchelli E, Bennetzen M, Magnes C, Sinner F, Pieber T, López-Otín C, Maiuri MC, Codogno P, Andersen JS, Hill JA, Madeo F, Kroemer G (2014b) Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell 53:710–725
Martin PM, Sutherland AE (2001) Exogenous amino acids regulate trophectoderm differentiation in the mouse blastocyst through an mTOR-dependent pathway. Dev Biol 240:182–193
Mates JM, Segura JA, Martin-Rufian M, Campos-Sandoval JA, Alonso FJ, Marquez J (2013) Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med 13:514–534
Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100:914–922
Mavrakis M, Lippincott-Schwartz J, Stratakis CA, Bossis I (2006) Depletion of type IA regulatory subunit (RIalpha) of protein kinase A (PKA) in mammalian cells and tissues activates mTOR and causes autophagic deficiency. Hum Mol Genet 15:2962–2971
McAlpine F, Williamson LE, Tooze SA, Chan EY (2013) Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy 9:361–373
Meijer AJ (2008) Amino acid regulation of autophagosome formation. Methods Mol Biol 445:89–109
Meijer AJ (2009) Autophagy research: lessons from metabolism. Autophagy 5:3–5
Meijer AJ, Codogno P (2008) Nutrient sensing: TOR’s Ragtime. Nat Cell Biol 10:881–883
Meijer AJ, Codogno P (2009) Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 46:210–240
Meijer AJ, Codogno P (2011) Autophagy: regulation by energy sensing. Curr Biol 21:R227–R229
Meijer AJ, Baquet A, Gustafson L, van Woerkom GM, Hue L (1992) Mechanism of activation of liver glycogen synthase by swelling. J Biol Chem 267:5823–5828
Meijer AJ, Gustafson LA, Luiken JJ, Blommaart PJ, Caro LH, van Woerkom GM, Spronk C, Boon L (1993) Cell swelling and the sensitivity of autophagic proteolysis to inhibition by amino acids in isolated rat hepatocytes. Eur J Biochem 215:449–454
Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ (2006) AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 281:34870–34879
Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156:771–785
Mizushima N, Klionsky DJ (2007) Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 27:19–40
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741
Moazed B, Desautels M (2002) Control of proteolysis by norepinephrine and insulin in brown adipocytes: role of ATP, phosphatidylinositol 3-kinase, and p70 S6K. Can J Physiol Pharmacol 80:541–552
Morales CR, Pedrozo Z, Lavandero S, Hill JA (2014) Oxidative stress and autophagy in cardiovascular homeostasis. Antioxid Redox Signal 20:507–518
Mordier S, Deval C, Bechet D, Tassa A, Ferrara M (2000) Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin-independent signaling pathway. J Biol Chem 275:29900–29906
Mortimore GE, Mondon CE (1970) Inhibition by insulin of valine turnover in liver. Evidence for a general control of proteolysis. J Biol Chem 245:2375–2383
Mortimore GE, Poso AR, Kadowaki M, Wert JJ Jr (1987) Multiphasic control of hepatic protein degradation by regulatory amino acids. General features and hormonal modulation. J Biol Chem 262:16322–16327
Mortimore GE, Wert JJ Jr, Adams CE (1988) Modulation of the amino acid control of hepatic protein degradation by caloric deprivation. Two modes of alanine co-regulation. J Biol Chem 263:19545–19551
Naito T, Kuma A, Mizushima N (2013) Differential contribution of insulin and amino acids to the mTORC1-autophagy pathway in the liver and muscle. J Biol Chem 288:21074–21081
Nakajo T, Yamatsuji T, Ban H, Shigemitsu K, Haisa M, Motoki T, Noma K, Nobuhisa T, Matsuoka J, Gunduz M, Yonezawa K, Tanaka N, Naomoto Y (2005) Glutamine is a key regulator for amino acid-controlled cell growth through the mTOR signaling pathway in rat intestinal epithelial cells. Biochem Biophys Res Commun 326:174–180
Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ (2001) Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol 21:4347–4368
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136:521–534
Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, Zwartkruis FJ, Thomas G (2005) Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci USA 102:14238–14243
Noda T, Ohsumi Y (1998) Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273:3963–3966
Ohsumi Y (2014) Historical landmarks of autophagy research. Cell Res 24:9–23
Oshiro N, Rapley J, Avruch J (2013) Amino acids activate mTOR complex1 without changing Rag GTPase guanyl nucleotide charging. J Biol Chem 289:2658–2674
Patti ME, Brambilla E, Luzi L, Landaker EJ, Kahn CR (1998) Bidirectional modulation of insulin action by amino acids. J Clin Invest 101:1519–1529
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P (2000) Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 275:992–998
Petit CS, Roczniak-Ferguson A, Ferguson SM (2013) Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol 202:1107–1122
Pfeifer U (1977) Inhibition by insulin of the physiological autophagic breakdown of cell organelles. Acta Biol Med Ger 36:1691–1694
Pfeifer U (1978) Inhibition by insulin of the formation of autophagic vacuoles in rat liver. A morphometric approach to the kinetics of intracellular degradation by autophagy. J Cell Biol 78:152–167
Pfisterer SG, Mauthe M, Codogno P, Proikas-Cezanne T (2011) Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol Pharmacol 80:1066–1075
Polson HE, de LJ, Rigden DJ DJ, Reedijk M, Urbe S, Clague MJ, Tooze SA (2010) Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6:506–522
Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC (2013) Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 154:1285–1299
Quan W, Lee MS (2013) Role of autophagy in the control of body metabolism. Endocrinol Metab (Seoul) 28:6–11
Rahman M, Mofarrahi M, Kristof AS, Nkengfac B, Harel S, Hussain SN (2014) Reactive oxygen species regulation of autophagy in skeletal muscles. Antioxid Redox Signal 20:443–459
Ramanathan A, Schreiber SL (2009) Direct control of mitochondrial function by mTOR. Proc Natl Acad Sci USA 106:22229–22232
Rao VA, Klein SR, Bonar SJ, Zielonka J, Mizuno N, Dickey JS, Keller PW, Joseph J, Kalyanaraman B, Shacter E (2010) The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J Biol Chem 285:34447–34459
Rigbolt KT, Zarei M, Sprenger A, Becker AC, Diedrich B, Huang X, Eiselein S, Kristensen AR, Gretzmeier C, Andersen JS, Zi Z, Dengjel J (2014) Characterization of early autophagy signaling by quantitative phosphoproteomics. Autophagy 10:356–371
Roccio M, Bos JL, Zwartkruis FJ (2006) Regulation of the small GTPase Rheb by amino acids. Oncogene 25:657–664
Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730
Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y (2010) AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab 298:E751–E760
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15:741–750
Russell RC, Yuan HX, Guan KL (2014) Autophagy regulation by nutrient signaling. Cell Res 24:42–57
Sahani MH, Itakura E, Mizushima N (2014) Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 10:431–441
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168
Scherz-Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36:30–38
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26:1749–1760
Schieke SM, Phillips D, McCoy JP Jr, Aponte AM, Shen RF, Balaban RS, Finkel T (2006) The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem 281:27643–27652
Schink KO, Raiborg C, Stenmark H (2013) Phosphatidylinositol 3-phosphate, a lipid that regulates membrane dynamics, protein sorting and cell signalling. BioEssays 35:900–912
Schriever SC, Deutsch MJ, Adamski J, Roscher AA, Ensenauer R (2013) Cellular signaling of amino acids towards mTORC1 activation in impaired human leucine catabolism. J Nutr Biochem 24:824–831
Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD (1993) Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 260:88–91
Schworer CM, Mortimore GE (1979) Glucagon-induced autophagy and proteolysis in rat liver: mediation by selective deprivation of intracellular amino acids. Proc Natl Acad Sci USA 76:3169–3173
Seglen PO (1977) Inhibitor of protein degradation formed during incubation of isolated rat hepatocytes in a cell culture medium. Its identification as ammonia. Exp Cell Res 107:207–217
Seglen PO, Bohley P (1992) Autophagy and other vacuolar protein degradation mechanisms. Experientia 48:158–172
Seglen PO, Gordon PB (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 79:1889–1892
Seglen PO, Gordon PB, Poli A (1980) Amino acid inhibition of the autophagic/lysosomal pathway of protein degradation in isolated rat hepatocytes. Biochim Biophys Acta 630:103–118
Sener A, Malaisse WJ (1980) l-leucine and a nonmetabolized analogue activate pancreatic islet glutamate dehydrogenase. Nature 288:187–189
Settembre C, Di Malta C, Polito VA, Garcia AM, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31:1095–1108
Shah OJ, Anthony JC, Kimball SR, Jefferson LS (2000) 4E-BP1 and S6K1: translational integration sites for nutritional and hormonal information in muscle. Am J Physiol Endocrinol Metab 279:E715–E729
Shanware NP, Bray K, Abraham RT (2013) The PI3 K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Annu Rev Pharmacol Toxicol 53:89–106
Shen HM, Mizushima N (2014) At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci 39:61–71
Shibutani ST, Yoshimori T (2014) A current perspective of autophagosome biogenesis. Cell Res 24:58–68
Shigemitsu K, Tsujishita Y, Hara K, Nanahoshi M, Avruch J, Yonezawa K (1999a) Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J Biol Chem 274:1058–1065
Shigemitsu K, Tsujishita Y, Miyake H, Hidayat S, Tanaka N, Hara K, Yonezawa K (1999b) Structural requirement of leucine for activation of p70 S6 kinase. FEBS Lett 447:303–306
Simonsen A, Tooze SA (2009) Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 186:773–782
Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG (2005) The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem 280:18717–18727
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496:101–105
Stanley RE, Ragusa MJ, Hurley JH (2014) The beginning of the end: how scaffolds nucleate autophagosome biogenesis. Trends Cell Biol 24:73–81
Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK (2009) The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci USA 106:17049–17054
Stoll B, Gerok W, Lang F, Häussinger D (1992) Liver cell volume and protein synthesis. Biochem J 287:217–222
Sun Y, Chen J (2008) mTOR signaling: PLD takes center stage. Cell Cycle 7:3118–3123
Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y (2001) The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J 20:5971–5981
Tallóczy Z, Jiang W, Virgin HW 4th, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B (2002) Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99:190–195
Tan CY, Hagen T (2013) Post-translational regulation of mTOR complex 1 in hypoxia and reoxygenation. Cell Signal 25:1235–1244
Tardito S, Chiu M, Franchi-Gazzola R, Dall’Asta V, Comi P, Bussolati O (2012) The non-proteinogenic amino acids l-methionine sulfoximine and DL-phosphinothricin activate mTOR. Amino Acids 42:2507–2512
Tassa A, Roux MP, Attaix D, Bechet DM (2003) Class III phosphoinositide 3-kinase–Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J 376:577–586
Thevelein JM, de Winde JH (1999) Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol Microbiol 33:904–918
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284:8023–8032
Toyoda T, Hayashi T, Miyamoto L, Yonemitsu S, Nakano M, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Inoue G, Otaka A, Sato K, Fushiki T, Nakao K (2004) Possible involvement of the alpha1 isoform of 5′AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab 287:E166–E173
Tremblay F, Marette A (2001) Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 276:38052–38060
Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, Spooner E, Sabatini DM (2013) The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 52:495–505
Tzatsos A, Kandror KV (2006) Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol 26:63–76
Ueno T, Ezaki J, Kominami E (2012) Metabolic contribution of hepatic autophagic proteolysis: old wine in new bottles. Biochim Biophys Acta 1824:51–58
Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431:200–205
van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van BR, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, Verhagen LP, Groot Koerkamp MJ, Braat AK, Dansen TB, Holstege FC, Gebhardt R, Burgering BM, Coffer PJ (2012) Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol 14:829–837
van Sluijters DA, Dubbelhuis PF, Blommaart EF, Meijer AJ (2000) Amino-acid-dependent signal transduction. Biochem J 351:545–550
Wang X, Campbell LE, Miller CM, Proud CG (1998) Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem J 334:261–267
Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B (2012) Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338:956–959
Wang XJ, Yu J, Wong SH, Cheng AS, Chan FK, Ng SS, Cho CH, Sung JJ, Wu WK (2013) A novel crosstalk between two major protein degradation systems: regulation of proteasomal activity by autophagy. Autophagy 9:1500–1508
Wauson EM, Zaganjor E, Lee AY, Guerra ML, Ghosh AB, Bookout AL, Chambers CP, Jivan A, McGlynn K, Hutchison MR, Deberardinis RJ, Cobb MH (2012) The G protein-coupled taste receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol Cell 47:851–862
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30:678–688
Weidberg H, Shvets E, Elazar Z (2011) Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 80:125–156
Wiczer BM, Thomas G (2012) Phospholipase D and mTORC1: nutrients are what bring them together. Sci Signal 5:e13
Wilkinson DJ, Hossain T, Hill DS, Phillips BE, Crossland H, Williams J, Loughna P, Churchward-Venne TA, Breen L, Phillips SM, Etheridge T, Rathmacher JA, Smith K, Szewczyk NJ, Atherton PJ (2013) Effects of leucine and its metabolite beta-hydroxy-beta-methylbutyrate on human skeletal muscle protein metabolism. J Physiol 591:2911–2923
Wirth M, Joachim J, Tooze SA (2013) Autophagosome formation–the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol 23:301–309
Wong PM, Puente C, Ganley IG, Jiang X (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9:124–137
Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, Meng Q, Cheng Y, Gao X, Li J, Liu Y, Guo F (2011) Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes 60:746–756
Xu G, Kwon G, Marshall CA, Lin TA, Lawrence JC Jr, McDaniel ML (1998) Branched-chain amino acids are essential in the regulation of PHAS-I and p70 S6 kinase by pancreatic beta-cells. A possible role in protein translation and mitogenic signaling. J Biol Chem 273:28178–28184
Xu G, Kwon G, Cruz WS, Marshall CA, McDaniel ML (2001) Metabolic regulation by leucine of translation initiation through the mTOR-signaling pathway by pancreatic beta-cells. Diabetes 50:353–360
Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, Foster DA (2011) Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1). J Biol Chem 286:25477–25486
Yan L, Mieulet V, Burgess D, Findlay GM, Sully K, Procter J, Goris J, Janssens V, Morrice NA, Lamb RF (2010) PP2A T61 epsilon is an inhibitor of MAP4K3 in nutrient signaling to mTOR. Mol Cell 37:633–642
Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, Deberardinis RJ (2009) Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 69:7986–7993
Ye L, Varamini B, Lamming DW, Sabatini DM, Baur JA (2012) Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2. Front Genet 3:177
Yoon MS, Du G, Backer JM, Frohman MA, Chen J (2011) Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol 195:435–447
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465:942–946
Yuan HX, Russell RC, Guan KL (2013) Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 9:1983–1995
Zhang J, Kim J, Alexander A, Cai S, Tripathi DN, Dere R, Tee AR, Tait-Mulder J, Di Nardo A, Han JM, Kwiatkowski E, Dunlop EA, Dodd KM, Folkerth RD, Faust PL, Kastan MB, Sahin M, Walker CL (2013) A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat Cell Biol 15:1186–1196
Zhou J, Farah BL, Sinha RA, Wu Y, Singh BK, Bay BH, Yang CS, Yen PM (2014) Epigallocatechin-3-Gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS ONE 9:e87161
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334:678–683
Acknowledgments
PC is supported by institutional funding from INSERM, CNRS and University Paris Descartes and grants from ANR and INCa. SL is supported by funding from the University Paris-Sud.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Meijer, A.J., Lorin, S., Blommaart, E.F. et al. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 47, 2037–2063 (2015). https://doi.org/10.1007/s00726-014-1765-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-014-1765-4