
Abstract
Given the clear role of GBA in the pathogenesis of Parkinson’s disease (PD) and its impact on phenotypical characteristics, this review provides an overview of the current knowledge of GBA-associated PD with a special focus on clinical trajectories and the underlying pathological mechanisms. Importantly, differences and characteristics based on mutation severity are recognized, and current as well as potential future treatment options are discussed. These findings will inform future strategies for patient stratification and cohort enrichment as well as suitable outcome measures when designing clinical trials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Over the last decades, research in genetically defined forms of Parkinson’s disease (PD) led to the identification of specific pathways underlying the pathophysiology of the disease. Next to defects in vesicular trafficking, mitochondrial and importantly lysosomal dysfunction represent the most relevant pathways (Jankovic and Tan 2020). Studying these early events provide entry points to develop novel therapeutic targets for stratified patient groups as an important step towards precision neurology. The present article exemplifies such strategies focusing on PD patients with different variants in the glucocerebrosidase (GBA) gene (PDGBA). Also, obstacles of translational research into patient cohorts and study designs for clinical trials are discussed.
GBA and Parkinson
GBA variants are the most important genetic risk factor for PD
Biallelic variants in the GBA gene cause Gaucher’s disease (GD), the most common lysosomal storage disorder with tissue accumulation of glucosylceramides due to deficiency of the lysosomal enzyme glucocerebrosidase (GCase). Interestingly, about 25% of GD patients report a first- or second-degree relative to present with PD (Goker-Alpan et al. 2004; Halperin et al. 2006). This important clinical observation was the hint to the fact that heterozygous variants in the GBA gene are associated with PD. Subsequently, a large multi-centre study across four continents analysed 5691 PD patients of different ethnic origin compared to 4898 controls and confirmed that with an overall odds ratio (OR) of 5.43, heterozygous variants in the GBA gene represent the most important genetic risk factor for PD (Sidransky et al. 2009). This has now been confirmed across different ethnic populations with Caucasian, Asian (Japanese, Chinese, Taiwanese), Hispanic, and African ancestry (den Heijer et al. 2020; Neumann et al. 2009; Lesage et al. 2011; Chen et al. 2014; Mahungu et al. 2020).
To date, more than 100 different variants have been associated with the risk of PD. However, the pathogenicity of different variants varies largely (Table 1). Whereas variants classified as severe variants (e.g. p.L444P) show an odds ratio of 10–15 for developing PD and mild variants (e.g. p.N370S) have an odd ratio of ≤ 5 for PD, some variants that are non-pathogenic for GD have been proven to increase the risk for PD e.g. p.E326K and p.T369M (Iwaki et al. 2019; Zhang et al. 2018; Straniero et al. 2020). These variants show the lowest odds ratios and are thus classified as risk variants. Consequently, GBA-subgroup classification for PD patients is often based on variant severity according to established genotype risks reported for PD (PDGBA_severe, PDGBA_mild, PDGBA_risk). Interestingly, we see a huge variability of variant distribution among different ethnicities. About 20% of PD patients with Ashkenazi Jewish ancestry carry a GBA variant, with the large majority harbouring the mild p.N370S (> 70%), whereas the severe p.L444P variant is identified in about 5%. Together, the two variants account for about 80% of variants in Ashkenazi Jewish PD patients. In non-Ashkenazi Jewish PD patients, p.L444P is detected in about 30–40% of patients and p.N370S in about 20%, together accounting for 50–60% of variants (Sidransky et al. 2009), indicating that about 40% of variants could be missed if focusing solely on p.N370S and p.L444P. These findings highlight the need for full-gene sequencing and stratification according to variant severity. Moreover, penetrance and disease risk in PDGBA are age-dependent (Anheim et al. 2012; Straniero et al. 2020) and further modified by the composite PD-associated polygenetic risk score (PRS) and single-nucleotide polymorphisms in SNCA, CSTB and TMEM175, the two latter genes encoding proteins associated with lysosomal homeostasis and protein clearance (Blauwendraat et al. 2020b).
PDGBA: severe clinical trajectories with early cognitive decline
Detailed investigation of the phenotypical spectrum, longitudinal trajectories, and rate of progression of motor and non-motor symptoms is of utmost importance to estimate effect sizes and design clinical trials for disease-modifying therapies (duration, sample sizes, progression rates, expected spectrum of symptoms, etc.).
In general, PDGBA show an earlier age at onset compared to PD patients without GBA variants (PDGBA_wildtype) with a median onset in the early 50s (Sidransky et al. 2009; Blauwendraat et al. 2019). Of note, this effect is not only attributable to GBA variants per se, but is driven by GBA variant severity and variant burden with severe variants as well as homozygous and compound heterozygous variants predisposing to the youngest age at onset (Malek et al. 2018; Thaler et al. 2017). Moreover, age at onset is further reduced in PDGBA by non-coding variants in SNCA and TMEM175 (Blauwendraat et al. 2020b). Although younger, PDGBA present with a higher prevalence of cognitive impairment and more frequently suffer from additional non-motor symptoms including neuropsychiatric disturbances (depression, anxiety, and hallucination), autonomic dysfunction and sleep disturbances such as REM-sleep-behaviour disorder (RBD) when compared to PDGBA_wildtype, (Brockmann et al. 2011; Barrett et al. 2014). These findings have been replicated consistently over the following years in other PD cohorts worldwide, the latest large clinical genome-wide association study in 4093 PD patients (Iwaki et al. 2019). Importantly, GBA variants that are classified as severe (PDGBA_severe) have been associated with a more aggressive clinical phenotype suggesting a relevant effect depending on GBA variant severity (Cilia et al. 2016; Thaler et al. 2018; Petrucci et al. 2020; Lerche et al. 2021a).
Data from longitudinally investigated cohorts of PDGBA confirm findings from cross-sectional evaluations and revealed that PDGBA, although younger in age and age at onset, present with an accelerated disease progression in terms of motor impairment and cognitive decline as compared to PDGBA_wildtype. Moreover, survival rates are shorter when compared to PDGBA_wildtype (Brockmann et al. 2015b; Cilia et al. 2016). In a British cohort, after 10 years of disease duration, 46% of PDGBA remained dementia-free in comparison to 68% of PDGBA_wildtype. After 15 years, 64% of the surviving PDGBA_wildtype remained dementia-free. At that time point, all PDGBA had developed dementia or already died. Mean time to dementia was 8.3 years in PDGBA compared to 13.7 years in PDGBA_wildtype. Similarly, at 5 year disease duration, 67.5% of PDGBA had reached HY stadium 3, compared to 43% of PDGBA_wildtype. Mean time to Hoehn and Yahr staging 3 was 4.7 years in PDGBA compared to 6.8 years in PDGBA_wildtype (Stoker et al. 2020). Similar results were reported in a large longitudinal cohort of Italian patients with a clearly more aggressive pattern depending on GBA variant severity (Cilia et al. 2016). Interestingly, a recent study reports that PDGBA who are treated with deep brain stimulation (DBS) in the subthalamic nucleus (STN) showed an even more rapid cognitive decline compared to PDGBA without DBS as well as PDGBA_wildtype with and without DBS. This finding suggests that the additive effect of GBA variants and STN-DBS negatively impact cognition and that presurgical genetic screening should be considered (Pal et al. 2022). Further studies are needed for replication and to evaluate the underlying pathophysiological mechanisms.
The typical motor manifestation of PD is preceded by a prodromal phase that is characterized by a variety non-motor and early motor signs (Berg et al. 2015). Non-motor symptoms include among others hyposmia, autonomic dysfunction, and neuropsychiatric symptoms, whereas reduced arm swing and bradykinesia indicate early motor signs. However, type, prevalence, time of occurrence, and rate of progression of these prodromal symptoms are variable between patients. Given the findings from the manifest disease phase in PDGBA with the pronounced non-motor profile and more rapid disease progression, we retrospectively assessed patient’s perception of their individual prodromal phase before PD diagnosis. Comparing PDGBA and PDGBA_wildtype, we could show that: (i) PDGBA demonstrate a higher prevalence of prodromal symptoms and a shorter prodromal phase with almost parallel beginning of non-motor and early motor signs before PD diagnosis. Contrary, PDGBA_wildtype show a long prodromal interval starting with non-motor symptoms long before early motor signs manifested. (ii) PDGBA with severe variants reported the highest total amount of prodromal signs. These findings suggest that complexity of symptoms known from the manifest disease might be present already in the prodromal phase (Zimmermann et al. 2018). Similarly, prospective studies found that prodromal GBA variant carriers present with more pronounced deterioration of motor and non-motor symptoms, specifically cognitive decline and hyposmia when compared to healthy controls without GBA variant (Avenali et al. 2019; Beavan et al. 2015; Mullin et al. 2019). Another study in patients with REM-sleep behaviour disorder (RBD) reports that GBA variants are associated with accelerated phenoconversion to PD and/or dementia in this specific cohort (Honeycutt et al. 2019).
GBA variants are an important genetic risk factor for Dementia with Lewy Bodies (DLB)
The important finding that PDGBA shows pronounced and early development of dementia prompted the community to perform a large multicenter analysis across 11 centres evaluating GBA variants in 721 cases with DLB, which represents a clinico-pathological continuum to PD. With an even higher OR than seen in PD, GBA variants are also strongly associated with DLB (8.28). Similar to PD, GBA variants predispose to an earlier age at onset, more pronounced disease severity/progression and rather “pure” form of DLB without concomitant Alzheimer’s profile as defined by CSF p-tau/Aβ1-42 ratio (Nalls et al. 2013; van der Lee et al. 2021). This study further supports GBA variants as a significant genetic risk factor for synucleinopathies and confirmed the overall impression that GBA-associated Parkinsonism predisposes to an increased incidence of dementia (Fig. 1).

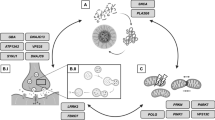
Pathogenic mechanisms underlying PDGBA. Loss of lysosomal GCase activity results in impaired autophagy affecting the degradation of both physiological (red dot) and misfolded α-synuclein (red dot complex) resulting in the aggregation of α-synuclein (red strains). GBA variants also cause the GCase protein to misfold in the ER (brown enzyme) with impaired trafficking to the lysosome which also affects α-synuclein degradation. Accumulation of GCase substrates (GlcCer and GlcSph, yellow) also causes α-synuclein misfolding and aggregation, as may changes in the lipid homeostasis (both sphingolipids and phospholipids) of cellular membranes (yellow) due to decreased lysosomal function. In PDGBA_wildtype, the trafficking of wild-type GCase (green enzyme) can be inhibited by increased levels of α-synuclein (red dot complex) and α-synuclein fibrils (red strains), and contribute GCase deficiency (brown enzyme) irrespective of a GBA mutation. This figure was adapted from “Brainstem with Callout” and “Structural Overview of an Animal Cell”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates
Pathomechanisms in PDGBA
Experimental evidence from cell models suggests that GBA variants result in disrupted protein folding of GCase in the endoplasmic reticulum (ER), impaired trafficking of GCase from the ER to Golgi and ultimately in lower lysosomal GCase enzyme activity. This in turn causes a build-up of glucosylceramides (GlcCer) and glucosylsphingosines (GlcSph) (Beutler 1992) and impairs lysosomal function and thereby the degradation of α-synuclein (Mazzulli et al. 2011).
GBA variants predispose to accelerated α-synuclein aggregation and Lewy-body pathology
Post-mortem studies show enhanced aggregation and propagation of α-synuclein not only in the substantia nigra and putamen but also wide-spread neocortical Lewy-body pathology in brain tissue of PDGBA and DLBGBA (Neumann et al. 2009; Gundner et al. 2019).
The field of PET imaging markers to assess the cerebral load of α-synuclein in-vivo is difficult. However, this month [03(2022)] first positive results were reported at the AD/PD Conference for a new PET tracer developed by AC Immune to distinguish multiple system atrophy (MSA) from healthy controls and patients with other forms of α-synuclein (PD, DLB). Therefore, research in PD has focused on CSF. Yet, it is unclear whether CSF profiles of α-synuclein species reflect brain pathology. Cross-sectional and longitudinal analyses in PDGBA_wildtype and PDGBA demonstrated decreased CSF levels of total α-synuclein compared to healthy controls with the highest decrease in PDGBA patients carrying severe variants (Malek et al. 2014; Mollenhauer et al. 2019; Lerche et al. 2020, 2021a). Correspondingly, the same pattern was also reported in patients with DLBGBA (Lerche et al. 2019a). However, a substantial inter-individual variability and overlap with healthy controls is seen, so that CSF levels of total α-synuclein are not ideal. Recently, the ultrasensitive assays real‐time quaking‐induced conversion (RT‐QuIC) and protein misfolding cyclic amplification (PMCA) have been successfully implemented. These assays exploit the seeding capacities of prion or prion-like proteins as an amplification strategy to reveal minute amounts of disease-specific protein aggregates in CSF (Fairfoul et al. 2016; Shahnawaz et al. 2017). Both methods are highly sensitive (88–96%) and specific (83–98%) for α-synuclein aggregates and Lewy-body pathology in PD and DLB as assessed in matched CSF/brain samples compared to healthy controls and other forms of dementia and parkinsonism (Rossi et al. 2020; Kang et al. 2019). However, histopathological findings in some genetic forms of PD are remarkably variable. While PDGBA show extensive Lewy-body pathology, most PD patients with bi-allelic mutations in the recessive gene PRKN (PDrecessive_bi-allelic) show nigral degeneration without Lewybodies (Schneider and Alcalay 2017). Also, histopathology in PD patients with LRRK2 mutations (PDLRRK2) is variable, including typical Lewy-body pathology, misfolded tau deposition, or nigral degeneration without Lewy-body (Zimprich et al. 2004; Heckman et al. 2016; Kalia et al. 2015). This prompted us to evaluate CSF α-synuclein seeding capacities with RT-QuIC in two large cohorts of PD and DLB patients enriched for genetic forms. Remarkably, PDGBA (93%) and DLBGBA (100%), especially those carrying severe variants, showed the highest percentage of positive α-synuclein seeding and the most pronounced α-synuclein seeding kinetics. In contrast, PDrecessive_bi-allelic did not show CSF α-synuclein seeding at all, whereas those carrying heterozygous mutations in these recessive genes showed less α-synuclein seeding than PDwildtype (91%) with a reduced positivity rate of 59%. Also, PDLRRK2 showed a reduced rate of α-synuclein seeding (78%) compared to PDwildtype (Brockmann et al. 2021). The heterogeneity in α-synuclein seeding activity among the different genetic forms mirrors histopathological findings in these cases and highlight the value of α-synuclein seeding activity as an in-vivo marker of Lewy-body pathology.
The accelerated cognitive decline PDGBA makes this subgroup of PD a good model to study CSF profiles that are associated with cognitive impairment. In general, limbic and/or cortical Lewy-body pathology is hypothesized to be the main substrate forcing driving cognitive decline in PD (Aarsland et al. 2005). In more recent years, it became clear that a considerable proportion of PD patients who developed dementia in their disease course show concomitant amyloid-beta and tau pathology at autopsy in addition to the typical Lewy-body pathology (Halliday et al. 2008; Compta et al. 2011). Correspondingly, reduced CSF levels of Amyloid-beta1-42 (Aβ1-42) and/or elevated CSF levels of total-Tau (t-Tau) and phospho-Tau (p-Tau) have been reported to be associated with cognitive impairment in PD (Brockmann et al. 2015a, 2017; Lerche et al. 2019b; Kang et al. 2016). However, this seems not to be the case in PDGBA as CSF levels of Aβ1-42, t-Tau, and p-Tau are similar to those seen in healthy control individuals. In light of the CSF profiles of reduced total levels of α-synuclein and the prominent α-synuclein seeding activity, the pronounced cognitive decline in PDGBA is driven by α-synuclein aggregation and cortical Lewy-body pathology.
Taken together, these histopathological and CSF characteristics of predominant and accelerated α-synuclein-driven Lewy-body pathology make PDGBA and DLBGBA a role model to study pathways leading to α-synuclein aggregation and highlight these patient cohorts as prime candidates for clinical trials targeting α-synuclein.
GCase deficiency and α-synuclein aggregation
Heterozygous variants in the GBA gene are associated with a reduction of GCase protein levels and GCase enzyme activity in cell and animal models as well as in a variety of patient-derived biomaterials (Lerche et al. 2021a; Alcalay et al. 2015, 2020; Schondorf et al. 2014; Paciotti et al. 2019). Again, the degree of reduction is dependent from variant severity. Interestingly, GCase activity is also reduced in PDGBA_wildtype, albeit to a lesser degree (Parnetti et al. 2017).
There is reasonable evidence from different cell models including induced pluripotent stem (IPS) cell-derived human dopaminergic midbrain neurons and human midbrain organoids that deficiency of the GCase enzyme is paralleled by increased levels of intracellular α-synuclein, specifically α-synuclein species susceptible to aggregation such as high molecular weight and decreased tetramer/monomer ratio (Schondorf et al. 2014; Kim et al. 2018; Magalhaes et al. 2016; Aflaki et al. 2016; Mazzulli et al. 2016; Jo et al. 2021). Correspondingly, post-mortem studies in PDGBA and DLBGBA and to a lesser degree also in PDGBA_wildtype and DLBGBA_wildtype revealed that reduced GCase protein levels and reduced GCase enzyme activity are accompanied by increased levels of intracellular α-synuclein. Notably, these findings are not restricted to the substantia nigra and putamen but also identified in cortical regions (Murphy et al. 2014; Gegg et al. 2012; Moors et al. 2019; Gundner et al. 2019).
More specifically, it is suggested that lysosomal GCase and α-synuclein are linked in a bidirectional pathogenic loop as shown in cell cultures and IPS cell-derived dopaminergic midbrain neurons: (I) functional loss of GCase activity compromises lysosomal degradation of α-synuclein and promotes its aggregation. (II) α-Synuclein itself inhibits the activity of GCase (Mazzulli et al. 2011; Schondorf et al. 2014). Consequently, PDGBA fulfill both conditions of this bidirectional loop in parallel leading to a self-reinforcing mechanism. Thereby, α-synuclein aggregation and propagation might be accelerated which possibly explains the wide-spread neocortical Lewy-body pathology and rapid clinical progression.
However, this bidirectional loop between GCase and α-synuclein might be oversimplified, since we have clear evidence for a more complex impairment of the autophagy–lysosomal pathway including disrupted macroautophagy with reduced fusion of autophagosomes with lysosomes and decreased expression/activity of other proteolytic lysosomal enzymes such as cathepsin B and D (Aflaki et al. 2020; Blauwendraat et al. 2020a; Lerche et al. 2021b).
Disturbance in sphingolipid homeostasis and α-synuclein aggregation
Adding to the complexity of the underlying pathophysiology are additional alterations of intracellular and membrane-associated sphingolipid homeostasis. In GD patients, GD post-mortem brain studies, and IPS cell-derived human dopaminergic midbrain neurons with bi-allelic and heterozygous GBA variants, GCase deficiency results in accumulation of the GCase substrates GlcCer and GlcSph. PDGBA patients, specifically PDGBA with severe variants, show not only elevated levels of the GCase substrates GlcCer and GlcSph in CSF and plasma but also increased CSF levels of downstream-products (Cer) and by-products (SPA, S1P) when compared to healthy controls and PDGBA_wildtype (Lerche et al. 2021a; Surface et al. 2022). Assessments in plasma from PDGBA_wildtype as well as in aging mouse models further support findings that with decreasing GCase, activity levels of downstream/by-products are also elevated in addition to upstream substrates (Hallett et al. 2018; Mielke et al. 2013). In GD with pronounced GCase deficiency, GlcCer are alternatively processed to GlcSph and exit the lysosome into the cytosol (Hein et al. 2007; Elleder 2006; Ferraz et al. 2016). Cytosolic GlcSph is further hydrolyzed to ceramides, sphingosine, and sphingosine-1-phosphate. Recent studies highlight the role of ceramides and sphingosine-1-phosphate as key players in the regulation of cell death and survival with involvement in ER stress, autophagy, protein and lipid transport, exosome secretion with neurotoxic protein spreading, neuroinflammation, and mitochondrial dysfunction (Wang and Bieberich 2018). Data from α-synuclein/GBA transgenic mice and HEK cell cultures show that GlcCer, GlcSph, sphingosine, and sphingosine-1-phosphate promote the formation of oligomeric α-synuclein (Taguchi et al. 2017). Expanding these findings, recent data in human dopaminergic midbrain neurons suggest that conformational changes of α-synuclein towards an aggregation-prone pattern can be even induced by the presence of glycosphingolipids alone irrespective of GCase deficiency due to variants in the GBA gene (Zunke et al. 2018). Post-mortem studies show increased levels of GlcCer, GlcSph and ceramides in the substantia nigra and frontal cortex of PDGBA_wildtype (Rocha et al. 2015a; Huebecker et al. 2019; Kurzawa-Akanbi et al. 2021). However, no differences were seen in the putamen of PDGBA and PDGBA_wildtype compared to controls (Gegg et al. 2012). More post-mortem studies with uniformly assessed brain regions and cell types as well as stratification according to GBA variant severity are needed to shed light on these seemingly discrepancies.
Enhanced activation of phosphocholine cytidyltransferase resulting in increased synthesis of phosphatidylcholine as major component of phospholipid cell membranes was reported in GD (Bodennec et al. 2002). Interestingly, alterations in the lipid bilayer composition of membranes cause impaired α-synuclein membrane binding and enhance aggregation-prone fibril formation (Piccinini et al. 2010). Combined 1H and 31P magnetic resonance spectroscopic imaging revealed that PDGBA patients display a disturbed membrane phospholipid metabolism in the putamen and midbrain with reduced levels of the precursor choline and increased levels of the membrane-related phospholipid degradation product glycerophosphoethanolamine. These changes were accompanied by neuronal loss in these brain regions as measured by reduced levels of the neuronal marker N-acetyl-aspartate (Brockmann et al. 2012).
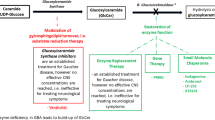
Therapeutic targets in GBA-associated PD
Based on the knowledge of the molecular mechanisms underlying PDGBA, pathway-specific treatment options are beginning to emerge.
GCase
The significant reduction of GCase protein levels and GCase enzyme activity offer a plausible therapeutic rational to either increase GCase protein levels or enhance enzyme activity. Unfortunately, intravenous enzyme replacement therapy is not possible due to insufficient central nervous system penetration.
Gene therapy with adeno-associated virus (AAV)-based vectors promoting GBA overexpression. This approach reduced α-synuclein accumulation, improved lysosomal function and lipid turnover, and attenuated deficits in working memory and fine motor performance in α-synuclein mutant/overexpressing wild-type and GD rodent models (Rocha et al. 2015b; Glajch et al. 2021; Sardi et al. 2011). The AAV9-based vector PR001 increased GCase activity, reduced glycolipid substrate accumulation, and improved motor deficits in two mouse models of GCase deficiency (Abeliovich et al. 2021). Based on these results, a phase 1/2a non-randomized clinical trial with a single administration of PR001 into the cisterna magna is currently under investigation in PD patients with at least one pathogenic GBA variant. The study duration is 5 years. During the first year, patients will be evaluated for safety, tolerability, immunogenicity, biomarkers, and clinical efficacy measures. Patients will continue to be followed for an additional 4 years to monitor safety and selected biomarker and efficacy measures (NCT04127578).
GCase-enhancing small-molecule chaperones refold misfolded GCase in the ER and promote proper trafficking, thereby increasing lysosomal GCase protein levels. Interestingly, experimental data in cell and animal models with GBA variants suggest that the expectorant Ambroxol increases GCase availability via such mechanism (Kopytova et al. 2021; Ambrosi et al. 2015; Maegawa et al. 2009; Magalhaes et al. 2018; McNeill et al. 2014; Yang et al. 2022; Migdalska-Richards et al. 2016). These findings led to a proof-of-principle phase 2 open-label study with Ambroxol in 17 PD patients with and without GBA variants. Ambroxol was well tolerated and CSF GCase protein levels as well as CSF levels of α-synuclein increased by 35% and 13%, respectively. However, CSF GCase enzyme activity decreased by 19% which might be explained by an inhibitory effect of Ambroxol on GCase activity within acellular human CSF with a neutral pH (Mullin et al. 2020).
More strikingly, a recent publication could show that the small-molecule S-181 increases wild-type GCase activity in iPSC-derived dopaminergic neurons not only from PDGBA but also from PDwildtype as well as from patients with other PD-related gene mutations in LRRK2, DJ-1, and PARKN who also had decreased levels of GCase activity. S-181 treatment of these PD iPSC-derived dopaminergic neurons partially restored lysosomal function and lowered accumulation of oxidized dopamine, GlcCer, and α-synuclein (Burbulla et al. 2019). These recent findings highlight not only the importance of lysosomal dysfunction in the pathophysiology of the prototype PDGBA but also the significance of this pathway, possibly in concert with additional pathways such as mitochondrial dysfunction, for PD in general.
Substrate reduction therapy
Substrate reduction therapy to reduce GlcCer production with penetration into the central nervous system is available for oral application in GD. Venglustat has been evaluated in a phase 2 randomized trial (MOVES-PD, NCT02906020) in PDGBA. The compound clearly reduced CSF levels of GlcCer in a dose-dependent manner in plasma and CSF. However, the study was stopped prematurely, since patients in the verum group showed enhanced clinical deterioration suggesting an off-target effect with possible anti-dopaminergic activity.
Alpha-synuclein-targeting compounds
Targeting alpha-synuclein also seems a reasonable treatment option given the predominant α-synuclein aggregation and wide-spread Lewy-body pathology in PDGBA.
Conclusion and outlook
GBA-associated PD is remarkable for several reasons. The phenotypical trajectories show a faster disease progression with pronounced early cognitive decline and a clear dependency based on mutation severity. Importantly, the development of dementia is not associated with Amyloid-β pathology as shown instead in a relevant proportion of PD without GBA variants but rather due to predominant α-synuclein aggregation. The identified pathophysiological mechanisms highlight GCase deficiency and lysosomal dysfunction resulting in disrupted glycosphingolipid homeostasis and ultimately impaired α-synuclein degradation with enhanced aggregation. Again, these are dependent on mutation severity and offer different targets for individualized treatment options. However, the failure of the MOVES-PD trial (NCT02906020) demonstrates the challenges we are facing in translational research. Findings from GD as typical and clearly defined young-onset lysosomal storage lipid disorder due to bi-allelic mutations in GBA are not simply transferable into PD, a multifactorial disease of the elderly with possibly additional contributing factors (e.g., mitochondrial dysfunction and lifetime environmental exposure). Specifically, the pathophysiological mechanisms of impaired glycosphingolipid homeostasis leading to impaired α-synuclein degradation need more investigation. In this context, longitudinal patient cohorts with repeated collections of biomaterials, ideally starting in the prodromal stage followed up until death with brain donation, might inform us on biomarkers that reflect the underlying pathological processes and possible read-outs for target engagement.
Future clinical trials in PDGBA might incorporate the knowledge learned over the last years: (i) Patients should be stratified according to GBA variant severity with those carrying severe mutations to be preferentially included in proof-of-concept trials. (ii) The early cognitive decline based on predominant α-synuclein-driven pathology offers the opportunity to address PD-associated dementia with disease-modifying agents in a clearly defined prodromal phase preceding dementia and based on clear biological stratification.
References
Aarsland D, Perry R, Brown A, Larsen JP, Ballard C (2005) Neuropathology of dementia in Parkinson’s disease: a prospective, community-based study. Ann Neurol 58:773–776
Abeliovich A, Hefti F, Sevigny J (2021) Gene therapy for Parkinson’s disease associated with GBA1 mutations. J Parkinsons Dis 11:S183–S188
Aflaki E, Borger DK, Moaven N, Stubblefield BK, Rogers SA, Patnaik S, Schoenen FJ, Westbroek W, Zheng W, Sullivan P, Fujiwara H, Sidhu R, Khaliq ZM, Lopez GJ, Goldstein DS, Ory DS, Marugan J, Sidransky E (2016) A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and Parkinsonism. J Neurosci 36:7441–7452
Aflaki E, Stubblefield BK, McGlinchey RP, McMahon B, Ory DS, Sidransky E (2020) A characterization of Gaucher iPS-derived astrocytes: potential implications for Parkinson’s disease. Neurobiol Dis 134:104647
Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, Mazzoni P, Pauciulo MW, Nichols WC, Gan-Or Z, Rouleau GA, Chung WK, Wolf P, Oliva P, Keutzer J, Marder K, Zhang X (2015) Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138:2648–2658
Alcalay RN, Wolf P, Chiang MSR, Helesicova K, Zhang XK, Merchant K, Hutten SJ, Scherzer C, Caspell-Garcia C, Blauwendraat C, Foroud T, Nudelman K, Gan-Or Z, Simuni T, Chahine LM, Levy O, Zheng D, Li G, Sardi SP, Parkinson’s Progression Markers I (2020) Longitudinal Measurements of Glucocerebrosidase activity in Parkinson’s patients. Ann Clin Transl Neurol 7:1816–1830
Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F (2015) Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol Dis 82:235–242
Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, Pollak P, Bonaiti B, Bonaiti-Pellie C, Brice A, French Parkinson Disease Genetic G (2012) Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78:417–420
Avenali M, Toffoli M, Mullin S, McNeil A, Hughes DA, Mehta A, Blandini F, Schapira AHV (2019) Evolution of prodromal parkinsonian features in a cohort of GBA mutation-positive individuals: a 6-year longitudinal study. J Neurol Neurosurg Psychiatry 90:1091–1097
Barrett MJ, Shanker VL, Severt WL, Raymond D, Gross SJ, Schreiber-Agus N, Kornreich R, Ozelius LJ, Bressman SB, Saunders-Pullman R (2014) Cognitive and antipsychotic medication use in monoallelic GBA-related Parkinson disease. JIMD Rep 16:31–38
Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AH (2015) Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol 72:201–208
Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, Gasser T, Goetz CG, Halliday G, Joseph L, Lang AE, Liepelt-Scarfone I, Litvan I, Marek K, Obeso J, Oertel W, Olanow CW, Poewe W, Stern M, Deuschl G (2015) MDS research criteria for prodromal Parkinson’s disease. Mov Disord 30:1600–1611
Beutler E (1992) Gaucher disease: new molecular approaches to diagnosis and treatment. Science 256:794–799
Blauwendraat C, Heilbron K, Vallerga CL, Bandres-Ciga S, von Coelln R, Pihlstrom L, Simon-Sanchez J, Schulte C, Sharma M, Krohn L, Siitonen A, Iwaki H, Leonard H, Noyce AJ, Tan M, Gibbs JR, Hernandez DG, Scholz SW, Jankovic J, Shulman LM, Lesage S, Corvol JC, Brice A, van Hilten JJ, Marinus J, Andme Research T, Eerola-Rautio J, Tienari P, Majamaa K, Toft M, Grosset DG, Gasser T, Heutink P, Shulman JM, Wood N, Hardy J, Morris HR, Hinds DA, Gratten J, Visscher PM, Gan-Or Z, Nalls MA, Singleton AB, International Parkinson’s Disease Genomics C (2019) Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord 34:866–875
Blauwendraat C, Nalls MA, Singleton AB (2020a) The genetic architecture of Parkinson’s disease. Lancet Neurol 19:170–178
Blauwendraat C, Reed X, Krohn L, Heilbron K, Bandres-Ciga S, Tan M, Gibbs JR, Hernandez DG, Kumaran R, Langston R, Bonet-Ponce L, Alcalay RN, Hassin-Baer S, Greenbaum L, Iwaki H, Leonard HL, Grenn FP, Ruskey JA, Sabir M, Ahmed S, Makarious MB, Pihlstrom L, Toft M, van Hilten JJ, Marinus J, Schulte C, Brockmann K, Sharma M, Siitonen A, Majamaa K, Eerola-Rautio J, Tienari PJ, Andme Research T, Pantelyat A, Hillis AE, Dawson TM, Rosenthal LS, Albert MS, Resnick SM, Ferrucci L, Morris CM, Pletnikova O, Troncoso J, Grosset D, Lesage S, Corvol JC, Brice A, Noyce AJ, Masliah E, Wood N, Hardy J, Shulman LM, Jankovic J, Shulman JM, Heutink P, Gasser T, Cannon P, Scholz SW, Morris H, Cookson MR, Nalls MA, Gan-Or Z, Singleton AB (2020b) Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143:234–248
Bodennec J, Pelled D, Riebeling C, Trajkovic S, Futerman AH (2002) Phosphatidylcholine synthesis is elevated in neuronal models of Gaucher disease due to direct activation of CTP:phosphocholine cytidylyltransferase by glucosylceramide. FASEB J 16:1814–1816
Brockmann K, Srulijes K, Hauser AK, Schulte C, Csoti I, Gasser T, Berg D (2011) GBA-associated PD presents with nonmotor characteristics. Neurology 77:276–280
Brockmann K, Hilker R, Pilatus U, Baudrexel S, Srulijes K, Magerkurth J, Hauser AK, Schulte C, Csoti I, Merten CD, Gasser T, Berg D, Hattingen E (2012) GBA-associated PD. Neurodegeneration, altered membrane metabolism, and lack of energy failure. Neurology 79:213–220
Brockmann K, Schulte C, Deuschle C, Hauser AK, Heger T, Gasser T, Maetzler W, Berg D (2015a) Neurodegenerative CSF markers in genetic and sporadic PD: classification and prediction in a longitudinal study. Parkinsonism Relat Disord 21:1427–1434
Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W, Gasser T, Berg D (2015b) GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 30:407–411
Brockmann K, Lerche S, Dilger SS, Stirnkorb JG, Apel A, Hauser AK, Liepelt-Scarfone I, Berg D, Gasser T, Schulte C, Maetzler W (2017) SNPs in Abeta clearance proteins: Lower CSF Abeta1-42 levels and earlier onset of dementia in PD. Neurology 89:2335–2340
Brockmann K, Quadalti C, Lerche S, Rossi M, Wurster I, Baiardi S, Roeben B, Mammana A, Zimmermann M, Hauser AK, Deuschle C, Schulte C, Waniek K, Lachmann I, Sjodin S, Brinkmalm A, Blennow K, Zetterberg H, Gasser T, Parchi P (2021) Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun 9:175
Burbulla LF, Jeon S, Zheng J, Song P, Silverman RB, Krainc D (2019) A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci Transl Med 11:eaau6870
Chen J, Li W, Zhang T, Wang YJ, Jiang XJ, Xu ZQ (2014) Glucocerebrosidase gene mutations associated with Parkinson’s disease: a meta-analysis in a Chinese population. PLoS ONE 9:e115747
Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, Zecchinelli AL, Canesi M, Mariani CB, Meucci N, Sacilotto G, Zini M, Barichella M, Magnani C, Duga S, Asselta R, Solda G, Seresini A, Seia M, Pezzoli G, Goldwurm S (2016) Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann Neurol 80:662–673
Compta Y, Parkkinen L, O’Sullivan SS, Vandrovcova J, Holton JL, Collins C, Lashley T, Kallis C, Williams DR, de Silva R, Lees AJ, Revesz T (2011) Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain 134:1493–1505
den Heijer JM, Cullen VC, Quadri M, Schmitz A, Hilt DC, Lansbury P, Berendse HW, van de Berg WDJ, de Bie RMA, Boertien JM, Boon AJW, Contarino MF, van Hilten JJ, Hoff JI, van Mierlo T, Munts AG, van der Plas AA, Ponsen MM, Baas F, Majoor-Krakauer D, Bonifati V, van Laar T, Groeneveld GJ (2020) A large-scale full GBA1 gene screening in Parkinson’s disease in the Netherlands. Mov Disord 35:1667–1674
Elleder M (2006) Glucosylceramide transfer from lysosomes—the missing link in molecular pathology of glucosylceramidase deficiency: a hypothesis based on existing data. J Inherit Metab Dis 29:707–715
Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, Joachim C, Esiri M, Evetts SG, Rolinski M, Baig F, Ruffmann C, Wade-Martins R, Hu MT, Parkkinen L, Green AJ (2016) Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 3:812–818
Ferraz MJ, Marques AR, Appelman MD, Verhoek M, Strijland A, Mirzaian M, Scheij S, Ouairy CM, Lahav D, Wisse P, Overkleeft HS, Boot RG, Aerts JM (2016) Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett 590:716–725
Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, Schapira AH (2012) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72:455–463
Glajch KE, Moors TE, Chen Y, Bechade PA, Nam AY, Rajsombath MM, Mccaffery TD, Dettmer U, Weihofen A, Hirst WD, Selkoe DJ, Nuber S (2021) Wild-type GBA1 increases the alpha-synuclein tetramer-monomer ratio, reduces lipid-rich aggregates, and attenuates motor and cognitive deficits in mice. Proc Natl Acad Sci USA 118
Goker-Alpan O, Schiffmann R, Lamarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E (2004) Parkinsonism among Gaucher disease carriers. J Med Genet 41:937–940
Gundner AL, Duran-Pacheco G, Zimmermann S, Ruf I, Moors T, Baumann K, Jagasia R, van de Berg WDJ, Kremer T (2019) Path mediation analysis reveals GBA impacts Lewy body disease status by increasing alpha-synuclein levels. Neurobiol Dis 121:205–213
Hallett PJ, Huebecker M, Brekk OR, Moloney EB, Rocha EM, Priestman DA, Platt FM, Isacson O (2018) Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol Aging 67:189–200
Halliday G, Hely M, Reid W, Morris J (2008) The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol 115:409–415
Halperin A, Elstein D, Zimran A (2006) Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis 36:426–428
Heckman MG, Soto-Ortolaza AI, Contreras MYS, Murray ME, Pedraza O, Diehl NN, Walton R, Labbe C, Lorenzo-Betancor O, Uitti RJ, van Gerpen J, Ertekin-Taner N, Smith GE, Kantarci K, Savica R, Jones DT, Graff-Radford J, Knopman DS, Lowe VJ, Jack CR, Petersen RC, Parisi JE, Rademakers R, Wszolek ZK, Graff-Radford NR, Ferman TJ, Dickson DW, Boeve BF, Ross OA (2016) LRRK2 variation and dementia with Lewy bodies. Parkinsonism Relat Disord 31:98–103
Hein LK, Meikle PJ, Hopwood JJ, Fuller M (2007) Secondary sphingolipid accumulation in a macrophage model of Gaucher disease. Mol Genet Metab 92:336–345
Honeycutt L, Montplaisir JY, Gagnon JF, Ruskey J, Pelletier A, Gan-Or Z, Postuma RB (2019) Glucocerebrosidase mutations and phenoconversion of REM sleep behavior disorder to parkinsonism and dementia. Parkinsonism Relat Disord 65:230–233
Huebecker M, Moloney EB, van der Spoel AC, Priestman DA, Isacson O, Hallett PJ, Platt FM (2019) Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol Neurodegener 14:40
Iwaki H, Blauwendraat C, Leonard HL, Liu G, Maple-Grodem J, Corvol JC, Pihlstrom L, van Nimwegen M, Hutten SJ, Nguyen KH, Rick J, Eberly S, Faghri F, Auinger P, Scott KM, Wijeyekoon R, van Deerlin VM, Hernandez DG, Day-Williams AG, Brice A, Alves G, Noyce AJ, Tysnes OB, Evans JR, Breen DP, Estrada K, Wegel CE, Danjou F, Simon DK, Ravina B, Toft M, Heutink P, Bloem BR, Weintraub D, Barker RA, Williams-Gray CH, van de Warrenburg BP, van Hilten JJ, Scherzer CR, Singleton AB, Nalls MA (2019) Genetic risk of Parkinson disease and progression: an analysis of 13 longitudinal cohorts. Neurol Genet 5:e348
Jankovic J, Tan EK (2020) Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry 91:795–808
Jo J, Yang L, Tran HD, Yu W, Sun AX, Chang YY, Jung BC, Lee SJ, Saw TY, Xiao B, Khoo ATT, Yaw LP, Xie JJ, Lokman H, Ong WY, Lim GGY, Lim KL, Tan EK, Ng HH, Je HS (2021) Lewy body-like inclusions in human midbrain organoids carrying glucocerebrosidase and alpha-synuclein mutations. Ann Neurol 90:490–505
Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, Ross OA, van Deerlin VM, Trojanowski JQ, Hurtig HI, Alcalay RN, Marder KS, Clark LN, Gaig C, Tolosa E, Ruiz-Martinez J, Marti-Masso JF, Ferrer I, Munain AL, Goldman SM, Schule B, Langston JW, Aasly JO, Giordana MT, Bonifati V, Puschmann A, Canesi M, Pezzoli G, de Paula AM, Hasegawa K, Duyckaerts C, Brice A, Stoessl AJ, Marras C (2015) Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 72:100–105
Kang JH, Mollenhauer B, Coffey CS, Toledo JB, Weintraub D, Galasko DR, Irwin DJ, van Deerlin V, Chen-Plotkin AS, Caspell-Garcia C, Waligorska T, Taylor P, Shah N, Pan S, Zero P, Frasier M, Marek K, Kieburtz K, Jennings D, Tanner CM, Simuni T, Singleton A, Toga AW, Chowdhury S, Trojanowski JQ, Shaw LM, Parkinson’s Progression Marker I (2016) CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: the Parkinson’s Progression Markers Initiative study. Acta Neuropathol 131:935–949
Kang UJ, Boehme AK, Fairfoul G, Shahnawaz M, Ma TC, Hutten SJ, Green A, Soto C (2019) Comparative study of cerebrospinal fluid alpha-synuclein seeding aggregation assays for diagnosis of Parkinson’s disease. Mov Disord 34:536–544
Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, Rhee P, Karuppagounder SS, Kwon SH, Lee H, Mao X, Kim D, Pandey A, Lee G, Dawson VL, Dawson TM, Ko HS (2018) GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc Natl Acad Sci USA 115:798–803
Kopytova AE, Rychkov GN, Nikolaev MA, Baydakova GV, Cheblokov AA, Senkevich KA, Bogdanova DA, Bolshakova OI, Miliukhina IV, Bezrukikh VA, Salogub GN, Sarantseva SV, Usenko TC, Zakharova EY, Emelyanov AK, Pchelina SN (2021) Ambroxol increases glucocerebrosidase (GCase) activity and restores GCase translocation in primary patient-derived macrophages in Gaucher disease and Parkinsonism. Parkinsonism Relat Disord 84:112–121
Kurzawa-Akanbi M, Tammireddy S, Fabrik I, Gliaudelyte L, Doherty MK, Heap R, Matecko-Burmann I, Burmann BM, Trost M, Lucocq JM, Gherman AV, Fairfoul G, Singh P, Burte F, Green A, McKeith IG, Hartlova A, Whitfield PD, Morris CM (2021) Altered ceramide metabolism is a feature in the extracellular vesicle-mediated spread of alpha-synuclein in Lewy body disorders. Acta Neuropathol 142:961–984
Lerche S, Machetanz G, Wurster I, Roeben B, Zimmermann M, Pilotto A, Preische O, Stransky E, Deuschle C, Hauser AK, Schulte C, Lachmann I, Waniek K, Gasser T, Berg D, Maetzler W, Brockmann K (2019a) Dementia with lewy bodies: GBA1 mutations are associated with cerebrospinal fluid alpha-synuclein profile. Mov Disord 34:1069–1073
Lerche S, Wurster I, Roben B, Machetanz G, Zimmermann M, Bernhard F, Stransky E, Deuschle C, Schulte C, Hansson O, Zetterberg H, Gasser T, Berg D, Maetzler W, Brockmann K (2019b) Parkinson’s disease: evolution of cognitive impairment and CSF Abeta1-42 profiles in a prospective longitudinal study. J Neurol Neurosurg Psychiatry 90:165–170
Lerche S, Wurster I, Roeben B, Zimmermann M, Riebenbauer B, Deuschle C, Hauser AK, Schulte C, Berg D, Maetzler W, Waniek K, Lachmann I, Liepelt-Scarfone I, Gasser T, Brockmann K (2020) Parkinson’s disease: glucocerebrosidase 1 mutation severity is associated with CSF alpha-synuclein profiles. Mov Disord 35:495–499
Lerche S, Schulte C, Wurster I, Machetanz G, Roeben B, Zimmermann M, Deuschle C, Hauser AK, Bohringer J, Krageloh-Mann I, Waniek K, Lachmann I, Petterson XT, Chiang R, Park H, Wang B, Liepelt-Scarfone I, Maetzler W, Galasko D, Scherzer CR, Gasser T, Mielke MM, Hutten SJ, Mollenhauer B, Sardi SP, Berg D, Brockmann K (2021a) The mutation matters: CSF profiles of GCase, sphingolipids, alpha-synuclein in PDGBA. Mov Disord 36:1216–1228
Lerche S, Sjodin S, Brinkmalm A, Blennow K, Wurster I, Roeben B, Zimmermann M, Hauser AK, Liepelt-Scarfone I, Waniek K, Lachmann I, Gasser T, Zetterberg H, Brockmann K (2021b) CSF protein level of neurotransmitter secretion, synaptic plasticity, and autophagy in PD and DLB. Mov Disord 36:2595–2604
Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, Viallet F, Lohmann E, Corvol JC, Honore A, Rivaud S, Vidailhet M, Durr A, Brice A, FRENCH PARKINSON’S DISEASE GENETICS STUDY G (2011) Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum Mol Genet 20:202–210
Maegawa GH, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D, Tang L, Kornhaber GJ, Hamuro Y, Clarke JT, Mahuran DJ (2009) Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem 284:23502–23516
Magalhaes J, Gegg ME, Migdalska-Richards A, Doherty MK, Whitfield PD, Schapira AH (2016) Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet 25:3432–3445
Magalhaes J, Gegg ME, Migdalska-Richards A, Schapira AH (2018) Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci Rep 8:1385
Mahungu AC, Anderson DG, Rossouw AC, van Coller R, Carr JA, Ross OA, Bardien S (2020) Screening of the glucocerebrosidase (GBA) gene in South Africans of African ancestry with Parkinson’s disease. Neurobiol Aging 88:156 e11-156 e14
Malek N, Swallow D, Grosset KA, Anichtchik O, Spillantini M, Grosset DG (2014) Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease - a systematic review. Acta Neurol Scand 130:59–72
Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, Bajaj N, Barker RA, Burn DJ, Foltynie T, Hardy J, Wood NW, Ben-Shlomo Y, Williams NW, Grosset DG, Morris HR, Consortium PRC (2018) Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J Neurol Neurosurg Psychiatry 89:702–709
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52
Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D (2016) alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci USA 113:1931–1936
McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, Foltynie T, Cooper JM, Abramov AY, Gegg M, Schapira AH (2014) Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137:1481–1495
Mielke MM, Maetzler W, Haughey NJ, Bandaru VV, Savica R, Deuschle C, Gasser T, Hauser AK, Graber-Sultan S, Schleicher E, Berg D, Liepelt-Scarfone I (2013) Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS ONE 8:e73094
Migdalska-Richards A, Daly L, Bezard E, Schapira AH (2016) Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol 80:766–775
Mollenhauer B, Caspell-Garcia CJ, Coffey CS, Taylor P, Singleton A, Shaw LM, Trojanowski JQ, Frasier M, Simuni T, Iranzo A, Oertel W, Siderowf A, Weintraub D, Seibyl J, Toga AW, Tanner CM, Kieburtz K, Chahine LM, Marek K, Galasko D, Study P (2019) Longitudinal analyses of cerebrospinal fluid alpha-Synuclein in prodromal and early Parkinson’s disease. Mov Disord 34:1354–1364
Moors TE, Paciotti S, Ingrassia A, Quadri M, Breedveld G, Tasegian A, Chiasserini D, Eusebi P, Duran-Pacheco G, Kremer T, Calabresi P, Bonifati V, Parnetti L, Beccari T, van de Berg WDJ (2019) Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurobiol 56:1344–1355
Mullin S, Beavan M, Bestwick J, McNeill A, Proukakis C, Cox T, Hughes D, Mehta A, Zetterberg H, Schapira AHV (2019) Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov Disord 34:1365–1373
Mullin S, Smith L, Lee K, D’Souza G, Woodgate P, Elflein J, Hallqvist J, Toffoli M, Streeter A, Hosking J, Heywood WE, Khengar R, Campbell P, Hehir J, Cable S, Mills K, Zetterberg H, Limousin P, Libri V, Foltynie T, Schapira AHV (2020) Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol 77:427–434
Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, Cooper A, Garner B, Halliday GM (2014) Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137:834–848
Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, Morris CM, Theuns J, Crosiers D, Cras P, Engelborghs S, de Deyn PP, van Broeckhoven C, Mann DM, Snowden J, Pickering-Brown S, Halliwell N, Davidson Y, Gibbons L, Harris J, Sheerin UM, Bras J, Hardy J, Clark L, Marder K, Honig LS, Berg D, Maetzler W, Brockmann K, Gasser T, Novellino F, Quattrone A, Annesi G, de Marco EV, Rogaeva E, Masellis M, Black SE, Bilbao JM, Foroud T, Ghetti B, Nichols WC, Pankratz N, Halliday G, Lesage S, Klebe S, Durr A, Duyckaerts C, Brice A, Giasson BI, Trojanowski JQ, Hurtig HI, Tayebi N, Landazabal C, Knight MA, Keller M, Singleton AB, Wolfsberg TG, Sidransky E (2013) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70:727–735
Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, Li A, Holton J, Guerreiro R, Paudel R, Segarane B, Singleton A, Lees A, Hardy J, Houlden H, Revesz T, Wood NW (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132:1783–1794
Paciotti S, Gatticchi L, Beccari T, Parnetti L (2019) Lysosomal enzyme activities as possible CSF biomarkers of synucleinopathies. Clin Chim Acta 495:13–24
Pal G, Mangone G, Hill EJ, Ouyang B, Liu Y, Lythe V, Ehrlich D, Saunders-Pullman R, Shanker V, Bressman S, Alcalay RN, Garcia P, Marder KS, Aasly J, Mouradian MM, Link S, Rosenbaum M, Anderson S, Bernard B, Wilson R, Stebbins G, Nichols WC, Welter ML, Sani S, Afshari M, Verhagen L, de Bie RMA, Foltynie T, Hall D, Corvol JC, Goetz CG (2022) Parkinson disease and subthalamic nucleus deep brain stimulation: cognitive effects in GBA mutation carriers. Ann Neurol 91:424–435
Parnetti L, Paciotti S, Eusebi P, Dardis A, Zampieri S, Chiasserini D, Tasegian A, Tambasco N, Bembi B, Calabresi P, Beccari T (2017) Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in Parkinson’s disease patients. Mov Disord 32:1423–1431
Petrucci S, Ginevrino M, Trezzi I, Monfrini E, Ricciardi L, Albanese A, Avenali M, Barone P, Bentivoglio AR, Bonifati V, Bove F, Bonanni L, Brusa L, Cereda C, Cossu G, Criscuolo C, Dati G, de Rosa A, Eleopra R, Fabbrini G, Fadda L, Garbellini M, Minafra B, Onofrj M, Pacchetti C, Palmieri I, Pellecchia MT, Petracca M, Picillo M, Pisani A, Vallelunga A, Zangaglia R, di Fonzo A, Morgante F, Valente EM (2020) GBA-related parkinson’s disease: dissection of genotype-phenotype correlates in a large Italian cohort. Mov Disord 35:2106–2111
Piccinini M, Scandroglio F, Prioni S, Buccinna B, Loberto N, Aureli M, Chigorno V, Lupino E, Demarco G, Lomartire A, Rinaudo MT, Sonnino S, Prinetti A (2010) Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol Neurobiol 41:314–340
Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P, Isacson O (2015a) Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol 2:433–438
Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, Beagan J, McLean JR, Izen SC, Perez-Torres E, Hallett PJ, Isacson O (2015b) Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis 82:495–503
Rossi M, Candelise N, Baiardi S, Capellari S, Giannini G, Orru CD, Antelmi E, Mammana A, Hughson AG, Calandra-Buonaura G, Ladogana A, Plazzi G, Cortelli P, Caughey B, Parchi P (2020) Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol 140:49–62
Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, Passini MA, Grabowski GA, Schlossmacher MG, Sidman RL, Cheng SH, Shihabuddin LS (2011) CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci USA 108:12101–12106
Schneider SA, Alcalay RN (2017) Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord 32:1504–1523
Schondorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, Sardi SP, Valsecchi M, Hoffmann S, Schwarz LK, Hedrich U, Berg D, Shihabuddin LS, Hu J, Pruszak J, Gygi SP, Sonnino S, Gasser T, Deleidi M (2014) iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 5:4028
Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, Mollenhauer B, Soto C (2017) Development of a biochemical diagnosis of Parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 74:163–172
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, de Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661
Stoker TB, Camacho M, Winder-Rhodes S, Liu G, Scherzer CR, Foltynie T, Evans J, Breen DP, Barker RA, Williams-Gray CH (2020) Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J Neurol Neurosurg Psychiatry 91:695–702
Straniero L, Asselta R, Bonvegna S, Rimoldi V, Melistaccio G, Solda G, Aureli M, Della Porta M, Lucca U, di Fonzo A, Zecchinelli A, Pezzoli G, Cilia R, Duga S (2020) The SPID-GBA study: sex distribution, penetrance, incidence, and dementia in GBA-PD. Neurol Genet 6:e523
Surface M, Balwani M, Waters C, Haimovich A, Gan-Or Z, Marder KS, Hsieh T, Song L, Padmanabhan S, Hsieh F, Merchant KM, Alcalay RN (2022) Plasma glucosylsphingosine in gba1 mutation carriers with and without Parkinson’s disease. Mov Disord 37:416–421
Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, Keutzer J, Mistry PK, Chandra SS (2017) Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson’s disease. J Neurosci 37:9617–9631
Thaler A, Gurevich T, Bar Shira A, Gana Weisz M, Ash E, Shiner T, Orr-Urtreger A, Giladi N, Mirelman A (2017) A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat Disord 36:47–51
Thaler A, Bregman N, Gurevich T, Shiner T, Dror Y, Zmira O, Gan-Or Z, Bar-Shira A, Gana-Weisz M, Orr-Urtreger A, Giladi N, Mirelman A (2018) Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat Disord 55:45–49
van der Lee SJ, van Steenoven I, van de Beek M, Tesi N, Jansen IE, van Schoor NM, Reinders MJT, Huisman M, Scheltens P, Teunissen CE, Holstege H, van der Flier WM, Lemstra AW (2021) Genetics contributes to concomitant pathology and clinical presentation in dementia with Lewy bodies. J Alzheimers Dis 83:269–279
Wang G, Bieberich E (2018) Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv Biol Regul 70:51–64
Yang SY, Taanman JW, Gegg M, Schapira AHV (2022) Ambroxol reverses tau and alpha-synuclein accumulation in a cholinergic N370S GBA1 mutation model. Hum Mol Genet
Zhang Y, Shu L, Sun Q, Zhou X, Pan H, Guo J, Tang B (2018) Integrated genetic analysis of racial differences of common GBA variants in Parkinson’s disease: a meta-analysis. Front Mol Neurosci 11:43
Zimmermann M, Gaenslen A, Prahl K, Srulijes K, Hauser AK, Schulte C, Csoti I, Berg D, Brockmann K (2018) Patient’s perception: shorter and more severe prodromal phase in GBA-associated PD. Eur J Neurol 26:694–698
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, Toker NJ, Jeon S, Fredriksen K, Mazzulli JR (2018) Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron 97(92–107):e10
Acknowledgements
Kathrin Brockmann has received a research grants from the University of Tuebingen (Clinician Scientist), the German Society of Parkinson’s disease (dpv), the Michael J. Fox Foundation (MJFF), the German Centre for Neurodegenerative Diseases (DZNE, MIGAP), and the German Federal Ministry of Education and Research (BMBF) in the frame of ERACoSysMed2 (FKZ 031L0137B). She received Speaker honoraria from Abbvie, Lundbeck, UCB, and Zambon.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Günther Höglinger was supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy—ID 390857198) and within the Hannover Cluster RESIST (EXC 2155—ID 390874280), DFG grants (HO2402/6-2, HO2402/18-1 MSAomics), the German Federal Ministry of Education and Research (BMBF, 01KU1403A EpiPD; 01EK1605A HitTau; 01DH18025 TauTherapy), the German Center for Neurodegenerative Diseases e.V. (DZNE), Niedersächsisches Ministerium für Wissenschaft und Kunst (MWK, ZN3440.TP): REBIRTH—Forschungszentrum für translationale regenerative Medizin; VolkswagenStiftung (Niedersächsisches Vorab); Petermax-Müller Foundation (Etiology and Therapy of Synucleinopathies and Tauopathies); the German Parkinson Society (DPG; ProAPS), the German PSP Association (PSP Gesellschaft; ProPSP); participated in industry-sponsored research projects from Abbvie, Biogen, Biohaven, Novartis, Roche, Sanofi, UCB. Claudia Schulte has nothing to disclose. Wolfgang H. Jost is/was speaker and/or advisor Abbvie, Bial, Kyowa Kirin, Merz, UCB, Zambon. Alexander Storch has received funding from the Deutsche Forschungsgemeinschaft (DFG) and the Helmholtz-Association. He received honoraria for presentations/lectures/consultancies or advisory boards from AbbVie, Bayer Healthcare, Desitin, Bial, GKC, Grünenthal, UCB, Zambon, Ca, AbbVie, TEVA, Lundbeck, and UCB Pharma, outside the submitted work. He has served on the editorial boards of Stem Cells and Stem Cells International and received royalties from Kohlhammer Verlag and Elsevier Press. Dirk Woitalla has nothing to disclose. Rejko Krüger has nothing to disclose. Björn Falkenburger has nothing to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Höglinger, G., Schulte, C., Jost, W.H. et al. GBA-associated PD: chances and obstacles for targeted treatment strategies. J Neural Transm 129, 1219–1233 (2022). https://doi.org/10.1007/s00702-022-02511-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-022-02511-7