
Abstract
Background
Remarkable advances have been reached in the understanding of the genetic basis of Parkinson’s disease (PD), with the identification of monogenic causes (mPD) and a plethora of gene loci leading to an increased risk for idiopathic PD. The expanding knowledge and subsequent identification of genetic contributions fosters the understanding of molecular mechanisms leading to disease development and progression. Distinct pathways involved in mitochondrial dysfunction, oxidative stress, and lysosomal function have been identified and open a unique window of opportunity for individualized treatment approaches. These genetic findings have led to an imminent progress towards pathophysiology-targeted clinical trials and potentially disease-modifying treatments in the future.
Main body of the manuscript
In this review article we will summarize known genetic contributors to the pathophysiology of Parkinson’s disease, the molecular mechanisms leading to disease development, and discuss challenges and opportunities in clinical trial designs.
Conclusions
The future success of clinical trials in PD is mainly dependent on reliable biomarker development and extensive genetic testing to identify genetic cases. Whether genotype-dependent stratification of study participants will extend the potential application of new drugs will be one major challenge in conceptualizing clinical trials. However, the latest developments in genotype-driven treatments will pave the road to individualized pathophysiology-based therapies in the future.
Similar content being viewed by others
Background
Parkinson’s disease (PD) is an age-related neurodegenerative disorder characterized by the progressive degeneration of nigrostriatal dopaminergic neurons and extranigral brain regions due to the accumulation of certain α-synuclein species (Fujita et al. 2014; Moore et al. 2005; Schapira and Jenner 2011). The underlying pathophysiology is complex and involves a variety of molecular processes. Despite extensive research in unraveling the molecular mechanisms of PD, no disease-modifying treatments are available to date, and the standard patient care mainly relies on purely symptomatic therapies. Even though monogenic causes of PD (mPD) only account for a minority of PD cases, the investigation of genetic alterations sheds further light on individual disease causes and provides a unique opportunity for drug development and, subsequently, genotype-driven therapies (Blauwendraat et al. 2020). Biological findings and disease traits of mPD can also be useful to identify common disease mechanisms in idiopathic PD (IPD). Selected IPD patients may share molecular pathways with mPD which would be the pre-requisite for pathway-targeted therapies (Redenšek et al. 2017). The identification of such IPD patients requires the detection of reliable biomarkers that enable the stratification of individual IPD patients according to their underlying pathology and the development of appropriate clinical trial designs. Furthermore, the search for patients with mPD by genotyping is an important pre-requisite to gather a substantial number of potential study participants for genotype-driven treatment options and clinical trials. Genetic testing of PD patients is, however, not routinely applied in clinical practice, and consortium research efforts are needed to meet future recruitment targets. In contrast to mPD, IPD is a complex genetic disease where the interaction of genetic risk factors with environmental factors plays a pivotal role in the disease development. The individual identification of risk variants can help gain insights into particular disease pathophysiology independent of a monogenic trait. To date, three approaches have been or are presently evaluated for the stratification of patients with IPD: polygenic risk scoring, blood-based biomarkers (e.g., by measuring enzyme activity in peripheral tissue), or neuroimaging methods (e.g., by studying brain energy metabolism).
This review article will summarize the current understanding of PD genetics, challenges in the development of individualized treatments, current genotype-driven therapies under investigation, and the critical need for reliable biomarkers for patient stratification and treatment monitoring.
Main body
Genetics of monogenic Parkinson’s disease
PD is caused by complex interactions between genetic and environmental factors. Mutations in SNCA (α-synuclein), LRRK2 (Leucine Rich Repeat Kinase 2), and VPS35 (VPS35 Retromer Complex Component) cause an autosomal dominant form of mPD whereas mutations in PRKN (Parkin), PINK1 (PTEN-induced kinase 1), and PARK7 (oncogene DJ-1) (among others) are associated with autosomal-recessive mPD (Lill 2016). Although SNCA was the first identified PD-related gene almost 25 years ago it is very rare whereas mutations in LRRK2 are the most frequent cause of mPD (Cookson 2015). The identification of the autosomal-recessive genes PRKN, PINK1, and PARK7 linked the proposed role of mitochondrial dysfunction in the etiology of PD to genetic causes (Exnre et al. 2012). Monogenic causes are often summarized under the umbrella term of mPD even if parkinsonism is only one of the presenting symptoms and only part of a more complex or atypical phenotype (e.g., in DNAJC6 mutation carriers, Table 1) (Puschmann 2013). Whether molecular insights of causative genes for atypical phenotypes provide translatable findings to IPD needs to be critically evaluated (Grünewald et al. 2013; Klein et al. 2007). In addition, several genes were not yet replicated in independent families or populations. Furthermore, most forms are exceedingly rare making it unlikely that specific therapies are being developed. In addition to mPD that follows the rules of Mendelian inheritance, variants in the GBA (Glucosylceramidase Beta) gene are an unequivocal and frequent risk factor for the development of PD and a promising future drug target.
Drug targets in monogenic Parkinson’s disease
Most mPD genes and GBA converge to distinct molecular mechanisms and can be divided into (i) α-synuclein aggregation (Dehay et al. 2015), (ii) endosome-related involvement (Hafner Česen et al. 2012; Dehay et al. 2013; Smolders and Van Broeckhoven 2020), and (iii) those leading to mitochondrial impairment (Exnre et al. 2012). These pathways indicate potential drug targets for the disease-modifying treatment of mPD (Brüggemann and Klein 2019). Even though mPD helped identify these target pathways in the past decades, genetic stratification in clinical trials started only recently (Dehay et al. 2015; Shults et al. 2004).
The role of α-synuclein aggregation and the SNCA gene
α-Synuclein aggregation is the pathophysiological hallmark of IPD, which has been extensively demonstrated in post-mortem studies (Moore et al. 2005; Kellie et al. 2014). Mutations in the SNCA gene predispose to an increased α-synuclein accumulation and aggregation as the main driver of cell-to-cell propagation of α-synuclein pathology in PD (Xu and Pu 2016). Duplications or triplications result in an increased expression of the wildtype allele with a gene dosage effect whereas point mutations have an impact on the aggregative properties of α-synuclein (Book et al. 2018). α-Synucleinopathy has not only been found in IPD and SNCA mutation carriers but also in other forms of mPD (Poulopoulos et al. 2012). The histopathological changes in mPD, however, are more variable and include tau pathology in some LRRK2 carriers and the absence of α-synucleinopathy in most carriers of PRKN mutations (Henderson et al. 2019; Schneider and Alcalay 2017). The overall evidence of neuropathologic changes in mPD is still limited due to the rarity of autopsied cases.
Formed α-synuclein oligomers are unsuccessfully cleared by the lysosomal or ubiquitin–proteasome systems (UPS) resulting in the formation of Lewy bodies (Volpicelli-Daley et al. 2016). Rodent models demonstrated that the injection of α-synuclein fibrils into the brain activates prolonged α-synuclein aggregates and propagation in interconnected brain regions of model organisms (Luk et al. 2012; Dehay et al. 2016) and human subjects (McCann et al. 2016). The ascending propagation of α-synuclein pathology along neural structures are represented by the Braak stages, which have substantially shaped our current understanding of PD pathophysiology (McCann et al. 2016). Different target mechanisms are currently discussed to counteract the progressive propagation of α-synuclein pathology: (i) decreased α-synuclein production, (ii) decreased intracellular α-synuclein aggregation, (iii) enhanced intracellular α-synuclein degradation, (iv) enhanced extracellular α-synuclein degradation, (v) and the blockage of neural uptake of extracellular α-synuclein (Dehay et al. 2015). These target mechanisms may also expand to mPD forms with α-synuclein pathology but are thus not specific and do not interact with more upstream gene or pathway-related changes.
In animal models, the use of viral vectors successfully mediated the in-vivo production of siRNA (small-interfering RNA; double-stranded, non-coding RNA molecules, that typically lead to the targeted degradation of complementary mRNA molecules) against SNCA in the substantia nigra by employing numerous methods to reduce α-synuclein expression (Volpicelli-Daley et al. 2016). A decrease in α-synuclein levels could also be achieved by varying histone acetylation at the α-synuclein gene promoter and enhancer regions and by administering ß-2-adrenergic agonists (e.g., clenbuterol and salbutamol) (Mittal et al. 2017). Intrabodies bind with monomeric α-synuclein and inhibit oligomerization (Bhatt et al. 2013). Thus, in rodents with viral vector-mediated α-synuclein overexpression, an increase of intracellular α-synuclein aggregation as the cause for subsequent nigral neurodegeneration could be prevented (Volpicelli-Daley et al. 2016). The substance NPT200-11 (NPT200-11 trial, NCT02606682) was furthermore able to block the α-synuclein interface with cell membranes and slowed the oligomerization of aggregates in a mouse model (Bhatt et al. 2013). The phosphorylation of a rapamycin inhibitor was capable of promoting autophagy and reduction of α-synuclein pathology in model systems (Boyd et al. 2013). Nilotinib, a Tyrosine-protein kinase ABL1 inhibitor, was shown to inhibit protein aggregation, neurodegeneration, mitochondrial pyruvate carriers, and posttranslational modifications of α-synuclein in in mice with safety data already available for human use (PD Nilotinib, NCT02954978) (Pagan et al. 2016; Karuppagounder et al. 2014). The first preclinical trials have observed a reduction in extracellular α-synuclein or α-synuclein aggregation due to immunotherapy (Lindström et al. 2014; Tran et al. 2014; Spencer et al. 2017). The first α-synuclein immunotherapy used in a clinical PD trial was PRX002, a humanized IgG1 monoclonal antibody that acts against epitopes of the α-synuclein C-terminus (Brundin et al. 2017). An ascending-dose study in healthy volunteers proved the safety and tolerability at doses up to 30 mg/kg with a plasma half-life of 18.2 days, which was maintained for two to four weeks after a single infusion(BP39529, NCT03100149) (Schenk et al. 2017). In addition, optimized antibodies have been developed in preclinical models against different forms of mono-, oligomeric, fibrils, and aggregated forms to target different stages in α-synuclein related pathophysiology (Wang et al. 2019). Moreover, the recent development of the SNCA gene by antisense-oligonucleotides (ASOs; short-chain, synthetic, single-stranded oligonucleotides that bind to the complementary mRNA and modify/hinder their respective translation) has already been proven useful in in-vitro, rodent, and primate models (Uehara et al. 2019; Alarcón-Arís et al. 2018; Choong and Mochizuki 2017).
Targeting lysosomal dysfunction in PD: the role of the GBA gene
The GBA gene encodes the protein glucocerebrosidase (GCase), a lysosomal hydrolase, which converts glucosylceramide to ceramide and glucose (Goker-Alpan et al. 2004; Neudorfer et al. 1996). The accumulation of undegraded substrates by compound heterozygous or homozygous GBA mutations has been linked to the lysosomal storage disorder Gaucher's disease (GD). Interestingly, GD patients have a higher incidence of parkinsonism, and heterozygous mutations in the GBA gene increase the risk of developing PD (Goker-Alpan et al. 2004). Subsequently, heterozygous GBA mutations are considered the most common genetic risk factor for PD, and at least one putative damaging mutation can be present in up to 10% of PD patients (Robak et al. 2017). There is a crucial delineation between ‘pathogenic’ GBA variants (such as those causing GD in a compound heterozygous/homozygous carrier state) and ‘PD risk factor’ GBA variants, which show an association with PD risk but are not considered causative for GD (Skrahina et al. 2020). Another classification of GBA variants embraces the distinction of ‘mild’ and ‘severe’ variants (in patients with type 1 or type 2/3 GD) (Stirnemann et al. 2017). Age at onset, phenotype and disease course of carriers of mild GBA variants (mGBA-PD) are comparable with IPD whereas carriers of severe GBA variants (sGBA-PD) have a clearly increased risk of dementia with an earlier onset and a more rapid cognitive decline (Davis et al. 2016). Disease progression of carriers of mild mutations is usually slower than in those with severe mutations but still faster than in non-carriers (Cilia et al. 2016). The GCase activity is reduced in sGBA-PD and, to a lesser extent, in mGBA-PD, whereas there is only a slight reduction in IPD (Alcalay et al. 2015). Collectively, an increase of GCase activity by targeted therapies may only be beneficial for selected IPD patients as there is currently no persuasive evidence for increased accumulation of glycosylceramides in IPD patients (Niimi et al. 2020) and no efficacy and safety data are yet available on supraphysiological GCase levels. On the other hand, several, mainly preclinical, studies have shown that GBA deficiency predisposes to α-synuclein pathology. Here, reduced GCase activity causes increased levels of ubiquitin/α-synuclein aggregates and is related to motor and cognitive problems in a rodent model (Sardi et al. 2013). An inverse relationship between GCase activity and oligomeric α-synuclein levels can be explained by a pathological feedback loop (Mazzulli et al. 2011). Changes in glycosphingolipid homeostasis can affect the membrane composition and impair lysosomal function and vesicular transport, thus enhancing α-synuclein aggregation. This process results in selective synaptic dysfunction and neuronal degeneration (Schapira 2015).
Different treatment options are discussed in GBA-PD but also selected patients with IPD: (i) substrate reduction (as often considered for the treatment of GD), (ii) external GCase augmentation, and (iii) the enhancement of GCase activity (e.g., by ambroxol). Substrate reduction can be achieved by different mechanisms: Glucosylceramide synthase inhibitors decrease glycosphingolipid levels and are used to treat the hematological and visceral presentations of GD patients. The clinical trial GZ/SAR402671 (NCT02906020) showed that the glucosylceramide synthase inhibitor venglustat sufficiently crossed the blood brain barrier (BBB) in humans (Davis et al. 2016). In mouse models of GD-related synucleinopathy and alpha-synuclein overexpression, venglustat led to a decrease in alpha-synuclein expression (Mazzulli et al. 2011). Whether this approach may also be suitable for GBA-PD is currently under investigation in a clinical phase II trial in patients (Moves PD, NCT02906020).
The external augmentation of GCase in the brain requires the penetration of the enzyme through the blood brain barrier which cannot be sufficiently achieved with enzymes used in GD treatment (Sun et al. 2020). One approach is therefore a gene therapy-mediated viral overexpression of exogenous GCase in the brain that was shown to reverse behavioral and pathological abnormalities by restoring the membrane glycosphingolipid balance (Sardi et al. 2011; Rockenstein et al. 2016). This observation supports gene therapy aiming to increase GCase levels in the brain. In keeping, adeno-associated viruses are safe and biologically active vectors that target GCase augmentation, reverse cognitive problems, and reduce α-synuclein in an SNCAA53T mouse model (Hafner Česen et al. 2012). Moreover, a previous investigation showed that increased lysosomal GCase activity could be achieved by optimizing the delivery route, vector subtypes, small molecules, small molecular chaperones, and brain distribution in critical brain regions (Gegg and Schapira 2018). The third proposed approach for GBA-targeted therapies is the enhancement of GCase activity, e.g., by the repurposed drug ambroxol in order to enhance GCase activity and to reduce α-synuclein and S129-phosphorylated α-synuclein protein levels as shown in nonhuman primates. At present, the clinical trials UCL 15/0118 (NCT02941822) and R15-006 (NCT02914366) tested the safety, efficacy, and tolerability of ambroxol in PD (Migdalska-Richards et al. 2016, 2017). Preliminary data of human subjects are already available, supporting sufficient BBB crossing and molecular target site enrichment (Mullin et al. 2020). Another investigational drug class are noninhibitory GCase chaperones such as NCGC758 and NCGC607 that can bind to GCase at the active site and lead to conformational changes that enhance GCase activity. These chaperones penetrate the brain, increase lysosomal activity, GCase translocation to lysosomes, and reduce substrate and α-synuclein accumulation (Dehay et al. 2013). Another clinical trial of afegostat tartrate in GD (AT2101, NCT00433147) showed increased GCase activity and enzyme stabilization but did not show significant clinical improvement in GD patients, and the trial was discontinued (Boyd et al. 2013).
Challenges and opportunities of LRRK2-targeted clinical trials
The first gene mutation in LRRK2 was identified in a family series of autosomal-dominant parkinsonism (Paisán-Ruíz et al. 2004). The LRRK2G2019S accounts for the vast majority of LRRK2-associated PD worldwide and is highly frequent in certain populations of PD patients, e.g. in Israel and North Africa (Trinh et al. 2018). Symptomatic LRRK2G2019S mutation carrier usually present with a postural instability and gait difficulty (PIGD) phenotype, but show a relatively mild cognitive and motor decline during the overall disease course (Saunders-Pullman et al. 2018). Together with other LRRK2 variants, the LRRK2G2019S result in a gain of function (GOF) with an increase of LRRK2 kinase activity. The LRRK2 protein has a complex multidomain structure and belongs to the family of protein kinases, which play a fundamental role in the control and regulation of complex cellular processes by transferring phosphate groups to target proteins. The kinase domain of LRRK2 shares similarities with mitogen-activated protein (MAP) kinases, which play a central role in mediating cellular stress. Even though the precise mechanism of LRRK2 is poorly understood, the disease-causing GOF mutations allows heuristic treatments by inhibition of its activity. Currently, two main strategies exist for LRRK2-targeted treatment strategies: (i) pharmacological inhibition of LRRK2 activity and (ii) silencing of the LRRK2 gene by the use of ASOs. Both options aim to reduce LRRK2 activity in the CNS whereby ASOs may bypass potential peripheral adverse effects of kinase inhibitors due to its intrathecal application (Cookson 2015). DNL201, a small molecule LRRK2 inhibitor, reduced LRRK2 activity levels by more than 90% in a phase I study in healthy volunteers (DNLI-B-0001, NCT04551534). Ras-related protein Rab10 substrate phosphorylation and LRRK2 S935 phosphorylation were used to measure treatment response (by a decrease in peripheral LRRK2 activity) in blood. A trial with DNL151, a second LRRK2 inhibitor, is still actively recruiting healthy volunteers (DNLI-C-0001, NCT04557800) (Zeuner et al. 2019). Preclinical data suggested that inhibition of LRRK2 could be associated with pulmonary morphological changes in nonhuman primates, resulting in potential safety concerns. Here, targeting LRRK2 with a high dose of three compounds resulted in accumulating lamellar bodies in type-II pneumocytes (Fuji et al. 2015). However, these morphological abnormalities could be reversed after two weeks off dose, and no pulmonary abnormalities were found at the highest doses. Furthermore, there was no association of loss of function variants with a putatively decreased LRRK2 kinase activity and a specific phenotype or disease state in human databases (Whiffin et al. 2020).
Improving mitochondrial bioenergetics and antioxidative treatment strategies: PRKN and PINK1
Mitochondrial dysfunction is one of the main concepts in PD pathogenesis. Mitochondria play a fundamental role for a plethora of cellular processes relevant for the supply of energy, the overall cellular homeostasis, and neuronal survival. The first evidence for a role in PD derived from environmental studies illustrating the effect of neurotoxic agents in inhibiting the mitochondria's electron transport chain (ETC) (Schapira and Jenner 2011). The discovery of the autosomal recessively inherited genes PRKN and PINK1 provided further evidence to support a strong contribution of mitochondrial dysfunction to PD (Exnre et al. 2012). Under physiological conditions, PINK1 recruits Parkin to damaged mitochondria and leads to the clearance of mitochondria via the UPS, a process referred to as mitophagy (Park et al. 2018). Parkin and PINK1, therefore, jointly serve as a molecular quality control system (McWilliams and Muqit 2017). Possible therapeutic or preventive approaches in carriers of PRKN or PINK1 mutations are thus (i) the enhancement of PRKN or PINK1 expression, (ii) the prevention of Parkin or PINK1 inactivation, and (iii) the control of the downstream Parkin/PINK1 signaling pathway (Gaki and Papavassiliou 2014). However, mitochondrial dysfunction extends to a variety of pathological mechanisms including impaired mitochondrial biogenesis, fusion and fission functions, trafficking, metal ion and calcium homeostasis, neuroinflammation, and pro-apoptotic signaling (Dextera and Jenner 2013). These different aspects may be suitable as potential treatment targets. Alternative strategies for genotype-driven therapies in mitochondrial dysfunction consists of the enhanced clearance of dysfunctional mitochondria via mitophagy or other mitochondrial stress response pathways (Aman et al. 2020), the improvement of mitochondrial biogenesis (e. g., by glucagon-like peptide 1 [GLP1] receptor agonist exenatide exposition) (Athauda et al. 2017), gene therapies targeting the mitochondrial or nuclear genome (Choong and Mochizuki 2017), addressing mitochondrial calcium and metal ion dyshomeostasis (e.g., by iron chelators) (Sun et al. 2018; Rani and Mondal 2020), targeting the intersection to neuroinflammation (e.g., by disruption of interleukin 6 [IL-6] signaling) (Borsche et al. 2020), and stem cell therapies (Cheng et al. 2020). However, bioenergetic depletion and increased reactive oxygen species [ROS] production are common to all types of mitochondrial dysfunction, they do most likely recapitulate one of the earliest pathophysiological events not only in mPD but also IPD, and were thus the primary target mechanisms for most of the recent studies addressing mitochondrial pathology (Prasuhn et al. 2021). The most extensively investigated compound has been coenzyme Q10, a mitochondrial enhancer that, however, failed to show efficacy in most studies (see Table 2). One potential explanation for the negative outcome is the lack of genetic stratification of PD patients to enrich for patients with a strong contribution of mitochondrial dysfunction (e.g., biallellic PRKN or PINK1 mutation carriers) as listed in Table 2. To our knowledge, only two clinical trials (MitoPD [DRKS00015880] and PD-K2 [DRKS00019932]) are actively recruiting to date that use a combination of genetic stratification and treatment-response monitoring by neuroimaging (Prasuhn et al. 2019; Prasuhn et al. 2021). In the MitoPD study, groups are defined by a varying degree of predicted mitochondrial dysfunction: homozygous/compound heterozygous PRKN/PINK1 mutation carriers, heterozygous PRKN/PINK1 mutation carriers, and two IPD groups defined by the statistical extrema as determined by a mitochondrial PRS (Prasuhn et al. 2019). In PD-K2, homozygous/compound heterozygous PRKN/PINK1 mutation carriers, IPD patients, and healthy controls are included (DRKS00019932). Both studies have multimodal neuroimaging in common that will be applied as a surrogate marker for examining beneficial effects of in-vivo brain energy metabolism, i.e., by determining the change of energy equivalents using 31Phosphorus Magnetic Resonance Spectroscopy Imaging (31P-MRSI).
The interconnectedness of pathophysiological pathways in monogenic PD: one therapy, many targets
One key motivation for the study of mPD is the translation of molecular insights into the pathogenesis of IPD (Lin and Farrer 2014). The idea that molecular processes can be pinpointed down towards a single gene variant may, however, be a substantial oversimplification (Blauwendraat et al. 2020) as molecular processes are interwoven and complex, also in mPD. Based on our current understanding of PD pathophysiology, α-synuclein aggregation is the primary disease mechanism present in IPD, GBA-PD, and many but not all mPD cases. The histopathological findings in mPD are more heterogeneous and include tau pathology in LRRK2 and the absence of α-synuclein deposition in most autopsied PRKN mutation carriers. As mentioned above, α-synuclein reduction is a main target for IPD. Thus, mPD patients who also exhibit relevant α-synuclein pathology, most importantly GBA-PD, should also benefit from this pathophysiologically based therapy. For the other forms of mPD, the effect of these therapies is less predictable. It is, therefore, necessary to establish reliable biomarkers, e.g. α-synuclein PET, to sufficiently stratify PD patients largely independent of their genotype.
The enhancement of GCase leads to a reduction in α-synuclein pathology and may be a target of interest for other α-synuclein-related forms of mPD and the majority of IPD patients (Schapira 2015; Gan-Or et al. 2017). Recent developments of selective inhibitors of glycosphingolipid biosynthesis and noninhibitory pharmacological chaperones of glycosphingolipid processing enzymes are therefore promising treatment approaches. Current limitations with respect to BBB penetration or off-target-effects are limiting their clinical usability (Sybertz and Krainc 2014).
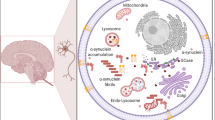
Third, there is supporting evidence that almost all mPD-causing gene variants are somewhat related to an impairment of mitochondrial function (Shadrina et al. 2010). Whether these patient groups may benefit from mitochondrial enhancers is still under debate. Figure 1 provides an overview of the interconnected character of highlighted genes in mPD.

Converging pathways in Parkinson’s disease pathophysiology and relevant genes associated. The main disease mechanisms and current drug targets for mPD and GBA-PD are summarized. The links between key pathophysiological aspects are highlighted with double arrows indicating that translational therapies targeting related pathways may also be of use for a plethora of mPD, GBA-PD and IPD cases. Panel A depicts the aggregation of monomeric to oligomeric α-synuclein aggregates resulting in the formation of Lewy's bodies. Panel B.I and B.II symbolize endosomal disturbances, resulting in impaired neurotransmitter release (in particular VPS35) or impaired degradation of complex molecule structures by autophagy. Panel C illustrates mitochondrial damage, e.g., caused by oxidative stress (highlighted with thunderbolts), which can result in impaired mitochondrial dynamics (fusion and fission processes) among other downstream effects. ATP13A2: ATPase Cation Transporting 13A2. DNAJC13: DnaJ Heat Shock Protein Family (Hsp40) Member C13. DNAJC6: DnaJ Heat Shock Protein Family (Hsp40) Member C6. FBXO7: F-Box only protein 7. GBA: Glucosylceramidase Beta. GBA-PD: GBA-associated Parkinson’s disease. LRRK2: Leucine Rich Repeat Kinase 2. mPD: monogenic Parkinson’s disease. PARK7: oncogene DJ-1. PINK1: PTEN-induced kinase 1. PLA2G6: Phospholipase A2 Group VI. POLG: Mitochondrial Polymerase Gamma. PRKN: Parkin. SNCA: α-synuclein. SYNJ1: Synaptojanin 1. VPS13C: Vacuolar Protein Sorting 13 Homolog C. VPS35: VPS35 Retromer Complex Component
Fourth, evidence from epidemiological (Gao and Chen 2011), neuroimaging (Wilson et al. 2019a), post-mortem (McGeer et al. 1988), and preclinical studies (Lindestam Arlehamn et al. 2020) suggest that neuroinflammation may be a shared pathophysiological hallmark of PD etiology. Epidemiological evidence points towards potential benefits of non-steroidal anti-inflammatory drugs, but published meta-analyses yield conflicting results (Bornebroek et al. 2007; Gagne and Power 2010; Samii et al. 2009). The long-term use of TNFα-targeted antibodies in patients with inflammatory bowel disease leads to a significant reduction in PD risk (Peter et al. 2018). The interconnectedness of the proposed pathomechanisms of PD (α-synuclein aggregation, endosome-related pathologies, and mitochondrial impairment) to neuroinflammation has been demonstrated for α-synuclein pathology (Li et al. 2019), increased LRRK2 activity (Lee et al. 2017), GCase alterations (Sanyal et al. 2020), and PRKN/PINK1-related mitochondrial dysfunction (Borsche et al. 2020). Therefore, targeting neuroinflammation could provide another substantial treatment opportunity for PD. However, the precise cellular and humoral driver of neuroinflammation in PD is still unclear, limiting yet the translation to clinical trials (Hirsch and Standaert 2020).
The ongoing discovery of disease mechanisms will possibly result in a combination of tailored disease-modifying therapies for individual PD patients. This will be perhaps somehow comparable with the symptomatic treatment of PD patients where the optimized combination of anti-Parkinsonian drugs is used to treat the individual disease burden of a given patient.
The role of targeted therapies in idiopathic PD patients
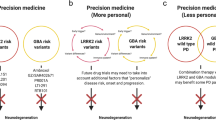
The potential causes of lacking disease modification in PD are manifold and include too advanced neurodegeneration, insufficient target engagement, a varying contribution of individual disease mechanisms across IPD patients and a too short observation interval. The enrichment of study cohorts by genetically well-defined participants is crucial for developing targeted therapies, and genotype-driven therapies are currently under investigation (see Table 2). Advancements in the genetic screening of PD patients have shown that mPD and GBA-PD cases may be more frequent than previously suspected (Skrahina et al. 2020). Genetic testing is, however, still not routinely applied in the diagnostic workup or clinical trial recruitment (Billingsley et al. 2018). This now becomes increasingly relevant due to the recent progress in translational therapies that should not be withheld from individuals with an unknown but potentially treatment-qualifying genotype. Genetic testing should be considered early in PD patients' diagnostic management to meet the narrowing window of opportunity for disease-modifying treatments. In other fields, e.g., cancer treatment, the consideration of genetic variants has already entered clinical practice and led to the development of more efficient adaptive clinical trial designs (Li et al. 2007; Chow and Chang 2008; Berry 2012). Genetic testing will increasingly become important for the therapeutic management of neurological patients. As an example, the recently FDA-approved oligonucleotide drug Nusinersen requires genetic testing of patients with spinal muscular atrophy to clarify the genetic diagnosis and to evaluate patients for their eligibility to participate in clinical trials (Chiriboga 2017). The development of trial designs for neuroprotective treatments itself faces significant challenges: Long interventional periods are needed to demonstrate the disease-modifying effects of investigational drugs. While considering the long-lasting prodromal phase of PD, it is desirable to identify not yet diseased individuals for neuroprotective therapies (Heinzel et al. 2019). However, there is currently a gap of knowledge concerning one individual's conversion to PD.
To extend the promising approach of targeted therapies in mPD to IPD cases, biomarkers are needed to group patients based on their underlying disease etiology (e.g., identifying those with critical mitochondrial impairment). The disease-modifying management of PD is herewith more challenging due to the absence of validated and dynamic mechanism-based biomarkers.
Need for reliable (para-)clinical biomarkers in the design of clinical trials for disease-modifying approaches
Biomarkers in clinical PD trials are mainly required to demonstrate target engagement and to quantify disease progression. Stratification of PD patients based on their primarily involved disease mechanisms is one substantial prerequisite for targeted therapies (Redenšek et al. 2017). Two main concepts are of importance: state and trait biomarkers. State biomarkers often refer to the genetic background of one's individual. This also includes monogenic gene variants (mPD) and the complex genetic architecture of IPD patients (e.g., by PRS referring to a predominant disease mechanism) (Heinzel et al. 2019). Trait biomarkers should recapitulate the pathophysiological processes caused by the aforementioned genetic variants and be responsive to interventions. To date, two main concepts for dynamic biomarkers are under evaluation: biomarkers based on peripheral tissues (e.g., blood or CSF based assays) and neuroimaging methods (Burciu et al. 2017; Postuma and Berg 2016; Bloem et al. 2019; Parnetti et al. 2019). There is one general concern about biomarkers of peripheral tissues: The negligible biomass of affected brain regions compared to the human body's remaining tissue requires hypersensitive analytical methods and can still be overshadowed by physiological background noise (Davis et al. 2020). Peripheral biomarkers for the converging pathophysiological mechanisms (i) α-synuclein aggregation, (ii) endosome-related, and (iii) mitochondrial impairment have already been evaluated and yielded contradictory results (Parnetti et al. 2019; Sharma et al. 2013). In most reports, their capability to treatment responses has not yet been evaluated. Neuroimaging offers the opportunity for mostly non-invasive analyses of affected brain tissue. Neuroimaging studies performed on SNCA (Si et al. 2019), GBA (Greuel et al. 2020), LRRK2 (Simuni et al. 2020), and PRKN/PINK1 (Van Nuenen et al. 2009; Anders et al. 2012; Nuenen et al. 2009) have been successfully used to illustrate neuroanatomical and functional group differences. These studies include brain mapping of the serotoninergic system in (pre-)symptomatic SNCAA53T mutation carrier (Wilson et al. 2019), or PET studies investigating the serotoninergic, dopaminergic, and cholinergic neurotransmitter systems in (pre-)symptomatic LRRK2 mutation carriers (Wile et al. 2017; Liu et al. 2018). In addition, a recent study has shown that brain metabolic networks in GBA or LRRK2 mutation carrier showed a distinct network pattern (Schindlbeck et al. 2020). In summary, these studies suggest that the plethora of specific neuroimaging methods can illustrate genotype-specific brain changes and can also be applied to investigate pre-manifest mutation carriers. The latter is of high translational relevance as these neuroimaging biomarkers will open a unique window of opportunity for pre-manifest, targeted, and neuroprotective treatment strategies. Even though these promising results on genetically defined PD patients, the used methods often lack specificity with respect to the potentially treatable disease mechanism. Consequently, imaging studies are often limited to the analysis of neurodegenerative changes and do not sufficiently take disease biology into account. One notable exception is the study of brain energy metabolism for the characterization of mitochondrial dysfunction. For example, the PET tracer [18F]BCPP‐EF has been used to investigate Complex I dysfunction in PD patients (Wilson et al. 2020). Non-invasive magnetic resonance spectroscopy imaging (MRSI) allows the in-vivo measurement of Lactate (1H-MRSI) and high energy phosphates such as ATP levels (31P-MRSI) (Bonvento et al. 2017). These methodologies have also been advanced to allow for dynamic measurements (such as ATP synthesis rate) (Clifford et al. 2020). In addition, the combination of functional MRI (by measuring the blood-oxygen-level-dependent signal, BOLD) and arterial spin labeling (ASL) can be used to study the cerebral oxygen consumption rate (Germuska et al. 2019), and near-infrared based spectroscopy is capable to quantify the redox state of Cytochrome c (Holper et al. 2019). The availability of these methods is restricted by different hardware setups and methodological limitations. Also, intra- and intersite reliability needs to be ensured and critically assessed before being applied in clinical trials. In summary,
Conclusion
The genetic discoveries in PD have aided the deepened understanding of clinical manifestations, underlying pathogenesis, and the potential for targeted therapies (Brüggemann and Klein 2019). Even though our current understanding of disease biology is continuously expanding, existing knowledge gaps need to be addressed in the future. Reliable biomarkers are needed that specifically recapitulate pathophysiological hallmarks for patient stratification and the monitoring of treatment responses. Genetic testing in 'idiopathic' or 'sporadic' PD patients is the prerequisite to identify individuals for genotype-driven therapies. Genotype-dependent stratification of study participants will extend the potential application of targeted drugs. Biomarker-assisted clinical trials will substantially benefit from new adaptive designs. However, the latest developments in genotype-driven treatments will, in the midterm, hopefully provide substantial benefits for PD patients and result in the first disease-modifying therapies.
Availability of data and materials
Not applicable.
Abbreviations
- 1H-MRSI:
-
Proton magnetic resonance spectroscopy imaging
- 31P-MRSI:
-
31Phosphorus resonance spectroscopy imaging
- ASL:
-
Arterial spin labelling
- ASO:
-
Anti-sense oligonucleotides
- BBB:
-
Blood brain barrier
- BOLD:
-
Blood-oxygen-level-dependent
- DNAJC6:
-
DnaJ heat shock protein family (Hsp40) member C6
- ETC:
-
Electron transport chain
- GBA-PD:
-
GBA-associated Parkinson’s disease
- GBA:
-
Glucosylceramidase Beta
- GCase:
-
Glucosylceramidase
- GD:
-
Gaucher’s disease
- GLP1:
-
Glucagon-like peptide 1
- GOF:
-
Gain of function
- GWAS:
-
Genome-wide association studies
- IgG1:
-
Immunoglobulin G type 1
- IL-6:
-
Interleukin 6
- IPD:
-
Idiopathic Parkinson’s disease
- LRRK2:
-
Leucine rich repeat kinase 2
- MAP:
-
Mitogen-activated protein
- mGBA-PD:
-
Carrier of a mild GBA variant with associated Parkinson’s disease
- mPD:
-
Monogenic Parkinson’s disease
- MRI:
-
Magnetic resonance imaging
- PD:
-
Parkinson’s disease
- PARK7:
-
Oncogene DJ-1
- PET:
-
Positron emission tomography
- PINK1:
-
PTEN-induced kinase 1
- PRKN:
-
Parkin
- PRS:
-
Polygenic Risk Score
- ROS:
-
Reactive oxygen species
- sGBA-PD:
-
Carrier of a severe GBA variant with associated Parkinson’s disease
- siRNA:
-
Small interfering ribonucleic acid
- SNCA:
-
α-Synuclein
- TNFα:
-
Tumor necrosis factor alpha
- UPS:
-
Ubiquitin–proteasome system
- VPS35:
-
VPS35 retromer complex component
References
Alarcón-Arís D, Recasens A, Galofré M, Carballo-Carbajal I, Zacchi N, Ruiz-Bronchal E, et al. Selective α-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: potential therapy for Parkinson’s disease. Mol Ther. 2018;26(2):550–67.
Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138(9):2648–58.
Aman Y, Ryan B, Torsetnes SB, Knapskog AB, Watne LO, McEwan WA, et al. Enhancing mitophagy as a therapeutic approach for neurodegenerative diseases. Int Rev Neurobiol. 2020;155:169–202.
Anders S, Sack B, Pohl A, Münte T, Pramstaller P, Klein C, et al. Compensatory premotor activity during affective face processing in subclinical carriers of a single mutant Parkin allele. Brain. 2012;135(4):1128–40.
Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10103):1664–75.
Berry DA. Adaptive clinical trials in oncology. Nat Rev Clin Oncol. 2012;9:199–207.
Bhatt MA, Messer A, Kordower JH. Can intrabodies serve as neuroprotective therapies for Parkinson’s disease? beginning thoughts. J Parkinson’s Dis. 2013;3(4):581–91.
Billingsley KJ, Bandres-Ciga S, Saez-Atienzar S, Singleton AB. Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 2018;373:9–20.
Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19:170–8.
Bloem BR, Marks WJ, Silva De Lima AL, Kuijf ML, Van Laar T, Jacobs BPF, et al. The personalized Parkinson project: examining disease progression through broad biomarkers in early Parkinson’s disease. BMC Neurol. 2019;19(1):160.
Bonvento G, Valette J, Flament J, Mochel F, Brouillet E. Imaging and spectroscopic approaches to probe brain energy metabolism dysregulation in neurodegenerative diseases. J Cereb Blood Flow Metab. 2017;37:1927–43.
Book A, Guella I, Candido T, Brice A, Hattori N, Jeon B, et al. A meta-analysis of α-synuclein multiplication in familial Parkinsonism. Front Neurol. 2018;9:1021.
Bornebroek M, De Lau LML, Haag MDM, Koudstaal PJ, Hofman A, Stricker BHC, et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Neuroepidemiology. 2007;11(1):1875.
Borsche M, König IR, Delcambre S, Petrucci S, Balck A, Bruggemann N, et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain. 2020;143(10):3041–51.
Boyd RE, Lee G, Rybczynski P, Benjamin ER, Khanna R, Wustman BA, et al. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J Med Chem. 2013;56:2705–25.
Brüggemann N, Klein C. Will genotype drive treatment options? Mov Disord. 2019;34(9):1294–9.
Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol. 2017;298:225–35.
Burciu RG, Ofori E, Archer DB, Wu SS, Pasternak O, McFarland NR, et al. Progression marker of Parkinson’s disease: a 4-year multi-site imaging study. Brain. 2017;140(8):2183–92.
Cheng XY, Biswas S, Li J, Mao CJ, Chechneva O, Chen J, et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl Neurodegen. 2020;9(1):13.
Chiriboga CA. Nusinersen for the treatment of spinal muscular atrophy. Expert Rev Neurother. 2017;17(10):955–62.
Choong CJ, Mochizuki H. Gene therapy targeting mitochondrial pathway in Parkinson’s disease. J Neural Trans. 2017;124:193–207.
Chow SC, Chang M. Adaptive design methods in clinical trials—a review. Orphanet J Rare Dis. 2008;3:11.
Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann Neurol. 2016;80(5):662–73.
Clifford B, Gu Y, Liu Y, Kim K, Huang S, Li Y, et al. High-resolution dynamic 31P-MR spectroscopic imaging for mapping mitochondrial function. IEEE Trans Biomed Eng. 2020;67(10):2745–53.
Cookson MR. LRRK2 pathways leading to neurodegeneration. Curr Neurol Neurosci Rep. 2015;15:42.
Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. 2016;73(10):1217–24.
Davis RL, Wong SL, Carling PJ, Payne T, Sue CM, Bandmann O. Serum FGF-21, GDF-15, and blood mtDNA copy number are not biomarkers of Parkinson disease. Neurol Clin Pract. 2020;10(1):40–6.
Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, et al. Lysosomal impairment in Parkinson’s disease. Mov Disord. 2013;28:725–32.
Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14:855–66.
Dehay B, Vila M, Bezard E, Brundin P, Kordower JH. Alpha-synuclein propagation: new insights from animal models. Mov Disord. 2016;31:161–8.
Dextera DT, Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med. 2013;62:132–44.
Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson′s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31:3038–62.
Fuji RN, Flagella M, Baca M, Baptista MAS, Brodbeck J, Chan BK, et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci Transl Med. 2015;7(273):273ra15.
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, et al. Integrating pathways of Parkinson’s disease in a molecular interaction map. Mol Neurobiol. 2014;49:88–102.
Gagne JJ, Power MC. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 2010;74(12):995–1002.
Gaki GS, Papavassiliou AG. Oxidative stress-induced signaling pathways implicated in the pathogenesis of Parkinson’s disease. NeuroMol Med. 2014;16(2):217–30.
Gan-Or Z, Liong C, Alcalay RN. GBA-associated Parkinson’s disease and other synucleinopathies. Curr Neurol Neurosci Rep. 2017;18:44.
Gao X, Chen H, Schwarzschild MA, Ascherio A. Use of ibuprofen and risk of Parkinson disease. Neurology. 2011;76(10):863–9.
Gegg ME, Schapira AHV. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018;285:3591–603.
Germuska M, Chandler HL, Stickland RC, Foster C, Fasano F, Okell TW, et al. Dual-calibrated fMRI measurement of absolute cerebral metabolic rate of oxygen consumption and effective oxygen diffusivity. Neuroimage. 2019;184:717–28.
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41(12):937–40.
Greuel A, Trezzi JP, Glaab E, Ruppert MC, Maier F, Jäger C, et al. GBA Variants in Parkinson’s disease: clinical, metabolomic, and multimodal neuroimaging phenotypes. Mov Disord. 2020;35:2201–10.
Grünewald A, Kasten M, Ziegler A, Klein C. Next-generation phenotyping using the Parkin example: time to catch up with genetics. JAMA Neurol. 2013;70:1186–91.
Hafner Česen M, Pegan K, Špes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res. 2012;318:1245–51.
Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–90.
Heckman MG, Soto-Ortolaza AI, Aasly JO, Abahuni N, Annesi G, Bacon JA, Bardien S, et al. Population-specific frequencies for LRRK2 susceptibility variants in the genetic epidemiology of Parkinson’s disease (GEO-PD) consortium. Mov Disord. 2013;28(12):1740–4.
Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB. Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2019;34:1464–70.
Henderson MX, Sengupta M, Trojanowski JQ, Lee VMY. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol Commun. 2019. https://doi.org/10.1186/s40478-019-0836-x.
Hirsch EC, Standaert DG. Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov Disord. 2020;36(1):16–24.
Holper L, Lan MJ, Brown PJ, Sublette EM, Burke A, Mann JJ. Brain cytochrome-c-oxidase as a marker of mitochondrial function: a pilot study in major depression using NIRS. Depress Anxiety. 2019;36(8):766–79.
Karuppagounder SS, Brahmachari S, Lee Y, Dawson VL, Dawson TM, Ko HS. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci Rep. 2014. https://doi.org/10.1038/srep04874.
Kellie JF, Higgs RE, Ryder JW, Major A, Beach TG, Adler CH, et al. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci Rep. 2014;4:1–10.
Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6:652–62.
Lee H, James WS, Cowley SA. LRRK2 in peripheral and central nervous system innate immunity: its link to Parkinson’s disease. Biochem Soc Trans. 2017;45(1):131–9.
Lesage S, Dürr A, Tazir M, Lohmann E, Leutenegger A, Janin S, et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006;354(4):422–3.
Li Y, Niu M, Zhao A, Kang W, Chen Z, Luo N, et al. CXCL12 is involved in α-synuclein-triggered neuroinflammation of Parkinson’s disease. J Neuroinflamm. 2019;16(1):263.
Li W, He S ju, Wang Y, Cheng X ru, Jia X. Adaptive designs for clinical trial. Zhonghua Liu Xing Bing Xue Za Zhi. 2007;28(6):605–7.
Lill CM. Genetics of Parkinson’s disease. Mol Cell Probes. 2016;30(6):386–96.
Lin MK, Farrer MJ. Genetics and genomics of Parkinson’s disease. Genome Med. 2014;6:48.
Lindestam Arlehamn CS, Dhanwani R, Pham J, Kuan R, Frazier A, Rezende Dutra J, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun. 2020. https://doi.org/10.1038/s41467-020-15626-w.
Lindström V, Fagerqvist T, Nordström E, Eriksson F, Lord A, Tucker S, et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol Dis. 2014;69:134–43.
Liu SY, Wile DJ, Fu JF, Valerio J, Shahinfard E, McCormick S, et al. The effect of LRRK2 mutations on the cholinergic system in manifest and premanifest stages of Parkinson’s disease: a cross-sectional PET study. Lancet Neurol. 2018;17:309–16.
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–53.
Maiese K, Chong ZZ, Shang YC, Wang S. MTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. 2013;19:51–60.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52.
McCann H, Cartwright H, Halliday GM. Neuropathology of α-synuclein propagation and braak hypothesis. Mov Disord. 2016;31:152–60.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the: substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–91.
McWilliams TG, Muqit MM. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol. 2017;45:83–91.
Migdalska-Richards A, Daly L, Bezard E, Schapira AHV. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann Neurol. 2016;80(5):766–75.
Migdalska-Richards A, Ko WKD, Li Q, Bezard E, Schapira AHV. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse. 2017;71(7):e21967.
Mittal S, Bjørnevik K, Im DS, Flierl A, Dong X, Locascio JJ, et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science. 2017;357(6354):891–8.
Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Ann Rev Neurosci. 2005;28:57–87.
Mullin S, Smith L, Lee K, D’Souza G, Woodgate P, Elflein J, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. 2020;77(4):427–34.
Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Achai E, et al. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM Mon J Assoc Phys. 1996;89(9):691–4.
Niimi Y, Mizutani Y, Akiyama H, Watanabe H, Shiroki R, Hirabayashi Y, et al. Cerebrospinal fluid profiles in Parkinson’s disease: no accumulation of glucosylceramide, but significant downregulation of active complement C5 fragment. J Parkinsons Dis. 2020. https://doi.org/10.3233/JPD-202310.
Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, et al. Nilotinib effects in Parkinson’s disease and dementia with lewy bodies. J Parkinsons Dis. 2016;6(3):503–17.
Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, Van Der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600.
Park JS, Davis RL, Sue CM. Mitochondrial dysfunction in Parkinson’s disease: new mechanistic insights and therapeutic perspectives. Curr Neurol Neurosci Rep. 2018;18:21.
Parnetti L, Gaetani L, Eusebi P, Paciotti S, Hansson O, El-Agnaf O, et al. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019;18:573–86.
Peter I, Dubinsky M, Bressman S, Park A, Lu C, Chen N, et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 2018;75(8):939–46.
Postuma RB, Berg D. Advances in markers of prodromal Parkinson disease. Nat Rev Neurol. 2016;12:622–34.
Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson’s disease. Mov Disord. 2012;27:831–42.
Prasuhn J, Brüggemann N, Hessler N, Berg D, Gasser T, Brockmann K, et al. An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol Res Pract. 2019;1(1):1–7.
Prasuhn J, Davis RL, Kumar KR. Targeting mitochondrial impairment in Parkinson’s disease: challenges and opportunities. Front Cell Dev Biol. 2021;8:1704.
Prasuhn J, Kasten M, Vos M, König IR, Schmid SM, Wilms B, et al. The use of vitamin K2 in patients with Parkinson’s disease and mitochondrial dysfunction (PD-K2): a theranostic pilot study in a placebo-controlled parallel group design. Front Neurol. 2021;11(January):1–11.
Puschmann A. Monogenic Parkinson’s disease and Parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord. 2013;19:407–15.
Rani L, Mondal AC. Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: pathogenic and therapeutic implications. Mitochondrion. 2020;50:25–34.
Redenšek S, Trošt M, Dolžan V. Genetic determinants of Parkinson’s disease: can they help to stratify the patients based on the underlying molecular defect? Front Aging Neurosci. 2017;9:20.
Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. 2017;140(12):3191–203.
Rockenstein E, Clarke J, Viel C, Panarello N, Treleaven CM, Kim C, et al. Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum Mol Genet. 2016;25(13):2645–60.
Samii A, Etminan M, Wiens MO, Jafari S. NSAID use and the risk of Parkinsons disease: systematic review and meta-analysis of observational studies. Drugs Aging. 2009;26(9):769–79.
Sanyal A, DeAndrade MP, Novis HS, Lin S, Chang J, Lengacher N, et al. Lysosome and inflammatory defects in GBA1-mutant astrocytes are normalized by LRRK2 inhibition. Mov Disord. 2020;35(5):760–73.
Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci USA. 2011;108(29):12101–6.
Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for Parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci USA. 2013;110(9):3537–42.
Saunders-Pullman R, Mirelman A, Alcalay RN, Wang C, Ortega RA, Raymond D, et al. Progression in the LRRK2-asssociated Parkinson disease population. JAMA Neurol. 2018;75(3):312–9.
Schapira AHV. Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci. 2015;66:37–42.
Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson’s disease. Mov Disord. 2011;26:1049–55.
Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W, et al. First-in-human assessment of PRX002, an anti–α-synuclein monoclonal antibody, in healthy volunteers. Mov Disord. 2017;32(2):211–8.
Schindlbeck KA, Vo A, Nguyen N, Tang CC, Niethammer M, Dhawan V, et al. LRRK2 and GBA variants exert distinct influences on Parkinson’s disease-specific metabolic networks. Cereb Cortex. 2020;30(5):2867–78.
Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord. 2017;32:1504–23.
Shadrina MI, Slominsky PA, Limborska SA. Molecular mechanisms of pathogenesis of Parkinson’s disease. Int Rev Cell Mol Biol. 2010;281(C):229–66.
Sharma S, Moon CS, Khogali A, Haidous A, Chabenne A, Ojo C, et al. Biomarkers in Parkinson’s disease (recent update). Neurochem Int. 2013;63:201–29.
Shults CW, Beal MF, Song D, Fontaine D. Pilot trial of high dosages of coenzyme Q10 in patients with Parkinson’s disease. Exp Neurol. 2004;188(2):491–4.
Si QQ, Yuan YS, Zhi Y, Wang M, Wang JW, Shen YT, et al. SNCA rs11931074 polymorphism correlates with spontaneous brain activity and motor symptoms in Chinese patients with Parkinson’s disease. J Neural Transm. 2019;126(8):1037–45.
Simuni T, Brumm MC, Uribe L, Caspell-Garcia C, Coffey CS, Siderowf A, et al. Clinical and dopamine transporter imaging characteristics of leucine rich repeat kinase 2 (LRRK2) and glucosylceramidase beta (GBA) Parkinson’s disease participants in the Parkinson’s progression markers initiative: a cross-sectional study. Mov Disord. 2020;35(5):833–44.
Skrahina V, Gaber H, Vollstedt EJ, Förster TM, Usnich T, Curado F, et al. The Rostock International Parkinson’s Disease (ROPAD) study: protocol and initial findings. Mov Disord. 2020. https://doi.org/10.1002/mds.28416.
Smolders S, Van Broeckhoven C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol Commun. 2020;8:1–28.
Spencer B, Valera E, Rockenstein E, Overk C, Mante M, Adame A, et al. Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun. 2017;5(1):7.
Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18020441.
Sun Y, Pham AN, Waite TD. Mechanism underlying the effectiveness of deferiprone in alleviating Parkinson’s disease symptoms. ACS Chem Neurosci. 2018;9(5):1118–27.
Sun Y, Liou B, Chu Z, Fannin V, Blackwood R, Peng Y, et al. Systemic enzyme delivery by blood-brain barrier-penetrating SapC-DOPS nanovesicles for treatment of neuronopathic Gaucher disease. EBioMedicine. 2020;55:102735.
Sybertz E, Krainc D. Development of targeted therapies for Parkinson’s disease and related synucleinopathies. J Lipid Res. 2014;55:1996–2003.
Tran HT, Chung CHY, Iba M, Zhang B, Trojanowski JQ, Luk KC, et al. α-Synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014;7(6):2054–65.
Trinh J, Zeldenrust FMJ, Huang J, Kasten M, Schaake S, Petkovic S, et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord. 2018;33:1857–70.
Uehara T, Choong CJ, Nakamori M, Hayakawa H, Nishiyama K, Kasahara Y, et al. Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting α-synuclein as a novel therapy for Parkinson’s disease. Sci Rep. 2019. https://doi.org/10.1038/s41598-019-43772-9.
Van Nuenen BFL, Weiss MM, Bloem BR, Reetz K, Van Eimeren T, Lohmann K, et al. Heterozygous carriers of a Parkin or PINK1 mutation share a common functional endophenotype. Neurology. 2009;72(12):1041–7.
Van Nuenen BFL, Van Eimeren T, Van Der Vegt JPM, Buhmann C, Klein C, Bloem BR, et al. Mapping preclinical compensation in Parkinson’s disease: an imaging genomics approach. Mov Disord. 2009;24:S703-10.
Volpicelli-Daley LA, Kirik D, Stoyka LE, Standaert DG, Harms AS. How can rAAV-α-synuclein and the fibril α-synuclein models advance our understanding of Parkinson’s disease? J Neurochem. 2016;139:131–55.
Wang Z, Gao G, Duan C, Yang H. Progress of immunotherapy of anti-α-synuclein in Parkinson’s disease. Biomed Pharmacother. 2019;115:108843.
Whiffin N, Armean IM, Kleinman A, Marshall JL, Minikel EV, Goodrich JK, et al. The effect of LRRK2 loss-of-function variants in humans. Nat Med. 2020;26(6):869–77.
Wile DJ, Agarwal PA, Schulzer M, Mak E, Dinelle K, Shahinfard E, et al. Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: cross-sectional studies. Lancet Neurol. 2017;16(5):351–9.
Wilson H, Dervenoulas G, Pagano G, Koros C, Yousaf T, Picillo M, et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T α-synuclein parkinsonism: a cross-sectional study. Lancet Neurol. 2019;18(8):748–59.
Wilson H, Dervenoulas G, Pagano G, Tyacke RJ, Polychronis S, Myers J, et al. Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson’s disease: an in vivo 11C-BU99008 PET study. Brain. 2019;142(10):3116–28.
Wilson H, Pagano G, de Natale ER, Mansur A, Caminiti SP, Polychronis S, et al. Mitochondrial complex 1, sigma 1, and synaptic vesicle 2A in early drug-naive Parkinson’s disease. Mov Disord. 2020;35(8):1416–27.
Xu L, Pu J. Alpha-synuclein in Parkinson’s disease: from pathogenetic dysfunction to potential clinical application. Parkinson’s Dis. 2016. https://doi.org/10.1155/2016/1720621.
Zeuner KE, Schäffer E, Hopfner F, Brüggemann N, Berg D. Progress of pharmacological approaches in Parkinson’s disease. Clin Pharmacol Therap. 2019;105:1106–20.
Acknowledgements
Not applicable.
Funding
JP received funding from the Parkinson’s Foundation (Miami, Florida, USA), the Deutsche Parkinsongesellschaft (Berlin, Germany), and the Deutsche Forschungsgemeinschaft via the Clinician Scientist School Lübeck (DFG-GEPRIS 413535489). NB received funding from the DFG (BR4328.2-1 (FOR2488), GRK1957).
Author information
Authors and Affiliations
Contributions
JP drafted the outline of the manuscript, performed literature search, and wrote the manuscript. NB drafted the outline and reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests to report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prasuhn, J., Brüggemann, N. Genotype-driven therapeutic developments in Parkinson’s disease. Mol Med 27, 42 (2021). https://doi.org/10.1186/s10020-021-00281-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10020-021-00281-8