
Abstract
Purpose
We investigated whether twice-daily administration of a bilayer tablet formulation of tramadol (35% immediate-release [IR] and 65% sustained-release) is as effective as four-times-daily IR tramadol capsules for managing cancer pain.
Methods
This randomized, double-blind, double-dummy, active-comparator, non-inferiority study enrolled opioid-naïve patients using non-steroidal anti-inflammatory drugs or acetaminophen (paracetamol) to manage cancer pain and self-reported pain (mean value over 3 days ≥ 25 mm on a 100-mm visual analog scale [VAS]). Patients were randomized to either bilayer tablets or IR capsules for 14 days. The starting dose was 100 mg/day and could be escalated to 300 mg/day. The primary endpoint was the change in VAS (averaged over 3 days) for pain at rest from baseline to end of treatment/discontinuation.
Results
Overall, 251 patients were randomized. The baseline mean VAS at rest was 47.67 mm (range: 25.6–82.7 mm). In the full analysis set, the adjusted mean change in VAS was − 22.07 and − 19.08 mm in the bilayer tablet (n = 124) and IR capsule (n = 120) groups, respectively. The adjusted mean difference was − 2.99 mm (95% confidence interval [CI] − 7.96 to 1.99 mm). The upper 95% CI was less than the predefined non-inferiority margin of 7.5 mm. Other efficacy outcomes were similar in both groups. Adverse events were reported for 97/126 (77.0%) and 101/125 (80.8%) patients in the bilayer tablet and IR capsule groups, respectively.
Conclusion
Twice-daily administration of bilayer tramadol tablets was as effective as four-times-daily administration of IR capsules regarding the improvement in pain VAS, with comparable safety outcomes.
Clinical trial registration
JapicCTI-184143/jRCT2080224082 (October 5, 2018).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pain is a common symptom in cancer patients; some studies suggested that almost half of patients experience pain at least 3 months after completing curative treatment, and nearly a third experience moderate to severe pain [1, 2]. Cancer pain may be caused by the cancer itself or metastases, or may be related to the treatments (e.g., surgical pain, neuropathic pain after chemotherapy) [3]. Despite its high prevalence and significant impact on patient well-being, it was reported that cancer pain is under-treated in approximately one-third of patients [3], constituting an important unmet need in clinical practice.
Clinical guidelines for managing cancer pain, including those developed by the World Health Organization (WHO) [4], National Comprehensive Cancer Network [5], American Society of Clinical Oncology [6], European Society of Medical Oncology [7], and the Japanese Society for Palliative Medicine [8], suggest that pain should be managed according to the patient’s pain intensity, and that treatment may include an opioid, such as tramadol. In particular, the WHO guidelines position opioids as drugs that should be used according to the clinical assessment and pain intensity for rapid, effective, and safe pain management from the initiation of pain management, even if not based on the conventional three-step analgesia ladder. The guidelines also state that any opioid may be selected for cancer-related pain. Patients may require stronger opioids, other analgesics, or adjuvant therapies, the choice of which will depend on their clinical condition [4,5,6,7,8].
Tramadol is a weak μ-opioid receptor agonist that also inhibits norepinephrine and serotonin reuptake, with proven efficacy for managing chronic pain. Oral administration is preferred, with a regular dosing frequency every 4 or 12 h depending on the formulation prescribed (e.g., immediate-release or extended-release formulations). However, another administration route may be required in some patients.
With a view to improving the pharmacokinetic profile of orally administered tramadol, Nippon Zoki developed a new tramadol formulation as bilayer sustained-release (SR) tablets (hereafter bilayer tablets) in which the top layer comprises 35% of the dose as an immediate-release (IR) formulation and the lower layer comprises 65% of the dose as a SR formulation administered twice-daily (Twotram® tablets; Nippon Zoki Pharmaceutical Co., Ltd.) [9, 10]. This is the first twice-daily tramadol formulation to be developed and marketed in Japan [10]. To date, several Phase III clinical studies have demonstrated the efficacy of these bilayer tablets for managing chronic non-cancer pain associated with knee osteoarthritis [11] and postherpetic neuralgia [12], and the long-term efficacy and safety were demonstrated in a 52-week study [10]. To expand the potential indications for the bilayer tablet, we performed a randomized controlled study to examine its effectiveness in Japanese patients with cancer pain by testing its non-inferiority versus IR tramadol capsules as an active comparator.
Methods
The study was registered on the Japan Pharmaceutical Information Center clinical trial information (JapicCTI-184143) and Japan Registry of Clinical Trials (jRCT2080224082) (date registered: October 5, 2018).
Patients
Patients were eligible for this study if they had been diagnosed with cancer, had an estimated survival of ≥ 3 months from the start of study drug administration, were currently using non-opioid analgesics (nonsteroidal anti-inflammatory drugs [NSAIDs] or acetaminophen [paracetamol]), had not previously used an opioid analgesic, and the physician deemed it necessary to start tramadol to manage cancer pain. Patients used a 100-mm visual analog scale (VAS) to assess their pain at rest on study days − 2, − 1, and 1 (where study day 1 was the day of starting treatment); only patients with a score of ≥ 25 mm averaged over the 3 days were eligible for the study. Other eligibility criteria included patients treated in an inpatient or outpatient setting, age ≥ 20 years, and adequate liver and renal functions. The major exclusion criteria are listed in the Supplementary Methods.
Study design
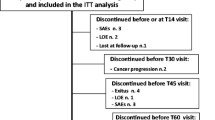
This was a randomized, double-blind, double-dummy, active-comparator non-inferiority study comprising three periods: screening period, treatment period, and follow-up period (Fig. 1). In the treatment period with a double-dummy procedure, the patients took bilayer tablets (active drug or placebo) twice daily (morning and evening) and IR capsules (placebo or active drug) four times per day (morning, noon, evening, and before bed) according to the random allocation method in a blinded manner for up to 14 days (study days 1–15). The rationale for the 14-day treatment period is described in the Supplementary Methods. The dosing of study drugs and use of rescue medication are summarized in Fig. 1 and described in detail in the Supplementary Methods. Patients were randomized centrally using a dynamic allocation method in which study site and the patient’s mean score for VAS at rest (averaged over study days − 2, − 1, and 1) before the start of study drug administration were used as the allocation factors. After the 14-day treatment period, the patients entered a 7-day follow-up period, during which they could be prescribed tramadol, NSAIDs, or acetaminophen at the discretion of the investigator/subinvestigator. Approved and prohibited therapies are summarized in the Supplementary Methods. The investigator/subinvestigator at each study site was responsible for enrolling the patients using a web-based registration system. The allocation manager was responsible for assigning the study drugs, maintaining blinding, and storing the blinding code. Blinding was maintained until the database was locked.

Study design. The patient’s eligibility was checked during the screening period (up to 15 days). Patients used a 100-mm visual analog scale (VAS) to assess their pain at rest on study days − 2, − 1, and 1 (where study day 1 was the day of starting treatment); only patients with a score of ≥ 25 mm averaged over the 3 days were eligible. The dosing procedure in each group is described in more detail in the Supplementary Methods. Patients received a dose of 50 mg/day in the evening on study day 1, followed by 100 mg/day on study day 2. On study days 2–10, the tramadol dose could be escalated by 100 mg/day to a maximum of 300 mg/day. The dose could not be escalated between study days 11 and 14. Rescue medication was permitted throughout the treatment period. IR immediate-release
Endpoints
Every evening, just before administering the study drug, the patients evaluated their pain at rest and during movement over the previous 24-h period using a 100-mm VAS. This information was used to determine the primary endpoint—the change in the VAS for pain at rest from baseline (averaged over the 3 days before starting treatment) to the end of treatment (EOT; averaged over study days 12–14) or at discontinuation (averaged over the 3 days before discontinuation). A clinically relevant change in the VAS for pain was defined as a moderate or greater improvement during treatment relative to the baseline score using the chart shown in Supplementary Table 1; this definition was developed and utilized in prior studies in Japan [13, 14]. The Supplementary Methods describes the secondary endpoints and safety assessments. There were no changes to the study design after the first patient had been enrolled.
Statistical analyses
In consideration of the sample size calculation (Supplementary Methods), it was planned to enroll 120 patients per group.
For this study, we defined three analysis populations. The full analysis set (FAS) comprised all patients who received at least one dose of study drug and for whom the primary endpoint (i.e., change in pain VAS at rest from baseline to the EOT or discontinuation) could be calculated for modified intention-to-treat analyses. The per-protocol set (PPS) comprised all patients in the FAS, excluding those with major protocol deviations (e.g., eligibility criteria, randomization/blinding violations, or non-compliance with study drug administration). The safety analysis set (SAF) comprised all patients who received at least one dose of the study drug.
The primary endpoint was analyzed using the FAS and verified using the PPS by analysis of covariance with treatment group as a fixed factor and the baseline VAS score as a covariate to estimate the adjusted mean change in each group and the between-group difference in adjusted mean change with 95% confidence intervals (CI). Non-inferiority was established if the upper limit of the CI for the between-group difference did not exceed the non-inferiority margin (7.5 mm). Descriptive statistics were also calculated for VAS scores at each visit. Other analyses are described in the Supplementary Methods. SAS version 9.4 (SAS Institute, Cary, NC, USA) was used for all data analyses.
Results
Patients
A total of 281 patients initially provided consent, of which 251 were randomized (126 to the bilayer tablet group and 125 to the IR capsule group) (Fig. 2). Of these, 105 completed the study in the bilayer tablet group and 91 in the IR capsule group (Fig. 2).

Patient disposition. A total of 251 patients were randomized to either the bilayer tablet or IR capsule groups. All of these patients received the allocated drugs, as randomized, and were included in the safety analysis set. Twenty-one patients discontinued treatment in the bilayer tablet group and 34 discontinued treatment in the IR capsule group. The most common reason for discontinuation was an adverse event, accounting for 10 patients in the bilayer tablet group and 21 patients in the IR capsule group. IR immediate-release
The baseline characteristics of patients in both groups (SAF) were similar (Table 1). At baseline, patients in both groups typically reported moderate–high levels of pain, with a mean VAS at rest of 47.67 mm, ranging from 25.6 to 82.7 mm. Most patients (81.7%) were treated as outpatients. The most common cancer site was the gastrointestinal tract (37.1%) followed by the bile duct/liver/pancreas (20.3%). The most common metastatic sites were bone (36.7%), liver (32.3%), and lower lymph nodes (29.1%). The main site of pain was the abdomen (43.4%) followed by the dorsal region (29.5%) and low back (23.9%). All of the patients were using concomitant drugs, including non-opioid analgesics in 98.8% and NSAIDs in 76.1%. Anti-cancer drugs were used in 68.9% of patients.
The primary endpoint could not be determined due to missing values at EOT/discontinuation for 2 patients in the bilayer tablet group and 5 patients in the IR capsule group. Therefore, the FAS comprised 244 patients (bilayer tablet group, 124; IR capsule group, 120).
Treatment adherence, which was assessed using the FAS, was high, with mean ± standard deviation (SD) medication compliance rates of 99.26% ± 2.44% in the bilayer tablet group and 99.15% ± 3.78% in the IR capsule group.
VAS for pain at rest and during movement
The adjusted mean change in VAS for pain at rest from baseline to EOT/discontinuation (FAS) was − 22.07 mm for the bilayer tablet group and − 19.08 mm for the IR capsule group, corresponding to a between-group adjusted mean difference of − 2.99 mm (95% CI − 7.96 to 1.99 mm). The upper 95% CI bound was less than the predefined non-inferiority margin of 7.5 mm, demonstrating non-inferiority of the bilayer tablets to the IR capsules (Fig. 3A). In the supplementary analysis using the PPS, the adjusted mean difference between the two groups was − 2.98 mm (95% CI − 8.16 to 2.20 mm), which was also less than the non-inferiority margin. Figure 3B shows the mean values for VAS for pain at rest at baseline and at EOT/discontinuation in both groups. Figure 3C shows the corresponding data for the VAS for pain during movement. The adjusted mean change in the VAS for pain during movement was − 20.43 and − 19.06 mm in the bilayer tablet and IR capsule groups, respectively, with an adjusted mean difference of − 1.38 mm (95% CI − 6.79 to 4.03 mm). The improvements in VAS scores for pain at rest and during movement on each day showed strong similarity in both groups (Fig. 4).

(A) Non-inferiority analysis of the adjusted mean change in VAS for pain at rest from baseline to EOT (primary endpoint). (B) Change in VAS for pain at rest from baseline to EOT. (C) Change in VAS for pain during movement from baseline to EOT. Values in B and C are mean ± standard deviation. CI confidence interval, EOT end of treatment, FAS full analysis set, IR immediate-release, PPS per-protocol set, VAS visual analog scale (100 mm)

Changes in VAS over time for pain at rest (A) and pain during movement (B). Values are mean ± standard deviation. aAveraged over 3 days before starting study drug administration. IR immediate-release, VAS visual analog scale (100 mm)
The proportion of patients with a clinically relevant improvement in pain at rest (at EOT/discontinuation) was numerically greater in the bilayer tablet group (87/124, 70.2%) than in the IR capsule group (69/120, 57.5%). Furthermore, a slightly greater proportion of patients in the bilayer tablet group experienced a clinically relevant improvement in pain during movement (71/124, 57.3% vs 60/120, 50.0%).
Estimated total duration of pain per day
The estimated total duration of pain per day was assessed using a five-item scale on study days 2–14 of the treatment period. On study day 2, 50.0% (62/124) of patients in the bilayer tablet group and 54.6% (65/119) of patients in the IR capsule group reported that their duration of pain was < 4 h. This percentage increased slightly in both groups to 59.6% (62/104) in the bilayer tablet group and 60.4% (55/91) in the IR capsule group on study day 14 (Supplementary Table 2).
Sleep
The majority of patients reported that their sleep was good during the treatment period. The percentage of patients who reported that they “slept well” or “slept moderately well” ranged from 77% to 86% in the bilayer tablet group and from 77% to 87% in the IR capsule group (Supplementary Fig. 1). The percentage of patients who reported that they “slept well” varied from 25% to 38% in the bilayer tablet group and from 24% to 41% in the IR capsule group.
Use of rescue medications
Rescue medications (one or more doses of tramadol capsule) were used by 14.8%–22.3% of patients in the bilayer tablet group and by 10.3%–22.8% in the IR capsule group (Supplementary Fig. 2A, B). The frequency of rescue medication use remained broadly stable throughout the treatment period. The majority of patients who used rescue medication took a single dose in each group, with percentages that ranged from 9.8% to 18.5% in the bilayer tablet group and from 6.8% to 19.6% in the IR capsule group (Supplementary Fig. 2C, D).
Quality of life
There were no marked changes in the quality of life (QOL) scores determined using the EuroQOL 5-dimension, 5-level questionnaire (EQ-5D-5L) (Supplementary Fig. 3), or in the individual domains, during the treatment period in either group.
Safety
Treatment period
During the 14-day treatment period, adverse events (AEs) were reported for 97 (77.0%) patients in the bilayer tablet group and 101 (80.8%) of patients in the IR capsule group (Table 2). This included severe AEs in 4.8% and 5.6% of patients, respectively, and serious AEs in 8.7% and 13.6% of patients, respectively. However, few of these AEs were thought to be related to the study drugs because most of the AEs corresponded to exacerbations of the primary or metastatic cancer. In the bilayer tablet group, one patient experienced a severe adverse drug reaction (ADR) and two patients experienced serious ADRs. No severe or serious ADRs were reported in the IR capsule group. AEs resulted in death in 4 (3.2%) patients in the bilayer tablet group and 3 (2.4%) patients in the IR capsule group, but none of these events were considered related to the study drugs. ADRs resulted in discontinuation of the study drug for 10 (7.9%) patients in the bilayer tablet group and 11 (8.8%) patients in the IR capsule group. No ADRs resulted in a reduction in the doses of the study drugs. The three most common AEs in both treatment groups were nausea, constipation, and vomiting (Table 2). The frequencies and types of ADRs were generally similar between the two treatment groups (Supplementary Table 3). ADRs that occurred in ≥ 2% of patients in the bilayer tablet group were nausea (bilayer tablet group and IR capsule group: 27.8% and 32.0%), constipation (19.8% and 16.0%), vomiting (16.7% and 16.8%), somnolence (14.3% and 9.6%), dizziness (7.1% and 4.8%), decreased appetite (6.3% and 0.8%), and malaise (2.4% and 0.8%). There were no consistent trends or notable findings regarding vital signs or 12-lead electrocardiography.
Follow-up period
During the follow-up period, AEs were reported for 43 (34.1%) patients in the bilayer tablet group and 46 (36.8%) patients in the IR capsule group, indicating no difference in safety during this period (Supplementary Table 4). ADRs were reported for 3 (2.4%) patients in the bilayer tablet group and 2 (1.6%) patients in the IR capsule group. One AE resulted in death in the bilayer tablet group, but the event was not considered related to the study drug. Severe and serious AEs were reported in both groups, but were not considered related to the study drugs. The most frequent AEs during the follow-up period were constipation, nausea, and vomiting in the bilayer tablet group and nausea, decreased appetite, and constipation in the IR capsule group. There were no reported cases of drug dependency based on the standardized MedDRA query “Drug abuse and dependence.”
Discussion
Our aim was to investigate the non-inferiority of a bilayer tablet formulation of tramadol, comprising IR and SR layers, versus an IR capsule formulation in terms of managing cancer pain. The two treatments achieved similar improvements in the VAS for pain at rest, satisfying the criterion for non-inferiority, which was confirmed in the PPS analysis. Additionally, the changes in VAS for pain at rest and during movement on each study day, percentages of patients who slept well or moderately well, use of rescue medication, and EQ-5D-5L QOL scores were highly comparable, indicating highly similar effects of both formulations on pain control. The improvement in pain was rapid, from within 2 days of starting administration, and showed good stability throughout the study in both groups. Overall, these findings indicate that twice-daily administration of the bilayer tablets is as effective as four-times-daily IR tramadol for managing cancer pain.
Opioids are frequently used to manage cancer pain [15,16,17,18,19,20,21], due to their effectiveness and inclusion in clinical guidelines/recommendations [4,5,6,7,8]. Furthermore, studies have shown that opioids can improve QOL by alleviating cancer-related pain [22,23,24,25,26,27]. Here, we have shown that two formulations of tramadol can achieve a clinically relevant improvement in cancer pain at rest and during movement, and both formulations were comparable in terms of other outcomes, including sleep quality, use of rescue medications, and QOL. Therefore, our findings provide further support for using tramadol to manage cancer pain, and that physicians could choose an administration regimen (e.g., twice-daily or four-times-daily) that might be most suitable for the individual patient.
We also investigated the safety of both study drugs in terms of AEs/ADRs during the 14-day treatment period and 7-day follow-up period. During the treatment period, AEs were reported for 77.0% and 80.8% of patients in the bilayer tablet and IR capsule groups, respectively, while ADRs were reported for 58.7% and 53.6%, respectively. These values seem reasonable when we consider the frequencies of AEs reported in the initial open-label treatment escalation periods (80.6% and 78.7% in the knee osteoarthritis and postherpetic neuralgia studies, respectively) of two previous dose-withdrawal studies using the bilayer tablet formulation [11, 12]. We enrolled opioid-naïve patients, which may increase the risk of opioid-related AEs and ADRs. Additionally, all of the patients were using concomitant drugs, such as non-opioid analgesics, two-thirds were receiving anti-cancer therapies, and nearly half were using a corticosteroid. Thus, the frequencies of AEs/ADRs are within expected ranges. The most common types of AEs and ADRs were nausea, constipation, vomiting, and somnolence, which are known to be associated with tramadol [5, 6, 28]. Nevertheless, there were few moderate or severe ADRs that were likely to interfere with daily activities, and only two serious ADRs and one severe ADR were reported. Overall, physicians should take appropriate care when prescribing tramadol while monitoring its safety, especially in opioid-naïve patients.
Clinical guidelines position opioids, including tramadol, as options for managing cancer pain [4,5,6,7,8]. If acetaminophen or NSAIDs do not provide sufficient pain control, it may be possible to switch to these bilayer tramadol tablets, which have already shown good long-term efficacy and tolerability in patients with chronic non-cancer pain [10,11,12]. These bilayer tablets could be started early in the patient’s clinical course and stepped down when no longer required, in accordance with WHO recommendations for the initiation, maintenance, and cessation of opioids [4].
Several preparations of tramadol, including IR and SR formulations, have been developed and are used to manage cancer pain. However, there are some potential disadvantages of the available formulations related to their pharmacokinetic properties. In particular, the pharmacokinetics of once-daily SR formulations may not be sufficient to maintain effective pain relief over the 24-h period between each dose [29]. As such, patients may require frequent use of rescue medications to maintain adequate pain relief. By comparison, the pharmacokinetics of IR formulations may provide adequate efficacy, but the frequent administration (four-times-daily) may pose a pill burden, which was associated with decreased treatment satisfaction and reduced medication adherence in other settings [30,31,32,33,34]. Further, studies in other settings suggested that patients were less adherent to a four-times-daily regimen than a twice-daily regimen [35,36,37]. Thus, patients may show better adherence to a twice-daily regimen, especially one that provides a rapid onset of action through the IR component and prolonged action through the SR component. Accordingly, we hypothesize that these bilayer tramadol tablets could offer greater compliance and at least comparable effectiveness to alternative tramadol regimens requiring more frequent administration.
Limitations
There are some limitations of this study that warrant mention. In particular, the treatment period was relatively short (14 days), which prevented us from assessing the longer-term effectiveness of tramadol. This period was selected based on an earlier study in Japan of the same length [14] and in consideration of the potential impact of anti-cancer therapy in longer-term studies. We should also consider the possibility that safety assessments may have been influenced by the use of concomitant drugs, including anti-cancer therapies, that might have inflated the frequency of AEs in this study. However, this risk seems low because the types of AEs were generally consistent with the known safety profile of tramadol. Because the study lacked a placebo group, we cannot exclude the possibility of a placebo or trial effect. However, this was deemed unethical because the patients all reported clinically significant pain despite treatment with non-opioid analgesics that would have necessitated other treatments or high rates of rescue medication. Furthermore, a placebo group was deemed unnecessary because both study drugs had already been evaluated in placebo-controlled trials of other indications [11, 12, 38, 39]. We did not use a cross-over design, which may have been useful to evaluate whether patients had preferences regarding formulation and administration frequency.
Conclusions
Twice-daily administration of bilayer tramadol tablets comprising 35% immediate-release and 65% sustained-release tramadol was as effective as four-times-daily IR capsules regarding the improvement in the VAS for pain at rest. We also observed strong similarity in the other effectiveness outcomes, including the improvements in VAS for pain at rest and during movement on each study day, sleep quality, use of rescue medications, and EQ-5D-5L QOL scores. Furthermore, the safety profiles of both study groups were consistent with the known safety profile for tramadol. Overall, these findings indicate that bilayer tramadol tablets are an effective and tolerable treatment option for managing cancer pain, comparable to four-times-daily administration of IR capsules.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Haenen V, Evenepoel M, De Baerdemaecker T, Meeus M, Devoogdt N, Morlion B, Dams L, Van Dijck S, Van der Gucht E, De Vrieze T, Vyvere TV, De Groef A (2023) Pain prevalence and characteristics in survivors of solid cancers: a systematic review and meta-analysis. Support Care Cancer 31:85. https://doi.org/10.1007/s00520-022-07491-8
Snijders RAH, Brom L, Theunissen M, van den Beuken-van Everdingen MHJ (2023) Update on prevalence of pain in patients with cancer 2022: a systematic literature review and meta-analysis. Cancers (Basel) 15:591. https://doi.org/10.3390/cancers15030591
Haroun R, Wood JN, Sikandar S (2022) Mechanisms of cancer pain. Front Pain Res (Lausanne) 3:1030899. https://doi.org/10.3389/fpain.2022.1030899
World Health Organization (2018) WHO guidelines for the pharmacological and radiotherapeutic management of cancer pain in adults and adolescents. Available at: https://www.who.int/publications/i/item/9789241550390. Accessed 30 Jun 2023
National Comprehensive Cancer Network (2023) NCCN guidelines, adult cancer pain, version 1.2023. Available at: https://www.nccn.org/professionals/physician_gls/pdf/pain.pdf. Accessed 30 Jun 2023
Paice JA, Bohlke K, Barton D, Craig DS, El-Jawahri A, Hershman DL, Kong LR, Kurita GP, LeBlanc TW, Mercadante S, Novick KLM, Sedhom R, Seigel C, Stimmel J, Bruera E (2023) Use of opioids for adults with pain from cancer or cancer treatment: ASCO guideline. J Clin Oncol 41:914–930. https://doi.org/10.1200/jco.22.02198
Fallon M, Giusti R, Aielli F, Hoskin P, Rolke R, Sharma M, Ripamonti CI; ESMO Guidelines Committee (2018) Management of cancer pain in adult patients: ESMO Clinical Practice Guidelines. Ann Oncol 29(Suppl 4):iv166‒iv191. https://doi.org/10.1093/annonc/mdy152
Mawatari H, Shinjo T, Morita T, Kohara H, Yomiya K (2022) Revision of pharmacological treatment recommendations for cancer pain: clinical guidelines from the Japanese Society of Palliative Medicine. J Palliat Med 25:1095–1114. https://doi.org/10.1089/jpm.2021.0438
Ishitsubo N, Oguro S, Shimahashi H, Kawanishi M, Adachi T, Mitsuda K, Ishibashi N (2023) Development, physicochemical characteristics and pharmacokinetics of a new sustained-release bilayer tablet formulation of tramadol with an immediate-release component for twice-daily administration. Eur J Drug Metab Pharmacokinet. Published online December 8, 2023. https://doi.org/10.1007/s13318-023-00865-1
Nippon Zoki Pharmaceutical Co., Ltd. Twotram® (tramadol) tablets, 25 mg, 50 mg, 100 mg, 150 mg. Interview Form, December 2022 (9th Edition). Available at: https://www.info.pmda.go.jp/go/interview/1/530288_1149038G2026_1_101_1F.pdf. Accessed May 15, 2023 (in Japanese)
Kawai S, Sobajima S, Jinnouchi M, Nakano H, Ohtani H, Sakata M, Adachi T (2022) Efficacy and safety of tramadol hydrochloride twice-daily sustained-release bilayer tablets with an immediate-release component for chronic pain associated with knee osteoarthritis: a randomized, double-blind, placebo-controlled, treatment-withdrawal study. Clin Drug Investig 42:403–416. https://doi.org/10.1007/s40261-022-01139-5
Kawai S, Hasegawa J, Ito H, Fukuuchi Y, Nakano H, Ohtani H, Sasaki K, Adachi T (2023) Efficacy and safety of twice-daily tramadol hydrochloride bilayer sustained-release tablets with an immediate release component for postherpetic neuralgia: results of a Phase III, randomized, double-blind, placebo-controlled, treatment-withdrawal study. Pain Pract 23:277–289. https://doi.org/10.1111/papr.13190
Hiraga K, Ohashi Y (1999) Efficacy evaluation of analgesic agents used for cancer pain management by visual analogue scale – a survey by questionnaire to physicians, nurses and patients. Pain Res 14:9–19 (in Japanese)
Hiraga K, Ohkuma S, Asano H, Honmura T, Nishimura T, Takeda F (2010) A Phase III clinical trial of NS-315 (tramadol hydrochloride), a weak opioid analgesic, in patients with cancer pain – randomized, double-blind, parallel, comparative study with morphine. Clin Med 26:569–584 (in Japanese)
Birke H, Ekholm O, Sjøgren P, Fredheim O, Clausen T, Skurtveit S (2019) Tramadol use in Norway: a register-based population study. Pharmacoepidemiol Drug Saf 28:54–61. https://doi.org/10.1002/pds.4626
Haider A, Zhukovsky DS, Meng YC, Baidoo J, Tanco KC, Stewart HA, Edwards T, Joy MP, Kuriakose L, Lu Z, Williams JL, Liu DD, Bruera E (2017) Opioid prescription trends among patients with cancer referred to outpatient palliative care over a 6-year period. J Oncol Pract 13:e972–e981. https://doi.org/10.1200/jop.2017.024901
Hurtado I, Robles C, Peiró S, García-Sempere A, Llopis-Cardona F, Sánchez-Sáez F, Rodríguez-Bernal C, Sanfélix-Gimeno G (2022) Real-world patterns of opioid therapy initiation in Spain, 2012–2018: a population-based, retrospective cohort study with 957,080 patients and 1,509,488 initiations. Front Pharmacol 13:1025340. https://doi.org/10.3389/fphar.2022.1025340
Keto J, Heiskanen T, Hamunen K, Kalliomäki ML, Linna M (2022) Opioid trends in Finland: a register-based nationwide follow-up study. Sci Rep 12:7261. https://doi.org/10.1038/s41598-022-10788-7
Rueter M, Baricault B, Lapeyre-Mestre M (2022) Patterns of opioid analgesic prescribing in cancer outpatients during the last year of life in France: a pharmacoepidemiological cohort study based on the French health insurance database. Therapie 77:703–711. https://doi.org/10.1016/j.therap.2022.01.019
Thinh DHQ, Sriraj W, Mansor M, Tan KH, Irawan C, Kurnianda J, Nguyen YP, Ong-Cornel A, Hadjiat Y, Moon H, Javier FO (2018) Analgesic prescription patterns and pain outcomes in Southeast Asia: findings from the Analgesic Treatment of Cancer Pain in Southeast Asia study. J Glob Oncol 4:1–10. https://doi.org/10.1200/jgo.17.00055
Wang Y, Wu D, Chan A, Chang CH, Lee VWY, Nichol MB (2022) Temporal trend of opioid and nonopioid pain medications: results from a national in-home survey, 2001 to 2018. Pain Rep 7:e1010. https://doi.org/10.1097/pr9.0000000000001010
Arbaiza D, Vidal O (2007) Tramadol in the treatment of neuropathic cancer pain: a double-blind, placebo-controlled study. Clin Drug Investig 27:75–83. https://doi.org/10.2165/00044011-200727010-00007
Hao X, Zhou Y, Ling Y, Miyoshi H, Sumitani M, Chan KY, Park HJ, Feng Z, Rao Y (2022) Effects of high-dose opioid analgesia on survival, pain relief, quality of life and adverse drug reactions in cancer and neuropathic pain patients: a retrospective cohort study in real-world clinical practice. Ann Transl Med 10:998. https://doi.org/10.21037/atm-22-4242
Jung JY, Chon HJ, Choi YJ, Yeon SE, Choi SY, Lee KH (2022) A prospective, multicenter, open-label study of the clinical efficacy of tapentadol extended-release in the treatment of cancer-related pain and improvement in the quality of life of opioid-naïve or opioid-resistant patients. Support Care Cancer 30:6103–6112. https://doi.org/10.1007/s00520-022-06992-w
Leppert W, Nosek K (2019) Comparison of the quality of life of cancer patients with pain treated with oral controlled-release morphine and oxycodone and transdermal buprenorphine and fentanyl. Curr Pharm Des 25:3216–3224. https://doi.org/10.2174/1381612825666190717091230
Schikowski A, Krings D, Schwenke K (2015) Tapentadol prolonged release for severe chronic cancer-related pain: effectiveness, tolerability, and influence on quality of life of the patients. J Pain Res 8:1–8. https://doi.org/10.2147/jpr.S72150
Shen WC, Hou MM, Huang TL, Wang CH, Huang YM, Chen JS, Chen ML (2023) Transdermal buprenorphine improves overall quality of life and symptom severity in cancer patients with pain. J Clin Nurs 32:539–547. https://doi.org/10.1111/jocn.16303
Subedi M, Bajaj S, Kumar MS, Yc M (2019) An overview of tramadol and its usage in pain management and future perspective. Biomed Pharmacother 111:443–451. https://doi.org/10.1016/j.biopha.2018.12.085
Kanzaki H, Tetsunaga T (2018) Use of tramadol for chronic pain in rheumatic and inflammatory diseases and other conditions. Jpn Med J 4900:38–45 (in Japanese)
Blüher M, Kurz I, Dannenmaier S, Dworak M (2015) Pill burden in patients with type 2 diabetes in Germany: subanalysis from the prospective, noninterventional PROVIL study. Clin Diabetes 33:55–61. https://doi.org/10.2337/diaclin.33.2.55
Hauber AB, Han S, Yang JC, Gantz I, Tunceli K, Gonzalez JM, Brodovicz K, Alexander CM, Davies M, Iglay K, Zhang Q, Radican L (2013) Effect of pill burden on dosing preferences, willingness to pay, and likely adherence among patients with type 2 diabetes. Patient Prefer Adherence 7:937–949. https://doi.org/10.2147/ppa.S43465
Ishida N, Tokumoto Y, Suga Y, Noguchi-Shinohara M, Abe C, Yuki-Nozaki S, Mori A, Horimoto M, Hayashi K, Iwasa K, Yokogawa M, Ishimiya M, Nakamura H, Komai K, Matsushita R, Ishizaki J, Yamada M (2021) Factors associated with self-reported medication adherence in Japanese community-dwelling elderly individuals: the Nakajima study. Yakugaku Zasshi 141:751–759. https://doi.org/10.1248/yakushi.20-00254
Sims TJ, Boye KS, Robinson S, Kennedy-Martin T (2022) Treatment-related attributes of diabetes therapies and how people with type 2 diabetes report their impact on indicators of medication-taking behaviors. Patient Prefer Adherence 16:1919–1939. https://doi.org/10.2147/ppa.S367046
Pergolizzi JV Jr, Taylor R Jr, Raffa RB (2011) Extended-release formulations of tramadol in the treatment of chronic pain. Expert Opin Pharmacother 12:1757–1768. https://doi.org/10.1517/14656566.2011.576250
Cramer J, Vachon L, Desforges C, Sussman NM (1995) Dose frequency and dose interval compliance with multiple antiepileptic medications during a controlled clinical trial. Epilepsia 36:1111–1117. https://doi.org/10.1111/j.1528-1157.1995.tb00469.x
Cramer JA, Mattson RH, Prevey ML, Scheyer RD, Ouellette VL (1989) How often is medication taken as prescribed? A novel assessment technique. JAMA 261:3273–3277. https://doi.org/10.1001/jama.1989.03420220087032
Mann M, Eliasson O, Patel K, ZuWallack RL (1992) A comparison of the effects of bid and qid dosing on compliance with inhaled flunisolide. Chest 101:496–499. https://doi.org/10.1378/chest.101.2.496
Ogawa S, Kikuchi H, Asano H, Tamada A, Takagaki K, Mino H (2013) A Phase III clinical trial of NS-315 (tramadol hydrochloride) in patients with chronic osteoarthritis – a randomized, double-blind, comparative study with placebo (enriched enrollment randomized withdrawal design). J Clin Ther Med 29:497–512 (in Japanese)
Ogawa S, Hosokawa T, Asano H, Tamada A, Takagaki K, Mino H (2013) A Phase III clinical trial of NS-315 (tramadol hydrochloride) in patients with chronic post-herpetic neuralgia – a randomized, double-blind, comparative study with placebo (enriched enrollment randomized withdrawal design). J Clin Ther Med 29:513–530 (in Japanese)
Acknowledgements
The authors express their gratitude to the patients, investigators, and research staff who were involved in this study. The study sponsor acknowledges Nippon Shinyaku Co., Ltd. for manufacturing and supplying the IR capsules. The authors thank Nicholas D. Smith (EMC K.K.) for medical writing support, which was funded by Nippon Zoki Pharmaceutical Co., Ltd.
Study investigators
The following investigators agreed to be mentioned: Hiroki Shomura (Japan Community Health Care Organization Hokkaido Hospital, Hokkaido), Yasunori Nishida (Keiyukai Sapporo Hospital, Hokkaido), Yasushi Tsuji (Tonan Hospital, Hokkaido), Osamu Sasaki (Miyagi Cancer Center, Miyagi), Naoya Sodeyama (Sendai City Hospital, Miyagi), Yasuhiro Sakamoto (Osaki Citizen Hospital, Miyagi), Yasuhiro Yanagita (Gunma Prefectural Cancer Center, Gunma), Hiroshi Kojima (Ibaraki Prefectural Central Hospital, Ibaraki), Naoto Miyanaga (Mito Saiseikai General Hospital, Ibaraki), Masahiro Kamiga (Hitachi, Ltd., Hitachinaka General Hospital, Ibaraki), Masaharu Shinkai (Tokyo Shinagawa Hospital, Tokyo), Hitoshi Arioka (Yokohama Rosai Hospital, Kanagawa), Kazuhiro Seike (Odawara Municipal Hospital, Kanagawa), Kazuhiro Sato (Nagaoka Red Cross Hospital, Niigata), Koichi Nishi (Ishikawa Prefectural Central Hospital, Ishikawa), Kazuhisa Yoshimoto (Fuji City General Hospital, Shizuoka), Kazutoshi Asano (Shizuoka Saiseikai General Hospital, Shizuoka), Keiji Aizu (Kasugai Municipal Hospital, Aichi), Hibiki Kanda (Omi Medical Center, Shiga), Yukito Adachi (Saiseikai Noe Hospital, Osaka), Hiroyuki Narahara (Hyogo Prefectural Nishinomiya Hospital, Hyogo), Keisuke Tomii (Kobe City Medical Center General Hospital, Hyogo), Tomoe Fukunaga (Japanese Red Cross Society Himeji Hospital, Hyogo), Nobukazu Fujimoto (JOHAS Okayama Rosai Hospital, Okayama), Shoichi Kuyama (National Hospital Organization Iwakuni Clinical Center, Yamaguchi), Hidenori Harada (Yamaguchi University Hospital, Yamaguchi), Ryo Katsuki (National Hospital Organization Ureshino Medical Center, Saga), Minoru Yoshida and Shima Uneda (Japanese Red Cross Kumamoto Hospital, Kumamoto), Kodai Kawamura (Saiseikai Kumamoto Hospital, Kumamoto), and Daisuke Himeji (Miyazaki Prefectural Miyazaki Hospital, Miyazaki).
Funding
This study was funded by Nippon Zoki Pharmaceutical Co., Ltd.
Author information
Authors and Affiliations
Contributions
Conceptualization: Noriyuki Katsumata, Shinichi Kawai, Hideshi Nakano, Hideaki Ohtani, Kazutaka Sasaki, Takeshi Adachi.
Data curation: Kazutaka Sasaki, Takeshi Adachi.
Formal analysis: Takeshi Adachi.
Investigation: Masaharu Shinkai, Shoichi Kuyama, Osamu Sasaki, Yasuhiro Yanagita, Minoru Yoshida, Shima Uneda, Yasushi Tsuji, Hidenori Harada, Yasunori Nishida, Yasuhiro Sakamoto, Daisuke Himeji, Hitoshi Arioka, Kazuhiro Sato, Ryo Katsuki, Hiroki Shomura.
Methodology: Noriyuki Katsumata, Shinichi Kawai, Hideshi Nakano, Hideaki Ohtani, Kazutaka Sasaki, Takeshi Adachi.
Project administration: Masaharu Shinkai, Hideaki Ohtani.
Resources: Masaharu Shinkai, Shoichi Kuyama, Osamu Sasaki, Yasuhiro Yanagita, Minoru Yoshida, Shima Uneda, Yasushi Tsuji, Hidenori Harada, Yasunori Nishida, Yasuhiro Sakamoto, Daisuke Himeji, Hitoshi Arioka, Kazuhiro Sato, Ryo Katsuki, Hiroki Shomura.
Supervision: Masaharu Shinkai, Noriyuki Katsumata, Shinichi Kawai, Hideshi Nakano, Hideaki Ohtani.
Visualization: Masaharu Shinkai, Hideaki Ohtani.
Writing – original draft: Masaharu Shinkai, Noriyuki Katsumata, Shinichi Kawai, Hideshi Nakano, Hideaki Ohtani, Kazutaka Sasaki, Takeshi Adachi.
Writing – review and editing: all authors.
Corresponding author
Ethics declarations
Competing interests
Masaharu Shinkai, Shoichi Kuyama, Osamu Sasaki, Yasuhiro Yanagita, Minoru Yoshida, Shima Uneda, Yasushi Tsuji, Hidenori Harada, Yasunori Nishida, Yasuhiro Sakamoto, Daisuke Himeji, Hitoshi Arioka, Kazuhiro Sato, Ryo Katsuki, and Hiroki Shomura received financial support (personal or to their institution) for this study under a clinical trial contract with Nippon Zoki. Masaharu Shinkai received writing fees from Nippon Zoki in relation to this manuscript. Noriyuki Katsumata received honoraria from Nippon Zoki as a medical expert for this study. Shinichi Kawai received research grants from Nippon Zoki and was involved in this study as a medical expert. Hideshi Nakano, Hideaki Ohtani, Kazutaka Sasaki, and Takeshi Adachi are employees of Nippon Zoki.
Ethics approval
This study was conducted in compliance with the Declaration of Helsinki, Good Clinical Practice, and Japanese ethical guidelines. The protocol was approved by the Institutional Review Board/Ethics Committee at all 49 institutions at which the study was conducted, including the Institutional Review Board of Tokyo Shinagawa Hospital, Medical Corporation Association, Tokyokyojuno-kai.
Consent to participate
All patients provided written informed consent to participate.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shinkai, M., Katsumata, N., Kawai, S. et al. Phase III study of bilayer sustained-release tramadol tablets in patients with cancer pain: a double-blind parallel-group, non-inferiority study with immediate-release tramadol capsules as an active comparator. Support Care Cancer 32, 69 (2024). https://doi.org/10.1007/s00520-023-08242-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00520-023-08242-z