
Abstract
During the early stages of the development of the living multiorgan systems, genome modifications other than sequence variation occur that guide cell differentiation and organogenesis. These modifications are known to operate as a fetal programming code during this period, and recent research indicates that there are some tissue-specific codes in organogenesis whose effects may persist after birth until adulthood. Consequently, the events that disrupt the pre-established epigenetic pattern could induce shifts in organ physiology, with implications on health from birth or later in adult life. Chronic kidney disease (CKD) is one of the main causes of mortality worldwide; its etiology is multifactorial, but diabetes, obesity, and hypertension are the main causes of CKD in adults, although there are other risk factors that are mainly associated with an individual’s lifestyle. Recent studies suggest that fetal reprogramming in the developing kidney could be implicated in the susceptibility to kidney disease in both childhood and adulthood. Some epigenetic modifications, such as genome methylation status, dysregulation of miRNA, and histone coding alterations in genes related to the regulation of the renin-angiotensin axis, a common denominator in CKD, may have originated during fetal development. This review focuses on epigenetic changes during nephrogenesis and their repercussions on kidney health and disease. In addition, the focus is on the influence of environmental factors during pregnancy, such as maternal metabolic diseases and dietary and metabolic conditions, as well as some sex differences in fetal kidney reprogramming during which dysregulation of the renin-angiotensin system is involved.
Similar content being viewed by others

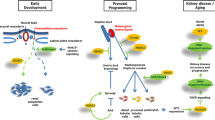

Introduction
Chronic kidney disease (CKD) is a major cause of morbidity and mortality in adulthood. High blood pressure, diabetes, family history, and ethnicity have been identified as the main risk factors associated with the onset of CKD. By contrast, in the pediatric population, a CKD prevalence of 15–75 cases per million children is reported, and the main causes include congenital anomalies of the kidney and urinary tract and hereditary nephropathies [1]. From the early developmental stages of living multiorgan systems, a series of finely coordinated cellular events take place for the determination of the patterns of growth, differentiation, and formation of the different organs and tissues, which is known as fetal programming [2, 3]. Both intrinsic and environmental factors during critical developmental stages can shift the pre-set cellular pathways with negative consequences not obvious at birth, but that could manifest themselves soon thereafter or even into adult life. This constitutes the core of the developmental origins of health and disease (DOHaD) hypothesis [3]. Studies in past decades have provided important clues that point to epigenetic modifications during kidney morphogenesis as a possible cause of kidney disease. Cumulative evidence suggests that certain stressing situations during early gestational stages could disturb the epigenomic pattern of a cell’s lineage, shifting the original fetal programming, which in turn could persist into adulthood and lead to disease [2]. These epigenetic changes, collectively referred to as kidney fetal reprogramming, include early molecular events that can confer increased susceptibility to common diseases such as CKD. Recent experimental and epidemiological studies indicate a dysregulation of the renin-angiotensin system (RAS), a pivotal kidney-function pathway, due to fetal reprogramming and whose consequences are not manifested until adulthood. This review will focus on recent advances in the understanding of the epigenetic modifications affecting RAS regulation. In addition, the focus is on the influence of environmental factors during pregnancy, such as maternal metabolic diseases and dietary and metabolic conditions, as well as some sex differences during fetal kidney reprogramming, during which dysregulation of the RAS is involved.
Nephrogenesis, fetal programming, and epigenetics
At birth, kidney development is complete. The establishment of more than one-half of the nephron pool occurs during the second trimester of pregnancy and ceases at 36 weeks [4]. The estimated total count of human nephrons ranges from 200,000 to more than 2.5 million [5], and the factors contributing to this wide variability are still poorly understood. During nephrogenesis, epigenetic modifications play a significant role in driving cell differentiation. These modifications include specific genome marks on the DNA molecule itself or on the proteins associated with it, such as the histones, the latter part of a shared code between progenitor nephrons and nascent nephrons for the development of proper cell-lineage fate. Currently, it is difficult to determine whether a specific epigenomic pattern found in a person with kidney disease was set up early during kidney development or whether there is a causal relationship among the DNA modifications, other than sequence variations and kidney damage in humans. The difficulties in determining these factors are mainly due to ethical and technical limitations, but experimental studies have provided some clues about the possible early origin of susceptibility to CKD.
Since the introduction of the term “fetal reprogramming” into the DOHaD hypothesis, during which the fetal environment impacts the health–disease process in childhood or adulthood [3, 6], many studies have provided evidence supporting it. In this regard, fetal programming is understood as all of the instructions of an organism driving the growth and development patterns for the formation of tissues and organs [2, 3, 6]. It has been proposed that there could exist fetal reprogramming, which refers to any modification during critical periods altering the original instruction manual with or without subsequent repercussions throughout life [2, 3].
Epigenetic dysregulation of the RAS
Epigenetic changes refer to modifications in chromatin structure that exert an influence on the phenotype, but do not involve alteration of the DNA nucleotide sequence (genotype). These changes can be heritable, established either from the early stages of embryonic development or even during adult life, and may be reversible [7]. These modifications mainly affect gene transcription, either in a generalized way in the organism, or they can be tissue-specific, cell lineage-specific, or even parental-specific [8]. Differences in gene expression may result from the direct modification of the chromatin structure, in which a chemical group binds to one of the DNA strands or proteins of the nucleosome (histones), or from DNA-RNA interactions, or from an increase or decrease in the accessibility of the transcriptional machinery to the DNA in response to different environmental stimuli, such as exposure to chemical or physical agents, chronic metabolic disturbances, and disease (Fig. 1). During the preceding decade, normal and abnormal kidney-specific epigenetic patterns were found to be related, and changes in kidney structure and function have been identified that were established either from early embryonic stages or later in postnatal life [9,10,11,12].

Epigenetic mechanisms involved in gene expression. Epigenetic mechanisms involved in gene expression include chromatin structure remodeling, histone modifications, and micro(mi)RNA-RNA interactions
The RAS is well known for its systemic effects on hemodynamic homeostasis, specifically in terms of controlling blood pressure, water, and electrolyte levels. The RAS pathway begins with the proteolytic cleavage of angiotensinogen (AGT) by renin (REN), an aspartyl protease, resulting in a decapeptide angiotensin I. The latter, in turn, is cleaved by the angiotensin-converting enzyme (ACE), resulting in the functional octapeptide angiotensin II, which performs its function through binding with two specific receptors (AT1R and AT2R). Paradoxically, these receptors exert opposite effects: while the AT1R have vasoconstrictor, sodium, water retention, proinflammatory, and pro-oxidant functions, the AT2R promote vasodilatation, sodium excretion, and anti-inflammatory and antioxidant responses [13].
The main known effects of the RAS are physiologically regulated by a hormonal feedback pathway, as well as by kidney osmoreceptors and baroreceptors that modulate its function. However, it has been proposed that the epigenetic regulation of the RAS is another important mechanism that could be involved in pathological processes [14]. Different modifiers of the fetal environment, specifically the maternal diet and the use of glucocorticoids, affect both the molecular and signaling pathways involved in nephrogenesis and converge in the dysregulation of the RAS (Fig. 2). For example, an intrarenal downregulation of RAS and a decreased number of glomeruli were found in the offspring of pregnant mice fed with a protein-restricted diet; even more so, these alterations were additionally associated with high blood pressure in adults [15]. Bogdarina et al. reported that maternal diet correlated with epigenetic modifications, which in turn influenced RAS gene expression in different tissues. These authors reported hypomethylation of the AT1b gene promoter, with the consequent increase in the expression of the AT1R in the adrenal glands in the offspring of pregnant rats fed with a low-protein diet (LPD) [16]. Subsequently, the downregulation of DNMT3A was shown as the cause of this hypomethylation, which in turn correlated with the elevation of hypothalamic angiotensin and a continuous activation of the autonomic nervous system (ANS) cycle involved in the development of hypertension in adults [17].

Environmental factors in renin-angiotensin system (RAS) fetal reprogramming. Major modifiers of the fetal environment, specifically the maternal diet, hypoxic conditions, maternal diseases, and use of glucocorticoids, affect both the molecular and signaling pathways involved in nephrogenesis and converge in the dysregulation of RAS. ACE, angiotensin-converting enzyme
Maternal dietary abnormalities and a prolonged protein deficit during postnatal life could be the causes of alterations in the RAS; specifically, nutritional zinc restrictions induced growth delay and dysregulation in kidney formation, as well as the activation of the angiotensin II-AT1R axis in male, but not in female progeny, after an increase of AT2R and ACE mRNA [18]. It is generally accepted that RAS-induced fetal reprogramming includes signaling pathways leading to alterations in the ANS, as well as in the circulatory system and the kidney by inducing the downregulation of ACE2, an imbalance in nitric oxide and sodium levels, and Ang II/Ang (1–7) expression at cerebral and circulatory levels [19]. In addition, decreased kidney expression was observed in the mitochondrial assembly 1 (Mas) receptor (MasR), which is related to vasodilatory and anti-inflammatory properties, counteracting the effects of AngII [19]. On the other hand, common variants in the RAS system have been associated with high blood pressure and nephropathy in humans, although the mechanisms explaining these associations are still unclear [20,21,22,23]. Variant AGT A[-6]G was associated with hypertension in humans because of a higher expression of AGT [20]. Male, but not female, progeny from transgenic mice carriers of this variant exhibited hypermethylation of the H19 gene cluster, a key regulator for nephrogenesis [21]. Notably in humans, the deletion of 287 base pairs (allele D) in intron 16 of the ACE gene (rs4340 variant) is associated with ACE hypomethylation and increased enzyme activity, although the role of the D allele on gene expression remains under discussion [22, 23]. These data suggest that the epigenetic changes caused by the RAS gene variants could explain, in part, the differential susceptibility to hypertension and CKD in association studies, although they may not be the main trigger.
Kidney reprogramming and pregnancy factors
During pregnancy, there are intrinsic and extrinsic factors influencing the maternal environment that are critical to the good growth and development of the fetus [3, 7]. The analysis of kidney fetal reprogramming requires a wider perspective that extends beyond the scope of this review. Therefore, we will only focus on aspects related to maternal nutrition and maternal diseases during pregnancy, among other factors related to the epigenetic modifications and fetal reprogramming affecting the RAS pathway as a possible mechanism contributing to kidney disease.
Maternal diet
During pregnancy, the quality of the maternal diet is key for good development of the fetus. Whether good or poor, the quality of the diet will have an important impact on fetal metabolism through the establishment of epigenetic marks that could in turn affect the physiology of the kidney from early childhood to adulthood. Studies have shown that a diet poor in essential nutrients and overfeeding in infants are both associated with chronic diseases in adulthood [24]. In addition, maternal LPD is now recognized as a risk factor for multifactorial diseases of progeny in adulthood, such as obesity, type 2 diabetes, insulin resistance, and kidney disease, all of these influenced by alterations in neurotrophic factors expressed in the hypothalamus [25], alterations in fertility [26, 27], and the risk of high blood pressure. It is noteworthy that low birth weight, in addition to fetoplacental insufficiency, infections, drug abuse, and preterm birth, is closely related to maternal nutrition, and the relationship between the decrease of nephron number and the percentage of glomeruli in newborns is well known [28, 29]. The impact of poor diet could be clearer in low- and middle-income economies such as those in some southern Asian, sub-Saharan African, and Latin American populations, in which clinical newborn registries demonstrated a mean prevalence of 26%, 14%, and 9% low-weight births, respectively, and coincidently with the reported prevalence of CKD in these populations [30,31,32]. Nevertheless, the possible evidence of maternal dietary involvement in offspring, the reprogramming of the renin-angiotensin axis in postnatal life, and the long-term effects of such reprogramming on the individual’s metabolism must be considered so that, on being deregulated at birth, they render it susceptible to environmental modifiers in adult life.
Studies in murine models fed with a LPD demonstrated alterations in RAS expression; the progeny of mothers with LPD revealed the increased expression of AT1b (angiotensin II type b receptor) in adrenal and kidney tissue, as well as alterations in other RAS genes caused by promoter hypomethylation in comparison with the progeny of mothers fed with a normal protein diet. The authors attributed these differences to the decrease in methyl donors associated with maternal malnutrition [16]. Additionally, LPD during pregnancy was associated with the increased sensitivity to salt of the progeny due to the hypermethylation of the prostaglandin E receptor 1 (Ptger1) gene, which in turn was the cause of the dysregulation of glomerular Na+ and water transport and increased renin secretion via the PGE2/EP1 signaling pathway [33]. In addition to low protein intake, a high-salt maternal diet is associated with alterations in the RAS. The progeny of mothers on a high sodium diet had increased renal AT1R and AT2R [34] but decreased glomerular number [35].
Mineral deficiencies in the maternal diet could also result in alterations in the RAS of the newborn. Gobetto and co-workers showed that male offspring appear to be more sensitive to maternal zinc deficiencies through an increase in the expression of components of the RAS [18]. As metalloproteinases, ACE and ACE2 are susceptible to the activating effect of zinc. Alternatively, epigenetic modifications induced by failures in zinc homeostasis could lead to fetal deregulation and reprogramming as well, probably due to the presence of zinc binding sites within the protein structure of DNA methyltransferases (DNMT) and histone acetyltransferases (HAT), histone deacetylases (HDAC), and histone demethylases (HDM) [36]. Therefore, it would be reasonable to consider zinc as an important modulator element in the activity of ACE and ACE2 due to epigenetic modifications. More research is needed on the early effects of the deficiencies (or excesses) of zinc or other diet-derived metal ions and their possible influence on RAS and kidney health.
All of the above data point to a close relationship between maternal diet and epigenetic modifications, which could affect the RAS in offspring. The lack of studies confirming these observations in humans and interventional approaches to prevent or revert alterations in fetal kidney programming do not allow their inclusion in clinical practice. Notwithstanding these limitations, the involvement of maternal diet in offspring as related to the reprogramming of the RAS in postnatal life and its long-term effects on the individual’s metabolism must be considered. For example, it is known that low birth weight was associated with a higher risk for dyslipidemia, as well as cardiovascular and metabolic alterations in the offspring of mothers exposed to a high-fat diet [37, 38]. The relationship between a high-fat diet and kidney damage is now known, but determining whether this relationship originates in prenatal life requires more research.
Maternal disease
Common maternal diseases, such as diabetes and obesity, have important effects during development that could be the result of fetal reprogramming. The long-term impact of maternal diabetes on offspring has been studied using different animal diabetes models, with special interest in cardiovascular and kidney effects. Intrauterine insults induced by maternal hyperglycemia converge in an altered inflammatory response. The dysregulation in the production of inflammatory mediators is a hallmark of metabolic and vascular diseases in adulthood [39]. Hyperglycemia and maternal diabetes can induce the dysfunction of the pancreatic islets (islets of Langerhans) during the early fetal stage, which is related to the overactivation of transcription factors, miRNA, and of the growth factors controlling the maturation of the pancreatic islets in formation, in addition to the downregulation of inflammatory response genes [40], as well as an influence on gene regulation through histone modifications [41]. At the clinical level, there is evidence in terms of the putative influence of both pre-pregnancy and gestational maternal diabetes on kidney volume in offspring, where the kidney parenchyma of the offspring of mothers with gestational diabetes is smaller than in controls [42]. In addition, hyperglycemia is a strong promoter of epigenomic modifications in offspring. Fetuses of mothers with type 1 diabetes exhibited widespread hypomethylation, although not in particular genes of embryogenesis or under a specific imprinting phenomenon. Among the groups of genes associated with this phenomenon, the hypomethylation of DNMT1 was found to be important because of its significant role in fetal reprogramming [43]. However, studies on maternal hyperglycemia or maternal diabetes and RAS are scarce, and the majority of these have been association studies [44, 45], which, due to their methodological nature, do not allow causality to be determined. Because of the increasing incidence of diabetes in young women, further efforts will be needed to understand the biological and epigenetic mechanisms related to CKD susceptibility both for themselves as well as in their future progeny.
Obesity is usually a comorbidity in gestational diabetes, in that it can give rise to alterations in fetal kidney hemodynamics, albuminuria, increased inflammation, and oxidative stress, among others [46,47,48]. Pro-oxidant molecules could be transmitted to the progeny’s kidneys, as well as the hypermethylation of genes associated with kidney fibrosis such as RASAL1, and alterations in the expression of DNMT1 are the main suggested mechanisms underlying kidney fibrosis in the offspring of obese mothers [47,48,49]. A renoprotective effect of hydralazine, a peripheral vasodilator often employed for the management of high blood pressure in pregnancy, appears to be associated with decreased genome-wide hypermethylation at the renal level in the product associated with maternal obesity, as well as ameliorated albuminuria and creatinine levels [50]. Also, the combined administration of insulin and metformin in murine models with gestational diabetes exerted a renoprotective effect on the fetal kidneys, which was observed at the proteome level [51]. Despite the presence of studies supporting the idea that some epigenetic modifications, such as histone methylation, are reversible, the specific molecular mechanisms that could allow for the design of targeted therapeutics remain unknown, and, to our knowledge, there are no long-term controlled follow-up studies conducted in these patients.
Other emerging factors
In addition to the epigenetic modifications occurring due to fetus-uterus interactions, there are recent and exciting findings that compel us to broaden our point of view, and although the evidence remains inconclusive to date, it is worth considering. Specifically, we refer to the role of the gut microbiota and the elements of response to hypoxia. Currently, it is well known that the microbiota is a key player in health and disease [52] and intestinal microorganism-derived uremic toxins can contribute to the development of CKD [53, 54]. The gut microbiota has been shown to interact with the RAS, both at the gastrointestinal and the systemic level, in a bidirectional way [55]. First, the intraluminal ACE inhibitory peptides or lipopolysaccharides promote the blockade of ACE1 and the activation of AT1 and ANG II [56]; likewise, short-chain fatty acids or tryptophan block prorenin and its receptor at the kidney level, which was reverted by prebiotics and probiotics by means of activating ANG (1–7), ACE2, and MAS [57]. On the other hand, the overactivation of ANG II generates intraluminal dysbiosis [58].
The aforementioned has allowed hypothesizing the role of the maternal gut microbiota on fetal kidney structure and function. The available studies focus on the relationship of the gut maternal microbiota with hypertension in offspring [59]. On the other hand, several studies have shown certain microbiota-derived chemical molecules, such as hydrogen sulfide (H2S), to possess beneficial effects for the offspring [60, 61]. Although the mechanism of action of H2S is still not completely clear, evidence points to a role played by the epigenome modifications, specifically on RAS genes. A study showed that prenatal or postnatal administration of H2S improved blood pressure values. The authors attributed this effect to a possible increase of the methylation of AT1b and the downregulation of AT1R expression [62]. Another study reported the possible involvement in reprogramming toward the prevention of hypertension and kidney damage [59], but concise evidence supporting the effect of H2S itself or other maternal microbiota-derived molecules in human kidney reprogramming is scarce. A deeper review of the advances in this fascinating research area can be found elsewhere [63].
Hypoxia is a strong inducer of differentiation, and several hypoxia-responsive transcription factors (HIF) have been found. The lack or low levels of these HIF during the development of the kidney results in a decrease in both nephron number and kidney mass [64]. Studies point to epigenetic regulation as a mechanism underlining the decrease in nephron number and to its possibly being sex-specific. In pregnant mice with global deletion of miRNA-210, a main hypoxia-induced miRNA, Hemker et al. found the increased expression of LIM homeobox 1 (Lhx1) in kidney tissue only from male offspring [65]. Likewise, fetal hypoxia could function as a susceptibility factor for certain nephrotoxins or other insults that can induce kidney damage in later postnatal life [66]. Little is known regarding the existence of a hypoxia-susceptible miRNA specific for kidney biogenesis, whether the effects of miRNA-210 form part of a global inflammatory response not exclusive to kidney tissue, or whether there is a sex-related miRNA profile in mesonephric development, as observed in lung development [67].
A possible sex-biased mechanism in fetal reprogramming for adult kidney disease?
Sex differences in the normal decline of kidney function due to the natural aging process are well recognized [68, 69], but knowledge concerning the mechanisms explaining these differences and whether they are preferentially “reprogrammed” from the embryonic stages are scarce. Studies have revealed that, under controlled conditions, such as maternal diet, or medication during pregnancy, a biased pattern of kidney programming can be induced, in which the RAS axis is primarily involved in a sex-specific manner [18, 69]. Also, prenatal steroid administration in different biological models is associated with increased blood pressure in males and experimental kidney damage in adulthood [70, 71]. Recent studies evaluating the effect of steroid use during gestation reported sex-biased alterations in the transcriptome, with specifically decreased Agt mRNA and increased Agtr2 and Mas1-receptor expression in female vs. male mice [72]. In addition to the idea that estrogen/testosterone hormonal differences are more favorable in females, the renoprotective effect, due to a more efficient ability to upregulate the anti-inflammatory and vasodilator responses, could also be more advantageous in females [73]. Under this hypothesis, several authors point out that the increase of testosterone in males induces an increase in blood pressure through RAS activation. This is supported by experimental studies showing that testosterone increases the expression of intrarenal angiotensinogen mRNA in males, which is prevented after castration [68]. Likewise, testosterone exerts a stimulatory effect on Ang II at the proximal-tubule level, mainly due to its mineralocorticoid action of sodium reabsorption [74].
In addition to the male disadvantage due to testosterone, there are other hormonal axes that could contribute to the upregulation of the RAS axis, such as the hormonal peak of the luteal phase; however, these elevated levels have no known influence on the increase in blood pressure [71, 72]. In this regard, there are sex-related differences in the kidney response to Ang-II, enhanced by the intrinsic metabolism of Ang-II and by the greater sensitivity to the effects of Ang1-7 in women [19]. Likewise, the hormonal influence during pregnancy may activate the non-classical RAS pathway. A higher placental expression of the metallopeptidase neprilysin during pregnancy has been shown as responsible for the increase in Ang1-7 [19]. Neprilysin is important in the kidney proteolytic process of Ang1-9 cleavage and in the regulation of blood pressure [75]. In this respect, the sex-related differences between the AngII/Ang1-7 ratio are enhanced in pregnancy and are related to the activation of the MasR, which is more involved in the regulation of nitric oxide (NO) and prostaglandins [19].
In the setting of prenatal steroid administration, the placentas of female embryos exhibit a higher expression of the glucocorticoid receptor and of 11-β-hydroxysteroid dehydrogenase type 2, which could inactivate or decrease the number of glucocorticoids in contrast to male fetuses [76]. Biased fetal programming has also been associated with infertility in males and a decreased testicular tissue mass [27]. The estrogenic difference between females and males may include the major mechanism for the increase in angiotensinogen, possibly mediated by estrogen response elements in the AGT gene promoter. However, there are other endocrine pathways that are poorly studied, such as increased progesterone and enhanced metabolic differences during pregnancy, in addition to considering further research on the role of the non-canonical pathway of RAS activation in fetal kidney reprogramming.
Conclusions and perspectives
Increasing evidence supports the notion that the uterine environment exerts a determining influence on kidney programming from early developmental stages, which could affect kidney physiology in both childhood and adulthood. These events could converge in the alteration of RAS. Also, sex differences in hormonal load and responses to changes in the uterine environment could affect the regulatory mechanisms of gene expression, but the relationship between a specific methylation pattern and kidney disease remains to be explored. Knowledge of the factors influencing the early epigenetic modifications of RAS genes and other fundamental pathways to embryonic kidney development will contribute to a better understanding of the pathogenesis of CKD and will supply guidelines in the search for therapeutic approaches aimed at correcting these modifications, even from the embryonic stage.
References
Harambat J, Van Stralen KJ, Kim JJ, Tizard EJ (2012) Epidemiology of chronic kidney disease in children. Pediatr Nephrol 27:363–373. https://doi.org/10.1007/s00467-011-1939-1
Simeoni U, Armengaud JB, Siddeek B, Tolsa JF (2018) Perinatal origins of adult disease. Neonatology 113:393–399. https://doi.org/10.1159/000487618
Morrison JL, Ayonrinde OT, Care AS, Clarke GD, Darby JRT, David AL, Dean JM, Hooper SB, Kitchen MJ, Macgowan CK, Melbourne A, McGillick EV, McKenzie CA, Michael N, Mohammed N, Sadananthan SA, Schrauben E, Regnault TRH, Velan SS (2021) Seeing the fetus from a DOHaD perspective: discussion paper from the advanced imaging techniques of DOHaD applications workshop held at the 2019 DOHaD World Congress. J Dev Orig Health Dis 12:153–167. https://doi.org/10.1017/S2040174420000884
Bertram JF, Douglas-Denton RN, Diouf B, Hughson MD, Hoy WE (2011) Human nephron number: implications for health and disease. Pediatr Nephrol 26:1529–1533. https://doi.org/10.1007/s00467-011-1843-8
Ryan D, Sutherland MR, Flores TJ, Kent AL, Dahlstrom JE, Puelles VG, Bertram JF, McMahon AP, Little MH, Moore L, Black MJ (2018) Development of the human fetal kidney from mid to late gestation in male and female infants. EBioMedicine 27:275–283. https://doi.org/10.1016/j.ebiom.2017.12.016
Barker DJ (1998) In utero programming of chronic disease. Clin Sci (Lond) 95:115–128
Sarkies P (2020) Molecular mechanisms of epigenetic inheritance: possible evolutionary implications. Semin Cell Dev Biol 97:106–115. https://doi.org/10.1016/j.semcdb.2019.06.005
Law PP, Holland ML (2019) DNA methylation at the crossroads of gene and environment interactions. Essays Biochem 63:717–726. https://doi.org/10.1042/EBC20190031
McLaughlin N, Wang F, Saifudeen Z, El-Dahr SS (2014) In situ histone landscape of nephrogenesis. Epigenetics 9:222–235. https://doi.org/10.4161/epi.26793
Franczyk B, Gluba-Brzozka A, Olszewski R, Parolczyk M, Rysz-Gorzynska M, Rysz J (2022) miRNA biomarkers in renal disease. Int Urol Nephrol 54:575–588. https://doi.org/10.1007/s11255-021-02922-7
Guan Y, Liu H, Ma Z, Li SY, Park J, Sheng X, Susztak K (2020) Dnmt3a and Dnmt3b-decommissioned fetal enhancers are linked to kidney disease. J Am Soc Nephrol 31:765–782. https://doi.org/10.1681/ASN.2019080797
Chu AY, Tin A, Schlosser P, Ko YA, Qiu C, Yao C, Joehanes R, Grams ME, Liang L, Gluck CA, Liu C, Coresh J, Hwang SJ, Levy D, Boerwinkle E, Pankow JS, Yang Q, Fornage M, Fox CS, Susztak K, Köttgen A (2017) Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat Commun 8:1286. https://doi.org/10.1038/s41467-017-01297-7
Takimoto-Ohnishi E, Murakami K (2019) Renin-angiotensin system research: from molecules to the whole body. J Physiol Sci 69:581–587. https://doi.org/10.1007/s12576-019-00679-4
Elgazzaz M, Lazartigues E (2021) Epigenetic modifications of the renin-angiotensin system in cardiometabolic diseases. Clin Sci (Lond) 135:127–142. https://doi.org/10.1042/CS20201287
Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R (2001) Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res 49:460–467. https://doi.org/10.1203/00006450-200104000-00005
Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ (2007) Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ Res 100:520–526. https://doi.org/10.1161/01.RES.0000258855.60637.58
Kawakami-Mori F, Nishimoto M, Reheman L, Kawarazaki W, Ayuzawa N, Ueda K, Hirohama D, Kohno D, Oba S, Shimosawa T, Marumo T, Fujita T (2018) Aberrant DNA methylation of hypothalamic angiotensin receptor in prenatal programmed hypertension. JCI Insight 3:21. https://doi.org/10.1172/jci.insight.95625
Gobetto MN, Mendes GarridoAbregú F, Caniffi C, Veiras L, Elesgaray R, Gironacci M, Tomat AL, Arranz C (2020) Fetal and postnatal zinc restriction: sex differences in the renal renin-angiotensin system of newborn and adult Wistar rats. J Nutr Biochem 81:108385. https://doi.org/10.1016/j.jnutbio.2020.108385
South AM, Shaltout HA, Washburn LK, Hendricks AS, Diz DI, Chappell MC (2019) Fetal programming and the angiotensin-(1–7) axis: a review of the experimental and clinical data. Clin Sci (Lond) 133:55–74. https://doi.org/10.1042/CS20171550
Inoue I, Nakajima T, Williams CS, Quackenbush J, Puryear R, Powers M, Cheng T, Ludwig EH, Sharma AM, Hata A, Jeunemaitre X, Lalouel JM (1997) A nucleotide substitution in the promoter of human angiotensinogen is associated with essential hypertension and affects basal transcription in vitro. J Clin Invest 99:1786–1797. https://doi.org/10.1172/JCI119343
DuPriest E, Hebert J, Morita M, Marek N, Meserve EEK, Andeen N, Houseman EA, Qi Y, Alwasel S, Nyengaard J, Morgan T (2020) Fetal renal DNA methylation and developmental programming of stress-induced hypertension in growth-restricted male mice. Reprod Sci 27:1110–1120. https://doi.org/10.1007/s43032-019-00121-5
Ajala AR, Almeida SS, Rangel M, Palomino Z, Strufaldi MW, Puccini RF, Araujo RC, Casarini DE, Franco MC (2012) Association of ACE gene insertion/deletion polymorphism with birth weight, blood pressure levels, and ACE activity in healthy children. Am J Hypertens 25:827–832. https://doi.org/10.1038/ajh.2012.50
He Q, Fan C, Yu M, Wallar G, Zhang ZF, Wang L, Zhang X, Hu R (2013) Associations of ACE gene insertion/deletion polymorphism, ACE activity, and ACE mRNA expression with hypertension in a Chinese population. PLoS One 1:e75870. https://doi.org/10.1371/journal.pone.0075870
Acevedo N, Alashkar Alhamwe B, Caraballo L, Ding M, Ferrante A, Garn H, Garssen J, Hii CS, Irvine J, Llinás-Caballero K, López JF, Miethe S, Perveen K, Pogge von Strandmann E, Sokolowska M, Potaczek DP, van Esch BCAM (2021) Perinatal and early-life nutrition, epigenetics, and allergy. Nutrients 25:724. https://doi.org/10.3390/nu13030724
Zheng J, Zhang L, Liu J, Li Y, Zhang J (2021) Long-term effects of maternal low-protein diet and post-weaning high-fat feeding on glucose metabolism and hypothalamic POMC promoter methylation in offspring mice. Front Nutr 16:657848. https://doi.org/10.3389/fnut.2021.657848
Winship AL, Gazzard SE, Cullen-McEwen LA, Bertram JF, Hutt KJ (2018) Maternal low-protein diet programmes low ovarian reserve in offspring. Reproduction 156:299–311. https://doi.org/10.1530/REP-18-0247
Ajuogu PK, Al-Aqbi MAK, Hart RA, McFarlane JR, Smart NA (2021) A low protein maternal diet during gestation has negative effects on male fertility markers in rats - a systematic review and meta-analysis. J Anim Physiol Anim Nutr (Berl) 105:157–166. https://doi.org/10.1111/jpn.13411
Esmeijer K, de Vries AP, Mook-Kanamori DO, de Fijter JW, Rosendaal FR, Rabelink TJ, Smit RAJ, de Mutsert R, Hoogeveen EK (2019) Low birth weight and kidney function in middle-aged men and women: the Netherlands epidemiology of obesity study. Am J Kidney Dis 74:751–760. https://doi.org/10.1053/j.ajkd.2019.05.007
Grillo MA, Mariani G, Ferraris JR (2021) Prematurity and low weight in neonates as a risk factor for obesity, hypertension, and chronic kidney disease in pediatric and adult age. Front Med (Lausanne) 8:769734. https://doi.org/10.3389/fmed.2021.769734
Rodríguez López S, Tumas N, Ortigoza A, de Lima Friche AA, Diez-Roux AV (2021) Urban social environment and low birth weight in 360 Latin American cities. BMC Public Health 21:795. https://doi.org/10.1186/s12889-021-10886-7
Blencowe H, Krasevec J, de Onis M, Black RE, An X, Stevens GA, Borghi E, Hayashi C, Estevez D, Cegolon L, Shiekh S, Ponce Hardy V, Lawn JE, Cousens S (2019) National, regional, and worldwide estimates of low birthweight in 2015, with trends from 2000: a systematic analysis. Lancet Glob Health 7:e849–e860. https://doi.org/10.1016/S2214-109X(18)30565-5
Liyanage T, Toyama T, Hockham C, Ninomiya T, Perkovic V, Woodward M, Fukagawa M, Matsushita K, Praditpornsilpa K, Hooi LS, Iseki K, Lin MY, Stirnadel-Farrant HA, Jha V, Jun M (2022) Prevalence of chronic kidney disease in Asia: a systematic review and analysis. BMJ Glob Health 7:e007525. https://doi.org/10.1136/bmjgh-2021-007525
Miyoshi M, Sato M, Saito K, Otani L, Shirahige K, Miura F, Ito T, Jia H, Kato H (2018) Maternal protein restriction alters the renal Ptger1 DNA methylation state in SHRSP offspring. Nutrients 10:10. https://doi.org/10.3390/nu10101436
Mao C, Liu R, Bo L, Chen N, Li S, Xia S, Chen J, Li D, Zhang L, Xu Z (2013) High-salt diets during pregnancy affected fetal and offspring renal renin-angiotensin system. J Endocrinol 218:61–73. https://doi.org/10.1530/JOE-13-0139
Tay S, Blache D, Gregg K, Revell D (2012) Consumption of a high-salt diet by ewes during pregnancy alters nephrogenesis in 5-month-old offspring. Animal 6:1803–1810. https://doi.org/10.1017/S1751731112000584
Brito S, Lee MG, Bin BH, Lee JS (2020) Zinc and its transporters in epigenetics. Mol Cells 43:323–330. https://doi.org/10.14348/molcells.2020.0026
Robinson SM, Batelaan SF, Syddall HE, Sayer AA, Dennison EM, Martin HJ, Barker DJ, Cooper C (2006) Combined effects of dietary fat and birth weight on serum cholesterol concentrations: the Hertfordshire Cohort Study. Am J Clin Nutr 84:237–244. https://doi.org/10.1093/ajcn/84.1.237
Simões-Alves AC, Arcoverde-Mello APFC, Campos JO, Wanderley AG, Leandro CVG, da Costa-Silva JH, de Oliveira Nogueira Souza V (2022) Cardiometabolic effects of postnatal high-fat diet consumption in offspring exposed to maternal protein restriction in utero. Front Physiol 10:829920. https://doi.org/10.3389/fphys.2022.829920
Pereira Júnior CD, Guimarães CS, da Silva AC, Rodrigues AR, da Glória MA, Teixeira VP, Câmara NO, Rocha LB, Dos Reis MA, Machado JR, Rocha LP, Helmo FR, Corrêa RR (2016) Influence of the expression of inflammatory markers on kidney after fetal programming in an experimental model of renal failure. J Immunol Res 2016:9151607. https://doi.org/10.1155/2016/9151607
Casasnovas J, Jo Y, Rao X, Xuei X, Brown ME, Kua KL (2019) High glucose alters fetal rat islet transcriptome and induces progeny islet dysfunction. J Endocrinol 240:309–323. https://doi.org/10.1530/JOE-18-0493
Strakovsky RS, Zhang X, Zhou D, Pan YX (2011) Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J Physiol 589:2707–2717. https://doi.org/10.1113/jphysiol.2010.203950
Brennan S, Kandasamy Y, Rudd DM, Schneider ME, Jones RE, Watson DL (2020) The effect of diabetes during pregnancy on fetal renal parenchymal growth. J Nephrol 33:1079–1089. https://doi.org/10.1007/s40620-020-00815-z
Abi Khalil C, Travert F, Fetita S, Rouzet F, Porcher R, Riveline JP, Hadjadj S, Larger E, Roussel R, Vexiau P, Le Guludec D, Gautier JF, Marre M (2010) Fetal exposure to maternal type 1 diabetes is associated with renal dysfunction at adult age. Diabetes 59:2631–2636. https://doi.org/10.2337/db10-0419
Chen YW, Chenier I, Tran S, Scotcher M, Chang SY, Zhang SL (2010) Maternal diabetes programs hypertension and kidney injury in offspring. Pediatr Nephrol 25:1319–1329. https://doi.org/10.1007/s00467-010-1506-1
Yan J, Li X, Su R, Zhang K, Yang H (2014) Long-term effects of maternal diabetes on blood pressure and renal function in rat male offspring. PLoS One 9:e88269. https://doi.org/10.1371/journal.pone.0088269
Glastras SJ, Chen H, Teh R, McGrath RT, Chen J, Pollock CA, Wong MG, Saad S (2016) Mouse models of diabetes, obesity and related kidney disease. PLoS One 11:e0162131. https://doi.org/10.1371/journal.pone.0162131
Larkin BP, Glastras SJ, Chen H, Pollock CA, Saad S (2018) DNA methylation and the potential role of demethylating agents in prevention of progressive chronic kidney disease. FASEB J 32:5215–5226. https://doi.org/10.1096/fj.201800205R
Glastras SJ, Chen H, Pollock CA, Saad S (2018) Maternal obesity increases the risk of metabolic disease and impacts renal health in offspring. Biosci Rep 38:2. https://doi.org/10.1042/BSR20180050
Bechtel W, McGoohan S, Zeisberg EM, Müller GA, Kalbacher H, Salant DJ, Müller CA, Kalluri R, Zeisberg M (2010) Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med 16:544–550. https://doi.org/10.1038/nm.2135
Larkin BP, Nguyen LT, Hou M, Glastras SJ, Chen H, Wang R, Pollock CA, Saad S (2021) Novel role of gestational hydralazine in limiting maternal and dietary obesity-related chronic kidney disease. Front Cell Dev Biol 9:705263. https://doi.org/10.3389/fcell.2021.705263
Kassab BM, Hussein HH, Mahmoud OM, Abdel-Alrahman G (2019) Effects of insulin and metformin on fetal kidney development of streptozotocin-induced gestational diabetic albino rats. Anat Cell Biol 52:161–175. https://doi.org/10.5115/acb.2019.52.2.161
Hou K, Wu ZX, Chen XY, Wang JQ, Zhang D, Xiao C, Zhu D, Koya JB, Wei L, Li J, Chen ZS (2022) Microbiota in health and diseases. Signal Transduct Target Ther 23:135. https://doi.org/10.1038/s41392-022-00974-4
Addi T, Dou L, Burtey S (2018) Tryptophan-derived uremic toxins and thrombosis in chronic kidney disease. Toxins (Basel) 10:412. https://doi.org/10.3390/toxins10100412
Velasquez MT, Centron P, Barrows I, Dwivedi R, Raj DS (2018) Gut microbiota and cardiovascular uremic toxicities. Toxins (Basel) 10:287. https://doi.org/10.3390/toxins10070287
Jaworska K, Koper M, Ufnal M (2021) Gut microbiota and renin-angiotensin system: a complex interplay at local and systemic levels. Am J Physiol Gastrointest Liver Physiol 321:G355–G366. https://doi.org/10.1152/ajpgi.00099.2021
Dave LA, Hayes M, Montoya CA, Rutherfurd SM, Moughan PJ (2016) Human gut endogenous proteins as a potential source of angiotensin- I-converting enzyme (ACE-I)-, renin inhibitory and antioxidant peptides. Peptides 76:30–44. https://doi.org/10.1016/j.peptides.2015.11.003
Wang L, Zhu Q, Lu A, Liu X, Zhang L, Xu C, Liu X, Li H, Yang T (2017) Sodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro) renin receptor and intrarenal renin–angiotensinsystem. J Hypertens 35:1899–1908. https://doi.org/10.1097/HJH.0000000000001378
Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S, Wild B, Camargo SMR, Singer D, Richter A, Kuba K, Fukamizu A, Schreiber S, Clevers H, Verrey F, Rosenstiel P, Penninger JM (2012) ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487:477–481. https://doi.org/10.1038/nature11228
Tain YL, Hsu CN (2022) Hypertension of developmental origins: consideration of gut microbiome in animal models. Biomedicines 10:875. https://doi.org/10.3390/biomedicines10040875
Lu M, Liu YH, Goh HS, Wang JJ, Yong QC, Wang R, Bian JS (2010) Hydrogen sulfide inhibits plasma renin activity. J Am Soc Nephrol 21:993–1002. https://doi.org/10.1681/ASN.2009090949
Feliers D, Lee HJ, Kasinath BS (2016) Hydrogen sulfide in renal physiology and disease. Antioxid Redox Signal 25:720–731. https://doi.org/10.1089/ars.2015.6596
Guo Q, Feng X, Xue H, Teng X, Jin S, Duan X, Xiao L, Wu Y (2017) Maternal renovascular hypertensive rats treatment with hydrogen sulfide increased the methylation of at1b gene in offspring. Am J Hypertens 30:1220–1227. https://doi.org/10.1093/ajh/hpx124
Gomaa EZ (2020) Human gut microbiota/microbiome in health and diseases: a review. Antonie Van Leeuwenhoek 113:2019–2040. https://doi.org/10.1007/s10482-020-01474-7
Dunwoodie SL (2009) The role of hypoxia in development of the mammalian embryo. Dev Cell 17:755–773. https://doi.org/10.1016/j.devcel.2009.11.008
Hemker SL, Cerqueira DM, Bodnar AJ, Cargill KR, Clugston A, Anslow MJ, Sims-Lucas S, Kostka D, Ho J (2020) Deletion of hypoxia-responsive microRNA-210 results in a sex-specific decrease in nephron number. FASEB J 34:5782–5799. https://doi.org/10.1096/fj.201902767R
Cargill KR, Chiba T, Murali A, Mukherjee E, Crinzi E, Sims-Lucas S (2020) Prenatal hypoxia increases susceptibility to kidney injury. PLoS One 15:e0229618. https://doi.org/10.1371/journal.pone.0229618
Lin NW, Liu C, Yang IV, Maier LA, DeMeo DL, Wood C, Ye S, Cruse MH, Smith VL, Vyhlidal CA, Kechris K, Sharma S (2022) Sex-specific differences in microRNA expression during human fetal lung development. Front Genet 13:762834. https://doi.org/10.3389/fgene.2022.762834
Baylis C (2005) Changes in renal hemodynamics and structure in the aging kidney; sexual dimorphism and the nitric oxide system. Exp Gerontol 40:271–278. https://doi.org/10.1016/j.exger.2005.01.008
Loria A, Reverte V, Salazár F, Saez F, Llinas MT, Salazár FJ (2007) Sex and age differences of renal function in rats with reduced ANG II activity during the nephrogenic period. Am J Physiol Renal Physiol 293:F506–F510. https://doi.org/10.1152/ajprenal.00066.2007
Ortiz LA, Quan A, Zarzar F, Weinberg A, Baum M (2003) Prenatal dexamethasone programs hypertension and renal injury in the rat. Hypertension 41:328–334. https://doi.org/10.1161/01.hyp.0000049763.51269.51
Tang L, Bi J, Valego N, Carey L, Figueroa J, Chappell M, Rose JC (2010) Prenatal betamethasone exposure alters renal function in immature sheep: sex differences in effects. Am J Physiol Regul Integr Comp Physiol 299:R793–R7803. https://doi.org/10.1152/ajpregu.00590.2009
Tain YL, Sheen JM, Yu HR, Chen CC, Tiao MM, Hsu CN, Lin YJ, Kuo KC, Huang LT (2015) Maternal melatonin therapy rescues prenatal dexamethasone and postnatal high-fat diet induced programmed hypertension in male rat offspring. Front Physiol 6:377. https://doi.org/10.3389/fphys.2015.00377
Reckelhoff JF, Yanes LL, Iliescu R, Fortepiani LA, Granger JP (2005) Testosterone supplementation in aging men and women: possible impact on cardiovascular-renal disease. Am J Physiol Renal Physiol 289:F941–F948. https://doi.org/10.1152/ajprenal.00034.2005
Yanes LL, Romero DG (2009) Dihydrotestosterone stimulates aldosterone secretion by H295R human adrenocortical cells. Mol Cell Endocrinol 303:50–56. https://doi.org/10.1016/j.mce.2008.12.020
Nalivaeva NN, Zhuravin IA, Turner AJ (2020) Neprilysin expression and functions in development, ageing and disease. Mech Ageing Dev 192:111363. https://doi.org/10.1016/j.mad.2020.111363
Clifton VL, Murphy VE (2004) Maternal asthma as a model for examining fetal sex-specific effects on maternal physiology and placental mechanisms that regulate human fetal growth. Placenta 25(Suppl A):S45–S52. https://doi.org/10.1016/j.placenta.2004.01.004
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pérez-Coria, M., Vázquez-Rivera, G.E., Gómez-García, E.F. et al. Sex differences in fetal kidney reprogramming: the case in the renin-angiotensin system. Pediatr Nephrol 39, 645–653 (2024). https://doi.org/10.1007/s00467-023-06112-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-023-06112-8