
Abstract
Endometriosis is a gynecological disease characterized by the presence of endometrial glandular epithelial and stromal cells growing in the extra-uterine environment. The disease afflicts 10%–15% of menstruating women causing debilitating pain and infertility. Endometriosis appears to affect every part of a woman’s reproductive system including ovarian function, oocyte quality, embryo development and implantation, uterine function and the endocrine system choreographing the reproductive process and results in infertility or spontaneous pregnancy loss. Current treatments are laden with menopausal-like side effects and many cause cessation or chemical alteration of the reproductive cycle, neither of which is conducive to achieving a pregnancy. However, despite the prevalence, physical and psychological tolls and health care costs, a cure for endometriosis has not yet been found. We hypothesize that endometriosis causes infertility via multifaceted mechanisms that are intricately interwoven thereby contributing to our lack of understanding of this disease process. Identifying and understanding the cellular and molecular mechanisms responsible for endometriosis-associated infertility might help unravel the confounding multiplicities of infertility and provide insights into novel therapeutic approaches and potentially curative treatments for endometriosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Endometriosis
Endometriosis is a gynecological disease characterized by the presence of endometrial glandular and stromal cells existing in the extra-uterine environment (Benagiano and Brosens 1991). These extra-uterine glands and stroma, called endometriotic lesions, can be found on the ovaries and on the surfaces of pelvic cavity organs (Koger et al. 1993; Remorgida et al. 2007). Endometriosis, although not malignant, occurs spontaneously in women and non-human primates that menstruate, causing pain and infertility.
Because the disease requires expensive and invasive surgical procedures to diagnose, estimates of the prevalence of endometriosis are difficult to establish. Estimates show that endometriosis affects 10%–15% of all women of reproductive age (Allaire 2006). These women often suffer for years before the diagnosis of endometriosis is made (Hadfield et al. 1996).
A significant economic burden is imposed by endometriosis. Recent estimates of the costs of surgical removal of endometriotic lesions are 17.3 billion dollars per year in the USA alone (Simoens et al. 2007). Indirect costs such as the loss of work productivity attributable to the debilitating chronic pelvic pain accounts for an estimated 4.7 billion dollars lost in the USA (Simoens et al. 2007). Without doubt, a more effective treatment for this disease needs to be developed.
Endometriosis (originally named adenomyoma) was described 150 years ago by Rokitansky as the occurrence of epithelial glands and stroma, resembling those found in the mucosal lining of the uterus, growing elsewhere in the peritoneal cavity (Benagiano and Brosens 1991). The way that the endometrium-like lesions became established outside of the uterus was unclear but several hypotheses were proposed.
In 1940, the current widely accepted explanation for endometriosis was put forward. Sampson posed a unique theory of the pathogenesis of endometriosis called retrograde menstruation. Retrograde menstruation occurs when naturally shed endometrium sloughs off but, instead of exiting though the cervix, it moves out of the oviducts and into the peritoneal cavity by retrograde movement. Up to 90% of all women are estimated to experience some amount of retrograde menstruation but only a portion of women develop endometriosis (Ozkan et al. 2008; Seli et al. 2003). A combination of retrograde menstruation, eutopic endometrial anomalies such as the altered synthesis and secretion of proteins and gene expression and abnormalities in the immune system might be involved in the pathogenesis of endometriosis (Chegini et al. 2003; Fowler et al. 2007; Sharpe-Timms 2001; Siristatidis et al. 2006; Ulukus et al. 2006; Wu and Ho 2003).
Other workers have hypothesized that an increase in telomerase production in the sloughed endometrium during retrograde menstruation has “enhanced replicative capacity” aiding the establishment of ectopic lesions (Hapangama et al. 2008). Additional hypotheses include: the induction theory in which, during menstruation, the sloughing endometrium gives off “factors” that cause changes in the surface epithelium of the ovary leading to the differentiation of endometrium-like tissue and migration to the peritoneal cavity; the in situ theory in which totipotent cells remain undifferentiated from the fetus surviving into adulthood and during the reproductive years, becoming activated and thus differentiating into endometriotic lesions (Nap et al. 2004). Endometrial stem cells might contribute to the pathogenesis of endometriosis and explain some rare and extreme cases of endometriosis (Sasson and Taylor 2008). Probably, a combination of mechanisms is involved during the development of the disease.
The main symptoms of endometriosis are pain and infertility (Allaire 2006; Berkley et al. 2005; Giudice and Kao 2004). The mechanisms of these symptoms in endometriosis are not known. Not all patients experience the same symptoms with endometriosis. Indeed, some women with endometriosis do not learn about their disease until after elective sterilization surgery. No cure is available for endometriosis and current treatments focus on reducing the pain associated with the disease often causing cessation or chemical alteration of the reproductive cycle. Treatments are not curative and may cause detrimental side effects. Further, many treatments are inappropriate for patients seeking treatment for infertility.
Infertility in women with endometriosis
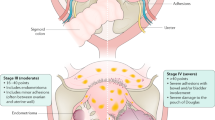
Historically, endometriosis-associated infertility in women has been associated with subtle, explicit, or multifaceted abnormalities (Cahill and Hull 2000; Doody et al. 1988; Garrido et al. 2002, 2003; Groll 1984; Hahn et al. 1986; Hull et al. 1998; Tanbo et al. 1995; Tummon et al. 1988). Indeed, endometriosis appears to affect every part of a woman’s reproductive tract (Fig. 1). Many women with minimal, mild, or moderate endometriosis experience difficulties conceiving and maintaining pregnancy, neither of which can be accounted for by anatomical obstructions (Burns and Schenken 1999). It is estimated that 50% of endometriosis patients are subfertile (Bulletti et al. 2010). The following information characterizes reproductive irregularities associated with endometriosis.

Factors associated with reduced fecundity in women with endometriosis
Pituitary-ovarian feedback
In the normal cycle of fertile women, the pituitary secretes follicle stimulated hormone (FSH) and luteinizing hormone (LH) to stimulate growing ovarian follicles. These follicles provide positive and negative feedback to the pituitary culminating in an LH surge to signal ovulation at the optimum time (Senger 2005). However, in women with endometriosis, a pituitary-ovarian axis dysfunction has been noted altering feedback pathways thereby preventing normal cyclic changes in the ovary. The length of the follicular phase is extended in endometriosis (Cahill et al. 1995; Cheesman et al. 1982) when compared with controls. Additionally, women with endometriosis seem to have abnormal patterns of LH secretion. The LH surge is delayed in endometriosis with lower levels of LH being present and occasionally biphasic surges occur leading to abnormal urinary hormone profiles (Bancroft et al. 1992; Cahill et al. 1995; Tummon et al. 1988; Williams et al. 1986). These problems can impair follicular growth, ovulation and corpus luteum development in the ovary specifically with respect to the timing of ovarian events.
Impact on the ovary
Folliculogenesis
During the normal follicular phase, follicular growth is controlled by a balance of hormones. When FSH causes follicles to grow and develop, these follicles produce estradiol, activin and inhibin, which, in turn, provide a feedback mechanism to control the hypothalamus-pituitary-ovarian axis. While the follicles are growing in size, the cells within the follicle are changing. Visibly, an antrum forms and is filled with follicular fluid. Within the follicle, follicular cells develop LH receptors, which prepare the follicle for ovulation (Senger, 2005).
Folliculogenesis is impaired in women with endometriosis. The number of preovulatory follicles, follicular growth, dominant follicle size and follicular estradiol concentrations are reduced in ovaries of endometriosis patients (Doody et al. 1988; Tummon et al. 1988; Cahill et al. 1995; Dlugi et al. 1989). The follicular fluid of patients with endometriosis have been reported to have altered hormone profiles including reduced estrogen, androgen and progesterone and increased activin (Cahill and Hull 2000). Further, the follicular fluid from patients has been shown to contain factors such as cytokines and growth factors that might promote the maintenance of endometriotic lesions and lead to a suboptimum follicular environment (Abae et al. 1994; Bahtiyar et al. 1998; Pellicer et al. 1998).
Ovulation
The process of ovulation is impaired in women with endometriosis. During normal ovulation, the LH surge starts a cascade of events in the follicle that leads to the expulsion of the oocyte-cumulus complex. Several protective layers including the granulosa, follicular basement membrane and theca must be overcome within each ovulating follicle. To achieve this, changes in proteolytic enzymes, cytokines, inflammatory molecules, steroid hormones and vasculature must occur (Espey 1980, 1994).
In women with endometriosis, mechanisms that facilitate normal ovulation are impaired. As mentioned before, the LH surge might be altered; however, others suggest that a deficiency in follicular LH receptors (Ronnberg et al. 1984). Additionally, lower levels of estrogen and progesterone have been noted in the serum and urine of women with endometriosis (Brosens et al. 1978; Cheesman et al. 1982; Cunha-Filho et al. 2003; Smith et al. 2002; Trinder and Cahill 2002; Tummon et al. 1988). Changes in proteolytic enzymes (Ebisch et al. 2007; Smedts et al. 2006; Wunder et al. 2005), cytokines (Carlberg et al. 2000; Garrido et al. 2000; Pellicer et al. 1998; Wunder et al. 2006), inflammatory molecules (Carlberg et al. 2000; Lachapelle et al. 1996; Wunder et al. 2006) and the vasculature (Abae et al. 1994; Garrido et al. 2000; Pellicer et al. 1998), all of which are required for normal ovulation, can also be found in the follicles of women with endometriosis. Collectively, these data provide evidence of mechanisms that could cause ovulatory dysfunction in endometriosis.
A phenomenon exists whereby oocytes become trapped in a luteinizing corpus hemorrhagicum. This failure of ovulation, defined as luteinized unruptured follicle syndrome (LUFs), has been associated with endometriosis and infertility in women (Donnez and Thomas 1982; Kaya and Oral 1999; Marik and Hulka 1978; Mio et al. 1992; Muse and Wilson 1982). Peritoneal concentrations of steroid hormones, including progesterone and estradiol, are reported to decrease in women with LUFs; however, whether this is a cause or consequence of the phenomenon is unclear (Koninckx et al. 1980). Whereas the mechanism causing this syndrome remains unknown, any one of the factors necessary for follicular rupture could contribute to failed ovulation.
Luteal function
After ovulation in normal cycles, the granulosa and theca cells of the ovulated follicle differentiate into luteal cells. The main function of this transformation is to produce progesterone to prepare the reproductive tract for successful implantation and pregnancy (Owen 1975). Altered luteal function has been noted in endometriosis patients and affects both large and small luteal cells (Cheesman et al. 1983; Cunha-Filho et al. 2003). Early luteal events, specifically patterns of estrogen and progesterone secretion, are altered in women with endometriosis (Cheesman et al. 1982). Indeed, endometriosis patients with luteal defects secrete less progesterone than those from healthy patients (Cunha-Filho et al. 2003). Women with endometriosis who have a luteal deficiency are more likely to experience infertility (Cunha-Filho et al. 2001).
Impact on oocyte quality
Women with endometriosis ovulate fewer oocytes than healthy women (Al-Fadhli et al. 2006; Bergqvist and D'Hooghe 2002; Cahill and Hull 2000; Kumbak et al. 2008; Mahutte and Arici 2001; Yanushpolsky et al. 1998) and those oocytes ovulated by women with endometriosis are sometimes compromised (Garrido et al. 2000, 2002, 2003; Navarro et al. 2003; Pellicer et al. 2000). A recent study has shown that women with endometriosis exhibit an increase in apoptosis of the cumulus cells surrounding the oocyte (Díaz-Fontdevila et al. 2009). Apoptosis in ovarian cells is a good indicator of poor oocyte quality (Nakahara et al. 1997). Death of cumulus cells probably leads to reduced oocyte quality and maturation attributable to the loss of the essential support that the cumulus cells give to the oocyte (e.g., pyruvate, hormones, growth factors; Russell and Robker 2007).
Morphology is one indicator of the potential for each oocyte to produce an embryo. Oocyte morphological characteristics includes extracytoplasmic and cytoplasmic defects. Extracytoplasmic defects that seem to impair fertilization rate include abnormal first polar body extrusion and a large perivitelline space (Rienzi et al. 2008). Cytoplasmic defects disrupting fertilization rate include cytoplasmic granularity, central location of cytoplasmic granularity and the presence of vacuoles (Rienzi et al. 2008). Morphological analysis is, however, a subjective evaluation and does not completely correlate with the outcome.
The potential of the development of biomarkers clearly to identify “good” versus “bad” oocytes is exciting. Potential targets recently investigated include nuclear export factor CRM1 in high-quality pig oocytes and components of the ubiquitin-proteasome pathway in low-quality pig oocytes (Powell et al. 2010). Despite this, current methods of visualizing these markers requires oocyte fixation rendering them unusable for use in artificial reproductive techniques. Investigators are trying to discover ways of using proteomic methodology to detect these biomarkers in oocyte maturation medium.
Assisted reproductive therapies can help restore fertility in women with endometriosis but unfortunately produce inconsistent results. Some studies have shown that the pregnancy outcome with use of in vitro fertilization (IVF) is similar in women with and without endometriosis (Bergendal et al. 1998; Geber et al. 1995; Huang et al. 1997). Women with endometriosis undergoing IVF treatments involving oocytes from a non-affected individual show normal implantation and pregnancy rates (Simon et al. 1994). However, other workers have reported that fertilization and/or embryo cleavage rates after IVF, both in stimulated and unstimulated cycles, are significantly lower in endometriosis compared with controls (Cahill and Hull 2000; Harlow et al. 1996; Hull et al. 1998; Tanbo et al. 1995). Fertilization and embryo cleavage rates remain impaired in women with endometriosis after spermatozoa from their partners are substituted with spermatozoa from donors (Groll 1984; Hull et al. 1998). Additionally, implantation rates of oocytes from donors with endometriosis are reduced in recipients who do not have endometriosis (Navarro et al. 2003).
Several factors from a woman with endometriosis contribute to the failure of a spermatozoon to fertilize a potentially compromised oocyte. An increase in peritoneal macrophages during endometriosis can lead to increased phagocytosis of healthy spermatozoa that might have otherwise been able to fertilize the ova (Muscato et al. 1983). Uterine/oviductal sperm transport is impaired in endometriosis (Kissler et al. 2005, 2006, 2007; Leyendecker et al. 1996). This impairment emerges in the early stages of endometriosis (Kissler et al. 2007). The peritoneal fluid of patients with endometriosis has a negative impact on sperm binding to the zona pellucida of the oocyte in vitro (Coddington et al. 1992). Peritoneal fluid of women with endometriosis has been shown to increase DNA fragmentation in sperm from healthy donors (Mansour et al. 2009b). Interleukin-6 (IL-6) and its soluble receptor, which are present in the peritoneal fluid of women with endometriosis (Harada et al. 1997), reduce sperm motility (Iwabe et al. 2002; Yoshida et al. 2004). Additionally, endometriosis negatively impacts sperm binding to the oviductal epithelium (Reeve et al. 2005).
Impact on embryo development
Endometriosis negatively impacts embryo development (Table 1). Because of the use of assisted reproductive techniques, data are available about embryo quality and rates of cleavage, implantation and pregnancy loss. Aberrant nuclear and cytoplasmic events in embryos from women with endometriosis are six times more likely compared with women without endometriosis (Brizek et al. 1995). These events include cytoplasmic fragmentation (Brizek et al. 1995), darkened cytoplasm (Brizek et al. 1995), reduced cell numbers (Garrido et al. 2002; Pellicer et al. 1995; Tanbo et al. 1995) and increased frequency of arrested embryos (Garrido et al. 2000; Yanushpolsky et al. 1998) leading to significantly fewer transferable blastocysts (Garrido et al. 2002; Pellicer et al. 1995). Additionally, the quality of embryos that develop from endometriosis patients has been shown to be reduced (Brizek et al. 1995; Cahill and Hull 2000; Garrido et al. 2000, 2002; Pellicer et al. 1995; Yanushpolsky et al. 1998). Treatment with a gonadotrophin-releasing hormone agonist that temporarily causes regression of the endometriotic lesions and cessation of reproductive cyclicity helps to improve embryo quality in these patients (Takahashi et al. 2004).
Inflammatory cytokines in the peritoneal fluid of women with endometriosis provide a plausible hypothesis to explain decreased embryo quality from such women. Exposure of the embryo to peritoneal fluid while in the reproductive tract can cause these defects (Esfandiari et al. 2005; Furukubo et al. 1998; Gomez-Torres et al. 2002). Murine embryos cultured in the presence of peritoneal fluid from women with endometriosis have decreased rates of development after the two-cell stage (Taketani et al. 1992). In a similar study, murine embryos cultured in the presence of human peritoneal fluid from women with endometriosis show increased rates of DNA fragmentation and apoptosis compared with treatment by control peritoneal fluid (Mansour et al. 2009a). Further, embryos cultured in the presence of IL-6 (found in the peritoneal fluid of women with endometriosis) arrest at the blastocyst stage or earlier (Iwabe et al. 2002). Even sera from women with endometriosis are embryo toxic to murine embryos in vitro (Abu-Musa et al. 1992; Ito et al. 1996; Tzeng et al. 1994).
Apoptosis or programmed cell death of the embryo can occur through several mechanisms associated with endometriotic lesions such as increased concentrations of inflammatory cytokines or reactive oxygen species (ROS; Agic et al. 2006; Jana et al. 2010; Zeller et al. 1987; Fig. 2). Inflammatory cytokines such as tumor necrosis factor-α can activate caspase-dependent signaling pathways to increase apoptosis (Hu 2003). ROS can cause mitochondrial damage and DNA strand breaks (Lao et al. 2009). This might also encourage the cell to undergo programmed cell death or apoptosis.

Mechanisms by which endometriosis affects apoptosis signaling in embryo development (TNF-α tumor necrosis factor-α)
ROS have been implicated as a potential source of endometriosis-related infertility (Augoulea et al. 2009). Early studies have shown increased concentrations of ROS and lipid peroxides in the peritoneal fluid from women with endometriosis (Murphy et al. 1998; Zeller et al. 1987). More recent studies have demonstrated no difference in the amount of ROS in the peritoneal fluid (Agarwal et al. 2003) but a decrease in the antioxidants present (Jackson et al. 2005). This suggests that antioxidant protection is decreased in the peritoneal fluid from women with endometriosis, an occurrence that could negatively affect embryo development (Augoulea et al. 2009).
Impact on uterine receptivity
Uterine receptivity, which allows the developing embryo to implant, is a complex process involving regulation by hormones, cytokines, adhesion molecules and other factors (Aghajanova et al. 2008). In women, uterine receptivity can be marked by the expression of integrins, specifically αVβ3 (Donaghay and Lessey 2007). Integrins are cell surface receptors that mediate intracellular signals. Notably, about 50% of women with endometriosis have decreased or, in some cases, absent expression of endometrial αVβ3 (Donaghay and Lessey 2007). These data are correlated to the ~50% of patients with endometriosis who, even with assisted reproductive technologies, cannot conceive (Donaghay and Lessey 2007; Lessey 2002).
HOXA10, which is known to be a potent stimulator of αVβ3 expression, is a transcription factor and member of the Homeobox family of genes expressed by the normal endometrium (Eun Kwon and Taylor 2004). Decreased endometrial expression and altered methylation of HOXA10 have been reported in women with endometriosis providing a potential mechanism for the deficiency of αVβ3 (Donaghay and Lessey 2007; Eun Kwon and Taylor 2004; Taylor et al. 1999; Vitiello et al. 2007). Other uterine biomarkers of implantation such as glycodelin A, osteopontin, leukemia inhibitory factor and lysophosphatidic acid receptor 3 are reduced in women with endometriosis (Giudice et al. 2002; Wei et al. 2009).
Together with a general decrease in the expression of key uterine receptivity factors, steroid hormone pathways are altered in endometriosis. Normally at the time of implantation, estrogen receptors are downregulated; however, women with endometriosis have an upregulation of endometrial estrogen receptors (Lessey et al. 1988). Aromatase is also aberrantly expressed by the endometrium of women with endometriosis, increasing the amount of active estradiol (Attar and Bulun 2006). To exacerbate altered estrogen during receptivity even further, 17β-hydroxysteroid dehydrogenase-2 is downregulated thereby inhibiting estradiol inactivation leading to a local increase in estrogen action (Giudice et al. 2002).
Conversely, progesterone actions, which are required for endometrial receptivity, are reduced (Bulun et al. 2006). An increase in progesterone compared with estrogen must occur for successful endometrial receptivity to the implanting blastocyst. Progesterone resistance has been reported in eutopic and ectopic endometrium (Giudice and Kao 2004). Differential expression of the isoforms of the progesterone receptor occurs in endometriosis: isoform A is present but B is not, most likely because of aberrant methylation of its promoter (Attia et al. 2000; Wu et al. 2006). Further, stromal cells of endometriotic lesions do not express 17 β-hydroxysteriod dehydrogenase type 2 thereby preventing the conversion of estradiol2 to estradiol1, usually induced by progesterone (Bulun et al. 2006). Reduced progesterone receptors and decreased levels of estrone lead to high levels of estradiol furthering the progesterone resistance. Collectively, these data provide evidence for mechanisms involved in reduced uterine receptivity.
Impact on embryo implantation
Quantifying embryo implantation in women with endometriosis is difficult and has led to inconsistent results. Women with endometriosis are reported to experience implantation failure more often than controls (Arici et al. 1996; Cahill and Hull 2000; Simon et al. 1994). However, others disagree (Geber et al. 1995; Sung et al. 1997). Much of the evidence regarding the success or failure of implantation originates from IVF data. Implantation rates are difficult to determine because of the variation in patient procedures, including differences in the numbers of embryos transferred and the selection of the most ideal sperm, oocyte and embryo. Nonetheless, a significant decrease in implantation per embryo transferred in IVF (Arici et al. 1996; Cahill and Hull 2000; Pellicer et al. 1995) has been found in association with endometriosis.
Decreased rates of embryo implantation are an additional aspect of infertility in women with endometriosis (Arici et al. 1996; Cahill and Hull 2000; Garrido et al. 2000; Yanushpolsky et al. 1998). Defects in embryo implantation might be associated with hormone level alterations, embryo anomalies and/or endometrial anomalies as described. For example, embryo anomalies can include slow growth and delayed blastocyst hatching, which are detrimental for implantation of the embryo in the uterine endometrium (Bazer et al. 2009).
Impact on the uterus: risk of miscarriage
Together with difficulty in establishing pregnancy, women with endometriosis can have an increased risk of miscarriage and even recurrent miscarriage (Tomassetti et al. 2006; Yanushpolsky et al. 1998). The mechanisms behind these spontaneous pregnancy losses are unknown but are probably multifaceted including but not limited to, B cell immunodeficiency and autoantibodies (Gleicher et al. 1989; Mahutte and Arici 2001).
Investigators disagree about the increased risk of spontaneous pregnancy loss after implantation (Al-Azemi et al. 2000; Diaz et al. 2000; Matalliotakis et al. 2008a, 2008b; Metzger et al. 1986; Olive et al. 1982; Pittaway et al. 1988; Wheeler et al. 1983; Yanushpolsky et al. 1998). Some studies suggest no increased risk of loss (Al-Azemi et al. 2000; Diaz et al. 2000; Pittaway et al. 1988). However, many of these investigations include women who have undergone IVF treatment with stimulated cycles and selection of the most favorable embryos to be transferred, both of which could have affected the outcome. Metzger et al. (1986) have however noted that abortion rates drop to zero after surgical intervention in women with endometriosis, suggesting that endometriosis itself does indeed play a role in these losses. Although definitive proof that endometriosis causes spontaneous pregnancy loss is lacking, women with endometriosis have an increased risk of spontaneous abortion.
Impact on peritoneal milieu
Endometriotic lesions secrete proteins and/or change the peritoneal environment in a way that has been hypothesized to affect the establishment, maintenance and symptoms of endometriosis. These substances include but are not limited to: prostaglandins (Chishima et al. 2007; Drake et al. 1981; Moon et al. 1983; Muzii et al. 1996; Sondheimer and Flickinger 1982); haptoglobin (Piva and Sharpe-Timms 1999; Sharpe-Timms 2005; Sharpe-Timms et al. 1998, 2002); cytokines such as IL-1, IL-6, IL-8 and IL-10; growth factors, such as vascular endothelial growth factor, nerve growth factor, transforming growth factor-β1 and 2, insulin-like growth factor-2 (Anaf et al. 2002; Gazvani and Templeton 2002; Sharpe-Timms 2001; Taylor et al. 2002); cellular remodeling enzymes, such as the matrix metalloproteinases (MMPs) and their inhibitors (tissue inhibitors of metalloproteinase, TIMPs; Chung et al. 2001; Osteen et al. 1996, 2003; Sharpe-Timms et al. 1995; Zhou and Nothnick 2005). Whereas the consequences of these and other molecules secreted from the lesions are not fully known, the altered milieu in the peritoneal fluid can clearly lead to changes in the reproductive tract.
Endometriosis is an heritable disease
Susceptibility to endometriosis is hypothesized to be heritable based on the increased risk of developing endometriosis if a family member is affected (Simpson et al. 2003). Retrospective studies have shown that women with a first-degree relative with endometriosis are 5%–8% more likely to have endometriosis (Simpson et al. 2003). Having a sister with endometriosis increases the risk of developing endometriosis by 5.2-fold (Stefansson et al. 2002).
Genome-wide studies have identified several potential loci that have mutations in women with endometriosis. Treloar et al. (2005) have found, in a genome-wide linkage study of over 1,000 sister-pair families, that women with endometriosis have a possible susceptibility locus on chromosome 10q26. This portion of the DNA includes coding regions for several reproductively important genes including EMX2, a gene required for reproductive tract development and PTEN, a tumor suppressor gene (Treloar et al. 2005). However, according to a review by Bischoff and Simpson (2004), genetic mutations in this region, or any other loci identified in population studies, of the DNA cannot alone account for the heritability of endometriosis.
Endometriosis is an epigenetic disease
Because of the lack of evidence to substantiate the idea of a common genetic mutation in endometriosis, the familial tendency of endometriosis might alternatively be attributable to epigenetic reprogramming during embryonic or fetal development (Dean et al. 2003). Epigenetics is a new exciting field that affects many disciplines of science from fetal origins of adult disease, assisted reproductive techniques, cancer biology, to other diseases without a link to a specific genetic anomaly (Dean et al. 2005). Epigenetics is the study of alterations to the cytosine base pairs and histone modifications that affect gene expression but are not mutations of the DNA itself.
In endometriosis, epigenetic changes might arise by several mechanisms (Fig. 3). Endometriotic lesion secretory products or inflammatory mediators from elevated numbers of peritoneal macrophages and other immune cells present in the peritoneal fluid might affect the methylation status of the genome of the embryo or fetus (Hill et al. 1988). This can occur by changing the gene expression of enzymes such as DNA methyltransferases (DNMTs) and histone-modifying enzymes such as histone deacetylases (Haaf 2006). One suggestion is that, in ectopic endometrium of women with endometriosis, DNMT1, DNMT3A and DNMT3B are over-expressed when compared with control levels (Wu et al. 2007).

Potential mechanisms of aberrant DNA methylation in endometriosis (DNMT DNA methyltransferase, HDAC histone deacetylase)
Inflammatory mediators might cause increased DNA methylation by a secondary mechanism (Ushijima and Okochi-Takada 2005). ROS associated with inflammation cause DNA damage such as halogenated pyrimidines, which mimic methylated cytosines (Lao et al. 2009; Valinluck and Sowers 2007). These halogenated pyrimidines cause DNMT1 to recognize the hemi-methylation of the DNA leading to the methylation of the opposite strand of DNA (Lao et al. 2009; Valinluck and Sowers 2007).
These aberrant methylation marks established during gametogenesis or gestation might persist through childhood and cause an increased risk for endometriosis. Aberrant epigenetic programming in endometriosis might begin during several events critical to the establishment of pregnancy such as oocyte maturation (Nafee et al. 2008), pre-implantation embryo development (Latham and Schultz 2001) and implantation (Paulson et al. 1990).
The methylation level of the oocyte genome remains low until the oocyte is activated during folliculogenesis (Nafee et al. 2008; Fig. 4). Upon follicular activation and recruitment, methylation marks are established (Nafee et al. 2008). No studies to date have focused on the effect of endometriosis on the establishment of methylation marks during oocyte maturation and follicular development.

Methylation dynamics during mammalian folliculogenesis and early mammalian embryo development (blue paternal genome, red maternal genome) adapted from Reik W. et al., 2001
Shortly after fertilization the paternal genome of the zygote in the mouse, rat and human undergoes active demethylation (Fig. 4; Dean et al. 2003; Zaitseva et al. 2007). The maternal zygotic genome undergoes a passive demethylation process from fertilization to the 8-cell stage in mice (Dean et al. 2003). Incomplete erasure of methylation marks can lead to increased incidence of disease later in life (Junien et al. 2005).
During embryonic development most of the epigenetic marks must be erased to allow for pluripotency. The growing embryo must make the transition from translating protein from maternally derived mRNA to transcribing its own mRNA for translation (Latham and Schultz 2001). The maternal to embryonic transition (MET) has been shown to occur at the 2-cell stage in mice, the 4-cell stage in rats and the 8-cell stage in human and bovine embryos (Telford et al. 1990). Within about two cell divisions from the MET, most maternal transcripts are degraded and the embryonic genome is transcriptionally active (Zeng et al. 2004). The time period immediately following this transition is ideal for studying the impact of endometriosis on embryo gene expression and epigenetic status, rather than maternal transcripts.
Another important part of embryo development is re-methylation of the embryonic genome to allow for differentiation of the cell lines (Fig. 4). By the blastocyst stage of development, methylation marks return to the genome as the blastomeres differentiate into various cell lineages including the trophoblast and inner cell mass (Reik et al. 2001). During this period of re-methylation, the embryo is hypothesized to be highly sensitive to stressors such as temperature changes and ROS exposure, which can cause aberrant methylation and possibly lead to embryo death or embryo growth problems such as those seen in endometriosis (Khosla et al. 2001).
Anomalous methylation during any part of embryo development might cause an arrested cell cycle and apoptosis of the blastomeres or inhibition of embryo implantation in the endometrium (Feil 2009). Whereas this aberrant methylation might not directly affect subsequent cell cycles, it might represent the embryonic origin of an adult disease such as endometriosis, as methylation marks are not easily removed once established (Nafee et al. 2008).
Human and rat embryo implantation is both an embryonic and maternal process (Paulson et al. 1990). Once embryos reach the blastocyst stage of development, they hatch from the zona pellucida and implant in the uterine endometrium. The maternal tissue must be correctly organized for implantation, which necessitates the patterning of gene expression of genes such as HOXA10 (Eun Kwon and Taylor 2004; Vitiello et al. 2007). For example, the suppression of HOXA10 by methylation might lead to failed implantation.
Evidence of epigenetic modifications in the eutopic endometrium has been described in adults with endometriosis. Genes important for implantation, such as HOXA10 and progesterone receptor isoform B (PR-B), are differentially methylated in the eutopic endometrium of women with endometriosis compared with controls (Lee et al. 2009; Wu et al. 2006). This aberrant methylation is correlated to the differential expression of these genes seen in the eutopic endometrium of women with endometriosis (Lee et al. 2009; Wu et al. 2006).
Animal models of endometriosis
Because of the ethical limitations of working with human embryos and experimentation in women, animal models of endometriosis are frequently used to study the anomalies associated with endometriosis (Sharpe-Timms 2002). Common rodent models of endometriosis include the rat (Vernon and Wilson 1985), rabbit (Schenken and Asch 1980) and mouse (Cummings and Metcalf 1995) models. These models have many advantages such as decreased cost and ethical limitations compared with working on primates (D'Hooghe et al. 2009; Grummer 2006; Sharpe-Timms 2002). Endometriosis is induced in rodents by autologous surgical transplantation of endometrial tissue from the animal’s own uterus into the arterial cascade of the small intestine (Sharpe-Timms 2002). These implants mimic human endometriotic lesions in that they establish a blood source, are influenced by the cycle stage and hormonal levels and show signs of causing decreased fertility (Vernon and Wilson 1985).
One advantage of the rat model is that the rat estrous cycle lasts 4-5 days, compared with the typical 28-day menstrual cycle in women, thereby allowing many studies to be completed in a short period of time (Sharpe-Timms 2002). Moreover, reproductive cycle stage can easily be monitored by using vaginal cytology (Sharpe-Timms 2002).
The rat model of endometriosis, because of its many similarities to endometriosis in women, has been used to understand mechanisms of subfertility (Table 1). Vernon and Wilson validated the rat model of endometriosis in 1985. In this model, the presence of endometrial implants in the peritoneum caused a decrease in fecundity by 28% at day 14 of pregnancy and by 48% at term (Vernon and Wilson 1985). Others have shown that the cytokine milieu of the peritoneal fluid changes in rats with surgically induced endometriosis in a similar fashion to that of humans (Umezawa et al. 2008). We have demonstrated that the peritoneal fluid components can enter the uterine horns via the oviduct and possibly affect embryonic or eutopic-endometrial quality (Stilley et al. 2009).
Rats with endometriosis have also been shown to experience more spontaneous abortions and to have a decreased litter size (Pal et al. 1999), an increased incidence of LUFS (Moon et al. 1993) and increased early embryonic mortality when compared with sham-operated controls (Stilley et al. 2009). This similarity to subfertility seen in human endometriosis makes the rat model a suitable alternative for studying the effects of endometriosis.
Based on the rat model, studies from our laboratory have shown that TIMP1 is increased in the ovarian theca of antral follicles, associated with decreased follicle numbers, LUFS and poor embryo quality (Stilley et al. 2009). Further, reducing levels of intraperitoneal fluid TIMP1 in Endo rats by a TIMP1-function-blocking antibody mitigates the impact of endometriosis on the ovary (Stilley et al. 2010). Conversely, increasing TIMP1 in rats by sham surgery decreases ovarian function to levels similar to those of Endo rats with fewer numbers of follicles and corpora lutea and poor embryo quality (Stilley et al. 2010). In addition to these observations, work at our laboratory has shown that TIMP1 is able to act independently of MMP action to impair the ovulatory function through changes to pathways involved in extracellular matrix production, angiogenesis and apoptosis (Stilley and Sharpe-Timms 2011).
Interestingly, research at our laboratory has also demonstrated that daughters of rats with endometriosis have similar embryo anomalies as their mothers (Stilley et al. 2009). By combining these findings suggesting an epigenetic inheritance of endometriosis-like embryo anomalies in a rat model of endometriosis (Stilley et al. 2009), the recent advances in the field of epigenetics (Burdge and Lillycrop 2010) and the development of possible treatments to prevent these aberrant epigenetic marks during development (Waterland et al. 2008), we are presently testing the hypothesis that endometriosis-associated subfertility is multigeneration with an epigenetic mode of inheritance in offspring from mothers with endometriosis. Epigenetic heritability of subfertility in endometriosis is a unique idea that has not been previously postulated.
Concluding remarks
Endometriosis seems to impact, in a negative manner, every part of the reproductive process subtly but significantly (Fig. 1). However, to date, a cause and effect relationship between endometriosis and reduced fecundity has not been established. Infertility associated with endometriosis can be even more puzzling, as not every patient experiences the same symptoms. Therefore, not all patients respond to therapies in the same way, making treatments particularly difficult to develop. Nonetheless, research into therapeutic modalities for subfertility associated with endometriosis needs to be continued, particularly with regard to targeting the molecular mechanisms. Animal models have proven to be valuable in providing insights into principles of mechanisms underlying subfertility in endometriosis, when such studies in women are ethically restricted.
References
Abae M, Glassberg M, Majercik MH, Yoshida H, Vestal R, Puett D (1994) Immunoreactive endothelin-1 concentrations in follicular fluid of women with and without endometriosis undergoing in vitro fertilization-embryo transfer. Fertil Steril 61:1083–1087
Abu-Musa A, Takahashi K, Kitao M (1992) Effect of serum from patients with endometriosis on the development of mouse embryos. Gynecol Obstet Invest 33:157–160
Agarwal A, Saleh RA, Bedaiwy MA (2003) Role of reactive oxygen species in the pathophysiology of human reproduction. Fertil Steril 79:829–843
Aghajanova L, Hamilton AE, Giudice LC (2008) Uterine receptivity to human embryonic implantation: histology, biomarkers, and transcriptomics. Semin Cell Dev Biol 19:204–211
Agic A, Xu H, Finas D, Banz C, Diedrich K, Hornung D (2006) Is endometriosis associated with systemic subclinical inflammation? Gynecol Obstet Invest 62:139–147
Al-Azemi M, Bernal AL, Steele J, Gramsbergen I, Barlow D, Kennedy S (2000) Ovarian response to repeated controlled stimulation in in-vitro fertilization cycles in patients with ovarian endometriosis. Hum Reprod 15:72–75
Al-Fadhli R, Kelly SM, Tulandi T, Tanr SL (2006) Effects of different stages of endometriosis on the outcome of in vitro fertilization. J Obstet Gynaecol Can 28:888–891
Allaire C (2006) Endometriosis and infertility: a review. J Reprod Med 51:164–168
Anaf V, Simon P, El Nakadi I, Fayt I, Simonart T, Buxant F, Noel JC (2002) Hyperalgesia, nerve infiltration and nerve growth factor expression in deep adenomyotic nodules, peritoneal and ovarian endometriosis. Hum Reprod 17:1895–1900
Arici A, Oral E, Bukulmez O, Duleba A, Olive DL, Jones EE (1996) The effect of endometriosis on implantation: results from the Yale University in vitro fertilization and embryo transfer program. Fertil Steril 65:603–607
Attar E, Bulun SE (2006) Aromatase and other steroidogenic genes in endometriosis: translational aspects. Hum Reprod Update 12:49–56
Attia GR, Zeitoun K, Edwards D, Johns A, Carr BR, Bulun SE (2000) Progesterone receptor isoform A but not B is expressed in endometriosis. J Clin Endocrinol Metab 85:2897–2902
Augoulea A, Mastorakos G, Lambrinoudaki I, Christodoulakos G, Creatsas G (2009) The role of the oxidative-stress in the endometriosis-related infertility. Gynecol Endocrinol 25:75–81
Bahtiyar MO, Seli E, Oral E, Senturk LM, Zreik TG, Arici A (1998) Follicular fluid of women with endometriosis stimulates the proliferation of endometrial stromal cells. Hum Reprod 13:3492–3495
Bancroft K, Williams CAV, Elstein M (1992) Pituitary–ovarian function in women with minimal or mild endometriosis and otherwise unexplained infertility. Clin Endocrinol 36:177–181
Bazer F, Spencer T, Johnson G, Burghardt R, Wu G (2009) Comparative aspects of implantation. Reproduction 138:195–209
Benagiano G, Brosens I (1991) The history of endometriosis: identifying the disease. Hum Reprod 6:963–968
Bergendal A, Naffah S, Nagy C, Bergqvist A, Sjoblom P, Hillensjo T (1998) Outcome of IVF in patients with endometriosis in comparison with tubal-factor infertility. J Assist Reprod Genet 15:530–534
Bergqvist A, D'Hooghe T (2002) Mini symposium on pathogenesis of endometriosis and treatment of endometriosis-associated subfertility. Introduction: the endometriosis enigma. Hum Reprod Update 8:79–83
Berkley KJ, Rapkin AJ, Papka RE (2005) The pains of endometriosis. Science 308:1587–1589
Bischoff F, Simpson JL (2004) Genetics of endometriosis: heritability and candidate genes. Best Pract Res Clin Obstet Gynaecol 18:219–232
Brizek CL, Schlaff S, Pellegrini VA, Frank JB, Worrilow KC (1995) Increased incidence of aberrant morphological phenotypes in human embryogenesis—an association with endometriosis. J Assist Reprod Genet 12:106–112
Brosens IA, Koninckx PR, Corveleyn PA (1978) A study of plasma progesterone, oestradiol-17beta, prolactin and LH levels, and of the luteal phase appearance of the ovaries in patients with endometriosis and infertility. Br J Obstet Gynaecol 85:246–250
Bulletti C, Coccia ME, Battistoni S, Borini A (2010) Endometriosis and infertility. J Assist Reprod Genet 27:441–447
Bulun SE, Cheng YH, Yin P, Imir G, Utsunomiya H, Attar E, Innes J, Julie Kim J (2006) Progesterone resistance in endometriosis: link to failure to metabolize estradiol. Mol Cell Endocrinol 248:94–103
Burdge GC, Lillycrop KA (2010) Nutrition, epigenetics, and developmental plasticity: implications for understanding human disease. Annu Rev Nutr 30:315–339
Burns WN, Schenken RS (1999) Pathophysiology of endometriosis-associated infertility. Clin Obstet Gynecol 42:586–610
Cahill DJ, Hull MG (2000) Pituitary-ovarian dysfunction and endometriosis. Hum Reprod Update 6:56–66
Cahill DJ, Wardle PG, Maile LA, Harlow CR, Hull MG (1995) Pituitary-ovarian dysfunction as a cause for endometriosis-associated and unexplained infertility. Hum Reprod 10:3142–3146
Carlberg M, Nejaty J, Froysa B, Guan Y, Soder O, Bergqvist A (2000) Elevated expression of tumour necrosis factor alpha in cultured granulosa cells from women with endometriosis. Hum Reprod 15:1250–1255
Cheesman KL, Ben N, Chatterton RT Jr, Cohen MR (1982) Relationship of luteinizing hormone, pregnanediol-3-glucuronide, and estriol-16-glucuronide in urine of infertile women with endometriosis. Fertil Steril 38:542–548
Cheesman KL, Cheesman SD, Chatterton RT Jr, Cohen MR (1983) Alterations in progesterone metabolism and luteal function in infertile women with endometriosis. Fertil Steril 40:590–595
Chegini N, Roberts M, Ripps B (2003) Differential expression of interleukins (IL)-13 and IL-15 in ectopic and eutopic endometrium of women with endometriosis and normal fertile women. Am J Reprod Immunol 49:75–83
Chishima F, Hayakawa S, Yamamoto T, Sugitani M, Karasaki-Suzuki M, Sugita K, Nemoto N (2007) Expression of inducible microsomal prostaglandin E synthase in local lesions of endometriosis patients. Am J Reprod Immunol 57:218–226
Chung HW, Wen Y, Chun SH, Nezhat C, Woo BH, Lake Polan M (2001) Matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-3 mRNA expression in ectopic and eutopic endometrium in women with endometriosis: a rationale for endometriotic invasiveness. Fertil Steril 75:152–159
Coddington CC, Oehninger S, Cunningham DS, Hansen K, Sueldo CE, Hodgen GD (1992) Peritoneal fluid from patients with endometriosis decreases sperm binding to the zona pellucida in the hemizona assay: a preliminary report. Fertil Steril 57:783–786
Cummings AM, Metcalf JL (1995) Induction of endometriosis in mice: a new model sensitive to estrogen. Reprod Toxicol 9:233–238
Cunha-Filho JS, Gross JL, Lemos NA, Brandelli A, Castillos M, Passos EP (2001) Hyperprolactinemia and luteal insufficiency in infertile patients with mild and minimal endometriosis. Horm Metab Res 33:216–220
Cunha-Filho JS, Gross JL, Bastos de Souza CA, Lemos NA, Giugliani C, Freitas F, Passos EP (2003) Physiopathological aspects of corpus luteum defect in infertile patients with mild/minimal endometriosis. J Assist Reprod Genet 20:117–121
D'Hooghe TM, Kyama CM, Chai D, Fassbender A, Vodolazkaia A, Bokor A, Mwenda JM (2009) Nonhuman primate models for translational research in endometriosis. Reprod Sci 16:152–161
Dean W, Santos F, Reik W (2003) Epigenetic reprogramming in early mammalian development and following somatic nuclear transfer. Semin Cell Dev Biol 14:93–100
Dean W, Lucifero D, Santos F (2005) DNA methylation in mammalian development and disease. Birth Defects Res C Embryo Today 75:98–111
Diaz I, Navarro J, Blasco L, Simon C, Pellicer A, Remohi J (2000) Impact of stage III-IV endometriosis on recipients of sibling oocytes: matched case-control study. Fertil Steril 74:31–34
Díaz-Fontdevila M, Pommer R, Smith R (2009) Cumulus cell apoptosis changes with exposure to spermatozoa and pathologies involved in infertility. Fertil Steril 91:2061–2068
Ding GL, Chen XJ, Luo Q, Dong MY, Wang N, Huang HF (2010) Attenuated oocyte fertilization and embryo development associated with altered growth factor/signal transduction induced by endometriotic peritoneal fluid.Fertil Steril 93:2538–2544
Dlugi AM, Loy RA, Dieterle S, Bayer SR, Seibel MM (1989) The effect of endometriomas on in vitro fertilization outcome. J In Vitro Fert Embryo Transf 6:338–341
Dodds WG, Miller FA, Friedman CI, Lisko B, Goldberg JM, Kim MH (1992)The effect of preovulatory peritoneal fluid from cases of endometriosis on murine in vitro fertilization, embryo development, oviduct transport, and implantation.Am J Obstet Gynecol 166:219–224
Donaghay M, Lessey BA (2007) Uterine receptivity: alterations associated with benign gynecological disease. Semin Reprod Med 25:461–475
Donnez J, Thomas K (1982) Incidence of the luteinized unruptured follicle syndrome in fertile women and in women with endometriosis. Eur J Obstet Gynecol Reprod Biol 14:187–190
Doody MC, Gibbons WE, Buttram VC Jr (1988) Linear regression analysis of ultrasound follicular growth series: evidence for an abnormality of follicular growth in endometriosis patients. Fertil Steril 49:47–51
Drake TS, O'Brien WF, Ramwell PW, Metz SA (1981) Peritoneal fluid thromboxane B2 and 6-keto-prostaglandin F1 alpha in endometriosis. Am J Obstet Gynecol 140:401–404
Ebisch IMW, Steegers-Theunissen RPM, Sweep FCGJ, Zielhuis GA, Geurts-Moespot A, Thomas CMG (2007) Possible role of the plasminogen activation system in human subfertility. Fertil Steril 87:619–626
Esfandiari N, Falcone T, Goldberg JM, Agarwal A, Sharma RK (2005) Effects of peritoneal fluid on preimplantation mouse embryo development and apoptosis in vitro. Reprod Biomed Online 11:615–619
Espey LL (1980) Ovulation as an inflammatory reaction—a hypothesis. Biol Reprod 22:73–106
Espey LL (1994) Current status of the hypothesis that mammalian ovulation is comparable to an inflammatory reaction. Biol Reprod 50:233–238
Eun Kwon H, Taylor HS (2004) The role of HOX genes in human implantation. Ann N Y Acad Sci 1034:1–18
Feil R (2009) Epigenetic asymmetry in the zygote and mammalian development. Int J Dev Biol 53:191–201
Fowler PA, Tattum J, Bhattacharya S, Klonisch T, Hombach-Klonisch S, Gazvani R, Lea RG, Miller I, Simpson WG, Cash P (2007) An investigation of the effects of endometriosis on the proteome of human eutopic endometrium: a heterogeneous tissue with a complex disease. Proteomics 7:130–142
Furukubo M, Fujino Y, Umesaki N, Ogita S (1998) Effects of endometrial stromal cells and peritoneal fluid on fertility associated with endometriosis. Osaka City Med J 44:43–54
Garrido N, Navarro J, Remohi J, Simon C, Pellicer A (2000) Follicular hormonal environment and embryo quality in women with endometriosis. Hum Reprod Update 6:67–74
Garrido N, Navarro J, Garcia-Velasco J, Remoh J, Pellice A, Simon C (2002) The endometrium versus embryonic quality in endometriosis-related infertility. Hum Reprod Update 8:95–103
Garrido N, Pellicer A, Remohi J, Simon C (2003) Uterine and ovarian function in endometriosis. Semin Reprod Med 21:183–192
Gazvani R, Templeton A (2002) Peritoneal environment, cytokines and angiogenesis in the pathophysiology of endometriosis. Reproduction 123:217–226
Geber S, Paraschos T, Atkinson G, Margara R, Winston RM (1995) Results of IVF in patients with endometriosis: the severity of the disease does not affect outcome, or the incidence of miscarriage. Hum Reprod 10:1507–1511
Giudice LC, Kao LC (2004) Endometriosis. Lancet 364:1789–1799
Giudice LC, Telles TL, Lobo S, Kao L (2002) The molecular basis for implantation failure in endometriosis. Ann N Y Acad Sci 955:252–264
Gleicher N, el-Roeiy A, Confino E, Friberg J (1989) Reproductive failure because of autoantibodies: unexplained infertility and pregnancy wastage. Am J Obstet Gynecol 160:1376-1380
Gomez-Torres MJ, Acien P, Campos A, Velasco I (2002) Embryotoxicity of peritoneal fluid in women with endometriosis. Its relation with cytokines and lymphocyte populations. Hum Reprod 17:777–781
Groll M (1984) Endometriosis and spontaneous abortion. Fertil Steril 41:933–935
Grummer R (2006) Animal models in endometriosis research. Hum Reprod Update 12:641–649
Haaf T (2006) Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr Top Microbiol Immunol 310:13–22
Hadfield R, Mardon H, Barlow D, Kennedy S (1996) Delay in the diagnosis of endometriosis: a survey of women from the USA and the UK. Hum Reprod 11:878–880
Hahn DW, Carraher RP, Foldesy RG, McGuire JL (1986) Experimental evidence for failure to implant as a mechanism of infertility associated with endometriosis. Am J Obstet Gynecol 155:1109–1113
Hapangama DK, Turner MA, Drury JA, Quenby S, Saretzki G, Martin-Ruiz C, Von Zglinicki T (2008) Endometriosis is associated with aberrant endometrial expression of telomerase and increased telomere length. Hum Reprod 23:1511–1519
Harada T, Yoshioka H, Yoshida S, Iwabe T, Onohara Y, Tanikawa M, Terakawa N (1997) Increased interleukin-6 levels in peritoneal fluid of infertile patients with active endometriosis. Am J Obstet Gynecol 176:593–597
Harlow CR, Cahill DJ, Maile LA, Talbot WM, Mears J, Wardle PG, Hull MG (1996) Reduced preovulatory granulosa cell steroidogenesis in women with endometriosis. J Clin Endocrinol Metab 81:426–429
Hill JA, Faris HM, Schiff I, Anderson DJ (1988) Characterization of leukocyte subpopulations in the peritoneal fluid of women with endometriosis. Fertil Steril 50:216–222
Hu X (2003) Proteolytic signaling by TNFalpha: caspase activation and IkappaB degradation. Cytokine 21:286–294
Huang HY, Lee CL, Lai YM, Chang MY, Chang SY, Soong YK (1997) The outcome of in vitro fertilization and embryo transfer therapy in women with endometriosis failing to conceive after laparoscopic conservative surgery. J Am Assoc Gynecol Laparosc 4:299–303
Hull MG, Williams JA, Ray B, McLaughlin EA, Akande VA, Ford WC (1998) The contribution of subtle oocyte or sperm dysfunction affecting fertilization in endometriosis-associated or unexplained infertility: a controlled comparison with tubal infertility and use of donor spermatozoa. Hum Reprod 13:1825–1830
Ito F, Fujino Y, Ogita S (1996) Serum from endometriosis patients impairs the development of mouse embryos in vitro—comparison with serum from tubal obstruction patient and plasmanate. Acta Obstet Gynecol Scand 75:877–880
Iwabe T, Harada T, Terakawa N (2002) Role of cytokines in endometriosis-associated infertility. Gynecol Obstet Invest 53:19–25
Jackson LW, Schisterman EF, Dey-Rao R, Browne R, Armstrong D (2005) Oxidative stress and endometriosis. Hum Reprod 20:2014–2020
Jana SK, K NB, Chattopadhyay R, Chakravarty B, Chaudhury K (2010) Upper control limit of reactive oxygen species in follicular fluid beyond which viable embryo formation is not favorable. Reprod Toxicol 29:447–451
Junien C, Gallou-Kabani C, Vige A, Gross MS (2005) Nutritional epigenomics of metabolic syndrome (in French). Med Sci (Paris) 21 (Spec No.):44-52
Kaya H, Oral B (1999) Effect of ovarian involvement on the frequency of luteinized unruptured follicle in endometriosis. Gynecol Obstet Invest 48:123–126
Khosla S, Dean W, Brown D, Reik W, Feil R (2001) Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol Reprod 64:918–926
Kissler S, Hamscho N, Zangos S, Gatje R, Muller A, Rody A, Dobert N, Menzel C, Grunwald F, Siebzehnrubl E, et al (2005) Diminished pregnancy rates in endometriosis due to impaired uterotubal transport assessed by hysterosalpingoscintigraphy. BJOG 112:1391–1396
Kissler S, Hamscho N, Zangos S, Wiegratz I, Schlichter S, Menzel C, Doebert N, Gruenwald F, Vogl TJ, Gaetje R, et al (2006) Uterotubal transport disorder in adenomyosis and endometriosis—a cause for infertility. BJOG 113:902–908
Kissler S, Zangos S, Wiegratz I, Kohl J, Rody A, Gaetje R, Doebert N, Wildt L, Kunz G, Leyendecker G, et al (2007) Utero-tubal sperm transport and its impairment in endometriosis and adenomyosis. Ann N Y Acad Sci 1101:38–48
Koger KE, Shatney CH, Hodge K, McClenathan JH (1993) Surgical scar endometrioma. Surg Gynecol Obstet 177:243–246
Koninckx PR, Moor PD, Brosens IA (1980) Diagnosis of the luteinized unruptured follicle syndrome by steroid hormone assays on peritoneal fluid. BJOG 87:929–934
Kumbak B, Kahraman S, Karlikaya G, Lacin S, Guney A (2008) In vitro fertilization in normoresponder patients with endometriomas: comparison with basal simple ovarian cysts. Gynecol Obstet Invest 65:212–216
Lachapelle MH, Hemmings R, Roy DC, Falcone T, Miron P (1996) Flow cytometric evaluation of leukocyte subpopulations in the follicular fluids of infertile patients. Fertil Steril 65:1135–1140
Lao VV, Herring JL, Kim CH, Darwanto A, Soto U, Sowers LC (2009) Incorporation of 5-chlorocytosine into mammalian DNA results in heritable gene silencing and altered cytosine methylation patterns. Carcinogenesis 30:886–893
Latham KE, Schultz RM (2001) Embryonic genome activation. Front Biosci 6:D748–D759
Lee B, Du H, Taylor HS (2009) Experimental murine endometriosis induces DNA methylation and altered gene expression in eutopic endometrium. Biol Reprod 80:79–85
Lessey BA (2002) Implantation defects in infertile women with endometriosis. Ann N Y Acad Sci 955:265-280
Lessey BA, Killam AP, Metzger DA, Haney AF, Greene GL, McCarty KS Jr (1988) Immunohistochemical analysis of human uterine estrogen and progesterone receptors throughout the menstrual cycle. J Clin Endocrinol Metab 67:334–340
Leyendecker G, Kunz G, Wildt L, Beil D, Deininger H (1996) Uterine hyperperistalsis and dysperistalsis as dysfunctions of the mechanism of rapid sperm transport in patients with endometriosis and infertility. Hum Reprod 11:1542–1551
Mahutte NG, Arici A (2001) Endometriosis and assisted reproductive technologies: are outcomes affected? Curr Opin Obstet Gynecol 13:275–279
Mansour G, Abdelrazik H, Sharma RK, Radwan E, Falcone T, Agarwal A (2009a) L-carnitine supplementation reduces oocyte cytoskeleton damage and embryo apoptosis induced by incubation in peritoneal fluid from patients with endometriosis. Fertil Steril 91:2079–2086
Mansour G, Aziz N, Sharma R, Falcone T, Goldberg J, Agarwal A (2009b) The impact of peritoneal fluid from healthy women and from women with endometriosis on sperm DNA and its relationship to the sperm deformity index. Fertil Steril 92:61–67
Marik J, Hulka J (1978) Luteinized unruptured follicle syndrome: a subtle cause of infertility. Fertil Steril 29:270–274
Matalliotakis I, Cakmak H, Dermitzaki D, Zervoudis S, Goumenou A, Fragouli Y (2008a) Increased rate of endometriosis and spontaneous abortion in an in vitro fertilization program: no correlation with epidemiological factors. Gynecol Endocrinol 24:194–198
Matalliotakis I, Cakmak H, Fragouli Y, Goumenou A, Mahutte N, Arici A (2008b) Epidemiological characteristics in women with and without endometriosis in the Yale series. Arch Gynecol Obstet 277:389–393
Metzger DA, Olive DL, Stohs GF, Franklin RR (1986) Association of endometriosis and spontaneous abortion: effect of control group selection. Fertil Steril 45:18–22
Mio Y, Toda T, Harada T, Terakawa N (1992) Luteinized unruptured follicle in the early stages of endometriosis as a cause of unexplained infertility. Am J Obstet Gynecol 167:271–273
Moon CE, Bertero MC, Curry TE, London SN, Muse KN, Sharpe KL, Vernon MW (1993) The presence of luteinized unruptured follicle syndrome and altered folliculogenesis in rats with surgically induced endometriosis. Am J Obstet Gynecol 169:676–682
Moon YS, Gomel V, Yuen BH, Nickerson KG (1983) The role of prostaglandin F in the symptoms of endometriosis. Can Med Assoc J 129:458–459
Murphy AA, Palinski W, Rankin S, Morales AJ, Parthasarathy S (1998) Evidence for oxidatively modified lipid-protein complexes in endometrium and endometriosis. Fertil Steril 69:1092–1094
Muscato JJ, Haney AF, Weinberg JB (1983) Sperm phagocytosis by human peritoneal macrophages: a possible cause of infertility in endometriosis. Obstet Gynecol Surv 38:177–178
Muse KN, Wilson EA (1982) How does mild endometriosis cause infertility? Fertil Steril 38:145–152
Muzii L, Marana R, Brunetti L, Romanini ME, Vavala VV, Mancuso S, Vacca M (1996) Production of prostaglandin F2alpha by the different forms of endometriosis. J Am Assoc Gynecol Laparosc 3:S33
Nafee TM, Farrell WE, Carroll WD, Fryer AA, Ismail KM (2008) Epigenetic control of fetal gene expression. BJOG 115:158–168
Nakahara K, Saito H, Saito T, Ito M, Ohta N, Takahashi T, Hiroi M (1997) The incidence of apoptotic bodies in membrana granulosa can predict prognosis of ova from patients participating in in vitro fertilization programs. Fertil Steril 68:312–317
Nap AW, Groothuis PG, Demir AY, Evers JLH, Dunselman GAJ (2004) Pathogenesis of endometriosis. Best Pract Res Clin Obstet Gynaecol 18:233–244
Navarro J, Garrido N, Remohi J, Pellicer A (2003) How does endometriosis affect infertility? Obstet Gynecol Clin North Am 30:181–192
Olive DL, Franklin RR, Gratkins LV (1982) The association between endometriosis and spontaneous abortion. A retrospective clinical study. J Reprod Med 27:333–338
Osteen KG, Bruner KL, Sharpe-Timms KL (1996) Steroid and growth factor regulation of matrix metalloproteinase expression and endometriosis. Semin Reprod Endocrinol 14:247–255
Osteen KG, Yeaman GR, Bruner-Tran KL (2003) Matrix metalloproteinases and endometriosis. Semin Reprod Med 21:155–164
Owen JA (1975) Physiology of the menstrual cycle. Am J Clin Nutr 28:333–338
Ozkan S, Murk W, Arici A (2008) Endometriosis and infertility: epidemiology and evidence-based treatments. Ann N Y Acad Sci 1127:92–100
Pal AK, Biswas S, Goswami SK, Kabir SN (1999) Effect of pelvic endometrial implants on overall reproductive functions of female rats. Biol Reprod 60:954–958
Paulson RJ, Sauer MV, Lobo RA (1990) Factors affecting embryo implantation after human in vitro fertilization: a hypothesis. Am J Obstet Gynecol 163:2020–2023
Pellicer A, Oliveira N, Ruiz A, Remohi J, Simon C (1995) Exploring the mechanism(s) of endometriosis-related infertility: an analysis of embryo development and implantation in assisted reproduction. Hum Reprod 10 (Suppl 2):91–97
Pellicer A, Albert C, Mercader A, Bonilla-Musoles F, RemohI J, Simón C (1998) The follicular and endocrine environment in women with endometriosis: local and systemic cytokine production. Fertil Steril 70:425–431
Pellicer A, Albert C, Garrido N, Navarro J, Remohi J, Simon C (2000) The pathophysiology of endometriosis-associated infertility: follicular environment and embryo quality. J Reprod Fertil Suppl 55:109–119
Pittaway DE, Vernon C, Fayez JA (1988) Spontaneous abortions in women with endometriosis. Fertil Steril 50:711–715
Piva M, Sharpe-Timms KL (1999) Peritoneal endometriotic lesions differentially express a haptoglobin-like gene. Mol Hum Reprod 5:71–78
Powell MD, Manandhar G, Spate L, Sutovsky M, Zimmerman S, Sachdev SC, Hannink M, Prather RS, Sutovsky P (2010) Discovery of putative oocyte quality markers by comparative ExacTag proteomics. Proteomics Clin Appl 4:337–351
Reeve L, Lashen H, Pacey AA (2005) Endometriosis affects sperm-endosalpingeal interactions. Hum Reprod 20:448–451
Reik W, Dean W, Walter J (2001) Epigenetic reprogramming in mammalian development. Science 293:1089–1093
Remorgida V, Ferrero S, Fulcheri E, Ragni N, Martin DC (2007) Bowel endometriosis: presentation, diagnosis, and treatment. Obstet Gynecol Surv 62:461–470
Rienzi L, Ubaldi FM, Iacobelli M, Minasi MG, Romano S, Ferrero S, Sapienza F, Baroni E, Litwicka K, Greco E (2008) Significance of metaphase II human oocyte morphology on ICSI outcome. Fertil Steril 90:1692–1700
Ronnberg L, Kauppila A, Rajaniemi H (1984) Luteinizing hormone receptor disorder in endometriosis. Fertil Steril 42:64–68
Russell DL, Robker RL (2007) Molecular mechanisms of ovulation: co-ordination through the cumulus complex. Hum Reprod Update 13:289–312
Sasson IE, Taylor HS (2008) Stem cells and the pathogenesis of endometriosis. Ann N Y Acad Sci 1127:106–115
Schenken RS, Asch RH (1980) Surgical induction of endometriosis in the rabbit: effects on fertility and concentrations of peritoneal fluid prostaglandins. Fertil Steril 34:581–587
Seli E, Berkkanoglu M, Arici A (2003) Pathogenesis of endometriosis. Obstet Gynecol Clin North Am 30:41–61
Senger PL (2005) Pathways to pregnancy and parturition, 2nd edn. Current Conceptions, Pullman
Sharpe-Timms KL (2001) Endometrial anomalies in women with endometriosis. Ann N Y Acad Sci 943:131–147
Sharpe-Timms KL (2002) Using rats as a research model for the study of endometriosis. Ann N Y Acad Sci 955:318–327
Sharpe-Timms KL (2005) Haptoglobin expression by shed endometrial tissue fragments found in peritoneal fluid. Fertil Steril 84:22–30
Sharpe-Timms KL, Penney LL, Zimmer RL, Wright JA, Zhang Y, Surewicz K (1995) Partial purification and amino acid sequence analysis of endometriosis protein-II (ENDO-II) reveals homology with tissue inhibitor of metalloproteinases-1 (TIMP-1). J Clin Endocrinol Metab 80:3784–3787
Sharpe-Timms KL, Piva M, Ricke EA, Surewicz K, Zhang YL, Zimmer RL (1998) Endometriotic lesions synthesize and secrete a haptoglobin-like protein. Biol Reprod 58:988–994
Sharpe-Timms KL, Zimmer RL, Ricke EA, Piva M, Horowitz GM (2002) Endometriotic haptoglobin binds to peritoneal macrophages and alters their function in women with endometriosis. Fertil Steril 78:810–819
Simoens S, Hummelshoj L, D'Hooghe T (2007) Endometriosis: cost estimates and methodological perspective. Hum Reprod Update 13:395–404
Simon C, Gutierrez A, Vidal A, Santos MJ de los, Tarin JJ, Remohi J, Pellicer A (1994b) Outcome of patients with endometriosis in assisted reproduction: results from in-vitro fertilization and oocyte donation. Hum Reprod 9:725–729
Simpson JL, Bischoff FZ, Kamat A, Buster JE, Carson SA (2003) Genetics of endometriosis. Obstet Gynecol Clin North Am 30:21-40
Siristatidis C, Nissotakis C, Chrelias C, Iacovidou H, Salamalekis E (2006) Immunological factors and their role in the genesis and development of endometriosis. J Obstet Gynaecol Res 32:162–170
Smedts AM, Lele SM, Modesitt SC, Curry TE (2006) Expression of an extracellular matrix metalloproteinase inducer (basigin) in the human ovary and ovarian endometriosis. Fertil Steril 86:535–542
Smith M, Keay S, Margo F, Harlow C, Wood P, Cahill D, Hull M (2002) Total cortisol levels are reduced in the periovulatory follicle of infertile women with minimal and mild endometriosis. Am J Reprod Immunol 47:52–56
Sondheimer SJ, Flickinger G (1982) Prostaglandin F2 alpha in the peritoneal fluid of patients with endometriosis. Int J Fertil 27:73–75
Stefansson H, Geirsson RT, Steinthorsdottir V, Jonsson H, Manolescu A, Kong A, Ingadottir G, Gulcher J, Stefansson K (2002) Genetic factors contribute to the risk of developing endometriosis. Hum Reprod 17:555–559
Stilley JA, Sharpe-Timms KL (2011) TIMP1 contributes to ovarian anomalies in both an MMP-dependent and independent manner in a rat model. Biol Reprod (in press)
Stilley JA, Woods-Marshall R, Sutovsky M, Sutovsky P, Sharpe-Timms KL (2009) Reduced fecundity in female rats with surgically induced endometriosis and in their daughters: a potential role for tissue inhibitors of metalloproteinase 1. Biol Reprod 80:649–656
Stilley JA, Birt JA, Nagel SC, Sutovsky M, Sutovsky P, Sharpe-Timms KL (2010) Neutralizing TIMP1 restores fecundity in a rat model of endometriosis and treating control rats with TIMP1 causes anomalies in ovarian function and embryo development. Biol Reprod 83:185–194
Sung L, Mukherjee T, Takeshige T, Bustillo M, Copperman AB (1997) Endometriosis is not detrimental to embryo implantation in oocyte recipients. J Assist Reprod Genet 14:152–156
Takahashi K, Mukaida T, Tomiyama T, Goto T, Oka C (2004) GnRH antagonist improved blastocyst quality and pregnancy outcome after multiple failures of IVF/ICSI-ET with a GnRH agonist protocol. J Assist Reprod Genet 21:317–322
Taketani Y, Kuo TM, Mizuno M (1992) Comparison of cytokine levels and embryo toxicity in peritoneal fluid in infertile women with untreated or treated endometriosis. Am J Obstet Gynecol 167:265–270
Tanbo T, Omland A, Dale PO, Abyholm T (1995) In vitro fertilization/embryo transfer in unexplained infertility and minimal peritoneal endometriosis. Acta Obstet Gynecol Scand 74:539–543
Taylor HS, Bagot C, Kardana A, Olive D, Arici A (1999) HOX gene expression is altered in the endometrium of women with endometriosis. Hum Reprod 14:1328–1331
Taylor RN, Lebovic DI, Mueller MD (2002) Angiogenic factors in endometriosis. Ann N Y Acad Sci 955:89–100
Telford NA, Watson AJ, Schultz GA (1990) Transition from maternal to embryonic control in early mammalian development: a comparison of several species. Mol Reprod Dev 26:90–100
Tomassetti C, Meuleman C, Pexsters A, Mihalyi A, Kyama C, Simsa P, D'Hooghe TM (2006) Endometriosis, recurrent miscarriage and implantation failure: is there an immunological link? Reprod Biomed Online 13:58–64
Treloar SA, Wicks J, Nyholt DR, Montgomery GW, Bahlo M, Smith V, Dawson G, Mackay IJ, Weeks DE, Bennett ST, et al (2005) Genomewide linkage study in 1,176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26. Am J Hum Genet 77:365–376
Trinder J, Cahill DJ (2002) Endometriosis and infertility: the debate continues. Hum Fertil (Camb) 5:S21–S27
Tummon IS, Maclin VM, Radwanska E, Binor Z, Dmowski WP (1988) Occult ovulatory dysfunction in women with minimal endometriosis or unexplained infertility. Fertil Steril 50:716–720
Tzeng CR, Chien LW, Chang SR, Chen AC (1994) Effect of peritoneal fluid and serum from patients with endometriosis on mouse embryo in vitro development. Zhonghua Yi Xue Za Zhi (Taipei) 54:145–148
Ulukus M, Cakmak H, Arici A (2006) The role of endometrium in endometriosis. J Soc Gynecol Investig 13:467–476
Umezawa M, Sakata C, Tanaka N, Kudo S, Tabata M, Takeda K, Ihara T, Sugamata M (2008) Cytokine and chemokine expression in a rat endometriosis is similar to that in human endometriosis. Cytokine 43:105–109
Ushijima T, Okochi-Takada E (2005) Aberrant methylations in cancer cells: where do they come from? Cancer Sci 96:206–211
Valinluck V, Sowers LC (2007) Inflammation-mediated cytosine damage: a mechanistic link between inflammation and the epigenetic alterations in human cancers. Cancer Res 67:5583–5586
Vernon MW, Wilson EA (1985) Studies on the surgical induction of endometriosis in the rat. Fertil Steril 44:684–694
Vitiello D, Kodaman PH, Taylor HS (2007) HOX genes in implantation. Semin Reprod Med 25:431–436
Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S (2008) Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obes Relat Metab Disord 32:1373–1379
Wei Q, St. Clair JB, Fu T, Stratton P, Nieman LK (2009) Reduced expression of biomarkers associated with the implantation window in women with endometriosis. Fertil Steril 91:1686–1691
Wheeler JM, Johnston BM, Malinak LR (1983) The relationship of endometriosis to spontaneous abortion. Fertil Steril 39:656–660
Williams CA, Oak MK, Elstein M (1986) Cyclical gonadotrophin and progesterone secretion in women with minimal endometriosis. Clin Reprod Fertil 4:259–268
Wu MY, Ho HN (2003) The role of cytokines in endometriosis. Am J Reprod Immunol 49:285–296
Wu Y, Strawn E, Basir Z, Halverson G, Guo SW (2006) Promoter hypermethylation of progesterone receptor isoform B (PR-B) in endometriosis. Epigenetics 1:106–111
Wu Y, Strawn E, Basir Z, Halverson G, Guo SW (2007) Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometriosis. Fertil Steril 87:24–32
Wunder DM, Mueller MD, Birkhäuser MH, Bersinger NA (2005) Steroids and protein markers in the follicular fluid as indicators of oocyte quality in patients with and without endometriosis. J Assist Reprod Genet 22:257–264
Wunder DM, Mueller MD, Birkhäuser MH, Bersinger NA (2006) Increased ENA-78 in the follicular fluid of patients with endometriosis. Acta Obstet Gynecol Scand 85:336–342
Yanushpolsky EH, Best CL, Jackson KV, Clarke RN, Barbieri RL, Hornstein MD (1998) Effects of endometriomas on ooccyte quality, embryo quality, and pregnancy rates in in vitro fertilization cycles: a prospective, case-controlled study. J Assist Reprod Genet 15:193–197
Yoshida S, Harada T, Iwabe T, Taniguchi F, Mitsunari M, Yamauchi N, Deura I, Horie S, Terakawa N (2004) A combination of interleukin-6 and its soluble receptor impairs sperm motility: implications in infertility associated with endometriosis. Hum Reprod 19:1821–1825
Zaitseva I, Zaitsev S, Alenina N, Bader M, Krivokharchenko A (2007) Dynamics of DNA-demethylation in early mouse and rat embryos developed in vivo and in vitro. Mol Reprod Dev 74:1255–1261
Zeller JM, Henig I, Radwanska E, Dmowski WP (1987) Enhancement of human monocyte and peritoneal macrophage chemiluminescence activities in women with endometriosis. Am J Reprod Immunol Microbiol 13:78–82
Zeng F, Baldwin DA, Schultz RM (2004) Transcript profiling during preimplantation mouse development. Dev Biol 272:483–496
Zhou HE, Nothnick WB (2005) The relevancy of the matrix metalloproteinase system to the pathophysiology of endometriosis. Front Biosci 10:569–575
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Funding source in the manuscript is supported in part by NIH 057445 to KST.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Stilley, J.A.W., Birt, J.A. & Sharpe-Timms, K.L. Cellular and molecular basis for endometriosis-associated infertility. Cell Tissue Res 349, 849–862 (2012). https://doi.org/10.1007/s00441-011-1309-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-011-1309-0