
Abstract
Introduction
In recent decades, many theories have been proposed about the cause of hereditary diseases such as cancer. However, most studies state genetic and environmental factors as the most important parameters. It has been shown that gene expression data are valuable information about hereditary diseases and their analysis can identify the relationships between these diseases.
Objective
Identification of damaged genes from various diseases can be done through the discovery of cell-to-cell biological communications. Also, extraction of intercellular communications can identify relationships between different diseases. For example, gene disorders that cause damage to the same cells in both breast and blood cancers. Hence, the purpose is to discover cell-to-cell biological communications in gene expression data.
Methodology
The identification of cell-to-cell biological communications for various cancer diseases has been widely performed by clustering algorithms. However, this field remains open due to the abundance of unprocessed gene expression data. Accordingly, this paper focuses on the development of a semi-supervised ensemble clustering algorithm that can discover relationships between different diseases through the extraction of cell-to-cell biological communications. The proposed clustering framework includes a stratified feature sampling mechanism and a novel similarity metric to deal with high-dimensional data and improve the diversity of primary partitions.
Results
The performance of the proposed clustering algorithm is verified with several datasets from the UCI machine learning repository and then applied to the FANTOM5 dataset to extract cell-to-cell biological communications. The used version of this dataset contains 108 cells and 86,427 promoters from 702 samples. The strength of communication between two similar cells from different diseases indicates the relationship of those diseases. Here, the strength of communication is determined by promoter, so we found the highest cell-to-cell biological communication between “basophils” and “ciliary.epithelial.cells” with 62,809 promoters.
Conclusion
The maximum cell-to-cell biological similarity in each cluster can be used to detect the relationship between different diseases such as cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human body consists of hundreds of types of cells (Kayal et al. 2019; Peng et al. 2022). These cells are directly or indirectly dependent on each other and have the ability to communicate and influence each other. Therefore, an effective mechanism is needed to find the relationship between this astronomical number of cells. Finding these communications will help identify relationships between different diseases. The nucleus of each cell has the coded instructions necessary to direct the cell's activities and make the necessary proteins. A whole group of these instructions is called a genome (Sivadas et al. 2022). Human genome is the genetic set and genes inside the nucleus of human cells (de Souza et al. 2016). There are millions of genes on each of the chromosomes, each of which has a specific role in the cell. Let the gene expression associated with a cell be represented by promoters (Shahraki et al. 2023).
To date, many diagnostic models have been presented for different diseases such as cancer. Each model uses different tools based on a specific dataset for prediction work (Zhang et al. 2022a). In recent years, datasets have been created that include a wide range of diseases. Datasets based on gene expression such as FANTOM5 include 1836 different samples from 201,803 regions of different genes that simultaneously cover several diseases (Rezaeipanah and Ahmadi 2022). Each sample contains the information of one patient from one cell or tissue. Here, sampling has been done in the form of gene expression, which shows how many times a gene has produced itself (de Souza et al. 2016).
In general, damaged cells from the body due to a disease can also be observed for other diseases (Li et al. 2023). If the promoters of a cell from two or more diseases are high enough, then it can be said that these diseases have a similar effect on this cell. Since there is information related to cells/tissues for each person, this dataset can be used to detect the communications between cells and tissues in the expression of different genes (Forouzandeh et al. 2023). In general, the analysis of gene expression information in order to identify intercellular communications requires mapping the problem to a clustering problem. Clustering algorithms can find relationships between different diseases by finding the most similar damaged cells.
Clustering algorithms are one of the most important techniques in data mining, machine learning and pattern recognition and are known as an effective method in data visualization and analysis (Rezaeipanah et al. 2021). These algorithms have wide applications in image processing, image segmentation, document analysis, market research, etc. Data clustering is data analysis without any prior information to assign each sample of the dataset to a group as a cluster (Zhang et al. 2022b; Zhao et al. 2023a). Each clustering algorithm seeks to create groups of data with maximum similarity between samples in the same clusters and minimum similarity between samples in different clusters. These algorithms are known as unsupervised learning methods, because the class labels are not available in the data analysis process (Cao et al. 2022; Tang et al. 2023).
In general, the types of clustering algorithms include hierarchical and partitional (Zhang et al. 2018). Hierarchical algorithms use a similarity metric for the clustering task. In each step of these algorithms, the data are divided into two categories to finally create a tree structure as a dendrogram (Wang et al. 2022). Dendrogram is a tree-structured graph that visualizes the result of a clustering algorithm at different levels of partitions (Forouzandeh et al. 2023). Meanwhile, partitional algorithms directly put data into multiple clusters based on distance or similarity. Hard and soft are common types of partitional clustering algorithms (Cheng et al. 2023). In hard clustering, a sample belongs to only one cluster; while in soft clustering, the degree of belonging of a sample to each cluster is determined by a number between 0 and 1.
In many real-world applications, the number of features in a dataset is too large for clustering. In most cases, there are a large number of unrelated features for clustering (Hou et al. 2020). Also, some features may be less important than others. Therefore, applying clustering with a subset of features can lead to an increase in the quality of the final partition. Meanwhile, not all clustering algorithms perform best for all data (Mojarad et al. 2021). Ensemble clustering is very popular to improve the performance of individual clustering algorithms. In an ensemble clustering algorithm, several individual clustering algorithms are combined to cover each other's weaknesses (Zhang et al. 2018). According to this, it is expected that the use of ensemble clustering algorithms will perform better than individual clustering algorithms.
Combining individual clustering algorithms with fixed weight is a common approach in ensemble technique. However, using fixed weights in the whole clustering process leads to a decrease in efficiency. In recent years, approaches based on adaptive weights during the clustering process have been developed to solve this shortcoming (Hou et al. 2020). In general, the use of traditional clustering algorithms does not perform well in dealing with high-dimensional data due to the correlation of features, noise, and dispersion.
On the other hand, applying the information of paired constraints can increase the effectiveness of individual clustering algorithms (Wang et al. 2020; Zhang et al. 2022c). This information includes must-link and cannot-link constraints. The must-link constraint indicates that a pair of samples belong to the same cluster, and the cannot-link constraint indicates that a pair of samples belongs to two different clusters. Since effective clustering is challenging due to the lack of prior knowledge, using the constraints information as limited prior knowledge can improve the clustering performance. The use of constraint information in the clustering process has led to the emergence of clustering with semi-supervised learning (Mojarad et al. 2021; Bridges and Miller-Jensen 2022).
This paper proposes a semi-supervised ensemble clustering framework to discover relationships between diseases based on the extraction of cell-to-cell biological communications. The proposed semi-supervised framework uses prior knowledge in both parts of the ensemble, including the creation of primary partitions and the consensus function. Also, we present a stratified feature sampling mechanism to deal with high-dimensional data, which can reduce the risk of not selecting features to create primary partitions. In addition, the proposed clustering framework uses a new similarity metric developed based on the information of all primary partitions. Our method has medical applications for the treatment and prevention of cancer. In fact, we are looking to identify cells that may be destroyed in the same way in two different cancers.
The main contribution of this study is as follows:
-
A clustering framework is proposed by joining “semi-supervised learning” and “ensemble technique”, which is configured based on stratified feature sampling mechanism and a novel similarity metric
-
Identification of cells with the highest promoters in order to discover relationships between different diseases on the FANTOM5 dataset
-
Validation of the effectiveness of the proposed clustering framework on a wide range of UCI datasets
The remainder of this paper is organized as follows: The related work is summarized in “Related works”. The fundamental concepts related to the problem are given in “Background”. “Proposed clustering framework” explains the proposed clustering framework. The effectiveness of the proposed framework is discussed through numerical simulations in “Experiments”. Finally, the paper ends with conclusions in “Conclusions”.
Related works
Identification of intercellular communication from gene expression data with clustering algorithms is very common (Mojarad et al. 2021). Clustering is one of the data analysis techniques and so far, various solutions have been provided for it (Tan et al. 2022; Chang et al. 2022). For example, k-means, density-based spatial clustering of applications with noise (DBSCAN), multi-view spectral clustering, non-negative matrix factorization-based clustering, unsupervised deep embedding clustering, mean shift clustering, hierarchical clustering, etc. (Zhang et al. 2020; Lei et al. 2022). Compared to partitional clustering algorithms, many efforts have been reported for the improvements of hierarchical clustering algorithms in the last few decades.
Compared to classification, prior knowledge such as class labels is not available for clustering. Some studies use limited prior knowledge as constraint information in clustering (Hou et al. 2020). Zhang et al. (2018) used the pairwise constraints information to improve clustering performance and obtained some successes. Other semi-supervised clustering algorithms include constraints k-means, Constraint-based DBSCAN (C-DBSCAN), Pairwise Constrained k-means (PCKmeans), semi-supervised deep fuzzy c-mean clustering, semi-supervised denpeak clustering with pairwise constraints, semi-supervised deep embedded clustering, exhaustive and efficient constraint propagation, and semi-supervised maximum margin clustering (Mojarad et al. 2021).
Prades et al. (2020) proposed an agglomerative clustering approach to detect the number of endmembers in hyperspectral images. The authors follow this hypothesis in clustering that there is a cluster for each material different from image. Here, an approach based on principal component analysis applied to the centered image is used to reduce the dimensions. With reducing the dimensions of the image, the authors use a k-means algorithm to create primary clusters. Here, symmetric Kullback–Leibler (SKL) divergence is used as the distance calculation metric. SKL, also known as relative entropy, is a statistical metric from information theory to quantify the difference. This study uses principal component analysis to calculate the density of clusters. After that, a model-based agglomerative clustering approach is applied to provide a hierarchy of partitions. Eventually, the final partition of the hierarchy is determined by a validation algorithm. The number of clusters in the results of this model is considered as the number of materials.
Rezaeipanah and Ahmadi (2022) introduced multi-stage weights adjustment in the multi-layer perceptron (MWAMLP) for breast cancer detection. MWAMLP is an ensemble approach that uses three homogeneous multi-layer perceptron (MLP) neural networks for the classification task. The consensus function used in MWAMLP is developed based on the meta-classifier technique. The accuracy of this method on the WBCD dataset is 98.76% on average.
Mojarad et al. (2021) used an ensemble clustering algorithm to model inherited disease behavior (ECIDB). Here, cell-to-cell and tissue-to-tissue communications are extracted from the FANTOM5 dataset to identify cells with the highest disruption in each disease pair. The proposed algorithm uses the graph topological structure to represent the FANTOM5 dataset and uses an innovative similarity metric to calculate the cell-to-cell similarity matrix. An ensemble clustering is then applied to identify primary intercellular or intertissue communications. Finally, a friend recommender-based system considering clustering information and topological similarities is used to identify related cells.
Sangeetha and Prakash (2021) proposed using deep learning to improve breast cancer disease prediction. A stacked sparse auto encoder network (SSAE) is constructed to learn features effectively. The network consists of a softmax classifier and several sparse autoencoders. In addition to adjusting the parameters of the algorithm, deep learning models are required. The parameters of the stacked sparse autoencoder can, therefore, be adjusted using particle swarm optimization (PSO). Regarding feature learning and classification, the PSO improves the performance of the SSAE.
Kayal et al. (2019) conducted a study to provide a new advanced classification method using a deep neural network (DNN) to predict the survival of patients with hepatic cancer. In the proposed method, the authors selected 15 risk factors out of 49 risk factors which are significantly responsible for hepatocellular carcinoma and then applied their method. According to the results, the proposed method is more accurate than other methods.
Sivadas et al. (2022) attempted to investigate the impact of racial information and natural factors on the incidence and progression of cancer by employing a multi-omics data fusion breast cancer survival cycle marker detection prediction model. The primary objective of this research is to enhance the prediction of breast cancer survival cycles through the development of a multi-omics fusion prediction model based on ensemble learning. This model incorporates clinical data, transcriptomics data, and methylomics data derived from The Cancer Genome Atlas (TCGA) datasets. The experimental results demonstrate that the three-omics fusion approach (with an accuracy rate of 97.43%) outperforms single-omics experiments and other race-based multi-omics and single-omics experiments in the context of the three-omics experiments, considering racial disparities. This research offers technical support for the classification of breast cancer patient survival cycle predictions and introduces novel concepts for the study of breast cancer survival prognostics.
Talatian Azad et al. (2021) proposed an intelligent ensemble classification method based on multi-layer perceptron (IEC-MLP) for breast cancer detection. IEC-MLP uses genetic algorithm for feature selection and parameter settings of MLP neural network. Here, MLP is developed based on an ensemble classification approach with three classifiers. This method detects breast cancer with high accuracy on the WBCD dataset, where the average accuracy is reported to be 98.74%.
Background
In this section, some basic concepts about the research method are explained. These concepts include system model, hierarchical clustering, semi-supervised clustering, ensemble clustering, and feature sampling.
System model
An individual clustering algorithm is denoted by \(\pi\). Ensemble clustering consists of several individual clustering algorithms. We assume that \(\Pi =\left\{{\pi }_{1},{\pi }_{2},\dots ,{\pi }_{k},\dots ,{\pi }_{Z}\right\}\) is the set of \(Z\) individual clustering algorithms, where \({\pi }_{k}\) represents the \(k\)-th clustering algorithm. Each \({\pi }_{k}\in \Pi\) can be applied to a dataset. We assume that \(X=\left\{{x}_{1},{x}_{2},\dots ,{x}_{i},\dots ,{x}_{N}\right\}\) is a dataset with \(N\) samples, where \({x}_{i}=\langle {x}_{i,1},{x}_{i,2},\dots ,{x}_{i,j},\dots ,{x}_{i,M}\rangle\) represents the \(i\)-th sample with \(M\) features.
Applying each \({\pi }_{k}\) to \(X\) results in a partition with multiple clusters. We assume that \({p}_{k}=\left[{c}_{k,1},{c}_{k,2},\dots ,{c}_{k,l},\dots ,{c}_{k,\left|{p}_{k}\right|}\right]\) is the partition obtained by applying \({\pi }_{k}\) on \(X\) with \(\left|{p}_{k}\right|\) clusters. Here, \({c}_{k,l}\) represents the \(l\)-th cluster of the \(k\)-th partition. Considering ensemble clustering, applying set \(\Pi\) on \(X\) results in \(P=\left\{{p}_{1},{p}_{2},\dots ,{p}_{Z}\right\}\). We assume that \({p}_{*}=\Gamma \langle {p}_{1},{p}_{2},\dots ,{p}_{Z}\rangle\) is the final partition obtained by consensus of set \(P\). Here, \(\Gamma\) represents a consensus function such as majority vote. Let \({p}_{*}=\left[{c}_{1},{c}_{2},\dots ,{c}_{K}\right]\) be the details of the final partition, where \(K\) represents the total number of clusters.
Hierarchical clustering
Clustering is an unsupervised learning mechanism for grouping data, where samples belonging to each group have the highest similarity to each other and samples from different groups have the lowest similarity to each other. Partitional clustering and hierarchical clustering are two common types of clustering (Rostami et al. 2023). Partitional clustering clusters samples based on an objective function, where each sample belongs to only one cluster and the total number of clusters is known in advance. The k-means is one of the most common partitional clustering algorithms that performs clustering with the objective of minimizing the average distance to the center of each cluster (Torabi et al. 2022; Cao et al. 2023a). Meanwhile, hierarchical clustering can show a hierarchy of samples by dendrogram.
There are two general types of hierarchical clustering: (1) Divisive hierarchical clustering (DHC) or top-down approach where all samples belong to the same cluster at first. After that, each cluster is divided into smaller clusters so that finally each sample has its own cluster. (2) Agglomerative hierarchical clustering (AHC) or bottom-up approach where each sample represents a cluster at first. After that, each pair of clusters with the highest similarity are merged until finally all samples belong to the same cluster (Farahbakhsh et al. 2021). As shown in Fig. 1, the final result for both DHC and AHC is in the form of a dendrogram. Each level in the dendrogram represents a partition as the result of clustering.

An example of hierarchical clustering
Linkage-based metrics are one of the most common AHC methods, which are defined by inter-cluster distance metrics (Rostami et al. 2023). Single linkage, average linkage, centroid linkage, and complete linkage are examples of linkage-based AHC clustering. A summary of these methods is presented in Table 1. In this table, \(x\in {c}_{i}\) represents sample \(x\) from cluster \({c}_{i}\), \(\left|{c}_{i}\right|\) indicates the number of cluster members \({c}_{i}\) and \({d}_{x,y}\) indicates the distance between \(x\) and \(y\) based on a distance measure such as Euclidean (Sivadas et al. 2022). Basically, the difference between these methods is in the distance calculation metric.
Semi-supervised clustering
In unsupervised clustering, the learning algorithm has no knowledge about the labels of the samples. However, semi-supervised clustering can use prior knowledge such as labels of samples for clustering (Wang et al. 2023; Yue et al. 2023). Usually, the prior knowledge used by semi-supervised learning is known as constraint information (Sangeetha and Prakash 2021). Constraint information can include dependencies between samples or an additional set of labeled samples. Pairwise constraints information is the most common prior knowledge used for semi-supervised learning. Pairwise constraints include pairs of samples that are labeled as belonging to the same or different clusters. Therefore, the quality of the partition created by semi-supervised clustering should be improved compared to unsupervised clustering, because semi-supervised clustering uses prior knowledge.
Basically, the constraint information can be based on metrics, clusters, and samples (Rostami et al. 2023). Metric-based constraint information allows the use of different distance/similarity measures in the learning process. Cluster-based constraint information provides the possibility of using cluster characteristics such as shape, size, and diameter. Also, sample-based constraint information includes must-link and cannot-link parameters (Jannesari et al. 2023). Here, must-link indicates the possibility of assigning two samples to one cluster, while cannot-link indicates the impossibility of assigning two samples to one cluster. Selecting the most potential sample for semi-supervised clustering is an important challenge for using information constraints (Shahidinejad et al. 2021). Since the labels of samples are not available in clustering, dense groups should be identified in order to find samples that definitely belong to the same cluster.
According to the above, semi-supervised clustering simultaneously uses both labeled and unlabeled samples, as shown in Fig. 2. Typically, semi-supervised clustering is configured based on a small number of labeled samples compared to a large number of unlabeled samples. Constraint-based semi-supervised clustering and distance-based semi-supervised clustering are two common categories of semi-supervised clustering (Hayashi et al. 2018). The former uses constraint information to support the algorithm and improve clustering, while the latter includes adaptive distance metrics to extract constraints in supervised learning.

Example of clustering with unsupervised and semi-supervised learning
Ensemble clustering
It has been proven that no individual clustering method can provide the best performance for all contexts (Sivadas et al. 2022). Since each individual clustering method has its own advantages and disadvantages, combining several methods can provide more stable, scalable and accurate results compared to each of the individual methods. Ensemble clustering-based methods combine the results of several clustering methods to avoid the disadvantages of each of them and enable effective clustering for more datasets. As shown in Fig. 3, ensemble clustering consists of a number of individual homogeneous or heterogeneous clustering algorithms. These algorithms are considered as members of ensemble clustering. Selecting suitable members that can achieve quality and diversity in the final consensus is an important challenge in ensemble clustering.

Ensemble clustering architecture
Each individual clustering algorithm \({\pi }_{k}\) is applied as a weak method on the dataset and outputs a partition \({p}_{k}\). The partitions created in this step are merged by a consensus function \(\Gamma\) to create the final partition \({p}_{*}\). Although all partitions can participate in the consensus process, a subset of primary partitions or part of their associated clusters can be candidates for the consensus function. This is a major challenge to address in ensemble clustering. Therefore, ensemble clustering has two main phases: creating primary partitions and merging them by a consensus function (Forouzandeh et al. 2023). The consensus function is an important issue in ensemble clustering, for which various methods have been introduced so far. The most common consensus functions include simple voting, iterative voting, weighted similarity, mixture model, correlation matrix, meta-clustering, etc.
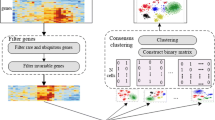
In various studies, the superiority of semi-supervised clustering algorithms against unsupervised clustering has been proven (Sangeetha and Prakash 2021). Meanwhile, ensemble clustering provides better performance than individual clustering. With this motivation, we focus on SSEC-based approaches. The use of constraint information in SSEC is a hot research topic in machine learning. Here, prior knowledge such as pairwise constraints and labels of samples are incorporated into ensemble clustering in order to improve efficiency. Most of the existing SSEC approaches use constraint information only to create primary partitions, while the use of this information is ignored in the consensus function (Rezaeipanah and Ahmadi 2022). Figure 4 shows a schematic framework of SSEC-based approaches considering prior knowledge.

SSEC framework considering prior knowledge
Feature sampling
Today, the number of large-scale datasets has increased significantly due to the growth of data collection devices (Zhao et al. 2023b; Cao et al. 2023b). Machine learning algorithms for effective analysis of these datasets face serious challenges. Meanwhile, clustering algorithms face issues such as feature correlation, noise, sparseness, and computational complexity when processing big data, which may lead to their failure. Reducing the dimensions of the data by selecting a subset of the original features is one of the most common solutions to address this problem (Rezaeipanah and Ahmadi 2022).
Techniques based on randomization such as random projection (Rostami et al. 2023) and random feature sampling (Sangeetha and Prakash 2021) are among the most common methods for selecting the subset of suitable features. However, randomization-based techniques do not consider correlations between features and cannot select effective features for clustering. Stratified feature sampling mechanism was introduced by Jing et al. (2015) to address this issue. This mechanism uses the k-means algorithm to cluster features into a specified number of groups. After that, a number of features are randomly selected from each cluster with the same proportion to obtain several subsets of features. The ensemble clustering architecture considering feature sampling is shown in Fig. 5.

Ensemble clustering architecture considering feature sampling
Proposed clustering framework
The proposed clustering framework has four general phases. In the first phase, stratified feature sampling mechanism is applied. This mechanism clusters the features of the dataset using the K-means algorithm to create an independent subset of features for each individual clustering algorithm. Here, feature selection probabilities are adjusted with the aim of reducing the risk of not selecting some features for the clustering task. The second phase is related to the generation of primary partitions by \(Z\) individual clustering algorithms. We use AHC-based algorithms for the clustering task, where each algorithm creates its own partition based on a subset of specified features. The output partition in each AHC-based algorithm is determined from the dendrogram by Bayesian PAC theory (Abddallah and Yousef 2018).
The third phase consists of presenting a new similarity metric that uses a wide range of information to calculate the similarity between each sample pair, cluster pair and meta-cluster pair. The consensus function is configured in the fourth phase. Since not all primary clusters and not all primary partitions have the same strength, we develop a weighting policy in which the merits of clusters and the strength of partitions are considered to contribute to the final consensus. Finally, the meta-clustering technique is applied as a consensus function to create the final partition. We configure each AHC-based clustering algorithm with semi-supervised learning and use the information of pairwise constraints to improve the clustering performance in both parts of creating primary partitions and the consensus function. An overview of the proposed clustering framework is shown in Fig. 6.

An overview of the proposed clustering framework
The proposed algorithm for large-scale data clustering uses the stratified feature sampling mechanism. In this mechanism, each \({\pi }_{k}\in \Pi\) performs clustering based on a subset of the main features. Let \({\pi }_{k}\) form an primary partition based on \({\mathcalligra{s}}_{k}\), where \({\mathcalligra{s}}_{k}\in \mathcal{S}\) represents the subset of the \(k\)-th selected feature. The mechanism of stratified feature sampling can provide the most suitable set \(\mathcal{S}\) for ensemble clustering. Here, the features of the dataset \(X\) are clustered by K-means, and then a number of features are sampled from each cluster to form \({\mathcalligra{s}}_{k}\). This process is applied to all \({\mathcalligra{s}}_{k}\in \mathcal{S}, \forall k=1, 2,\dots ,Z\).
To reduce the risk of not selecting some features, we calculate the probability of selecting the features by considering the sampling history. Let \({\mathcalligra{r}}_{j}\) refer to the sampling rate of the \(j\)th feature from the dataset \(X\). Here, the sampling rate for selecting the first subset is the same for all features, for example, \({\mathcalligra{r}}_{j}=1/M\). The sampling rate is updated to select the second subset, where the sampling rate of unselected features is halved. This process is repeated for other subsets to reduce the risk of not selecting features.
Let \({s}_{i,j}\in S\) be the similarity between samples \({x}_{i}\) and \({x}_{j}\). We use a new similarity metric considering a wide range of information to calculate the similarity matrix \(S\). The Eq. (1) defines the similarity for \({s}_{i,j}\).
where \(Z\) is the total number of partitions, \({p}_{k}\) is the detail of the kth partition, \(P\) is the set of all partitions, \(\left|{p}_{k}\right|\) is the number of clusters in \({p}_{k}\), \({c}_{k,l}\) is the detail of the \(l\)th cluster in \({p}_{k}\), \(\left|{c}_{k,l}\right|\) is the number of samples of \({c}_{k,l}\), \({d}_{i,j}\) is the Euclidean distance between \({x}_{i}\) and \({x}_{j}\), \({M}_{{c}_{k,l}}\) is the merit associated with \({c}_{k,l}\), \({W}_{{p}_{k}}\) is the strength/weight associated with \({p}_{k}\), and \(\beta\) is a damping factor to reduce the effect of large cluster sizes.
In addition to the similarity between each pair of samples, we calculate the similarity between each pair of clusters and each pair of meta-clusters. Let each meta-cluster be a set of several clusters. Equation (2) formulates the similarity between two clusters \({c}_{k,1}\) and \({c}_{k,2}\) as \({{\text{CS}}}_{{c}_{k,1},{c}_{k,2}}\). Also, Eq. (3) formulates the similarity between two meta-clusters \({\psi }_{1}=\left\{{c}_{\mathrm{1,1}},{c}_{\mathrm{1,2}},\dots ,{c}_{1,u},\dots ,{c}_{1,\left|{\psi }_{1}\right|}\right\}\) and \({\psi }_{2}=\left\{{c}_{\mathrm{2,1}},{c}_{\mathrm{2,2}},\dots ,{c}_{2,v},\dots ,{c}_{2,\left|{\psi }_{2}\right|}\right\}\) as \({{\text{MS}}}_{{\psi }_{1},{\psi }_{2}}\).
Finally, we use a consensus function based on the meta-clustering technique to create the final partition. According to this technique, candidate clusters are considered from all partitions in a set and then re-clustered by average linkage to create meta-clusters. Here, the number of meta-clusters represents the number of final clusters. Eventually, the final partition is created by assigning each sample of the dataset \(X\) to a meta-cluster with the highest similarity. In this paper, candidate clusters are selected to participate in the final consensus based on the merit of the primary clusters and the strength of the primary partitions. In extensive studies, normalized mutual information (NMI) is used to evaluate the partition generated from a clustering algorithm (Rezaeipanah and Ahmadi 2022). NMI can calculate the similarity between two partitions such as \({p}_{u}\) and \({p}_{v}\) by Eq. (4).
where \({N}_{ij}\) is the number of identical samples in \({c}_{u,i}\in {p}_{u}\) and \({c}_{v,j}\in {p}_{v}\) and \({N}_{iu}\) is the number of samples in \({c}_{u,i}\).
If \({p}_{v}\) is assumed as the reference partition, then \({\text{NMI}}\left({p}_{u},{p}_{v}\right)\) represents the strength of the partition \({p}_{u}\). Let the strength of partition \({p}_{u}\) be formulated as the weight of partition \({p}_{u}\) by \({W}_{{p}_{u}}\). In addition to robustness, we use the merit of the clusters to determine the candidate clusters in the final consensus. Law et al. (2004) developed the NMI criterion and used it to calculate the merit of clusters. The authors converted a cluster into a partition in order to use NMI for evaluation work. Let \({\overline{c} }_{k}\) be a cluster with all samples not in \({c}_{k}\). \({c}_{k}\) is considered a positive cluster if at least half of its samples are found in another cluster. According to these definitions, the cluster \({c}_{k}\) is considered as a partition \({\widehat{p}}_{k}=\left\{{c}_{k},{\overline{c} }_{k}\right\}\) with the union of all positive clusters. With converting \({c}_{k}\) to \({\widehat{p}}_{k}\), cluster merit of \({c}_{k}\) is formulated by Eq. (5). According to the aforementioned concepts, each \({c}_{k,l}\in {p}_{k}\) with a predefined threshold can participate in the final consensus. The Eq. (6) defines the condition for \({c}_{k,l}\) to be a candidate for participating in the final consensus.
where \({p}_{0}\) is defined as the reference partition. Also, \(\xi\) is the influence coefficient of the cluster level with respect to the partition level and \(\theta\) is a threshold for determining the consensus candidates.
Each \({\pi }_{k}\in \Pi\) is an individual clustering algorithm based on AHC such as average linkage. Here, all \({\pi }_{k}\in \Pi\) are configured using average linkage and based on semi-supervised learning. Also, the algorithm used in the consensus function is applied using average linkage and based on semi-supervised learning. Let \({d}_{i,j}\) be the distance between samples \({x}_{i}\) and \({x}_{j}\). We use the information of pairwise constraints such as must-link and cannot-link to define \({d}_{i,j}\) in semi-supervised learning. If the sample pair \(({x}_{i},{x}_{j})\) is covered by the must-link, then it belongs to the set \({\Delta }_{M}\). Meanwhile, if the sample pair \(({x}_{i},{x}_{j})\) is covered by cannot-link, then it belongs to the set \({\Delta }_{C}\). Let all members of sets \({\Delta }_{M}\) and \({\Delta }_{C}\) have symmetry and transitivity properties. The symmetry property is formulated by Eq. (7) and the transitivity property is formulated by Eq. (8). Considering semi-supervised learning in the average linkage algorithm, \({d}_{i,j}\) is formulated by pairwise constraints information with Eq. (9).
Experiments
We validate the performance of the proposed framework with several numerical experiments considering the UCI dataset and then use it to extract intercellular communication on the FANTOM5 dataset. The proposed clustering algorithm has been implemented using the MATLAB 2021a simulator on a personal computer with Intel® Core™ i7 Processor up to 3.4.00 GHz and 16 GB DDR3 Memory.
Datasets
The evaluations are based on 10 datasets from the UCI machine learning repository, as shown in Table 2. We use a mean replacement policy when dealing with missing values. All datasets used have class labels, which are used as reference partitions in clustering. Since the proposed clustering framework is based on semi-supervised learning, we consider 5% of the supervised samples as the constraint information.
In addition, we use the FANTOM5 dataset to analyze gene expression data and extract intercellular communication. FANTOM5 was compiled in collaboration with the University of Sydney, Australia. In addition to cell information, this dataset also contains tissue information, which is not considered in the current study. Details of this dataset are available at http://fantom.gsc.riken.jp/5. The full version of the FANTOM5 dataset contains 1836 samples per column, where each sample contains information related to a cell or tissue from a single patient. For each sample, 201,802 promoters from different regions of a gene from a specific cell are available. With filtering data related to tissues, we found 108 unique cells. Here, there are 702 examples related to cells.
Meanwhile, the rows in this dataset represent the numbers of promoters, which are identified using “entrezgene_id”. Some promoter values are not specified and specifically have the value “NA”. Unavailable promoter information is removed. After that, 86,428 promoters are available for each sample. The columns related to cells are taken from different samples of the human body and there may be several samples of the same cell. In general, the first 7 columns related to the promoter information have been sampled and the 8th columns are samples. In addition, the ID of each sample includes details such as disease type, time point, cell name and patient ID. For example, the ID of a sample from the FANTOM5 dataset is: “239SLAM rinderpest infection, 00hr, biol_rep1.CNhs14406.13541-145H4”. Here, “SLAM” represents a family of cell surface receptors and other coding are related to the patient. An overview of the FANTOM5 dataset for cells is shown in Table 3.
Evaluation metrics
A partition generated by a clustering algorithm is ideal if it has a maximum inter-cluster distance and a minimum intra-cluster distance. We use criteria such as NMI, Adjusted Rand Index (ARI) and silhouette coefficient to evaluate the clustering results (Talatian Azad et al. 2021). NMI is a common criterion for evaluating the performance of clustering algorithms that can measure the similarity between two independent partitions. NMI is defined according to Eq. (4). ARI is another criterion for evaluating the performance of clustering algorithms. ARI uses the Rand Index (RI) to calculate the similarity between two independent partitions. ARI can calculate the similarity between two partitions such as \({p}_{u}\) and \({p}_{v}\) by Eq. (10).
The silhouette coefficient is an internal index to calculate the performance of clustering algorithms, which performs the evaluation process based on density and separation characteristics. In silhouette, the validity of a partition is calculated based on the combination of intra-cluster and inter-cluster similarity for each pair of independent clusters. The obtained value of the silhouette coefficient is between − 1 and + 1, and a silhouette with a value of + 1 represents an ideal clustering. The silhouette coefficient for \({x}_{i}\in {c}_{l}\) from the \({p}_{k}\) partition is calculated by Eq. (11).
where \({a}_{i}\) and \({b}_{i}\) are calculated by Eqs. (12) and (13), respectively.
Analysis of results
The proposed clustering algorithm is compared with several equivalent algorithms such as MWAMLP (Rezaeipanah and Ahmadi 2022), ECIDB (Mojarad et al. 2021), SSAE (Sangeetha and Prakash 2021), and TCGA (Sivadas et al. 2022). Before the comparisons, we prove that the proposed clustering algorithm using the average linkage algorithm provides the best performance in both the creation of primary partitions and the consensus function. We compare the average linkage algorithm with other AHC-based algorithms such as single linkage, centroid linkage and complete linkage. Table 4 shows the results of this comparison. The results of this comparison are presented based on accuracy and the best results are bolded. Also, each row presents the results associated with a dataset, while the last row is the average of the results. The results clearly prove the superiority of the average linkage algorithm and its use in the proposed clustering framework.
The comparison of the proposed algorithm based on NMI and ARI criteria compared to MWAMLP, ECIDB, SSAE and TCGA is presented in Tables 5 and 6, respectively. The best results of these tables are highlighted in bold. The proposed algorithm performs better than all existing algorithms in many datasets. However, the simulation results show that ECIDB produces quite competitive results with the proposed algorithm. Among the 10 existing ECIDB datasets, the proposed algorithm outperforms the proposed algorithm considering the NMI criterion in the Iris and Colon datasets. Also, ECIDB performs best considering the ARI criterion on the Titanic, Banana and Splice datasets. On average, in the NMI criterion, the proposed algorithm is 8.8%, 1.7%, 12.9%, and 16.5% superior compared to MWAMLP, ECIDB, SSAE, and TCGA, respectively. This superiority for the ARI criterion is 4.6%, 1.8%, 11.5%, and 8.1%, respectively.
Although the proposed clustering algorithm performs better in terms of accuracy, NMI and ARI compared to equivalent algorithms, runtime analysis is also important. High-complexity clustering algorithms are not capable of processing large-scale datasets. The proposed clustering algorithm is equipped with a stratified feature sampling mechanism to deal with big data. This mechanism leads to the reduction of computational complexity and it is expected that the runtime in the proposed algorithm is lower than other algorithms. Figure 7 shows the runtime results of different clustering algorithms. The results clearly show that our algorithm has lower runtime in all datasets. On average, the proposed clustering algorithm provides 6.1%, 34.6%, 43.5%, and 30.8% less runtime compared to MWAMLP, ECIDB, SSAE, and TCGA algorithms, respectively.

Comparison of different algorithms in terms of running time
We proved that the proposed clustering framework has ideal performance for clustering real-world datasets. Hence, we apply it to clustering the FANTOM5 dataset and extracting cell-to-cell biological communications. The FANTOM5 dataset is multifaceted, where multiple samples from the same cell with multiple patients are available. Also, there are different samples of the same cell in different diseases. Therefore, each cell may be related to other cells through various diseases. The concept of communication in FANTOM5 is expressed with promoters. A high value of a promoter indicates the reproduction or disruption of a part of gene expression related to a cell. The activation threshold of promoters has a significant effect on the discovery of intercellular communication. Here, we cluster with different thresholds from 500 to 4000 samples of the FANTOM5 dataset and report the results in terms of the silhouette coefficient. We compare the presented results with ECIDB (Mojarad et al. 2021), as this algorithm was also applied to the FANTOM5 dataset. The results of this comparison are presented in Table 7. The results show the superiority of the proposed algorithm in most thresholds. Meanwhile, the best results are obtained for the silhouette factor with a threshold of 1000. Here, the proposed algorithm with a silhouette coefficient of 0.952 and 19 clusters of samples related to cells have been clustered. These results were obtained for ECIDB with silhouette coefficient equal to 0.809 and 20 clusters.
We analyzed the clustering of the FANTOM5 dataset with different thresholds. A suitable threshold is equal to 1000, considering it leads to the identification of strong communications between cells. In each cluster, the pair of cells with the strongest correlation may indicate a relationship between different diseases. We extracted pairs of cells from different clusters with the highest correlation, whose samples belong to different diseases. Table 8 shows some of the strongest cell-to-cell communications, along with disease names and genes sampled. It shows the hereditary behavior between which diseases, based on which genes and in which cells.
Conclusions
Gene expression data contain important information of various diseases. The gene expression data of some diseases may be similar. Indeed, some cells in different diseases may contain similar gene expression data. Therefore, discovering the relationships between diseases through the extraction of cell-to-cell biological communications is challenging and can change our understanding of how diseases such as cancer develop. The communication between two cells occurs when the number of promoters is significantly expressed in a number of cells. It is obvious that designing a method to discover cell-to-cell biological communications and identify the real communication between diseases is important for the medical society. A clustering algorithm based on semi-supervised learning and ensemble technique was proposed in the paper to identify intercellular communication. This framework is equipped with a stratified feature sampling mechanism to deal with high-dimensional data. Also, in this framework, a new similarity metric is developed that uses a wide range of primary partition information to estimate similarity. Our proposed framework uses the constraints information in both the phases of creating the primary partitions and the consensus function. The performance of the proposed framework has been validated through clustering of the UCI dataset. Therefore, the proposed framework for extracting intercellular communication was successfully applied to the FANTOM5 dataset. The results of the simulations show that the most promoters between cancer and diseases such as inflammation, monocytosis and aortic aneurysm occur on the “ABLIM1” gene.
Data availability
Data sharing not applicable to this manuscript as no datasets were generated or analyzed during the current study.
Code availability
There is no free code for this study.
References
Abddallah L, Yousef M (2018) Ensemble clustering based dimensional reduction. In: Database and expert systems applications: DEXA 2018 international workshops, BDMICS, BIOKDD, and TIR, Regensburg, Germany, September 3–6, 2018, Proceedings 29. Springer International Publishing, pp 115–125
Bridges K, Miller-Jensen K (2022) Mapping and validation of scRNA-Seq-derived cell-cell communication networks in the tumor microenvironment. Front Immunol 13:885267
Cao C, Wang J, Kwok D, Cui F, Zhang Z, Zhao D et al (2022) webTWAS: a resource for disease candidate susceptibility genes identified by transcriptome-wide association study. Nucleic Acids Res 50(D1):D1123–D1130
Cao Z, Niu B, Zong G, Xu N (2023a) Small-gain technique-based adaptive output constrained control design of switched networked nonlinear systems via event-triggered communications. Nonlinear Anal Hybrid Syst 47:101299
Cao Y, Xu N, Wang H, Zhao X, Ahmad AM (2023b) Neural networks-based adaptive tracking control for full-state constrained switched nonlinear systems with periodic disturbances and actuator saturation. Int J Syst Sci 54(14):2689–2704
Chang Y, Niu B, Wang H, Zhang L, Ahmad AM, Alassafi MO (2022) Adaptive tracking control for nonlinear system in pure-feedback form with prescribed performance and unknown hysteresis. IMA J Math Control Inf 39(3):892–911
Cheng F, Liang H, Niu B, Zhao N, Zhao X (2023) Adaptive neural self-triggered bipartite secure control for nonlinear MASs subject to DoS attacks. Inf Sci 631:256–270
de Souza PS, Faccion RS, Bernardo PS, Maia RC (2016) Membrane microparticles: shedding new light into cancer cell communication. J Cancer Res Clin Oncol 142:1395–1406
Farahbakhsh F, Shahidinejad A, Ghobaei-Arani M (2021) Multiuser context-aware computation offloading in mobile edge computing based on Bayesian learning automata. Trans Emerg Telecommun Technol 32(1):e4127
Forouzandeh S, Berahmand K, Sheikhpour R, Li Y (2023) A new method for recommendation based on embedding spectral clustering in heterogeneous networks (RESCHet). Expert Syst Appl 231:120699
Hayashi T, Ozaki H, Sasagawa Y, Umeda M, Danno H, Nikaido I (2018) Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat Commun 9(1):619
Hou R, Denisenko E, Ong HT, Ramilowski JA, Forrest AR (2020) Predicting cell-to-cell communication networks using NATMI. Nat Commun 11(1):5011
Jannesari V, Keshvari M, Berahmand K (2023) A novel nonnegative matrix factorization-based model for attributed graph clustering by incorporating complementary information. Expert Syst Appl 242:122799
Jing L, Tian K, Huang JZ (2015) Stratified feature sampling method for ensemble clustering of high dimensional data. Pattern Recogn 48(11):3688–3702
Kayal CK, Bagchi S, Dhar D, Maitra T, Chatterjee S (2019) Hepatocellular carcinoma survival prediction using deep neural network. In: Proceedings of international ethical hacking conference 2018: eHaCON 2018, Kolkata, India. Springer, Singapore, pp 349–358
Law MH, Topchy AP, Jain AK (2004) Multiobjective data clustering. In: Proceedings of the 2004 IEEE computer society conference on computer vision and pattern recognition, 2004. CVPR 2004, vol 2. IEEE, pp II-II
Lei X, Li Z, Zhong Y, Li S, Chen J, Ke Y et al (2022) Gli1 promotes epithelial–mesenchymal transition and metastasis of non-small cell lung carcinoma by regulating snail transcriptional activity and stability. Acta Pharm Sin B 12(10):3877–3890
Li X, Chen X, Rezaeipanah A (2023) Automatic breast cancer diagnosis based on hybrid dimensionality reduction technique and ensemble classification. J Cancer Res Clin Oncol 149:7609–7627
Mojarad M, Sarhangnia F, Rezaeipanah A, Parvin H, Nejatian S (2021) Modeling hereditary disease behavior using an innovative similarity criterion and ensemble clustering. Curr Bioinform 16(5):749–764
Peng L, Wang F, Wang Z, Tan J, Huang L, Tian X et al (2022) Cell–cell communication inference and analysis in the tumour microenvironments from single-cell transcriptomics: data resources and computational strategies. Brief Bioinform 23(4):bbac234
Prades J, Safont G, Salazar A, Vergara L (2020) Estimation of the number of endmembers in hyperspectral images using agglomerative clustering. Remote Sens 12(21):3585
Rezaeipanah A, Ahmadi G (2022) Breast cancer diagnosis using multi-stage weight adjustment in the MLP neural network. Comput J 65(4):788–804
Rezaeipanah A, Syah R, Wulandari S, Arbansyah A (2021) Design of ensemble classifier model based on MLP neural network for breast cancer diagnosis. Intel Artif 24(67):147–156
Rostami M, Oussalah M, Berahmand K, Farrahi V (2023) Community detection algorithms in healthcare applications: a systematic review. IEEE Access 11:30247–30272
Sangeetha K, Prakash S (2021) An early breast cancer detection system using stacked auto encoder deep neural network with particle swarm optimization based classification method. J Med Imaging Health Inform 11(12):2897–2906
Shahidinejad A, Ghobaei-Arani M, Masdari M (2021) Resource provisioning using workload clustering in cloud computing environment: a hybrid approach. Clust Comput 24(1):319–342
Shahraki K, Boroumand PG, Lotfi H, Radnia F, Shahriari H, Sargazi S et al (2023) An update in the applications of exosomes in cancer theranostics: from research to clinical trials. J Cancer Res Clin Oncol 149:8087–8116
Sivadas A, Kok VC, Ng KL (2022) Multi-omics analyses provide novel biological insights to distinguish lobular ductal types of invasive breast cancers. Breast Cancer Res Treat 193(2):361–379
Talatian Azad S, Ahmadi G, Rezaeipanah A (2021) An intelligent ensemble classification method based on multi-layer perceptron neural network and evolutionary algorithms for breast cancer diagnosis. J Exp Theor Artif Intell 34(6):949–969
Tan J, Liu L, Li F, Chen Z, Chen GY, Fang F et al (2022) Screening of endocrine disrupting potential of surface waters via an affinity-based biosensor in a rural community in the Yellow River Basin, China. Environ Sci Technol 56(20):14350–14360
Tang F, Wang H, Chang XH, Zhang L, Alharbi KH (2023) Dynamic event-triggered control for discrete-time nonlinear Markov jump systems using policy iteration-based adaptive dynamic programming. Nonlinear Anal Hybrid Syst 49:101338
Torabi E, Ghobaei-Arani M, Shahidinejad A (2022) Data replica placement approaches in fog computing: a review. Clust Comput 25(5):3561–3589
Wang J, Jiang X, Zhao L, Zuo S, Chen X, Zhang L et al (2020) Lineage reprogramming of fibroblasts into induced cardiac progenitor cells by CRISPR/Cas9-based transcriptional activators. Acta Pharm Sin B 10(2):313–326
Wang H, Sha Y, Wang D, Nazari H (2022) A gene expression clustering method to extraction of cell-to-cell biological communication. Intel Artif 25(69):1–12
Wang T, Zhang L, Xu N, Alharbi KH (2023) Adaptive critic learning for approximate optimal event-triggered tracking control of nonlinear systems with prescribed performances. Int J Control. https://doi.org/10.1080/00207179.2023.2250880
Yue S, Niu B, Wang H, Zhang L, Ahmad AM (2023) Hierarchical sliding mode-based adaptive fuzzy control for uncertain switched under-actuated nonlinear systems with input saturation and dead-zone. Robot Intell Autom 43(5):523–536
Zhang D, Jiao L, Bai X, Wang S, Hou B (2018) A robust semi-supervised SVM via ensemble learning. Appl Soft Comput 65:632–643
Zhang L, Deng S, Zhang Y, Peng Q, Li H, Wang P et al (2020) Homotypic targeting delivery of siRNA with artificial cancer cells. Adv Healthc Mater 9(9):1900772
Zhang H, Zhao X, Wang H, Zong G, Xu N (2022a) Hierarchical sliding-mode surface-based adaptive actor–critic optimal control for switched nonlinear systems with unknown perturbation. IEEE Trans Neural Netw Learn Syst. https://doi.org/10.1109/TNNLS.2022.3183991
Zhang H, Zhao X, Zhang L, Niu B, Zong G, Xu N (2022b) Observer-based adaptive fuzzy hierarchical sliding mode control of uncertain under-actuated switched nonlinear systems with input quantization. Int J Robust Nonlinear Control 32(14):8163–8185
Zhang H, Zou Q, Ju Y, Song C, Chen D (2022c) Distance-based support vector machine to predict DNA N6-methyladenine modification. Curr Bioinform 17(5):473–482
Zhao Y, Niu B, Zong G, Xu N, Ahmad AM (2023a) Event-triggered optimal decentralized control for stochastic interconnected nonlinear systems via adaptive dynamic programming. Neurocomputing 539:126163
Zhao H, Wang H, Xu N, Zhao X, Sharaf S (2023b) Fuzzy approximation-based optimal consensus control for nonlinear multiagent systems via adaptive dynamic programming. Neurocomputing 553:126529
Funding
This work was supported by the talent project of Weinan Normal University. No.18ZRRC07.
Author information
Authors and Affiliations
Contributions
All authors contributed to the design and implementation of the research, to the analysis of the results and to the writing of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shi, X., Yue, C., Quan, M. et al. A semi-supervised ensemble clustering algorithm for discovering relationships between different diseases by extracting cell-to-cell biological communications. J Cancer Res Clin Oncol 150, 3 (2024). https://doi.org/10.1007/s00432-023-05559-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00432-023-05559-4