
Abstract
Tumors of the endocrine glands are common. Knowledge of their molecular pathology has greatly advanced in the recent past. This review covers the main molecular alterations of tumors of the anterior pituitary, thyroid and parathyroid glands, adrenal cortex, and adrenal medulla and paraganglia. All endocrine gland tumors enjoy a robust correlation between genotype and phenotype. High-throughput molecular analysis demonstrates that endocrine gland tumors can be grouped into molecular groups that are relevant from both pathologic and clinical point of views. In this review, genetic alterations have been discussed and tabulated with respect to their molecular pathogenetic role and clinicopathologic implications, addressing the use of molecular biomarkers for the purpose of diagnosis and prognosis and predicting response to molecular therapy. Hereditary conditions that play a key role in determining predisposition to many types of endocrine tumors are also discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The endocrine system includes several organs all devoted to the physiologic role of maintaining homeostasis and mediating medium- to long-term reactions of the human body to adapt it to external modifications. Tumors of the main endocrine glands, anterior pituitary (adenohypophysis), thyroid and parathyroid glands, adrenal cortex, adrenal medulla, and paraganglia are the object of this review. Tumors of the diffuse neuroendocrine system are not included, excellent reviews have comprehensively covered the topic [1]. A variety of tumors and nodules develop in endocrine glands, with different pathologic features and clinical behavior. Some are very common. Indeed, small indolent foci of papillary carcinoma are found in ~ 35% of well-sampled thyroid glands at autopsy. Others, like parathyroid carcinoma, are very aggressive, but also very rare. A significant minority of endocrine gland tumors develop in the context of inherited syndromes and paraganglionic tumors of the adrenal medulla and paraganglia have the highest degree of hereditability among human neoplasms. Table 1 summarizes inherited syndromes of endocrine tumors [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18]. Our knowledge of the molecular pathogenesis of endocrine gland tumors has exploded in the recent past due to the application of high-throughput molecular analysis. These studies show a remarkable correlation between genotype and histologic phenotype. They are also allowing us to refine risk stratification for prognostic purposes, as well as providing targets for molecular therapy in the case of aggressive endocrine gland carcinomas. The purpose of this review is to summarize the principal findings and innovations in the field of endocrine gland tumors in order to provide a state-of-the-art outline of molecular alterations and their clinicopathologic relevance.
PitNET (pituitary adenoma): molecular pathology and correlation with clinicopathologic features
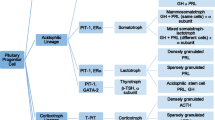
Pituitary adenomas, now termed pituitary neuroendocrine tumors (PitNET), originate from the six neuroendocrine hormone-secreting cell types derived from three main lineages: SF1-lineage gonadotrophs, TPIT-lineage corticotrophs, PIT1-lineage somatotrophs, lactotrophs, mammosomatotrophs, and thyrotrophs. Examples of PitNET are illustrated in Fig. 1 [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]. Table 2 is a summary of the main genes mutated in PitNET [19,20,21,22,23,24, 26].

PitNET (pituitary adenoma). GH-producing densely granulated PitNET (A, hematoxylin and eosin; B, GH immunohistochemistry): GNAS1 mutations occur in a subset of GH-producing pituitary adenoma, more frequently in densely granulated tumors. PRL-producing sparsely granulated PitNET with Golgi pattern PRL staining, the tumor was mitotically active and eventually metastasized to the brainstem and cerebellum (C, hematoxylin and eosin; D, PRL immunohistochemistry): the molecular pathogenesis of metastatic PitNET is still unclear
Although the majority of PitNET lacks known recurrent driver mutations, several — sometimes involving hormone synthesis pathways — have been identified in subsets of sporadic tumors. In addition, a small percentage of PitNET affects patients with inherited predisposition due to germline genetic alterations (Table 1). In some instances, these alterations can be inferred by immunohistochemistry but need to be confirmed by sequence analysis. An example is the immunohistochemical loss of Menin in the tumors of patients with MEN1 syndrome [38,39,40]. To date, morphologic classification, together with clinical and radiologic data, continues to be the best predictor of patient prognosis and therapy response. The relevance of genetic profiling for the diagnostic process is still being defined [19,20,21,22,23,24,25]. Epigenetic alterations, particularly those connected to chromatin remodeling and cell cycle regulation [25], play a major role in the development of PitNET, independently of the hormone-producing phenotype of the tumor. Genes frequently dysregulated by epigenetic modifications include the following: CDKN2A, RB1, DAPK1, GADD45G, THBS1, RASSF1A, FGFR2, MGMT, CASP8, TP73, HMGA1, HMGA2 [41]. In general, chromosomal abnormalities do not correlate with prognosis but are more common in hormone-producing tumors compared with those not associated with hormone production (silent PitNET) [23].
Activating GNAS mutations have been reported in 40–60% of sporadic densely granulated somatotroph PitNET and are present as a mosaic in the 10–15% of patients with McCune–Albright syndrome that have excess GH, usually due to GH-producing cell hyperplasia and less commonly to a GH-producing PitNET [42]. Mammosomatotrope PitNET also exhibits GNAS-related alterations [23]. Conversely, in sparsely granulated somatotroph tumors, somatic mutations of the GH receptor (GHR) altering GH autoregulation and STAT signaling have been reported [20, 25].
In corticotroph PitNET, ATRX mutations correlate with aggressive biological behavior and distant metastasis [43]. Densely granulated biochemically functioning corticotroph tumors harbor USP8 [44], USP48, and less frequently BRAF p.V600E mutations [22]. The role of these changes and their potential therapeutic implications are still controversial [24].
The distinctive molecular signature of lactotroph PitNET includes epigenomic alterations such as high expression of MYC targets and dopamine receptor D2 (DRD2) [23]. However, the SF3B1 p.R625H hotspot mutation has been recently discovered in some lactotroph tumors characterized by high prolactin levels and short progression-free survival [45]. Furthermore, somatic SDHA mutations and SDHD loss of heterozygosity have been reported in rare spontaneous PRL-producing macrotumors [16, 46].
The molecular pathogenesis of metastatic PitNETs is still unclear, due to the rarity of these tumors. ATRX [19, 47] and PTEN [43] mutations have all been reported in some metastatic PitNETs.
Thyroid tumors: molecular pathology and correlation with clinicopathologic features
Tumors of the thyroid gland enjoy a remarkable correlation between histologic phenotype and genotype. This correlation has contributed to refining the current classification scheme. The vast majority of tumors arising in the thyroid are of follicular cell derivation, most are benign, and when endowed with malignant potential, usually follow a very favorable clinical course. This generally favorable course is due to the first effective form of a molecularly targeted therapy, radioactive iodide treatment [48]. A small proportion of tumors are neuroendocrine, originating from parafollicular cells (C-cells). Since they always have malignant potential, they are classified as medullary carcinoma, which represents ~ 3–5% of all carcinomas of the thyroid gland. Up to 25% of medullary carcinoma is inherited in the context of MEN syndromes (Table 1).
Based on clinical outcome, malignant tumors of follicular cells are broadly divided into three groups: those that have a favorable prognosis, anaplastic (undifferentiated) thyroid carcinoma characterized by a very poor prognosis, and a third group of tumors that have intermediate prognosis. While tumors in the first group are histologically well differentiated with clearly defined papillary or follicular architecture or are composed of clearly recognizable oncocytic cells, tumors with very poor prognosis are undifferentiated (i.e., anaplastic). Tumors in the group with intermediate prognosis are often poorly differentiated but may also retain conventional histologic differentiation (papillary, follicular, oncocytic). Under the microscope, they have in common with the prognostically favorable tumor group at least some degree of histologic differentiation, while they share with anaplastic carcinoma high-grade features, i.e., the presence of high mitotic activity and/or tumor necrosis. This classification scheme for thyroid carcinoma of follicular cells based on prognosis is clinically relevant and has been endorsed by the latest 5th edition of the World Health Organization (WHO) scheme (Table 3). The group of tumors that are well differentiated is in turn histologically divided into three subgroups. The first subgroup is composed of tumors that are follicular patterned, which include follicular adenoma and follicular carcinoma (follicular carcinoma when there is the invasion of tumor capsule or of blood vessels), as well as tumors of the encapsulated follicular variant papillary carcinoma family: encapsulated follicular variant papillary carcinoma when there is the invasion of tumor capsule or of blood vessels, and NIFTP (non-invasive follicular thyroid neoplasm with papillary-like nuclear features) when no invasion can be identified [49]. These tumors have a RAS-like molecular signature following the 2014 TCGA molecular classification scheme [50] as discussed in the next paragraph. The second subgroup is that of conventional (i.e., not encapsulated follicular variant type) papillary carcinoma, characterized by the well-known alterations of nuclear morphology (nuclear clearing, irregular contours of the nuclear membrane, grooves, and pseudoinclusions) [49]. These tumors are characterized by infiltrative growth and typically make papillae, although sometimes they can have less typical features, such as infiltrative follicular or solid/trabecular growth, or other less common features that characterize the numerous papillary carcinoma subtypes [51]. These tumors have a BRAF p.V600E-like molecular signature following the 2014 TCGA molecular classification scheme [50], as discussed in the next paragraph. The third subgroup is that in which tumor cells are oncocytic and lack the nuclear alterations of papillary carcinoma. These tumors are characterized by homoplasmic mtDNA mutations [52] associated with dramatic DNA copy-number alterations with widespread loss of heterozygosity [53], as discussed in the next paragraph.
Medullary thyroid carcinoma is the primary neuroendocrine tumor of the thyroid gland. In spite of the remarkable variability of cell morphology and growth patterns (none of which is prognostically relevant), the only subtype recognized by the current WHO 5th edition is the medullary microcarcinoma, i.e., a tumor measuring less than 10 mm (or less than 5 mm according to some authors) scheme. The WHO 5th edition emphasizes the importance of proliferative grading for medullary carcinoma, following the International Medullary Thyroid Carcinoma Grading System (IMTCGS) [54]. The IMTCGS, based on the evaluation of mitotic count and tumor necrosis (Table 4), is in line with the classification framework of neuroendocrine neoplasms [55].
The molecular landscape of thyroid tumors, particularly that of follicular cell derivation, has come into focus also thanks to next-generation sequencing and other high-throughput methods. One of the forces driving these studies has been the need to identify genomic alterations that can be targeted by pathway-specific molecular drugs in aggressive carcinomas that do not respond to conventional radioiodine therapy [54, 56,57,58]. Table 5 is a summary of the main genes involved in thyroid tumor development and progression and of their clinicopathologic relevance. Overall, results are very consistent and converge on several important points:
-
i.
Genetic alterations include “Early/Driver” molecular changes and “Late/Progression associated” events [28, 50, 62, 63, 79]. These are illustrated in Figs. 2 and 3. Examples of tumors with “Early/Driver” alterations are shown in Fig. 4.
Fig. 2 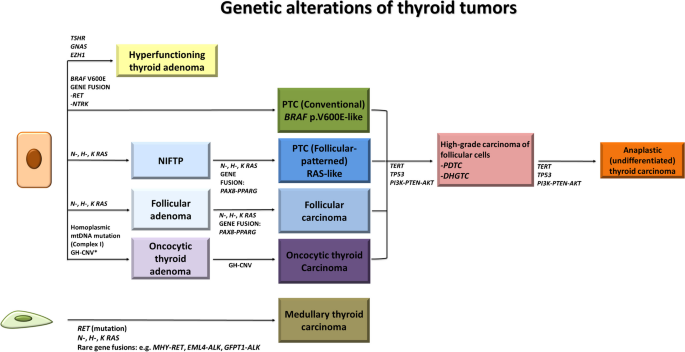
Modified from: Acquaviva G. et al. [28]
Genetic alterations of thyroid tumors. Genetic alterations include “Early/Driver” molecular changes (BRAF p.V600E-like for conventional papillary carcinoma, RAS-like for follicular patterned tumors, and coexistence of mtDNA mutations with severe DNA copy-number alterations for oncocytic tumors) as well as “Late/Progression-associated” molecular changes such as TERT promoter mutation, TP53 mutation, alterations of the PI3K/PTEN/AKT pathway in high-grade non-anaplastic carcinoma of follicular cells, and anaplastic thyroid carcinoma. PDTC, poorly differentiated thyroid carcinoma; DHGTC, differentiated high-grade non-anaplastic thyroid carcinoma; GH-CNV, genome haploidization-type DNA copy number variation leading to copy number neutral uniparental disomy.
Fig. 3 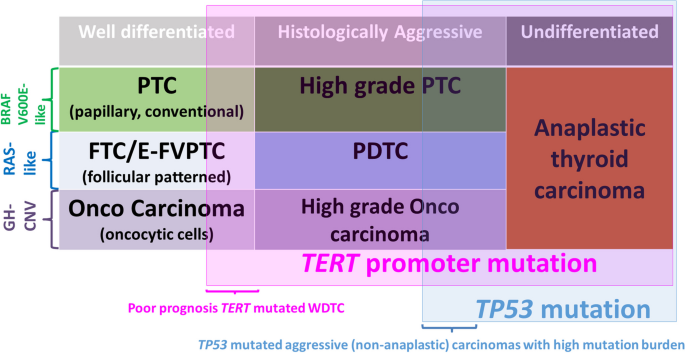
Modified from: Volante et al. [79]
Driver molecular alterations, tumor type, and progression in thyroid tumors of follicular cells. PTC, papillary thyroid carcinoma; E-FVPTC, encapsulated follicular variant papillary thyroid carcinoma; PDTC, poorly differentiated thyroid carcinoma; GH-CNV, genome haploidization-type DNA copy number variation leading to uniparental disomy/aneuploidy.
Fig. 4 
Thyroid tumors. NIFTP (A, hematoxylin and eosin): follicular patterned tumors such as NIFTP have RAS-like molecular signature. Papillary carcinoma (B, hematoxylin and eosin): conventional papillary carcinoma has BRAF p.V600E-like molecular signature, this case featuring glomeruloid papillae harbors a TPR::NTRK1 rearrangement. Oncocytic carcinoma (C, hematoxylin and eosin): oncocytic tumors have both mtDNA mutations and severe DNA copy number alterations. High-grade non-anaplastic papillary carcinoma with tumor necrosis (D, hematoxylin and eosin), poorly differentiated thyroid carcinoma (E, hematoxylin and eosin), and anaplastic thyroid carcinoma (F, hematoxylin and eosin): these aggressive high-grade tumors harbor early/driver molecular alterations and additional mutations associated with tumor progression affecting TERT promoter and PI3K/PTEN/AKT pathway genes; inactivating TP53 mutations are typically associated with anaplastic carcinoma
-
ii.
“Early/Driver” alterations are mutually exclusive [28, 50, 62, 63, 79]. They are commonly used for molecular analysis of preoperative fine needle aspiration specimens. The features as well as the pros and cons of the starting material for this type of analysis are summarized in Table 6.
Table 6 Starting material for preoperative molecular analysis of thyroid nodules -
iii.
“Late/Progression associated” alterations are found in combination with “Early/Driver” changes, consistent with a general model of multi-step progression from well-differentiated to undifferentiated carcinoma. In cases where poorly or undifferentiated areas are associated with a well-differentiated component, “Early/Driver” alterations are identified in both areas, while “Late/Progression associated” changes are restricted to the less differentiated portions of the tumor [80]. Thus, the number of mutations per tumor increases from well-differentiated to undifferentiated carcinoma. Mutation burden is highest in anaplastic carcinoma, lowest in conventional papillary carcinoma, and intermediate in aggressive/advanced papillary and follicular carcinoma [81]. Examples of tumors with “Late/Progression associated” alterations are shown in Fig. 4.
-
iv.
RAS mutations or equivalent molecular alteration (RAS-like tumors) are “Early/Driver” events (see paragraph (ii)) for follicular patterned tumors (Figs. 2, 3, and 4 and Table 5). RAS-like tumors have a homogeneous molecular profile, low MAPK-signaling (because of ERK to RAF monomer negative feedback), high differentiation score, and are malignant only if there is an invasion of the tumor capsule or blood vessels [50].
-
v.
BRAF p.V600E mutation or equivalent molecular alterations (BRAF p.V600E-like tumors) are “Early/Driver” events (see paragraph (ii)) for conventional papillary carcinoma (Figs. 2, 3, and 4 and Table 5). BRAF p.V600E-like tumors have a heterogeneous molecular profile, high MAPK-signaling (because of the lack of ERK to RAF monomer negative feedback), and low differentiation score (based on the level of expression of 16 thyroid metabolism and function genes, e.g., TG, TPO, PAX8).
-
vi.
Coexistence of mtDNA mutations with severe DNA copy-number alterations represents the “Early/Driver” event (see paragraph (ii)) for oncocytic tumors (Figs. 2, 3, and 4 and Table 5). Mutations of mitochondrial DNA (mtDNA) are homoplasmic mtDNA and mostly affect mitochondrial genes encoding Complex I of the respiratory chain [52]. DNA copy-number alterations are dramatic, with widespread loss of heterozygosity and loss of chromosomal DNA, following genome-wide DNA haploidization and copy-number-neutral uniparental disomy [53, 82, 83]. This pathway is unique to oncocytic tumors, which typically do not carry conventional BRAF-like or RAS-like alterations [82,83,84]. While mtDNA mutations are responsible for the oncocytic phenotype [52], the loss of chromosomal DNA is linked to tumor development, since genome haploidization-type DNA copy-number changes are more common in cases diagnosed histologically as oncocytic carcinoma as opposed to oncocytic adenomas and are rare in hyperplastic oncocytic nodules. This has potentially important implications for molecular testing of preoperative fine needle aspiration, since conventional BRAF-like or RAS-like alterations are commonly absent in oncocytic tumors, while Bethesda category III and IV with oncocytic changes have a higher prevalence of DNA copy-number alterations compared with the same categories without cytologically identified oncocytic morphology [85].
-
vii.
“Late/Progression-associated” alterations include mostly somatic mutations of TP53, TERT promoter, and dysregulation of the PI3K/PTEN/AKT pathway (Figs. 2 and 3 and Table 5). Mutations of CDKN2A, of SWI/SNF (switch/sucrose non-fermentable) chromatin remodeling complex genes (ARID1A, ARID1B, ARID2, ARID5B, SMARCB1, PBRM1, ATRX), of Histone methyltransferase genes (KMT2A, KMT2C, KMT2D, SETD2), and of DNA mismatch repair (MMR) genes (MSH2, MSH6, and MLH1) have also been reported [56, 62, 63]. “Late/Progression-associated” changes, in particular TERT promoter mutations, can be utilized for risk stratification, also using preoperative fine needle aspiration specimens [57].
-
viii.
TERT promoter mutations are more frequent and have higher mutated allelic fraction in poorly differentiated, anaplastic, and aggressive/advanced cancers (including high-grade papillary carcinoma) compared with well-differentiated carcinoma [56, 62, 63].
-
ix.
TERT promoter and particularly its co-mutation with BRAF p.V600E or RAS is a powerful marker of poor outcome. Aggressive/advanced papillary carcinomas, many of which are histologically high-grade, have at least one of three genetic alterations: duplication of chromosome 1q, duplication of chromosome 5 p harboring the TERT genomic locus, and TERT promoter mutation (THYT1 signature) [57].
-
x.
TP53 mutation has the highest prevalence in anaplastic carcinoma compared to all forms of advanced/aggressive thyroid carcinoma, including both poorly differentiated and high-grade papillary carcinoma [56, 62, 63, 66].
-
xi.
Rearrangements — such as RET/PTC, NTRK1, NTRK3, and PAX8-PPRG not rare in well-differentiated tumors — are uncommon [56, 62, 63].



Parathyroid tumors: molecular pathology and correlation with clinicopathologic features
The spectrum of parathyroid tumors includes adenoma, atypical tumor (neoplasm of uncertain malignant potential, previously defined as “atypical parathyroid adenoma”), and carcinoma. These tumors can arise in any gland including ectopic ones or areas where embryonic parathyroid remnants may be found [86]. The majority of them cause primary hyperparathyroidism, with adenomas accounting for at least 85% of cases [87, 88].
In general, immunohistochemistry is not necessary for diagnostic purposes. However, it may be useful to screen for hereditary conditions associated with inactivation of genes such as CDC73 and MEN1, causing hyperparathyroidism-jaw tumor (HPTJT) and MEN1 syndrome, respectively [89]. These hereditary conditions usually present with multiglandular involvement and/or multinodular tumors. Parathyroid tumors that arise in the context of hyperparathyroidism-jaw tumor (HPTJT) syndrome feature eosinophilic cytoplasm, perinuclear clearing, nuclear expansion, micro-cystic structures, and sheet-like growth pattern [90].
The main genes mutated in parathyroid tumors are summarized in Table 7 and illustrated in Fig. 5. Examples of parathyroid tumors are shown in Fig. 6. The molecular pathogenesis underlying the majority of sporadic parathyroid adenoma remains unknown. Syndromic parathyroid adenomas constitute ~ 10% of cases [91], found in MEN1, MEN2, MEN4, HPTJT, and isolated familial hyperparathyroidism (FIHP). These syndromes have recently been complemented by MEN5, associated with hereditary mutations of MAX [92] (Table 1). In these inherited conditions, the parathyroid glands contain multiple clonal adenomas which mimic the clinical appearance of cases traditionally diagnosed as parathyroid hyperplasia [93, 94].

Genetic alterations of parathyroid tumors. MEN1 loss of function represents the most common alteration in parathyroid adenoma. The most common alteration of parathyroid carcinoma is CDC73 inactivation, also found in a minority of atypical parathyroid tumors

Parathyroid carcinoma. There is mitotic activity, nuclear pleomorphism (A, hematoxylin and eosin), and parafibromin expression is lost (B, parafibromin immunohistochemistry)
In sporadic adenoma, the most common somatic alteration is inactivation of MEN1. This is caused by loss of heterozygosity (LOH) due to large deletions or genetic recombination at 11q13 (where MEN1 is located) found in ~ 35% of all parathyroid adenoma and/or MEN1 inactivating mutations found in up to ~ 20% of cases [105]. In addition to LOH and somatic mutations, other mechanisms can lead to the inactivation of MEN1, including epigenetic silencing. Interestingly, biallelic MEN1 inactivation occurs in approximately 50% of cases in which LOH at 11q13 is detected, raising the hypothesis that other genes on 11q may also play a role in tumor development.
Cyclin D1 (also located at 11q13) is overexpressed in 10–40% of parathyroid adenomas due to aberrant promoter methylation of different cyclin-dependent kinase inhibitors (CDKIs), while rearrangement of the Cyclin D1 gene (CCND1) occurs in up to ~ 10% of parathyroid adenomas [105].
Other somatic mutations involving CDKN1B (encoding p27), EZH2 (encoding the zinc-finger protein X-linked transcription factor), ASXL3, and MTOR have been reported in a small minority of parathyroid adenomas [105,106,107]. Somatic CDC73 mutations are rare in adenomas. They have been reported only in atypical parathyroid tumors, in some adenomas in the context of HPTJT, and in a small number of cystic adenomas [108]. Interestingly, parathyroid nodules in secondary or tertiary hyperparathyroidism — typically associated with chronic renal failure — harbor different somatic changes compared with those of adenomas in primary hyperparathyroidism [106].
Parathyroid carcinoma is rare and the majority of cases are sporadic. Parathyroid carcinoma develops in 10–15% of patients with HPJT and FIHP (Table 1), while it is uncommon in other inherited conditions [90, 109, 110]. Only a few studies have been conducted on the molecular pathogenesis of sporadic carcinomas. Contrary to parathyroid adenomas, parathyroid cancer rarely exhibits MEN1 mutations [111]. Inactivating CDC73 alterations have been reported in 40–80% of sporadic cases [111,112,113]. CDC73 alterations include truncating or frameshift mutations, as well as missense mutations leading to the loss of parafibromin immunoreactivity [114]. CDC73-mutant parathyroid carcinomas exhibit higher genomic instability with DNA copy number changes, greater mutational burden, and worse patient outcomes compared with wild-type cases [115].
Loss of TP53 and RB1 alleles, CCND1 (encoding Cyclin D1) amplification, and TERT promoter mutations have been reported [116, 117]. PTEN, NF1, KDR, and PIK3CA mutations may represent potential targets for molecular therapy [118]. Metastatic parathyroid carcinoma has a different expression profile compared with non-metastatic parathyroid carcinoma and parathyroid adenoma [119, 120]. Several epigenetic alterations have been discovered in parathyroid carcinomas, including aberrant methylation of APC and of the cell cycle regulators CDKN2A and CDKN2B [121].
Adrenal cortical tumors: molecular pathology and correlation with clinicopathologic features
The spectrum of endocrine tumors of the adrenal cortex includes adrenocortical nodular disease, adrenal cortical adenoma, and adrenal cortical carcinoma. Recently, molecular insights have led to modify the terminology related to adrenocortical nodular disease [122] which currently includes several types of clonal benign proliferations: sporadic nodular adrenocortical disease (a common condition), micronodular adrenocortical disease (a rare condition), and bilateral macronodular adrenocortical disease (a rare condition). Micronodular and bilateral macronodular adrenocortical diseases are often associated with germline pathogenic mutations of several genes [123,124,125]. Our understanding of genomic and hormonal landscapes of adrenal cortical adenoma has also advanced significantly [126], and genotype–phenotype correlations have been proposed for both aldosterone-producing [127, 128] and cortisol-secreting adenomas [129]. Importantly, it is not uncommon for a single alteration to affect different functional pathways, as has been demonstrated for KCNJ5 mutations [130]. Concerning the pathogenesis of cortical carcinoma, several main pathways of tumorigenesis have been discovered, involving cell cycle regulation, Wnt signaling, chromosome maintenance/chromatin remodeling, and the PKA pathway [131] (Fig. 7).

Genetic alterations of adrenal cortical tumors. Adenomas arise as a result of mutations affecting two main groups of genes: the aldosterone-producing adenomas harbor most frequently mutations for KCNJ5 or the ion channel encoding genes, while cortisol-producing adenomas often develop due to alterations in the PKA pathway, typically PRKACA. Genetic alterations of carcinomas mainly involve TP53 but also genes commonly mutated in non-functioning adenomas
Furthermore, integrated analysis of these findings with transcriptomic data, epigenetic findings, and copy number changes has led to the identification of three main classes of adrenal cortex carcinoma, with important clinical and prognostic implications [132,133,134].
Adrenocortical nodular disease
Nodular adrenocortical disease with bilateral involvement of the adrenal cortex rarely occurs in young patients, but when present, it is frequently associated with germline conditions (Table 1). Germline variants of PRKAR1A, PRKACA, PDE11A, PDE8B, and 2p16 CNC2 locus alterations are frequently reported in micronodular adrenocortical disease with bilateral involvement of adrenal cortex, which typically affects children and young adults [135] (Table 1). Germline PRKAR1A mutations (less frequently of PDE8B and PDE11A) cause Primary Pigmented Nodular Adrenocortical Disease, a distinct subtype of bilateral micronodular adrenocortical disease typically found in association with Carney’s complex [136]. The bilateral macronodular adrenocortical disease is caused by pathogenic ARMC5 variants (~ 50% of cases). Further alterations may involve the following genes: MEN1, FH (Hereditary Leiomyomatosis and Renal Cell Cancer), APC (Familial Adenomatosis Polyposis), GNAS (McCune Albright Syndrome), and the rarely mutated PDE11A, PDE8B, and 2p16 CNC2 locus [123, 125, 132, 136, 137]. Variable patterns of Cushing syndrome are typical clinical manifestations of both micro- and macronodular adrenocortical disease.
Adrenal cortical adenoma
Cortical adenomas are the most common tumors of the adrenal cortex.
Alterations of distinctive pathways (active under normal conditions) involved in the physiologic production of aldosterone and cortisol are typical of the corresponding hormone-producing adenoma. Indeed, functioning adenomas that cause primary aldosteronism harbor specific somatic mutations of several ion channel genes which lead to both cellular proliferation and increased aldosterone production in the cells of the zona glomerulosa [126]. They are mutually exclusive and involve KCNJ5 (K + channel) [138], ATP1A1 (Na + /K + channel) [139], ATP2B3 (Ca2 + channel) [139], CACNA1D (Ca2 + channel), CACNA1H (Ca2 + channel) [140], and CLCN2 (Cl- channel) [95, 141]. KCNJ5 mutated adenomas account for the large majority of the cases (~ 40% of aldosterone-producing adenomas) and tend to mainly affect young female patients [137, 142]. Recurrent phenotypic and clinical characteristics have been identified in ion channel gene mutated adenomas [127, 128]. As shown in Table 8, these include the expression of steroidogenic enzymes, cytomorphology, and lateralization index of the adrenal vein sampling (AVS). Interestingly, ion channel genes such as KCNJ5, CACNA1H, CACNA1D, or CLCN2 may also be mutated in the germline, causing familial aldosteronism [126]. Somatic mutations of CTNNB1 — encoding the Wnt-pathway effector beta-catenin — occur in ~ 5% of sporadic aldosterone-producing adenomas and have been correlated to delayed disease onset and female prevalence [96].
Functioning adenomas that produce cortisol feature genetic alterations of the protein kinase A (PKA) pathway active under normal conditions in the production of cortisol. The PKA pathway is physiologically activated by ACTH so that PKA catalytic subunits (PKA-C) can enter the nucleus of zona fasciculata cells enhancing transcription of genes that promote cell proliferation and synthesis of cortisol [126]. PKA pathway genetic alterations of cortisol-producing adenomas affect most frequently the following genes: PRKACA (~ 40% of cases), PRKAR1A, GNAS, or PRKACB [97, 98]. As for sexual steroid-producing adenomas, the molecular pathogenesis remains largely unknown. CTNNB1 mutations are the most frequent molecular alterations of cortical adenomas not associated with hormone production (silent adenomas), particularly in large-sized tumors [129].
Adrenal cortical carcinoma
Adrenal cortical carcinoma is rare and mostly associated with somatic genetic changes (Figs. 7 and 8). Common clinical presentations include Cushing or adrenogenital (virilization-feminization) syndromes due to hormone production [99, 122]. Hereditary cases typically affect children, with up to 80% of pediatric cases carrying germline mutations. The most common mutation affects TP53 (Li-fraumeni syndrome), followed by alterations of the mismatch repair system (Lynch syndrome) [100, 101]. Additional hereditary conditions include Beckwith-Wideman syndrome and MEN1 [100]. Genes frequently altered in benign conditions such as PRKAR1A, MSH2, APC, MEN1, and NF1 are mutated only in small subsets of carcinomas [132]. The most common genetic signatures affect the cell cycle, Wnt signaling, and chromatin remodeling. TP53 mutations found in ~ 20% of adrenal cortical carcinomas are the most common changes [143]. Recurrent somatic genetic alterations affect other cell cycle regulatory genes such as RB1, CDK2NA [132], MDM2, and CDK4 [131, 133]. Wnt signaling is dysregulated by CTNNB1 mutations, ZNRF3 mutations, and deletions [132, 143]. Importantly, TP53 or Wnt pathway mutations are typically mutually exclusive but similarly associated with poor prognosis [132, 133, 143]. Dysregulation of chromatin remodeling is caused by the alteration of several genes, such as ATRX and DAXX [132].

Adrenal cortical tumors. Adrenal cortical adenoma composed by lipid-rich cells resembling the zona fasciculata with low mitotic activity (A, hematoxylin and eosin): cortisol-producing adenoma such as the one shown in the picture has PKA pathway alterations. Adrenal cortical carcinoma with nuclear pleomorphism, mitotic activity, and trabecular growth pattern (B, hematoxylin and eosin): TP53 is the gene most commonly mutated
Furthermore, structural alterations (rearrangements and deletions) in tumor DNA at 11p15 are a frequent finding and can cause loss of imprinted H19 tumor suppressor gene and overexpression of the IGF2 oncogene [103]. IGF2 overexpression in adrenal cortical carcinoma can be identified by immunohistochemistry and may be useful in the differential diagnosis with adenoma [104].
Chromosomal alterations are heterogeneous and have been clustered into three groups: those with extensive chromosome loss (~ 50%), those with variable levels of ploidy (~ 40% — the group of chromosomal changes with worse prognosis), and those with limited chromosomal DNA alteration (~ 10%).
Comprehensive molecular classification of adrenal cortical carcinoma is evolving [126, 143]. Over the years, evidence provided by mutation analysis, chromosomal, methylation, and transcriptome profiling has been integrated to define prognostic groups for risk stratification [126, 133, 134, 144]. According to an important study by Assié et al., there are two main types of adrenal cortical carcinoma [132]. The set of “CpG island methylator phenotype-low” (CIMP-low) carcinomas has infrequent alterations in TP53 or Wnt pathways, mRNA expression pattern predictive of less severe prognosis, chromosome loss, and low rate of disease progression. “CIMP-high” carcinomas typically have alterations of TP53 or Wnt pathway, mRNA expression pattern predictive of poor prognosis, whole-genome duplication, and high rate of disease progression. The TCGA has built on this experience and has proposed an integrated molecular classification model based on DNA copy number, DNA methylation, mRNA expression, and miRNA expression profiles [132,133,134]. This classification model has three groups — termed Cluster-1, -2, -3 — which have been defined after Cluster of Cluster (CoC) analysis. Each CoC cluster is highly relevant for patient prognosis, with Cluster 3 being associated with worse outcome [132,133,134].
Paraganglionic tumors (tumors of the adrenal medulla and of extra-adrenal paraganglia): molecular pathology and correlation with clinicopathologic features
The main genes mutated in paraganglionic tumors are illustrated in Fig. 9. An example of a paraganglionic tumor is shown in Fig. 10.

Genetic alterations of paraganglionic tumors. Genetic alterations in PPGL have been grouped into three main groups reflecting three different mechanisms of tumorigenesis: the Cluster 1-pseudohypoxia pathway, characterized by genetic alterations of the HIF1-alpha-activated response to hypoxia pathway and of other similar hypoxia-inducible factor gene pathways; the Cluster 2-Kinase signaling, including the most common alterations identified in paraganglionic tumors such as NF1 mutations; the more recently described Cluster 3-Wnt/Sonic Hedgehog pathway. *Germline and somatic alterations. **Only somatic alterations

Paraganglionic tumors. Cells resembling normal chromaffin cells with abundant granular cytoplasm are arranged in well-defined nests (A, hematoxylin and eosin). Head and neck paraganglioma, such as the one shown in the picture, often has inactivating SDHD mutations. If any of the subunits of the SDH complex is altered, the entire complex becomes unstable and immunohistochemical SDHB expression is lost (B, SDHB immunohistochemistry)
Paraganglionic tumors are neuroendocrine neoplasms that develop from neural crest-derived progenitors in the adrenal medulla (Pheochromocytoma) and paraganglia (Paraganglioma), respectively. In the latest WHO classification of endocrine tumors, they are considered malignant, although the overall proportion of cases that metastasize is low, ~ 10% of cases. Paragangliomas are further classified in sympathetic and parasympathetic neoplasms according to cell origin and localization. In particular, sympathetic paraganglioma arises within sympathetic nerve plexuses, fibers, and pre- and paravertebral sympathetic chains — thus abdominal cavity, retroperitoneum, pelvis, and thorax are the most prevalent sites. The majority of parasympathetic paraganglioma develops from parasympathetic glomera, and tumors are typically found in the head and neck region, while pheochromocytoma develops in the adrenal medulla. Paraganglionic tumors share the same embryonic origin and are frequently associated with inherited germline mutations (Table 1). Indeed, paraganglionic tumors have the highest degree of hereditability among human neoplasms, with germline mutations in up to ~ 40–80% of cases (vs. ~ 10% in other tumor types) [145,146,147]. Most cases of sympathetic paraganglionic tumors are functional due to catecholamine production, causing hypertension in the majority of patients [148], while parasympathetic paraganglioma typically presents as asymptomatic masses. Sympathetic paraganglionic tumors in children account for up to 20% of cases [149, 150] and most are associated with germline mutations [102, 150]. Conversely, parasympathetic paragangliomas rarely affect children [102, 150, 151]. Germline mutations are an important risk factor for the development of all types of paraganglionic tumors, occurring in up to ~ 40 of adult cases and in up to 80% of pediatric ones. Patients with germline mutation often develop synchronous or metachronous multicentric tumors [102, 150, 152, 153].
Germline mutations mainly affect the following genes: RET, NF1, VHL, TMEM127, SDHA, SDHB, SDHC, SDHD, SDHAF2, FH, MAX, EPAS1, DLST, MDH2, GOT2, and DNMT3A (Table 1).
SDH (succinate dehydrogenase, complex II of the mitochondrial respiratory chain) consists of four subunits (SDHA, SDHB, SDHC, SDHD), and the genes encoding SDHB and SDHD are those more frequently mutated in the germline [153]. SDHD mutations cause the majority of paragangliomas of the head and neck region and typically present with single or multifocal tumors exclusively located in the head and neck. On the other hand, thoracoabdominal tumors more frequently harbor SDHB mutations. Paraganglionic tumors can also be found in association with gastrointestinal stromal tumors (GIST) and pulmonary chondromas in the so-called Carney triad, a nonhereditary condition characterized by epigenetic alterations of SDHC [154, 155]. Interestingly, somatic NF1, RET, and VHL are also found in sporadic tumors, with NF1 representing the gene most commonly mutated ( ~ 20% of cases) [156, 157]. Additional somatic mutations, not found in the germline, affect HRAS, BRAF, SETD2, FGFR1, TP53, ATRX, ARNT, IDH1, H3F3A, MET, and CSDE1 [146].
Paraganglionic tumors have been divided into three molecular clusters that are also recognized by the TCGA [145]. Cluster 1 tumors have a response to hypoxia pathway dysregulation (pseudohypoxia) characterized by increased transcription of genes targeted by hypoxia-inducible factors (HIF1-alpha and other factors) which promote angiogenesis, cell proliferation, survival, and epithelial-mesenchymal transition [158]. Of note, in SDH-/FH-deficient and mutant IDH tumors, oncometabolite accumulation induces DNA hypermethylation and other epigenetic changes. Cluster 2 tumors feature abnormal activation of RAS/RAF/ERK, PI3K/PTEN/AKT, and MYC/MAX/MXD1 pathways. They also exhibit a hypomethylated phenotype and frequent somatic copy number changes [146]. Cluster 3 tumors are characterized by dysregulation of Wnt and Sonic Hedgehog pathways. Indeed, sporadic MAML3 fusions and CSDE1 mutations in paraganglionic tumors that activate Wnt and Sonic Hedgehog pathways have been discovered to be major driving factors in tumor development [145]. Given the high hereditability of paraganglionic tumors, genetic screening is recommended, particularly in pediatric patients [150, 159]. In this respect, immunohistochemistry is a very useful screening test: if any of the subunits of the SDH complex is lost due to mutations or epigenetic alterations, the entire complex becomes unstable and the SDHB subunit is degraded in the cytoplasm. Loss of the SDHB protein can be demonstrated by SDHB immunohistochemistry, pointing to the need for SDH sequencing to confirm SDH subunit germline mutation [38]. This type of so-called “molecular-immunohistochemistry” can be applied not only to anticipate the genetic background of individual paraganglioma tumors but also to prevent erroneous diagnostic conclusions in the case of multiple lesions mimicking metastatic disease [146, 147].
Change history
26 January 2024
A Correction to this paper has been published: https://doi.org/10.1007/s00428-024-03738-3
References
Rindi G, Mete O, Uccella S et al (2022) Overview of the 2022 WHO classification of neuroendocrine neoplasms. Endocr Pathol 33:115–154. https://doi.org/10.1007/s12022-022-09708-2
Beckers A, Aaltonen LA, Daly AF, Karhu A (2013) Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev 34:239–277. https://doi.org/10.1210/ER.2012-1013
Masi G, Barzon L, Iacobone M et al (2008) Clinical, genetic, and histopathologic investigation of CDC73-related familial hyperparathyroidism. Endocr Relat Cancer 15:1115–1126. https://doi.org/10.1677/ERC-08-0066
Sarquis MS, Silveira LG, Pimenta FJ et al (2008) Familial hyperparathyroidism: surgical outcome after 30 years of follow-up in three families with germline HRPT2 mutations. Surgery 143. https://doi.org/10.1016/j.surg.2007.12.012
Alrezk R, Hannah-Shmouni F, Stratakis CA (2017) MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer 24:T195–T208. https://doi.org/10.1530/ERC-17-0243
Frederiksen A, Rossing M, Hermann P et al (2019) Clinical features of multiple endocrine neoplasia type 4: novel pathogenic variant and review of published cases. J Clin Endocrinol Metab 104:3637–3646. https://doi.org/10.1210/JC.2019-00082
de Kock L, Wu MK, Foulkes WD (2019) Ten years of DICER1 mutations: provenance, distribution, and associated phenotypes. Hum Mutat 40:1939–1953. https://doi.org/10.1002/HUMU.23877
Burnichon N, Cascón A, Schiavi F et al (2012) MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res 18:2828–2837. https://doi.org/10.1158/1078-0432.CCR-12-0160
Cascon A, Robledo M (2012) MAX and MYC: a heritable breakup. Cancer Res 72:3119–3124. https://doi.org/10.1158/0008-5472.CAN-11-3891
Brandi ML, Agarwal SK, Perrier ND et al (2021) Multiple endocrine neoplasia type 1: latest insights. Endocr Rev 42:133–170. https://doi.org/10.1210/ENDREV/BNAA031
Ferner RE, Huson SM, Thomas N et al (2007) Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 44:81–88. https://doi.org/10.1136/JMG.2006.045906
Stratakis CA (2016) Carney complex: a familial lentiginosis predisposing to a variety of tumors. Rev Endocr Metab Disord 17:367–371. https://doi.org/10.1007/S11154-016-9400-1
Wells SA, Asa SL, Dralle H et al (2015) Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25:567–610. https://doi.org/10.1089/THY.2014.0335
Moline J, Eng C (2011) Multiple endocrine neoplasia type 2: an overview. Genet Med 13:755–764. https://doi.org/10.1097/GIM.0B013E318216CC6D
Korpershoek E, Favier J, Gaal J et al (2011) SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab 96. https://doi.org/10.1210/JC.2011-1043
Gill AJ (2018) Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 72:106–116. https://doi.org/10.1111/HIS.13277
Lubensky IA, Pack S, Ault D et al (1998) Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol 153:223–231. https://doi.org/10.1016/S0002-9440(10)65563-0
Salama Y, Albanyan S, Szybowska M et al (2019) Comprehensive characterization of a Canadian cohort of von Hippel-Lindau disease patients. Clin Genet 96:461–467. https://doi.org/10.1111/CGE.13613
Casar-Borota O, Boldt H, Engström B et al (2021) Corticotroph aggressive pituitary tumors and carcinomas frequently harbor ATRX mutations. J Clin Endocrinol Metab 106:1183–1194. https://doi.org/10.1210/CLINEM/DGAA749
Asa SL, Ezzat S (2014) Genomic approaches to problems in pituitary neoplasia. Endocr Pathol 25:209–213. https://doi.org/10.1007/S12022-013-9276-5
Weinstein LS, Yu S, Warner DR, Liu J (2001) Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 22:675–705. https://doi.org/10.1210/EDRV.22.5.0439
Chen J, Jian X, Deng S et al (2018) Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat Commun 9. https://doi.org/10.1038/s41467-018-05275-5
Neou M, Villa C, Armignacco R et al (2020) Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell 37. https://doi.org/10.1016/j.ccell.2019.11.002
Hayashi K, Inoshita N, Kawaguchi K et al (2016) The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur J Endocrinol 174:213–226. https://doi.org/10.1530/EJE-15-0689
Asa SL, Mete O, Ezzat S (2021) Genomics and epigenomics of pituitary tumors: What Do Pathologists Need to Know? Endocr Pathol 32:3–16. https://doi.org/10.1007/S12022-021-09663-4
WHO Classification of Tumours Editorial Board. Endocrine and neuroendocrine tumours [Internet]. Lyon (France): International Agency for Research on Cancer; 2022 [cited 2023, October, 12]. (WHO classification of tumours series, 5th ed.; vol. 10). Available from: https://tumourclassification.iarc.who.int/chapters/53
Chong AS, Nikiforov YE, Condello V et al (2021) Prevalence and spectrum of DICER1 mutations in adult-onset thyroid nodules with indeterminate cytology. J Clin Endocrinol Metab 106. https://doi.org/10.1210/clinem/dgab025
Acquaviva G, Visani M, Repaci A et al (2018) Molecular pathology of thyroid tumours of follicular cells: a review of genetic alterations and their clinicopathological relevance. Histopathology 72:6–31. https://doi.org/10.1111/HIS.13380
González IA, Stewart DR, Schultz KAP et al (2022) DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma. Mod Pathol 35:4–22. https://doi.org/10.1038/s41379-021-00905-8
Rooper LM, Bynum JP, Miller KP et al (2020) Recurrent DICER1 hotspot mutations in malignant thyroid gland teratomas: molecular characterization and proposal for a separate classification. Am J Surg Pathol 44. https://doi.org/10.1097/PAS.0000000000001430
Calebiro D, Grassi ES, Eszlinger M et al (2016) Recurrent EZH1 mutations are a second hit in autonomous thyroid adenomas. J Clin Investig 126. https://doi.org/10.1172/JCI84894
Nikiforova MN, Nikitski A V., Panebianco F et al (2019) GLIS rearrangement is a genomic hallmark of hyalinizing trabecular tumor of the thyroid gland. Thyroid 29. https://doi.org/10.1089/thy.2018.0791
Krohn K, Führer D, Bayer Y et al (2005) Molecular pathogenesis of euthyroid and toxic multinodular goiter. Endocr Rev 26:504–524. https://doi.org/10.1210/ER.2004-0005
Lumbroso S, Paris F, Sultan C (2004) Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome–a European Collaborative Study. J Clin Endocrinol Metab 89:2107–2113. https://doi.org/10.1210/JC.2003-031225
Chu YH, Wirth LJ, Farahani AA et al (2020) Clinicopathologic features of kinase fusion-related thyroid carcinomas: an integrative analysis with molecular characterization. Mod Pathol 33. https://doi.org/10.1038/s41379-020-0638-5
Dogan S, Wang L, Ptashkin RN et al (2016) Mammary analog secretory carcinoma of the thyroid gland: a primary thyroid adenocarcinoma harboring ETV6-NTRK3 fusion. Mod Pathol 29. https://doi.org/10.1038/modpathol.2016.115
Pekova B, Sykorova V, Dvorakova S et al (2020) RET, NTRK, ALK, BRAF, and MET fusions in a large cohort of pediatric papillary thyroid carcinomas. Thyroid 30. https://doi.org/10.1089/thy.2019.0802
Oudijk L, Gaal J, de Krijger RR (2019) The role of immunohistochemistry and molecular analysis of succinate dehydrogenase in the diagnosis of endocrine and non-endocrine tumors and related syndromes. Endocr Pathol 30:64–73. https://doi.org/10.1007/S12022-018-9555-2
Asa SL, Mete O (2018) Immunohistochemical biomarkers in pituitary pathology. Endocr Pathol 29:130–136. https://doi.org/10.1007/S12022-018-9521-Z
Thakker RV (2014) Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 386:2–15. https://doi.org/10.1016/J.MCE.2013.08.002
Pease M, Ling C, Mack WJ et al (2013) The role of epigenetic modification in tumorigenesis and progression of pituitary adenomas: a systematic review of the literature. PLoS One 8. https://doi.org/10.1371/JOURNAL.PONE.0082619
Yang I, Park S, Ryu M et al (1996) Characteristics of gsp-positive growth hormone-secreting pituitary tumors in Korean acromegalic patients. Eur J Endocrinol 134:720–726. https://doi.org/10.1530/EJE.0.1340720
Guo F, Wang G, Wang F et al (2018) Identification of novel genes involved in the pathogenesis of an ACTH-secreting pituitary carcinoma: a case report and literature review. Front Oncol 8. https://doi.org/10.3389/FONC.2018.00510
Ballmann C, Thiel A, Korah HE et al (2018) USP8 mutations in pituitary cushing adenomas-targeted analysis by next-generation sequencing. J Endocr Soc 2:266–278. https://doi.org/10.1210/JS.2017-00364
Li C, Xie W, Rosenblum JS et al (2020) Somatic SF3B1 hotspot mutation in prolactinomas. Nat Commun 11. https://doi.org/10.1038/S41467-020-16052-8
Papathomas TG, Gaal J, Corssmit EPM et al (2013) Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC-PGL syndromes: a clinicopathological and molecular analysis. Eur J Endocrinol 170:1–12. https://doi.org/10.1530/EJE-13-0623
Tanizaki Y, Jin L, Scheithauer BW et al (2007) P53 gene mutations in pituitary carcinomas. Endocr Pathol 18:217–222. https://doi.org/10.1007/S12022-007-9006-Y
No authors listed (1949) Radio-iodine halts one type of cancer: radioactive chemical brings about history-making recovery of patient dying from thyroid tumors. Life Mag 27:54–56
Tallini G, Tuttle RM, Ghossein RA (2017) The history of the follicular variant of papillary thyroid carcinoma. J Clin Endocrinol Metab 102:15–22. https://doi.org/10.1210/JC.2016-2976
Cancer Genome Atlas Research N (2014) Integrated genomic characterization of papillary thyroid carcinoma. Cell 159:676–690. https://doi.org/10.1016/j.cell.2014.09.050
Baloch ZW, Asa SL, Barletta JA et al (2022) Overview of the 2022 WHO classification of thyroid neoplasms. Endocr Pathol 33:27–63. https://doi.org/10.1007/S12022-022-09707-3
Gasparre G, Porcelli AM, Bonora E et al (2007) Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci U S A 104:9001–9006. https://doi.org/10.1073/PNAS.0703056104
Corver WE, Ruano D, Weijers K et al (2012) Genome haploidisation with chromosome 7 retention in oncocytic follicular thyroid carcinoma. PLoS One 7. https://doi.org/10.1371/JOURNAL.PONE.0038287
Xu B, Fuchs TL, Ahmadi S et al (2022) International medullary thyroid carcinoma grading system: a validated grading system for medullary thyroid carcinoma. J Clin Oncol 40:96–103. https://doi.org/10.1200/JCO.21.01329
Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, Busam KJ, de Krijger RR, Dietel M, El-Naggar AK, Fernandez-Cuesta L, Kloppel G, McCluggage WG, Moch H, Ohgaki H, Rakha EA, Reed NS, Rous BA, Sasano H, Scarpa A, Scoazec JY, Travis WD, Tallini G, Trouillas J, van Krieken JH, Cree IA (2018) A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol 31:1770–1786
Landa I, Ibrahimpasic T, Boucai L et al (2016) Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 126:1052–1066. https://doi.org/10.1172/JCI85271
Gandolfi G, Ragazzi M, de Biase D et al (2017) Genome-wide profiling identifies the THYT1 signature as a distinctive feature of widely metastatic Papillary Thyroid Carcinomas. Oncotarget 9:1813–1825. https://doi.org/10.18632/ONCOTARGET.22805
Chen H, Luthra R, Routbort MJ et al (2018) Molecular profile of advanced thyroid carcinomas by next-generation sequencing: characterizing tumors beyond diagnosis for targeted therapy. Mol Cancer Ther 17:1575–1584. https://doi.org/10.1158/1535-7163.MCT-17-0871
Kelly LM, Barila G, Liu P et al (2014) Identification of the transforming STRN-ALK fusion asa potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A 111. https://doi.org/10.1073/pnas.1321937111
Chou A, Fraser S, Toon CW et al (2015) A detailed clinicopathologic study of ALK-translocated papillary thyroid carcinoma. Am J Surg Pathol 39. https://doi.org/10.1097/PAS.0000000000000368
Godbert Y, De Figueiredo BH, Bonichon F et al (2015) Remarkable response to crizotinib in woman with anaplastic lymphoma kinase-rearranged anaplastic thyroid carcinoma. J Clin Oncol 33:e84–e87. https://doi.org/10.1200/JCO.2013.49.6596
Xu B, Fuchs T, Dogan S et al (2020) Dissecting anaplastic thyroid carcinoma: a comprehensive clinical, histologic, immunophenotypic, and molecular study of 360 cases. Thyroid 30:1505–1517. https://doi.org/10.1089/THY.2020.0086
Xu B, David J, Dogan S et al (2022) Primary high-grade non-anaplastic thyroid carcinoma: a retrospective study of 364 cases. Histopathology 80:322–337. https://doi.org/10.1111/HIS.14550
Ciampi R, Knauf JA, Kerler R et al (2005) Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Investig 115. https://doi.org/10.1172/JCI23237
De Biase D, Acquaviva G, Visani M et al (2020) Molecular diagnostic of solid tumor using a next generation sequencing custom-designed multi-gene panel. Diagnostics (Basel) 10. https://doi.org/10.3390/DIAGNOSTICS10040250
de Biase D, Torricelli F, Ragazzi M et al (2018) Not the same thing: metastatic PTCs have a different background than ATCs. Endocr Connect 7:1370–1379. https://doi.org/10.1530/EC-18-0386
Subbiah V, Kreitman RJ, Wainberg ZA et al (2022) Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: updated analysis from the phase II ROAR basket study. Ann Oncol 33. https://doi.org/10.1016/j.annonc.2021.12.014
Cameselle-Teijeiro JM, Peteiro-González D, Caneiro-Gómez J et al (2018) Cribriform-morular variant of thyroid carcinoma: a neoplasm with distinctive phenotype associated with the activation of the WNT/?-catenin pathway. Mod Pathol 31:1168–1179. https://doi.org/10.1038/s41379-018-0070-2
Chernock RD, Rivera B, Borrelli N et al (2020) Poorly differentiated thyroid carcinoma of childhood and adolescence: a distinct entity characterized by DICER1 mutations. Mod Pathol 33. https://doi.org/10.1038/s41379-020-0458-7
Agaimy A, Witkowski L, Stoehr R et al (2020) Malignant teratoid tumor of the thyroid gland: an aggressive primitive multiphenotypic malignancy showing organotypical elements and frequent DICER1 alterations—is the term “thyroblastoma” more appropriate? Virchows Archiv 477. https://doi.org/10.1007/s00428-020-02853-1
Prasad ML, Vyas M, Horne MJ et al (2016) NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in northeast United States. Cancer 122. https://doi.org/10.1002/cncr.29887
Kroll TG, Sarraf P, Pecciarini L et al (2000) PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science 289:1357–1360. https://doi.org/10.1126/science.289.5483.1357
Liaw D, Marsh DJ, Li J et al (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16. https://doi.org/10.1038/ng0597-64
Harach HR, Soubeyran I, Brown A et al (1999) Thyroid pathologic findings in patients with Cowden disease. Ann Diagn Pathol 3. https://doi.org/10.1016/S1092-9134(99)80011-2
Ciampi R, Mian C, Fugazzola L et al (2013) Evidence of a low prevalence of ras mutations in a large medullary thyroid cancer series. Thyroid 23. https://doi.org/10.1089/thy.2012.0207
Park H, Shin HC, Yang H et al (2022) Molecular classification of follicular thyroid carcinoma based on TERT promoter mutations. Mod Pathol 35. https://doi.org/10.1038/s41379-021-00907-6
Evangelisti C, de Biase D, Kurelac I et al (2015) A mutation screening of oncogenes, tumor suppressor gene TP53 and nuclear encoded mitochondrial complex I genes in oncocytic thyroid tumors. BMC Cancer 15. https://doi.org/10.1186/s12885-015-1122-3
Parma J, Duprez L, Van Sande J et al (1993) Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365:649–651. https://doi.org/10.1038/365649a0
Volante M, Lam AK, Papotti M, Tallini G (2021) Molecular pathology of poorly differentiated and anaplastic thyroid cancer: what do pathologists need to know? Endocr Pathol 32:63–76. https://doi.org/10.1007/s12022-021-09665-2
Ragazzi M, Torricelli F, Donati B et al (2021) Coexisting well-differentiated and anaplastic thyroid carcinoma in the same primary resection specimen: immunophenotypic and genetic comparison of the two components in a consecutive series of 13 cases and a review of the literature. Virchows Arch 478. https://doi.org/10.1007/S00428-020-02891-9
Pozdeyev N, Gay LM, Sokol ES et al (2018) Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 24:3059–3068. https://doi.org/10.1158/1078-0432.CCR-18-0373
Tallini G, Hsueh A, Liu S et al (1999) Frequent chromosomal DNA unbalance in thyroid oncocytic (Hurthle cell) neoplasms detected by comparative genomic hybridization. Lab Investig 79:547–555
Gopal RK, Kübler K, Calvo SE et al (2018) Widespread chromosomal losses and mitochondrial DNA alterations as genetic drivers in Hürthle cell carcinoma. Cancer Cell 34. https://doi.org/10.1016/j.ccell.2018.06.013
Nikiforova MN, Lynch RA, Biddinger PW et al (2003) RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metab 88:2318–2326. https://doi.org/10.1210/JC.2002-021907
Abi-Raad R, Prasad ML, Adeniran AJ, Cai G (2022) Copy number variations identified in thyroid FNA specimens are associated with Hürthle cell cytomorphology. Cancer Cytopathol 130. https://doi.org/10.1002/cncy.22569
Xin Y, Zhao T, Wei B et al (2020) Intrapericardial parathyroid carcinoma: a case report. Endocrine 69:456–460. https://doi.org/10.1007/S12020-020-02283-8
Perren A, Mete O, Williams MD, Johnson SJ, Juhlin CC (2022) Parathyroid adenoma. In: WHO Classification of Tumours Editorial Board. Endocrine and neuroendocrine tumours, 5th ed. IARC, Lyon. Available from: https://tumourclassification.iarc.who.int/chapters/53
DeLellis RA (2011) Parathyroid tumors and related disorders. Mod Pathol 24(Suppl 2):S78-93
Erickson LA, Mete O (2018) Immunohistochemistry in diagnostic parathyroid pathology. Endocr Pathol 29:113–129. https://doi.org/10.1007/S12022-018-9527-6
Gill AJ, Lim G, Cheung VKY et al (2019) Parafibromin-deficient (HPT-JT Type, CDC73 Mutated) parathyroid tumors demonstrate distinctive morphologic features. Am J Surg Pathol 43:35–46. https://doi.org/10.1097/PAS.0000000000001017
Thakker RV (2016) Genetics of parathyroid tumours. J Intern Med 280:574–583. https://doi.org/10.1111/JOIM.12523
Seabrook AJ, Harris JE, Velosa SB et al (2021) Multiple endocrine tumors associated with germline MAX mutations: multiple endocrine neoplasia type 5? J Clin Endocrinol Metab 106:1163–1182. https://doi.org/10.1210/CLINEM/DGAA957
Turchini J, Gill AJ (2020) Hereditary parathyroid disease: sometimes pathologists do not know what they are missing. Endocr Pathol 31:218–230. https://doi.org/10.1007/S12022-020-09631-4
Brandi ML, Gagel RF, Angeli A et al (2001) Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86:5658–5671. https://doi.org/10.1210/JCEM.86.12.8070
Rege J, Nanba K, Blinder AR et al (2020) Identification of somatic mutations in CLCN2 in aldosterone-producing adenomas. J Endocr Soc 4:1–9. https://doi.org/10.1210/JENDSO/BVAA123
Åkerström T, Maharjan R, Sven Willenberg H et al (2016) Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep 6. https://doi.org/10.1038/SREP19546
Di Dalmazi G, Kisker C, Calebiro D et al (2014) Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab 99:E2093–E2100. https://doi.org/10.1210/JC.2014-2152
Beuschlein F, Fassnacht M, Assié G et al (2014) Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med 370:1019–1028. https://doi.org/10.1056/NEJMOA1310359
Fassnacht M, Dekkers OM, Else T et al (2018) European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 179:G1–G46. https://doi.org/10.1530/EJE-18-0608
Kamilaris CDC, Hannah-Shmouni F, Stratakis CA (2020) Adrenocortical tumorigenesis: lessons from genetics. Best Pract Res Clin Endocrinol Metab 34:101428
Raymond VM, Everett JN, Furtado LV et al (2013) Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol 31:3012–3018. https://doi.org/10.1200/JCO.2012.48.0988
Virgone C, Andreetta M, Avanzini S et al (2020) Pheochromocytomas and paragangliomas in children: data from the Italian Cooperative Study (TREP). Pediatr Blood Cancer 67. https://doi.org/10.1002/PBC.28332
Henry I, Jeanpierre M, Couillin P et al (1989) Molecular definition of the 11p15.5 region involved in Beckwith-Wiedemann syndrome and probably in predisposition to adrenocortical carcinoma. Hum Genet 81:273–277. https://doi.org/10.1007/BF00279003
Schmitt A, Saremaslani P, Schmid S, Rousson V, Montani M, Schmid DM, Heitz PU, Komminoth P, Perren A (2006) IGFII and MIB1 immunohistochemistry is helpful for the differentiation of benign from malignant adrenocortical tumours. Histopathology 49:298–307
Brewer K, Costa-Guda J, Arnold A (2019) Molecular genetic insights into sporadic primary hyperparathyroidism. Endocr Relat Cancer 26:R53–R72. https://doi.org/10.1530/ERC-18-0304
Hong YA, Park KC, Kim BK et al (2021) Analyzing genetic differences between sporadic primary and secondary/tertiary hyperparathyroidism by targeted next-generation panel sequencing. Endocr Pathol 32:501–512. https://doi.org/10.1007/S12022-021-09686-X
Costa-Guda J, Marinoni I, Molatore S et al (2011) Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J Clin Endocrinol Metab 96. https://doi.org/10.1210/JC.2010-1338
Sulaiman L, Haglund F, Hashemi J et al (2012) Genome-wide and locus specific alterations in CDC73/HRPT2-mutated parathyroid tumors. PLoS One 7. https://doi.org/10.1371/JOURNAL.PONE.0046325
Korpi-Hyövälti E, Cranston T, Ryhänen E et al (2014) CDC73 intragenic deletion in familial primary hyperparathyroidism associated with parathyroid carcinoma. J Clin Endocrinol Metab 99:3044–3048. https://doi.org/10.1210/jc.2014-1481
Bricaire L, Odou MF, Cardot-Bauters C et al (2013) Frequent large germline HRPT2 deletions in a french national cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab 98. https://doi.org/10.1210/jc.2012-2789
Haven CJ, Van Puijenbroek M, Tan MH et al (2007) Identification of MEN1 and HRPT2 somatic mutations in paraffin-embedded (sporadic) parathyroid carcinomas. Clin Endocrinol (Oxf) 67. https://doi.org/10.1111/j.1365-2265.2007.02894.x
Cetani F, Ambrogini E, Viacava P et al (2007) Should parafibromin staining replace HRTP2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma? Eur J Endocrinol 156:547–554. https://doi.org/10.1530/EJE-06-0720
Hunt JL, Carty SE, Yim JH et al (2005) Allelic loss in parathyroid neoplasia can help characterize malignancy. Am J Surg Pathol 29. https://doi.org/10.1097/01.pas.0000166368.68459.99
Gill AJ, Clarkson A, Gimm O et al (2006) Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidism-jaw tumor (HPT-JT) syndrome-related adenomas from sporadic parathyroid adenomas and hyperplasias. Am J Surg Pathol 30:1140–1149. https://doi.org/10.1097/01.PAS.0000209827.39477.4F
Hu Y, Zhang X, Wang O et al (2020) The genomic profile of parathyroid carcinoma based on whole-genome sequencing. Int J Cancer 147:2446–2457. https://doi.org/10.1002/IJC.33166
Cryns VL, Rubio MP, Thor AD et al (1994) p53 abnormalities in human parathyroid carcinoma. J Clin Endocrinol Metab 78. https://doi.org/10.1210/jcem.78.6.8200932
Pandya C, Uzilov A V., Bellizzi J et al (2017) Genomic profiling reveals mutational landscape in parathyroid carcinomas. JCI Insight 2. https://doi.org/10.1172/jci.insight.92061
Kang H, Pettinga D, Schubert AD et al (2019) Genomic profiling of parathyroid carcinoma reveals genomic alterations suggesting benefit from therapy. Oncologist 24. https://doi.org/10.1634/theoncologist.2018-0334
Haven CJ, van Puijenbroek M, Karperien M et al (2004) Differential expression of the calcium sensing receptor and combined loss of chromosomes 1q and 11q in parathyroid carcinoma. J Pathol 202:86–94. https://doi.org/10.1002/PATH.1489
Condello V, Cetani F, Denaro M et al (2021) Gene expression profile in metastatic and non-metastatic parathyroid carcinoma. Endocr Relat Cancer 28. https://doi.org/10.1530/ERC-20-0337
Starker LF, Svedlund J, Udelsman R et al (2011) The DNA methylome of benign and malignant parathyroid tumors. Genes Chromosomes Cancer 50:735–745. https://doi.org/10.1002/GCC.20895
Hodgson A, Pakbaz S, Mete O (2019) A diagnostic approach to adrenocortical tumors. Surg Pathol Clin 12:967–995. https://doi.org/10.1016/j.path.2019.08.005
Carney JA, Hruska LS, Beauchamp GD, Gordon H (1986) Dominant inheritance of the complex of myxomas, spotty pigmentation, and endocrine overactivity. Mayo Clin Proc 61. https://doi.org/10.1016/S0025-6196(12)61843-6
Assié G, Libé R, Espiard S et al (2013) ARMC5 mutations in macronodular adrenal hyperplasia with cushing’s syndrome. N Engl J Med 369. https://doi.org/10.1056/nejmoa1304603
Matyakhina L, Freedman RJ, Bourdeau I et al (2005) Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab 90. https://doi.org/10.1210/jc.2004-2377
Juhlin CC, Bertherat J, Giordano TJ et al (2021) What did we learn from the molecular biology of adrenal cortical neoplasia? From histopathology to translational genomics. Endocr Pathol 32:102–133. https://doi.org/10.1007/s12022-021-09667-0
Wannachalee T, Zhao L, Nanba K et al (2019) Three discrete patterns of primary aldosteronism lateralization in response to cosyntropin during adrenal vein sampling. J Clin Endocrinol Metab 104:5867–5876. https://doi.org/10.1210/JC.2019-01182
Azizan EAB, Lam BYH, Newhouse SJ et al (2012) Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J Clin Endocrinol Metab 97. https://doi.org/10.1210/JC.2011-2965
Faillot S, Foulonneau T, Néou M et al (2021) Genomic classification of benign adrenocortical lesions. Endocr Relat Cancer 28. https://doi.org/10.1530/ERC-20-0128
Mulatero P, Schiavi F, Williams TA et al (2016) ARMC5 mutation analysis in patients with primary aldosteronism and bilateral adrenal lesions. J Hum Hypertens 30:374–378. https://doi.org/10.1038/JHH.2015.98
de Krijger RE, Bertherat J (2016) 5th International ACC Symposium: Classification of adrenocortical cancers from pathology to integrated genomics: real advances or lost in translation? Horm Cancer 7:3–8. https://doi.org/10.1007/s12672-015-0242-1
Assié G, Letouzé E, Fassnacht M et al (2014) Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46. https://doi.org/10.1038/ng.2953
Zheng S, Cherniack AD, Dewal N et al (2016) Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 29:723–736. https://doi.org/10.1016/J.CCELL.2016.04.002
Assié G, Jouinot A, Fassnacht M et al (2019) Value of molecular classification for prognostic assessment of adrenocortical carcinoma. JAMA Oncol 5:1440–1447. https://doi.org/10.1001/JAMAONCOL.2019.1558
Juhlin CC, Villablanca A, Sandelin K et al (2007) Parafibromin immunoreactivity: its use as an additional diagnostic marker for parathyroid tumor classification. Endocr Relat Cancer 14. https://doi.org/10.1677/ERC-07-0021.
Espiard S, Drougat L, Libé R et al (2015) ARMC5 mutations in a large cohort of primary macronodular adrenal hyperplasia: clinical and functional consequences. J Clin Endocrinol Metab 100. https://doi.org/10.1210/jc.2014-4204
Espiard S, Vantyghem MC, Assié G et al (2020) Frequency and incidence of carney complex manifestations: a prospective multicenter study with a three-year follow-up. J Clin Endocrinol Metab 105. https://doi.org/10.1210/clinem/dgaa002
Choi M, Scholl UI, Yue P et al (2011) K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331:768–772. https://doi.org/10.1126/SCIENCE.1198785
Beuschlein F, Boulkroun S, Osswald A et al (2013) Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 45:440–444. https://doi.org/10.1038/NG.2550
Scholl UI, Stölting G, Nelson-Williams C et al (2015) Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 4:e06315. https://doi.org/10.7554/ELIFE.06315
Scholl UI, Stölting G, Schewe J et al (2018) CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 50:349–354. https://doi.org/10.1038/S41588-018-0048-5
Lenzini L, Rossitto G, Maiolino G et al (2015) A meta-analysis of somatic KCNJ5 K(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab 100:E1089–E1095. https://doi.org/10.1210/JC.2015-2149
Juhlin CC, Goh G, Healy JM et al (2015) Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J Clin Endocrinol Metab 100:E493–E502. https://doi.org/10.1210/JC.2014-3282
Giordano TJ, Kuick R, Else T et al (2009) Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res 15:668–676. https://doi.org/10.1158/1078-0432.CCR-08-1067
Fishbein L, Leshchiner I, Walter V et al (2017) Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 31:181–193. https://doi.org/10.1016/J.CCELL.2017.01.001
Papathomas TG, Suurd DPD, Pacak K et al (2021) What have we learned from molecular biology of paragangliomas and pheochromocytomas? Endocr Pathol 32:134–153. https://doi.org/10.1007/S12022-020-09658-7
Turchini J, Cheung VKY, Tischler AS et al (2018) Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology 72:97–105. https://doi.org/10.1111/HIS.13402
Ebbehoj A, Stochholm K, Jacobsen SF et al (2021) Incidence and clinical presentation of pheochromocytoma and sympathetic paraganglioma: a population-based study. J Clin Endocrinol Metab 106:E2251–E2261. https://doi.org/10.1210/CLINEM/DGAA965
Bholah R, Bunchman TE (2017) Review of pediatric pheochromocytoma and paraganglioma. Front Pediatr 5. https://doi.org/10.3389/FPED.2017.00155
de Tersant M, Généré L, Freyçon C et al (2020) Pheochromocytoma and paraganglioma in children and adolescents: experience of the French Society of Pediatric Oncology (SFCE). J Endocr Soc 4. https://doi.org/10.1210/JENDSO/BVAA039
Erickson D, Kudva YC, Ebersold MJ et al (2001) Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab 86:5210–5216. https://doi.org/10.1210/JCEM.86.11.8034
Dannenberg H, van Nederveen FH, Abbou M et al (2005) Clinical characteristics of pheochromocytoma patients with germline mutations in SDHD. J Clin Oncol 23:1894–1901. https://doi.org/10.1200/JCO.2005.07.198
Smith JD, Harvey RN, Darr OA et al (2017) Head and neck paragangliomas: a two-decade institutional experience and algorithm for management. Laryngoscope Investig Otolaryngol 2:380–389. https://doi.org/10.1002/LIO2.122
Bernardo-Castiñeira C, Valdés N, Sierra MI et al (2018) SDHC promoter methylation, a novel pathogenic mechanism in parasympathetic paragangliomas. J Clin Endocrinol Metab 103:295–305. https://doi.org/10.1210/JC.2017-01702
Settas N, Faucz FR, Stratakis CA (2018) Succinate dehydrogenase (SDH) deficiency, Carney triad and the epigenome. Mol Cell Endocrinol 469:107–111. https://doi.org/10.1016/J.MCE.2017.07.018
Burnichon N, Buffet A, Parfait B et al (2012) Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet 21:5397–5405. https://doi.org/10.1093/HMG/DDS374
Welander J, Larsson C, Bäckdahl M et al (2012) Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet 21:5406–5416. https://doi.org/10.1093/HMG/DDS402
Dahia PLM, Ross KN, Wright ME et al (2005) A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet 1:0072–0080. https://doi.org/10.1371/JOURNAL.PGEN.0010008
Boedeker CC, Hensen EF, Neumann HPH et al (2014) Genetics of hereditary head and neck paragangliomas. Head Neck 36:907–916. https://doi.org/10.1002/HED.23436
Funding
Ministero della Salute, RC-2022-2773478.
Author information
Authors and Affiliations
Contributions
Conception and design: GT, ADL; administrative support: GT; provision of study materials or patients: ADL, MR, DdB; collection and assembly of data: ADL, MR, TM, SC, AR; manuscript writing: ADL, MR, DdB, GT; final approval of manuscript: all authors.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Antonio De Leo and Martina Ruscelli share first authorship. Dario de Biase and Giovanni Tallini share senior authorship.
The original online version of this article was revised: The missing keywords has been added.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
De Leo, A., Ruscelli, M., Maloberti, T. et al. Molecular pathology of endocrine gland tumors: genetic alterations and clinicopathologic relevance. Virchows Arch 484, 289–319 (2024). https://doi.org/10.1007/s00428-023-03713-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-023-03713-4