
Abstract
Duchenne muscular dystrophy is a highly progressive muscle wasting disorder due to primary abnormalities in one of the largest genes in the human genome, the DMD gene, which encodes various tissue-specific isoforms of the protein dystrophin. Although dystrophinopathies are classified as primary neuromuscular disorders, the body-wide abnormalities that are associated with this disorder and the occurrence of organ crosstalk suggest that a multi-systems pathophysiological view should be taken for a better overall understanding of the complex aetiology of X-linked muscular dystrophy. This article reviews the molecular and cellular effects of deficiency in dystrophin isoforms in relation to voluntary striated muscles, the cardio-respiratory system, the kidney, the liver, the gastrointestinal tract, the nervous system and the immune system. Based on the establishment of comprehensive biomarker signatures of X-linked muscular dystrophy using large-scale screening of both patient specimens and genetic animal models, this article also discusses the potential usefulness of novel disease markers for more inclusive approaches to differential diagnosis, prognosis and therapy monitoring that also take into account multi-systems aspects of dystrophinopathy. Current therapeutic approaches to combat muscular dystrophy are summarised.
Similar content being viewed by others


Introduction
Contractile tissues in general, and skeletal muscle fibres in particular, occupy a special position in the physiological systems of the human body, making up approximately 40% of body weight. Voluntary contractile fibres and their associated cell types display a remarkable array of special features on various levels of biological organisation ranging from genotype to phenotype [226]. Although enormous progress has been made in the elucidation of the underlying mechanisms of myogenesis [44, 307] and muscle plasticity [262, 263], various fundamental questions of skeletal muscle physiology remain to be fully resolved [273]. Many of the functional and structural specialisations of the muscular system play body-wide roles in health and disease, affecting especially locomotion, posture, heat homeostasis and metabolic networks and their integration [50]. Given this context, the complexity and multifunctionality of the constituents of the skeletal muscle proteome is reflected by the diversity of muscular disorders [81]. In addition to neurological, metabolic and autoimmune diseases that indirectly affect the motor system, intrinsic disorders of skeletal muscles manifest as inflammatory myopathies, myotonias, congenital myopathies, pharmacogenetic myopathies and muscular dystrophies [71, 340]. The current list of neuromuscular disorders includes over 1,000 individual pathologies with over 600 identified genes that are associated with monogenic neuromuscular disorders [21], whereby many of these muscular disorders are already diagnosed at young age [70]. The most frequently inherited primary muscle disease of early childhood is Duchenne muscular dystrophy (DMD) [197], a highly progressive disorder of voluntary contractile fibres [88, 98, 215] that is associated with a high level of caregiver burden and illness costs [289].
On the level of the skeletal musculature, genetic defects in the DMD gene cause the almost complete loss of the membrane cytoskeletal protein dystrophin, which causes progressive symmetrical muscle wasting, in combination with sterile inflammation, fat substitution and reactive myofibrosis. A variety of recent reviews provide excellent details on the discovery of dystrophin [130], the genetic basis of dystrophinopathy [84, 117, 228], the complexity of pathophysiological mechanisms that underlie the muscle-related pathogenesis [6, 141, 300, 319, 340], diagnosis and clinical management of Duchenne patients [25,26,27, 215] and novel therapeutic strategies to treat progressive muscle degeneration and associated complications in X-linked muscular dystrophy [101, 298, 299]. This review builds on this accumulated knowledge on dystrophinopathy with a focus on the concept that Duchenne muscular dystrophy displays multi-systemic abnormalities. This biomedical idea is especially based on recent findings generated by large-scale analyses of both genetic disease models and patient specimens. Therefore, this article attempts to provide an inclusive overview of the molecular and cellular aspects that lead to highly complex skeletal muscle degeneration in association with a multi-systems pathogenesis and organ crosstalk in X-linked inherited muscular dystrophy [111, 324]. Following an introduction into the organisation of the DMD gene, its tissue-specific expression pattern and the tight interactions within dystrophin complexes (which is crucial for our understanding of the role of the various dystrophin isoforms in multiple tissue and organ systems), the structure and function of the various protein products of the dystrophin gene and their pathophysiological role are examined.
Individual sections of this review outline crucial aspects of the differential effects of dystrophin deficiency on different skeletal muscles, late-onset cardio-respiratory complications and associated multi-system abnormalities including aberrant functioning of the nervous system, the liver, the kidney, the gastrointestinal tract and the immune system. Since systems biological approaches have been extensively applied to studying the molecular and cellular pathogenesis of muscular dystrophy and have resulted in the identification of complex biomarker signatures, the suitability and robustness of novel diagnostic and prognostic biomarker candidates of dystrophinopathy and organ crosstalk are presented. This includes the discussion of cell, tissue and organ disease markers in association with biofluid-related markers, as well as the evaluation of the potential for establishing therapeutic biomarkers of pharmacological interventions, cell-based approaches and gene therapies to counter-act dystrophin deficiency.
The DMD gene, its expressed proteoforms and the genetic basis of dystrophinopathy
In skeletal muscle fibres, one of the largest genes in the human genome, the X-chromosomal 79-exon spanning Dmd gene [130, 164], exhibits the highest expression levels in form of the full-length Dp427-M isoform of the membrane cytoskeletal protein dystrophin [228]. The DMD gene is positioned on the short arm of the X-chromosome at the Xp21.2 band. As outlined in Fig. 1, the 79 exons encode an amino-terminal region with an actin-binding site, 4 proline-rich hinge regions, large central spectrin-like rod domains, a cysteine-rich domain and a carboxy-terminal domain with binding sites for various dystrophin-associated proteins [164,165,166]. The large number of distinct binding sites displayed by the protein product of the DMD gene provides the structural basis for a supramolecular dystrophin node at the sarcolemma [75], as outlined below. The tissue-specific expression of dystrophin isoforms is driven by seven different promoters. The protein products include three full-length dystrophins in brain, muscle and Purkinje cells, i.e. isoforms Dp427-B, Dp427-M and Dp427-P [228, 261] and shorter isoforms in the retina, brain/kidney, Schwann cells, the brain and a variety of other tissues, i.e. Dp260-R [349], Dp140-B/K [182, 183], Dp116-S [198, 206], Dp71-G [240] and Dp45 [278].

Overview of the DMD gene, its promoter structure and tissue-specific expression pattern of dystrophin isoforms. Abbreviations used: B, brain; B/K, brain/kidney; Dp, dystrophin protein; G, general; M, muscle; P, Purkinje cell; R, retina; S, Schwann cell
The Dp427-M isoform belongs to the class of giant proteins [233] and was identified in all major types of contractile tissues, including skeletal muscle, cardiac muscle and smooth muscle [75, 134, 162]. An overview of the basic structure of the various isoforms of dystrophin, as compared to its autosomal homologue utrophin and the dystrobrevin family of proteins, is provided in Fig. 2. In contrast to the X-chromosomal DMD gene, the genes UTRN, DTNA and DTNB are autosomal and encode the dystrophin-related proteins utrophin, alpha-dystrobrevin and beta-dystrobrevin with the chromosomal locations 6q24, 18q12 and 2p24, respectively. Full-length utrophin of apparent 395 kDa is highly enriched in the neuromuscular junction [115, 251] and exists in several isoforms, including A-Up395, B-Up395, Up140, G-Up113 and Up71 [260, 342, 372]. Dystrobrevins were shown to exist as four distinct isoforms, i.e. alpha-DYB-1, alpha-DYB-2, alpha-DYB-3 and beta-DYB [29, 241, 271].

Domain structure of full-length dystrophin, shorter dystrophin isoforms and the main types of dystrophin-related proteins. Abbreviations used: B, brain; B/K, brain/kidney; CT, carboxy-terminus; CR, cysteine-rich domain; Dp, dystrophin protein; G, general; DYB, dystrobrevin; H, proline-rich hinge region; M, muscle; NT, amino-terminus; P, Purkinje cell; R, retina; S, Schwann cell; SLR, spectrin-like rod domain; WW, conserved region with signature tryptophan residues; Up, utrophin
The complexity and enormous size of the Dmd gene with its 2.4-Mbp sequence requires considerable processing of the dystrophin 14-kb mRNA and a lengthy period for transcription [332, 346]. A large array of mutations and genetic rearrangements in the Dmd gene result in distinct effects on the various protein products, including abnormal size and/or amount of dystrophin isoforms in X-linked muscular dystrophy [84]. As listed in Fig. 1, primary genetic abnormalities in the DMD gene on the short arm of the X-chromosome include small and large deletions, small and large insertions, large duplications, missense point mutations, nonsense point mutations, splice site mutations and mid-intronic mutations [28, 99]. Diagnostic testing of these diverse primary abnormalities in the DMD gene can be routinely performed with a variety of genetic techniques [297], such as (i) diverse types of polymerase chain reaction assays [2] that mostly focus on the analysis of potential deletions [149], (ii) comparative genomic hybridisation array technology that can predict whether genetic changes may disrupt the reading frame [204], (iii) multiplex ligation-dependent probe amplification methods which are capable of swiftly assessing the copy number of exons and related genetic changes [292] and (iv) next-generation sequencing for the analysis of nonsense or missense types of point mutations, as well as small deletions [243, 255]. Genomic sequencing has also been applied to the detailed genetic characterisation of female carriers of the mutated DMD gene [368]. While the highly progressive Duchenne type of X-linked muscular dystrophy is characterised by genetic defects that result in the almost complete loss of the Dp427-M isoform of dystrophin in contractile tissues [30, 133], late-onset and less progressive Becker muscular dystrophy shows only reduced density and/or size of the dystrophin protein [170]. Hence, the difference between severe versus more benign forms of dystrophinopathy is based on the type of genetic alterations [297] and whether particular mutations cause the loss of dystrophin or the production of an abnormal but still semi-functional protein product [255].
Association of dystrophin with other proteins
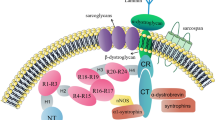
In the sub-sarcolemmal cytoskeleton of skeletal muscle fibres, the full-length isoform of dystrophin does not exist in isolation but forms a tightly associated membrane assembly [89, 90, 249, 252]. As recently reviewed, dystrophin can be considered an organising node of the muscle periphery [75]. A sub-complex consisting of dystrophin isoform Dp427-M and its associated integral glycoprotein beta-dystroglycan form the basis of a sarcolemma-spanning structure [147], which tightly interacts with the extracellular laminin-receptor alpha-dystroglycan, the integral glycoproteins alpha/beta/gamma/delta-sarcoglycan and the highly hydrophobic membrane component sarcospan, as well as the cytosolic proteins alpha/beta-dystrobrevin and alpha/beta-syntrophin [234]. This dystrophin core complex in turn provides a linkage to (i) the wider extracellular matrix (including laminin-211, fibronectin, biglycan and collagen isoforms COL-IV and COL-VI) [212, 230, 268], (ii) the intracellular cytoskeletal network (including cortical actin, cytokeratin, desmin, vimentin, tubulin, synemin and plectin) [23, 266, 274, 350], (iii) signalling proteins (such as the neuronal isoform nNOS of nitric oxide synthase, various kinases, the aquaporin water channel, the growth factor receptor-bound protein Grb2 and the insulin receptor, as well as crucial ion-regulatory proteins including Na+ channels, inward rectifier K+ channels, voltage-sensing L-type Ca2+ channels and transient receptor potential cation channels) [46, 103, 179, 290, 374] and (iv) the costamere structures (in conjunction with the mechano-sensing axis of integrin, vinculin and talin) of the fibre periphery [151].
Thus, the dystrophin node functions as a central integrator of fibre stability, cytoskeletal organisation, cellular signalling and lateral force transmission [6, 51, 75, 179]. The discovery that dystrophin interacts directly with tubulin puts this membrane-associated protein into the class of cytolinkers [266]. Since besides dystrophin, the dystrophin-associated protein complex also plays a key role in the pathogenesis of dystrophinopathy [137], its core structure and diverse interconnectivity with the actin cytoskeleton, sarcolemma membrane, ion-regulatory components and the extracellular matrix is shown diagrammatically in Fig. 3. Besides in skeletal muscles, where the dystrophin complex was shown to form a monomeric structure of apparent 1.2 MDa size [287], this membrane-associated protein assembly also exists in the heart [154] and smooth muscle cells, such as the outer layers of the stomach wall [77]. However, the structure and subcellular localisation is slightly different as compared to skeletal muscle fibres. In cardiac muscle, the dystrophin complex was shown to be also present in the transverse tubular membrane system [154, 162] in contrast to the almost exclusive subcellular localisation in the sarcolemma membrane in skeletal muscles [252, 366, 385].

Interaction sites of dystrophin isoform Dp427-M and overview of the dystrophin-glycoprotein complex at the sarcolemma of skeletal muscle fibres. Abbreviations used: ABD, actin-binding domain; CT, carboxy-terminus; CR, cysteine-rich domain; DG, dystroglycan; DYB, dystrobrevin; Dp427-M, muscle-specific dystrophin isoform of 427 kDa; H, proline-rich hinge region; nNOS, neuronal isoform of nitric oxide synthase; NT, amino-terminus; SG, sarcoglycan; SLR, spectrin-like rod domain; SSPN, sarcospan; SYN, syntrophin; WW, conserved region with signature tryptophan residues
Developmental stages of dystrophinopathy and skeletal muscle fibre degeneration
Duchenne muscular dystrophy is the most common neuromuscular disorder of early childhood with a prevalence of approximately 1 in 5,000 live male births [52, 197] and is often initially detected due to developmental delay in conjunction with contractile weakness, slower walking and Gower’s sign, indicating weakness of proximal muscles [84, 98, 111]. Major clinical milestones of the developmental stages of dystrophinopathy are summarised in Fig. 4. The presence of Gower’s sign in Duchenne patients relates to proximal muscular weakness in hip and thigh muscles, which requires the help of both hands and arms for rightening the body to reach a standing position. Muscular dystrophy-associated temporal and spatial variations in gait were shown to include changes in cadence, anterior pelvic tilt and dorsiflexion during swing [68]. The proper assessment of pathological gait patterns and functional ambulation are crucial for prediction of disease progression, as well as monitoring of drug treatment and physiotherapeutic interventions [157]. Detailed studies of gait abnormalities have established a drastic decrease in walking speed, stride length, step length, maximal power generation at the hip, maximal knee extension torque, maximal dorsiflexion torque and maximal power generation at the ankle in Duchenne children [112, 113]. Common complications are hip, knee and ankle joint plantar flexion contractures [47]. Progressive muscle weakness also affects bone strength due to low bone mineral density in Duchenne patients [285]. This may cause an increased risk of bone fragility [25, 221], especially in association with prolonged glucocorticoid therapy [327, 365]. Key developmental stages of the disease include initially frequent falls, difficulties with climbing stairs, toe walking and a waddling gait, followed by progressive limitations in general mobility, respiratory insufficiency and scoliosis [105].

Summary of developmental stages of dystrophinopathy and muscle symptoms
At a later stage, the loss of unassisted ambulation is followed by weakness of upper body musculature [25,26,27, 215]. Loss of ambulation as a clinical indicator of disease progression was shown to correlate with the type of genetic abnormality in the DMD gene. Of note, small mutations were associated with a younger age of loss of ambulation as compared to large deletions or duplications [19]. In the second decade of life, cardiomyopathy and respiratory failure requires ventilatory assistance and intervention with cardiac drug treatment [161, 216]. Duchenne patients also exhibit an increased susceptibility for fatty liver disease, gastrointestinal complications, renal failure, and bladder dysfunction [26, 200, 357]. Steady advances in cardiopulmonary care and pharmacological therapy have preserved quality of life and improved prognosis for survival of Duchenne patients in recent years. Premature death occurs usually at 20 to 40 years of age [27, 174]. However, cases of individual Duchenne patients living into their fifth and even sixth decade of life have been described [291, 360]. Especially the usage of oral corticosteroids has significantly prolonged ambulation in patients suffering from Duchenne muscular dystrophy, and therapy with angiotensin-converting enzyme inhibitors and beta-blockers was shown to delay the progression of cardiomyopathic complications [335]. The importance of ventilation is clearly supported by findings from a recent meta-analysis that has established differences in median life expectancy of 14–27 years versus 21–40 years in patients without versus with ventilatory support, respectively [174].
For the differential diagnostic evaluation of dystrophic patients, the above-described genetic tests are routinely utilised in conjunction with general physical examinations, motor and gait assessments, the evaluation of muscle biopsy specimens using histological and immunochemical tests [248] and serum assays focusing on the status of general muscle damage markers such as creatine kinase [237, 250]. In addition, magnetic resonance imaging presents a crucial non-invasive and multi-parametric assessment tool for the quantification of muscle pathology [4]. Muscle imaging correlates relatively well with histologic parameters [160] and can be employed for both diagnostic purposes and the extended monitoring of progressive alterations in the dystrophin-deficient skeletal musculature and clinical outcome measures [371]. Recently, the evaluation of increased collagen levels by multispectral optoacoustic tomography has been established and can be employed as an advanced imaging tool for the characterisation of reactive myofibrosis in association with dystrophinopathy [272]. The pathophysiological importance of fibrotic changes in dystrophinopathy is discussed in more detail in the below section.
In progressive X-linked muscular dystrophy, the decline in skeletal muscle strength is reflected on the histological level by characteristic changes in fibre size, a more roundly appearance of myofibres and a high degree of central nucleation, as well as myonecrosis, clusters of inflammatory cells, hypercontractility, fibre branching, fatty deposition and myofibrosis. Figure 5 illustrates the subcellular localisation of dystrophin isoform Dp427-M in skeletal muscle using immunofluorescence microscopy. Full-length dystrophin exhibits a peripheral localisation [252, 385] whereby this membrane cytoskeletal protein is proposed to form a stabilising lattice at the cytoplasmic face of the sarcolemma membrane [366]. A striking feature of dystrophinopathy is the almost complete loss of dystrophin and a drastic reduction in all dystrophin-associated proteins in contractile fibres [253]. Dystrophin deficiency renders myofibers more susceptible to micro-rupturing of its plasmalemma and contraction-induced injury. Leaky surface membrane systems and impaired luminal calcium buffering [53] were shown to trigger impaired calcium handling [13, 72, 74], abnormal excitation–contraction coupling [39] and activation of calcium-dependent proteolytic degradation of muscle proteins [3, 144, 202].

Absence of dystrophin isoform Dp427-M in X-linked muscular dystrophy and key histopathological features of dystrophinopathy. Shown is the immunofluorescence microscopical analysis (using monoclonal antibody NCL-DYS1 to dystrophin and Hoechst-33342 labelling of nuclei) and histological comparison (using haematoxylin and eosin staining; H&E) of skeletal muscle cryosections from wild type (wt) versus the mdx-4cv mouse model of Duchenne muscular dystrophy. Bar equals 40 μm
Physiological dysregulation and enhanced proteolysis in dystrophic fibres are accompanied by a sustained cellular stress response and the upregulation of various chaperoning proteins to counteract proteotoxic insults to dystrophin-deficient fibres [35], especially small heat shock proteins such as alphaB-crystallin and the muscle-specific chaperone cvHSP [73]. The comprehensive mass spectrometric profiling of muscle biopsy samples from Duchenne patients has confirmed severe cytoskeletal and extracellular dysregulation in Dp427-deficient skeletal muscle [38].
The importance of abnormal calcium homeostasis in X-linked muscular dystrophy is summarised in Fig. 6, which also highlights the pathophysiological interconnectivity between the innate immune response to muscle damage and resulting activation of myofibroblasts and their role in reactive myofibrosis. In addition to dysregulated ion homeostasis, sterile inflammation and myofibrosis, the drastic reduction of the dystrophin-associated nNOS isoform of nitric oxide synthase in dystrophic fibres affects signalling between contractile cells and their microvasculature causing use-dependent muscle ischemia [339]. A recent study using Doppler sonography established that the severity of the dystrophic phenotype correlates with a reduction in post-exercise blood flow in Duchenne patients [67]. Besides the central pathophysiological role of myofiber fragility, intrinsic satellite cell dysfunction also contributes to progressive muscle wasting via an impaired regenerative capacity of dystrophic muscles [45, 319].

Pathophysiological role of chronic inflammation, reactive myofibrosis and abnormal calcium handling in the molecular and cellular pathogenesis of dystrophinopathy. Abbreviations used: COL, collagen; DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; MMP, matrix metalloproteinase; PA, plasminogen activator; TIMP, tissue inhibitor of metalloproteinases; TGF, transforming growth factor; TNF, tumour necrosis factor; NF-κB, nuclear factor kappa B
In addition to a high frequency of micro-rupturing of the dystrophin-deficient sarcolemma and associated increases in cytosolic calcium levels and enhanced proteolytic degradation [144], the neuromuscular junction region was also shown to be more susceptible to contraction-induced injury, which results in impaired neuromuscular transmission [195, 244, 319]. Since the low-frequency versus high-frequency electro-stimulation pattern at the motor endplate dictates the slow-twitching versus fast-twitching properties within individual motor units [262, 263], a difference in the susceptibility of slow versus fast synaptic systems might play a key pathophysiological role in dystrophinopathy. Various neuromuscular disorders display a certain degree of fibre type specificity in muscle wasting [48, 81]. In Duchenne patients, subsets of fast myofibers appear to be more susceptible to initial degenerative processes as compared to a later onset of fibre wasting in slower-twitching fibre populations [367]. This correlates with an early decrease of fast myosin heavy-chain isoforms in dystrophic fibres [203, 259]. Indirect fast-to-slow transition processes were also observed in the DMD pig model of X-linked muscular dystrophy [104] and murine mdx skeletal muscle, whereby muscle transformation was shown to be linked to elevated levels of fibre respiration and enhanced protection from muscle damage [122]. Importantly, differential susceptibilities occur at the level of whole skeletal muscles. For example, extraocular muscles exhibit a very mild phenotype due to dystrophin deficiency [9].
Sparing of naturally protected extraocular muscles in dystrophinopathy
Although dystrophin deficiency is clearly associated with highly progressive skeletal muscle degeneration, different subtypes of skeletal muscles are affected in distinctive ways. In contrast to severely degenerative limb and trunk muscles, extraocular muscles are spared from severe dystrophic changes [80, 100]. Unusual cell biological features of extraocular muscles include anatomical, biomechanical and functional compartmentalisation, myogenic processes driven by specific upstream activators, a longitudinal distribution of multi-terminal motor endplates along contractile fibres and morphologically distinct muscle spindles [63, 355]. The large variety of contractile patterns, including extraordinary fatigue resistance of fast fibres, within the extraocular muscle system is probably based on a broad distribution and expression pattern of key sarcomeric proteins, such as a large variety of slow and fast isoforms of light and heavy myosin chains [135]. A recent proteomic survey of the extraocular muscle proteome has established MyHC14 and MyHC15 as new markers of this subtype of skeletal muscles [106], in addition to the already-well-established super-fast myosin isoform MyHC13 [135].
The naturally protected phenotype of dystrophin-deficient extraocular muscles has been linked to a highly efficient calcium extrusion system, a specialised stem cell niche that provides efficient cellular regeneration and an enhanced remodelling capacity, the non-junctional upregulation of the dystrophin homologue utrophin Up-395 and concomitant rescue of dystrophin-associated glycoproteins, an enhanced cellular stress response, metabolic adaptations, the lack of fibrotic scarring and the relatively low load bearing of extraocular fibres [74, 106, 355, 378]. The fact that the contractile system surrounding the eyeball stays functionally unaffected in Duchenne muscular dystrophy is of considerable biomedical importance [9]. A better comprehension of the molecular and cellular processes that underlie the sparing of extraocular muscles during the course of disease progression in dystrophinopathy could be helpful for the identification of new therapeutic targets to counteract dystrophin deficiency, i.e. manipulation of calcium handling, utrophin replacement therapy, the targeted upregulation of molecular chaperones or improving the capacity for cellular regeneration.
Reactive myofibrosis and chronic inflammation as key symptoms of dystrophinopathy
The disproportionate accumulation of proteins belonging to the extracellular matrix can be considered a key defining feature of dystrophinopathy [158, 254] and is probably closely linked to abnormal cellular signalling, extensive recruitment of the muscle repair machinery and chronic inflammation in the affected contractile tissues [309]. Chronic cycles of muscle tissue damage and fibre repair are triggered by dystrophin deficiency and cause a sustained immune response, which results in a chronic inflammatory phenotype of dystrophinopathy [280, 314, 315]. The innate immune response is accompanied by high levels of macrophage activity and the release of a variety of signalling factors, as well as the recruitment of myofibroblasts [31]. The potential interplay between chronic inflammation and myofibrosis in damaged fibres, as well as the role of abnormal Ca2+-handling in dystrophin-deficient fibres, is summarised in Fig. 6. Importantly, myofibroblasts exhibit an elevated synthetic capacity for the production of extracellular matrix proteins and therefore play a key role in reactive fibrotic changes in dystrophic muscle tissue.
The multi-functional cytokine named transforming growth factor TGF-β is released in large amounts from M2 macrophages and is involved in the activation of fibro-adipogenic progenitors [110] and other precursor cells in the muscle environment [20]. Enhanced activity of fibro-adipogenic progenitors plays a key role in dystrophinopathy. These precursors are resident in skeletal muscles and belong to the class of stromal cells that exhibit the potential to adapt to multiple cellular lineages [199]. Upon activation by fibre damage, fibro-adipogenic progenitors are involved in the generation of myofibroblasts, fibroblasts and adipocytes, as reviewed by Theret et al. [338]. Enhanced secretion of extracellular matrix components can then cause the excessive formation of fibro-fatty scars that surround contractile fibres and thereby negatively affect mechano-transduction and skeletal muscle elasticity [163]. In addition, adipogenic precursors were shown to be involved in interstitial remodelling, which is associated with disturbed adipogenesis [36]. This might explain the concomitant occurrence of reactive fibrosis and fat replacement in Duchenne muscular dystrophy causing major cellular complications for maintaining proper metabolic and contractile functions.
In one of the original descriptions of X-linked muscular dystrophy dating back to the year 1868, the French neurologist Guillaume-Benjamin-Amand Duchenne de Boulogne described an abundant production of fibrous tissue at the advanced stages of the disease and proposed to name this muscular disorder ‘paralysie myosclérosique’ (archived in [85]: Duchenne GB. Recherches sur la paralysie musculaire pseudo-hypertrophique ou paralysie myosclérosique. Archives générale médecine). The original medical description is certainly in agreement with the interstitial fibrotic phenotype of Duchenne muscular dystrophy. In normal skeletal muscles, the basal lamina and the extended layers of the extracellular matrix (consisting of the endomysium, perimysium and epimysium [379]) provide a supporting and signalling environment that forms protective sheets around contractile fibres [109]. Importantly, motor neurons and capillaries are embedded in the interstitial extracellular matrix for efficient neurotransmission within motor units and the steady supply of essential nutrients and oxygen to support the high bioenergetic needs of skeletal muscle metabolism.
Key proteins involved in structural maintenance of the extracellular matrix-sarcolemma axis via cell–matrix adhesion processes and the provision of mechanical support and lateral force transmission include various (i) collagen isoforms (Col I, III, IV, V and VI), (ii) a large number of proteoglycans (biglycan, prolargin, mimecan, decorin, asporin, fibromodulin, perlecan, syndecan, lumican, agrin and aggrecan), (iii) matrix crosslinkers (fibronectin), (iv) non-architectural matricellular proteins (periostin, osteopontin, dermatopontin and nidogen), (v) matrix metalloproteinases (MMP 1, 2, 9, 10 and 13) and their inhibitors named tissue inhibitors of metalloproteinases (TIMP) and plasminogen activators (PA), (vi) integrin (α7β1-integrin), (vii) laminin (α2β1γ1 laminin-211) of the basal lamina, (vii) signalling proteins (myokines and growth factors) and (vii) the dystroglycan complex with its laminin-binding component alpha-dystroglycan [109, 141, 254, 379]. Disturbances of adhesion receptors and rearrangements of structural fibres within the extracellular matrix play an important pathophysiological role in many neuromuscular pathologies, including X-linked muscular dystrophy [158]. Since the extracellular matrix is intrinsically involved in the maturation and differentiation of muscle fibres and adaptive fibre transitions, myofibrosis has a considerable influence on the loss of regenerative capacity in dystrophic muscles [163]. The loss of tissue elasticity and progressive cellular scarring causes decreased mechano-transduction and impaired skeletal muscle function, and may also affect crosstalk between muscle and tendon [375].
As indicated in Fig. 6, a large number of extracellular matrix proteins were shown to be drastically increased or modified in their isoform expression pattern in both patients and animal models of X-linked muscular dystrophy, including collagens, proteoglycans, adhesion receptors, matricellular proteins and matrix metalloproteinases [7, 92, 309, 344]. Systematic large-scale surveys using mass spectrometry–based proteomics identified a drastic increase in the matricellular proteins dermatopontin [41] and periostin [138], as well as the extracellular matrix stabilisers biglycan and fibronectin [230, 239] in conjunction with elevated collagen expression in the highly fibrotic- and dystrophin-deficient diaphragm muscle [141]. Especially striking are high levels of the cross-linking enzyme lysyl oxidase and concomitant increases in the level of cross-linked collagen clusters in the disturbed extracellular matrix of the dystrophic diaphragm [310]. These findings were confirmed by a longitudinal study of histological changes during progressive muscle wasting in Duchenne patients, which clearly established that endomysial fibrosis presents the most significant myopathological feature in correlation to the gradual loss of motor functions [64]. It is therefore not surprising that collagens detected by advanced imaging technology have been suggested as novel biomarkers for dystrophinopathy [272]. Primary fibroblasts isolated from Duchenne patients produce elevated levels of decorin and collagen, and are characterised by elevated proliferation rates [376, 377]. This pro-fibrotic phenotype shows high sensitivity to transforming growth factors, which agrees with the finding that muscular dystrophy-associated fibrosis is driven by the transforming growth factor TGF-β-related activation pathway [196, 208, 356]. Interestingly, the serum- and glucocorticoid-inducible kinase SGK1 appears to be a fibrosis-stimulating factor that plays a role in fibrotic remodelling and muscular weakness [321].
These findings of dysregulated matricellular proteins and elevated collagen fibrillogenesis due to hyperactive fibroblast populations are crucial for our general understanding of the detrimental role of myofibrosis in X-linked muscular dystrophy. Skeletal muscles appear to be capable to swiftly address minor acute injuries by upregulating repair mechanisms and the regenerative activation of myoblasts and protective involvement of fibroblasts [379]. However, chronic muscle wasting seems to overwhelm the beneficial effects of elevated activity levels of the connective tissue and causes instead fibrotic tissue scarring [141, 254]. As discussed in the below section on novel treatment options for dystrophinopathies, it will be crucial to counteract the progressive nature of fibrotic changes in order to increase the chance for the successful application of new cell-mediated or gene-based therapies [295]. However, a recent study on dystrophinopathy-associated fibrosis has shown different degrees of altered muscle stiffness and collagen amounts in extensor digitorum longus versus soleus muscles [34], suggesting the selective targeting of the alignment of large collagen structures rather than total collagen for the most efficient treatment of fibrotic stiffness in X-linked muscular dystrophy [309, 310].
The inflammatory phenotype of progressive skeletal muscle degeneration due to dystrophin deficiency is associated with a variety of cellular immune responses [280], especially interference by the innate immune system [31] but also acquired immune responses [314]. As already discussed above, there appears to be a close link between chronic inflammation and reactive myofibrosis in X-linked muscular dystrophy [20]. Resident immune cells play an important role in normal skeletal muscle homeostasis. Of note, a resident macrophage population associated with the epimysium and perimysium space between contractile fibres can swiftly act and remove cellular debris. In muscular dystrophy, the conversion of activated monocytes and the invasion by large numbers of additional macrophages is a key feature of muscle membrane lesions [358, 359]. Besides M1 and M2 macrophages, the promotion of the degenerative phenotype seen in dystrophic muscle fibres includes additional immune cells, including helper CD4 + T lymphocytes, cytotoxic CD8 + T lymphocytes, eosinophiles and neutrophiles as first responders of inflammation, as well as infiltration of the dystrophic muscle by myeloid cell populations [314, 315].
High levels of contraction-induced injury due to dystrophin deficiency cause fibre disintegration, and the cellular damage to the weakened sarcolemma is associated with damage-associated molecular patterns (DAMPs) and the release of typical DAMP components such as nucleic acids and ATP molecules [219]. DAMP molecules are recognised by the innate immune system and result in an inflammatory response with a central role played by the nuclear factor NF-κB and the inflammasome [280]. In addition, the shedding of peptides, protein fragments or proteins through the leaky muscle plasmalemma presents these muscle components as potential neoantigens to the adaptive immune system [341]. Importantly, a large number of signalling cytokines and chemokines promote the infiltration of dystrophic fibres by neutrophiles, macrophages and dendritic cell populations [31, 319].
The recent proteomic analysis of the role of the spleen in X-linked muscular dystrophy confirmed pathophysiological crosstalk between dystrophic muscles and the secondary lymphoid organ system [76]. The spleen, which is majorly involved in antigen detection, antibody production and the removal of abnormal erythrocytes, exhibits morphological adaptations of lymph nodes in its white pulp region due to dystrophin deficiency [293]. Importantly, muscular dystrophy is associated with changed numbers of splenic inflammatory monocytes and an increased migration pattern of immune cells from the splenic reservoir to damaged contractile fibres [219, 220, 256]. The movement of splenic monocytes and differentiation into macrophages seems to be crucial for promoting chronic inflammation in dystrophin-deficient skeletal muscles [277]. These major changes due to inflammatory mechanisms in dystrophic fibres are summarised in Fig. 6.
Late-onset cardiorespiratory pathophysiology in dystrophinopathy
Impaired cardiorespiratory function plays a key pathophysiological role in X-linked muscular dystrophy, especially in the second decade of life of Duchenne patients [25], and requires mechanical ventilatory support to manage respiratory insufficiency [32] and cardiac drug treatment including angiotensin-converting enzyme inhibitor therapy [161, 216]. Right and left diaphragmatic motions are reduced following inspiration [93], and progressive respiratory decline is already present during the early ambulatory phase in Duchenne patients [276]. Imaging studies of respiratory muscle movements revealed a drastically reduced thoracic cavity area and reduced chest wall contraction during inspiration-expiration patterns in muscular dystrophy, as well as increased fat infiltration in accessory respiratory muscles [17]. The progression of myocardial fibrosis in Duchenne and Becker muscular dystrophy patients is associated with poor prognosis. Thus, in analogy to skeletal muscle fibrosis and its detrimental effect on motor function [163, 272], excess accumulation of extracellular matrix components also plays a crucial role in progressive cardiomyopathic complications in dystrophinopathy.
Cardiomyocytes, which only exhibit limited regenerative capacity, do not undergo extensive degeneration-regeneration cycles in muscular dystrophy. The central importance of the cardio-respiratory syndrome and interconnectivity of body-wide effects of dystrophin deficiency is diagrammatically summarised in Fig. 7, which gives an overview of the complexity of abnormalities in the skeletal musculature, the central and peripheral nervous system, smooth muscles and the cardiovascular system, as well as potential organ crosstalk that is related to secondary dysfunctions in the liver and the renal-urinary tract. Interstitial fibrosis and myofiber necrosis cause cardiac weakness which in turn negatively affects efficient circulation. Thus, dystrophinopathy-associated cardiomyopathy is probably indirectly linked to a decreased whole-body supply of nutrients, oxygen and hormones in X-linked muscular dystrophy. Chronic impairment of circulation might be involved in fatty liver disease, as discussed in more detail in the below section on organ crosstalk. Both the long-term impairment of the circulatory system and chronic exposure to released intracellular molecules from damaged skeletal muscle fibres may also contribute to renal abnormalities in dystrophinopathy [172].

Overview of skeletal muscular degeneration, multi-systems pathophysiology and organ crosstalk in Duchenne muscular dystrophy
In analogy to skeletal muscle, full-length dystrophin also forms a tight complex with glycoproteins in the heart, where it occupies a major mechanical, protective and signalling role in the organisation and maintenance of the surface membrane system of cardiomyocytes [154, 162]. In conjunction with the talin-integrin system, the dystrophin complex associates with vinculin at costamers in a perpendicular direction to the longitudinal axis of cardiac muscle cells [56]. However, in contrast to skeletal muscle, the core cardiac dystrophin-glycoprotein complex exhibits a differential composition in relation to dystrobrevins and syntrophins and does not display a direct link to the nNOS isoform of nitric oxide synthase. The cardiac dystrophin complex features additional linkages to the molecular chaperone alphaB-crystallin, the large scaffolding phosphoprotein ahnak-1, the caveolae component cavin-1 and the cardiac cytoskeletal component cypher [152].
Important aspects of the molecular and cellular pathogenesis of dystrophinopathy-associated cardiomyopathy were determined by studying dystrophic mouse and pig models [139, 232, 331]. Systematic proteomic surveys of the dystrophin-deficient heart have revealed drastic changes in the dystrophin-associated glycoprotein complex [114], which in turn triggers an abnormal expression pattern of proteins involved in cytoskeletal networks, the extracellular matrix, the cardiac contractile apparatus, energy metabolism, signalling mechanisms and the cellular stress response [142]. On the subcellular and molecular level, the dystrophic heart is primarily characterised by sarcolemmal disintegration and significantly reduced levels of laminin, nidogen and annexin [232]. Pathophysiological patterns of cardiac fibre necrosis, interstitial inflammation and reactive myofibrosis result in heart disease in the majority of patients afflicted with Duchenne muscular dystrophy [216].
The fibrotic phenotype of the dystrophin-deficient heart resembles some of the alterations that occur during skeletal muscle fibrosis, including increased activity of pro-fibrotic genes, collagen accumulation, enhanced activity of connective tissue growth factors and heightened transforming growth factor TGF-β signalling via coronary endothelial cells [142, 148]. Workload-induced cardiomyocyte injury and the acute elevation of mechanical stress in the dystrophic heart is also linked to abnormal Ca2+ handling [175]. Fragility of the dystrophin-deficient plasma membrane and altered ion homeostasis is clearly associated with a lowered luminal Ca2+-buffering capacity of cardiomyocytes, hypersensitive excitation–contraction coupling and the activation of the stretch-activated Ca2+-influx pathway via transient receptor potential vanilloid channels of type TRPV2 [189, 192, 302].
Neurological complications in dystrophinopathy
The multi-system pathophysiology of Duchenne muscular dystrophy is characterised by a variety of neurological complications [210]. However, mental issues do not appear to be progressive and were shown not to correlate with the severe loss of motor function in dystrophic patients. Different degrees of neurodevelopmental, psychiatric, behavioural and emotional symptoms have been clearly established in a subset of individuals suffering from severe forms of dystrophinopathy [61] but can also occur in more benign cases of Becker muscular dystrophy [173]. Neurological issues are characterised by social, behavioural and emotional problems [129, 275], delayed milestones of language development [58], adaptive deficiencies [57], impaired working memory [336] and a variety of neuropsychiatric diseases such as hyperactivity, obsessive–compulsive behaviour, attention deficit and autism spectrum disorders [128, 257, 258]. However, it is important to emphasise that severe cognitive impairments and mental retardation occur only in a subgroup of patients and were shown to be secondary in nature to physical handicap [210, 311].
Non-progressive cognitive deficiencies in Duchenne patients were shown to correlate at least partially with the type of mutation in the DMD gene and accompanying effects on the expression levels of the various dystrophin isoforms in the central nervous system [69, 337]. Especially abnormalities in the dystrophin isoform Dp140 [16, 95] appear to be associated with severe forms of cognitive defects [18, 347]. Neuronal dystrophins include the short forms Dp45 and Dp71, the medium-sized isoform Dp140 and full-length Dp427 [228, 240]. Brain-associated proteoforms of Dp427 were shown to feature biochemical properties that are typical of cytoskeletal components and are similar to the actin-binding protein Dp427 present in skeletal muscles [96, 97]. The large Dp427 isoforms localise to neurons of the hippocampus and the cerebral cortex [159, 181, 348]. During the development of the central nervous system, the Dp140 isoform is most highly expressed in the brain [183]. However, following brain maturation, the most abundant dystrophin is represented by isoform Dp71 [240] that is mostly localised in neuronal and glia cells of the olfactory bulb and the dentate gyrus [54, 329]. In analogy to skeletal muscles, various dystrophin isoforms in the brain are linked to glycoproteins such as dystroglycans and sarcoglycans [55, 364]. Dystrophin-glycoprotein complexes were shown to be associated with key neuronal processes, including modulation of synaptic activity, excitability, plasticity and integration of signalling cascades [125, 260].
Abnormalities in brain white matter were recently established in children afflicted with dystrophinopathy by diffusion tensor imaging, a magnetic resonance imaging technique that is suitable to especially determine the axonal organisation within the brain [265]. These neurological complications seem to be connected to altered developmental pathways in the cerebellum resulting in disturbed cerebro-cerebellar loops [59]. Besides studying brains from Duchenne patients by mostly non-invasive technologies, crucial insights into complex neurological changes have been generated by studying molecular and cellular brain abnormalities in animal models of dystrophinopathy. One of the most widely employed dystrophinopathy models, the mdx mouse, exhibits deficits in long-term consolidation memory, characteristic changes in associative learning patterns and bioenergetic alterations in distinct brain regions [227, 343, 351] making it a suitable system for detailed neurochemical and cellular studies. Important findings include the establishment of disturbed interactions between glial and endothelial cells at the blood–brain barrier [246], an abnormal neuronal receptor density in Purkinje cells [169] and the central role of impaired cerebellar function as highlighted by disrupted circuit signals between Purkinje cells and the cerebellar nuclei in the dystrophin-deficient brain [320].
The altered blood–brain barrier function in the mdx brain is associated with a reduction in dystrophin-associated glycoproteins [247] and upregulation of the matrix-metalloproteinases MMP2 and MMP9 [245]. Interestingly, the proteomic analysis of the mdx-4cv mouse model revealed increases in vimentin and annexin that might be associated with the cytoskeletal stabilisation of dystrophin-lacking brain cells and enhanced membrane repair processes [235]. Elevated expression levels of the glial fibrillary acidic protein, which is an established marker of astrogliosis due to its unique localisation to astrocytes in the central nervous system [136], were also demonstrated to occur in the mdx-4cv brain by mass spectrometry, immunoblotting and immunofluorescence microscopy [235]. This agrees with the reported occurrence of multifocal glial nodules in the brain of a Duchenne patient with severe mental retardation [150]. Increases of glial fibrillary acidic protein indicate ongoing neurodegeneration-associated astrogliosis with accumulation of this intermediate filament component in the brain lacking certain dystrophin isoforms, such as Dp427 and Dp140.
Impaired energy metabolism, abnormal liver function and gastrointestinal abnormalities in dystrophinopathy
The recent comparative proteomic profiling of muscle biopsy samples from Duchenne patients, as compared to healthy controls, has demonstrated metabolic disturbances both at the level of anaerobic pathways and lipogenesis [38]. Since skeletal muscle tissue plays a crucial role in the interorgan crosstalk of metabolic regulation [10] and because of a close relationship between nutritional uptake by the gastrointestinal tract, liver metabolism and skeletal muscle function, dystrophinopathy-associated changes that affect the efficient crosstalk between these vital organs are collectively discussed in this section. The combined skeletal musculature is a key organ system involved in the integration of carbohydrate, lipid and protein metabolism [10]. The absorption of digested nutrients through the gastrointestinal tract leads to the transportation of vital biomolecular building blocks via the circulatory system towards muscle, adipose and liver tissue. Contractile fibre protein forms an essential primary amino acid reservoir for the regulation of whole-body protein. This is crucial for substrate provision to maintain protein synthesis during periods of starvation and disease [286]. In addition, muscle tissues serve as the most abundant primary location for insulin-dependent glucose uptake throughout the body and storage of glycogen for utilisation within skeletal muscles. The shuttling of lactate and glucose between muscle and liver, also known as the Cori cycle, is a key interorgan pathway that links anaerobic skeletal muscle glycolysis with liver metabolism [313]. The interrelationship between liver, adipose tissue and skeletal muscles also determines the rate of fatty acid transportation and the regulation of oxidative metabolism [87].
A variety of muscular dystrophies are associated with a considerably perturbed skeletal muscle metabolism in both the early and acute phase of these disorder [318], including changed lipid utilisation and impaired energy metabolism in Duchenne patients [60, 176, 317]. Pathological developments in Duchenne children are not only characterised by highly progressive muscle wasting, but also delayed growth resulting in relatively short stature and increased fat mass. The altered muscle-to-fat ratio and overall body mass alterations in dystrophinopathy cause a high prevalence of obesity in Duchenne children and these body mass changes are linked to metabolic disturbances [294]. This in turn has an impact on bioenergetic requirements and nutritional status. While weight gain due to corticosteroid therapy might require weight management in younger Duchenne patients, issues with malnutrition [94] could necessitate supplemental feeding regimes at more advanced stages of the disease [26, 62]. The implementation of nutritional interventions, including micronutrient supplements such as calcium, creatine and vitamins, is a potential way to address certain aspects of bioenergetic dysregulation and loss of motor function due to progressive fibre degeneration [288].
Disturbed metabolism in X-linked muscular dystrophy is associated with abnormal calcium handling and mitochondrial dysfunction, reduced ATP levels, enhanced phosphorylation of the AMP-activated protein kinase and accumulation of reactive oxygen species [126]. Abnormal bioenergetic processes are also observed in dystrophic animal models including defective regulation of metabolic pathways [91]. Substrate-selective limitation of biochemical reaction rates of anaerobic metabolism in fast muscles and aerobic metabolism in slow muscles were shown to be associated with decreased oxidative utilisation of both glucose and free fatty acids [184, 322]. Excellent indicators of disturbed fatty acid metabolism are the various isoforms of fatty acid binding protein (FABP) [78]. A large number of proteomic surveys have established a reduced abundance of the FABP3 (H-FABP) isoform in various dystrophic skeletal muscles and the heart [116, 138, 230, 232, 239] and concomitant increase in serum [123, 231, 316]. In contrast to contractile tissues, the liver exhibits increased levels of the FABP5 (E-FABP) isoform of fatty acid protein in X-linked muscular dystrophy [236]. Elevated amounts of liver-associated FABP5 suggests changes in the binding of long-chain fatty acids in dystrophinopathy and agrees with fatty liver disease and ectopic fat accumulation being associated with the dystrophic phenotype [78]. This agrees with elevated serum levels of the liver damage marker alanine aminotransferase in the majority of Duchenne patients [167].
Besides skeletal and cardiac muscle, the dystrophin isoform Dp427 and its associated glycoprotein complex are also present in smooth muscle cells [120, 134]. A recent proteomic survey of the stomach wall identified dystrophin Dp427-M in association with alpha/beta-dystroglycan, alpha/beta/delta-sarcoglycan, gamma/epsilon-sarcoglycan, alpha/beta-dystrobrevin and alpha1/beta1/beta2-syntrophin [77]. The interface between the pancreas and the stomach of the mdx-4cv mouse model of X-linked muscular dystrophy wall was confirmed to be characterised by a loss in dystrophin and reduced abundance of sarcoglycan and dystroglycan, in addition to lower expression levels of the extracellular matrix component laminin, the sarcomeric protein titin and the actin-binding protein filamin [77]. The DMD pig model of muscular dystrophy also exhibits abnormal digestion and absorption capacity in the gastrointestinal tract [384]. The associated patterns of malnutrition are in agreement with gastrointestinal dysfunction in the dystrophic phenotype [26, 177, 186, 200].
Abnormal kidney and bladder function in dystrophinopathy
The kidney contains the Dp140-B/K dystrophin isoform and the shorter Dp71 proteoform of dystrophin [182] and dystrophin-associated proteins are present at relatively high abundance in renal cells [83, 119]. Dystroglycans were shown to play a crucial role during kidney epithelial morphogenesis [86]. However, in contrast to contractile fibres, the dystrophin-related protein β-dystrobrevin appears to occupy the central position in the anchoring of dystrophin-associated proteins in non-muscle tissues instead of dystrophin [187]. These dystrobrevin complexes were found to be present in endothelial cells and the basal region of renal epithelial cells [188]. The smooth muscle system and afferent nerve fibres of the bladder express the short dystrophin isoforms Dp71 and Dp140 [185]. Dystrophin expression might therefore be primarily affected in the bladder and kidney of patients with certain mutations in the DMD gene [28, 99, 243], or these organ systems are altered due to secondary and body-wide adaptations.
With advancing age, the prevalence of urological manifestations increases in Duchenne patients [11, 172]. However, relatively little is known about the pathophysiological mechanisms of bladder smooth muscle dysfunction and urinary incontinence and their full clinical extend in patients afflicted with dystrophinopathy [200, 222]. In contrast, a variety of studies have characterised renal dysfunction in X-linked muscular dystrophy. Duchenne patients at advanced stages of muscular dystrophy are especially susceptible to kidney disease [223], including fatal cases of acute renal failure [207]. Kidney disease was shown to also correlate with cardiomyopathic complications causing in some cases the cardio-renal syndrome [357]. Symptomatic nephrolithiasis [303, 308], abnormal filtration rates [33] and impaired kidney perfusion [223] were shown to occur in large cohorts of non-ambulatory Duchenne patients.
It is possible to monitor the dysfunction of the kidney by routine clinical tests, such as the cystatin C–estimated glomerular filtration rate [357, 361]. Studies of the dystrophic mdx-type mouse models revealed cellular changes in the kidney [118] and reduced renal function [363] and were also instrumental to test for toxic side effects on the kidney due to drug treatment or experimental exon-skipping therapy [270, 381]. The systematic identification of proteome-wide changes in the kidney has demonstrated the increased expression levels of the FABP1 (L-FABP) isoform of fatty acid–binding protein [83]. Changes in this crucial metabolic protein [145] are most likely associated with ectopic fat deposition and chronic renal dysfunction [78, 373].
Disease processes occurring in the kidney and bladder can be studied completely non-invasively by analysing changes in biomarkers in urine specimens [153, 370]. Urine displays a highly complex metabolome and proteome with a large number of biomolecules that exhibit excellent diagnostic and prognostic properties [191, 369, 382]. However, it is difficult to establish bladder- or kidney-specific tissue damage markers [380]. The most drastic proteomic or metabolomic changes in urine samples from Duchenne patients are linked to muscle-specific or body-wide alterations, i.e. the presence of muscle titin fragments [107, 205, 283], high levels of ferritin [282] and an elevated concentration of the prostaglandin tetranor-PGDM [330]. An exception is the kidney-specific protein named uromodulin, which is exclusively produced in the ascending limb of the loop of Henle and in the distal tubular region of the nephron. Uromodulin is highly abundant in urine and a marker of chronic renal disease [143, 217] and was identified to be significantly increased in urine from Duchenne patients [283]. This indicates considerable renal injury in dystrophionapthy. Another striking feature is enhanced oxidative damage to urinary proteins, which is proposed to be generated by oxidant hypochlorous acid via the enzyme myeloperoxidase from neutrophils [333].
Novel proteomic biomarkers of the complex aetiology of Duchenne muscular dystrophy
X-linked muscular dystrophy is characterised by a highly complex process of skeletal muscle wasting and secondary pathophysiological effects that are reflected by considerable levels of multi-systemic change and organ crosstalk [84, 111, 213]. This body-wide aetiology requires multi-disciplinary approaches for the optimum diagnosis, prognosis and management of dystrophinopathy [25,26,27, 340]. Although standard serum enzyme assays used for the routine evaluation of skeletal muscle damage, such as creatine kinase or myoglobin tests, are useful indicators of tissue changes and the release of intracellular muscle content, these analytes are not specific enough for the monitoring of neuromuscular disease progression and therapeutic impact [250]. Thus, there is an urgent clinical need for the establishment of novel biomarker signatures that are suitable for the proper differentiation between muscle-related degeneration versus non-muscle-associated abnormalities in Duchenne patients, as well as monitoring of signalling events between different tissues and organ systems [79]. Improved diagnostic and prognostic tools would help to better predict the complexity of disease progression in relation to fibre necrosis, reactive myofibrosis, fat substitution and sterile inflammation. Therapeutic biomarkers should be able to evaluate both intended outcomes versus adverse effects of innovative therapeutic approaches at the genetic, pharmacological and cellular level [82, 237, 250]. As reviewed by Szigyarto and Spitali [328], biomarkers in the field of dystrophinopathy can be divided in relation to susceptibility, screening, diagnosis, prognosis, prediction, therapy monitoring and safety.
Clinical assays based on marker molecules, such as DNA, miRNA, metabolites, lipids and/or proteins [43, 146, 168], should on the one hand be highly specific, robust, sensitive and cost-effective and on the other hand exhibit ideally only a minimum susceptibility to interference by gender, age, ethnicity, nutrition, lifestyle and circadian rhythm [250]. User-friendly biomarker tests could be highly useful to improve prenatal analysis, new-born screening and estimating disease initiation in order to decisively reduce the time between observation of initial symptoms and consolidated differential diagnosis [156, 281]. The establishment of sets of prognostic and predictive biomarkers would allow the monitoring of pathological progression, potential adverse clinical events, the differential screening of patient populations and the determination of the sensitivity of individual patients to new treatment protocols [328]. While body-wide responses to new drug treatments can be examined by pharmacodynamic markers and potential cytotoxic effects by safety markers, the repeated assessment of improved disease status and possibly convalescence should be enabled by therapy-monitoring marker molecules [1, 237].
Besides systematic transcriptomic surveys [190, 306] and metabolomic research [60, 178, 345] in the field of dystrophinopathy, a major area of omics-based biomarker discovery employs mass spectrometric and proteomic screening protocols for the large-scale identification of new peptides, protein fragments and protein biomarker candidates [42, 82]. A few studies have also used multi-omics approaches for studying dystrophic changes at the various levels of biological organisation from gene to mRNA to protein expression [122, 224, 352]. The integration of genomics and proteomics for the in-depth proteogenomic characterisation of rare neuromuscular disorders is an attractive approach that combines advanced proteomic screening with gene discovery [279].
The initial experimental design of mass spectrometry-based proteomic workflows should ideally take into account the following: (i) an optimised experimental and bioanalytical design for tissue preparation, sample collection, specimen storage, subcellular fractionation and protein extraction prior to biochemical analysis; (ii) a suitable proteomic discovery strategy (i.e. a specific mass spectrometric technique and labelling method) following large-scale protein separation using either bottom-up (liquid chromatography) or top-down (two-dimensional gel electrophoresis) approaches; (iii) the unequivocal demonstration of disease/therapy-associated differential protein expression patterns using advanced bioinformatic analysis of proteomic data; (iv) the independent verification of the specificity, sensitivity and robustness of new protein biomarker candidates using standardised biochemical, immunochemical, molecular biological, cell biological and physiological assays; and finally (v) a preliminary prioritisation of the most promising biomarker signature for the tissue/biofluid-specific evaluation related to specific goals such as improved screening, diagnosis, prognosis, pharmacodynamics or therapy monitoring [82]. Following the identification of new peptides or proteins using patient biopsy or biofluid material, cell culture or animal models, new biomarker candidates undergo then an intense validation process using retrospective or prospective investigations to test their validity and significance prior to clinical usage [1].
The skeletal muscle proteome [37] consists of at least 10,000 proteoforms [65, 66, 229], and systematic screening of changes in both muscle-associated proteins and released proteoforms in biofluids has identified a large number of potential biomarker candidates [42, 82, 124, 237]. This includes significant expression changes in skeletal muscle proteins that are involved in ion homoeostasis, cellular signalling cascades, the regulation of excitation–contraction coupling, the sarcomeric units and the maintenance of contraction-relaxation cycles, the formation of the extracellular matrix, the stabilisation of the cytoskeletal network, the cellular stress response, metabolism and bioenergetics in X-linked muscular dystrophy [38, 137, 138, 140, 230, 239, 269, 353]. Abnormal protein expression was also documented in the heart, stomach, brain, liver, kidney and spleen of the dystrophic phenotype [77, 83, 232, 235, 236]. However, muscle tissue–associated markers are only of limited usefulness for routine and repeated sampling approaches. In order to avoid unnecessary complications due to highly invasive tissue biopsy procedures, instead suitable and representative biological fluids can be tested for the presence of disease markers.
Proteomic serum profiling has been successfully applied to study inflammation, mitochondrial abnormalities, membrane instability and fibrosis in muscular dystrophy [123, 124, 231, 237]. While serum and plasma specimens are harvested by minimally invasive methods, biofluids such as saliva or urine have the advantage of being sampled in a completely non-invasive way. Both biological fluids contain large and complex proteomes that are suitable for the routine diagnostic analysis of body-wide health status [107, 238]. Promising new biofluid markers of Duchenne muscular dystrophy include fibronectin, titin fragments, fatty acid–binding protein FABP3, malate dehydrogenase MDH2, the inflammation-inducible plasma marker haptoglobin, carbonic anhydrase CA3, myosin light-chain MLC3 and matrix metalloproteinase MMP9 [14, 168, 194, 231, 305, 323]. Some of these markers can now be validated in a number of new therapeutic applications [5].
Therapeutic implications and future perspectives
New treatment approaches to ameliorate the dystrophic phenotype include (i) pharmacological interventions using drugs that modulate the immune response and inflammation, abnormal ion homeostasis, impaired excitation–contraction coupling, cellular growth patterns, abnormal metabolic pathways, cholesterol metabolism, oxidative stress and cardio-respiratory complications [8, 132, 155, 209, 214]; (ii) myoblast transfer therapy [15, 225, 304]; (iii) stem cell therapy [24, 40, 325]; (iv) somatic genome editing using CRISPR/Cas9-mediated exon excision [12, 171, 218]; (v) heat shock protein induction to enhance the natural cellular stress response provided by molecular chaperones [108, 334]; (vi) stop codon read-through therapy [127, 264, 326]; (vii) vector transfer therapy [121, 242, 296]; (viii) exon-skipping therapy [49, 131, 180]; (ix) electrical nerve stimulation to induce muscle transitions [122]; and (x) utrophin substitution therapy [193, 312, 362]. An interesting approach is the repurposing of established pharmacological substances and testing of multi-drug combinations in experimental trials using genetic animal models of Duchenne muscular dystrophy [383].
Detailed discussions of the clinical advantages versus potential limitations of these new treatment options and their current validation status in preclinical or clinical studies have been published [101, 299, 354]. Recently, several novel compounds have emerged which have been approved or await final approval by medicines agencies. This includes the FDA-approved novel corticosteroid Deflazacort that was shown to be associated with improved muscle strength [267] and agents with conditional approval such as Casimersen for skipping exon 45 resulting in the elevated production of dystrophin in skeletal muscle [301] and Eteplirsen for skipping of exon 51 which causes delayed loss of ambulation in some patients [211] and Golodirsen and Viltolarsen for skipping exon 53 resulting in increased dystrophin levels [49, 102], as well as the oxadiazole drug named Ataluren, approved by the European Medicines Agency, which is supposed to help restore dystrophin by supressing nonsense mutations [22]. Ongoing clinical evaluations of further pharmacological interventions that focus on skeletal muscle abnormalities in dystrophinopathy include the anti-inflammatory substances Valmorolone and Cosyntropin, the anti-fibrotic drugs Givinostat and Pamrevlumab and the myostatin inhibitor RO7239361 [284, 298].
Since reactive myofibrosis plays such an important pathophysiological role in muscular dystrophy [64, 141, 254], the prevention, halting or reversal of fibrosis is a key factor for avoiding complications and providing optimised conditions prior to the application of novel therapeutic approaches, such as gene transfer or interventions with stem cells [163, 295]. A potential way to address the issue of excess tissue scarring would be to drastically decrease the expression levels of pro-fibrotic proteins via antisense oligonucleotide therapy [201].
Conclusions
Although Duchenne muscular dystrophy is primarily defined as a skeletal muscle wasting disorder, its pathophysiological changes also affect multiple non-muscle tissues and organ systems. This makes dystrophinopathy a multi-systems disorder with complex disturbances of whole-body homeostasis. Hallmarks of both sequential and overlapping alterations due to deficiency in dystrophin include (i) progressive skeletal muscle degeneration in association with fat replacement, chronic inflammation and reactive fibrosis; (ii) scoliosis, joint deformation and contractures; (iii) respiratory insufficiency; (iv) late-onset cardiomyopathy; (v) neurological deficiencies that may cause cognitive impairments, emotional issues and attention deficit; (vi) endocrine, metabolic and bioenergetic disturbances; (vii) gastrointestinal dysfunction; (viii) fatty liver disease; and (ix) renal and urinary tract dysfunction. Therefore, for the future development of new treatment approaches, such as gene therapy, as well as the establishment of novel biomarkers to improve differential diagnosis, prognosis and therapy monitoring of Duchenne muscular dystrophy, both multi-system pathology and organ crosstalk should be taken into account. In order to decisively increase the long-term survival of Duchenne patients, new combinations of pharmacological therapy, cellular interventions and gene substitution approaches should be designed that can be employed together with physiotherapy and optimum nutritional support to address the complex and body-wide pathology of dystrophinopathy.
Change history
14 August 2022
Missing Open Access funding information has been added in the Funding Note.
References
Aartsma-Rus A, Spitali P (2015) Circulating biomarkers for duchenne muscular dystrophy. J Neuromuscul Dis 2:S49–S58
Abildinova G, Abdrakhmanova Z, Tuchinsky H, Nesher E, Pinhasov A, Raskin L (2016) Fast detection of deletion breakpoints using quantitative PCR. Genet Mol Biol 39:365–369
Alderton JM, Steinhardt RA (2000) Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem 275:9452–9460
Alic L, Griffin JF 4th, Eresen A, Kornegay JN, Ji JX (2020) Using MRI to quantify skeletal muscle pathology in Duchenne muscular dystrophy: a systematic mapping review. Muscle Nerve 64:8–22
Al-Khalili Szigyarto C (2020) Duchenne Muscular Dystrophy: recent advances in protein biomarkers and the clinical application. Expert Rev Proteomics 17:365–375
Allen DG, Whitehead NP, Froehner SC (2016) Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev 96:253–305
Alvarez K, Fadic R, Brandan E (2002) Augmented synthesis and differential localization of heparan sulfate proteoglycans in Duchenne muscular dystrophy. J Cell Biochem 85:703–713
Amor F, Vu Hong A, Corre G, Sanson M, Suel L, Blaie S, Servais L, Voit T, Richard I, Israeli D (2021) Cholesterol metabolism is a potential therapeutic target in Duchenne muscular dystrophy. J Cachexia Sarcopenia Muscle 12:677–693
Andrade FH, Porter JD, Kaminski HJ (2000) Eye muscle sparing by the muscular dystrophies: lessons to be learned? Microsc Res Tech 48:192–203
Argilés JM, Campos N, Lopez-Pedrosa JM, Rueda R, Rodriguez-Mañas L (2016) Skeletal muscle regulates metabolism via interorgan crosstalk: roles in health and disease. J Am Med Dir Assoc 17:789–796
Askeland EJ, Arlen AM, Erickson BA, Mathews KD, Cooper CS (2013) Urological manifestations of Duchenne muscular dystrophy. J Urol 190:1523–1528
Aslesh T, Erkut E, Yokota T (2021) Restoration of dystrophin expression and correction of Duchenne muscular dystrophy by genome editing. Expert Opin Biol Ther 25:1–13
Avila G (2018) Disturbed Ca2+ homeostasis in muscle-wasting disorders. Adv Exp Med Biol 1088:307–326
Ayoglu B, Chaouch A, Lochmüller H, Politano L, Bertini E, Spitali P, Hiller M, Niks EH, Gualandi F, Pontén F, Bushby K, Aartsma-Rus A, Schwartz E, Le Priol Y, Straub V, Uhlén M, Cirak S, t’Hoen PA, Muntoni F, Ferlini A, Schwenk JM, Nilsson P, Al-Khalili Szigyarto C (2014) Affinity proteomics within rare diseases: a BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol Med 6:918–936
Bajek A, Porowinska D, Kloskowski T, Brzoska E, Ciemerych MA, Drewa T (2015) Cell therapy in Duchenne muscular dystrophy treatment: clinical trials overview. Crit Rev Eukaryot Gene Expr 25:1–11
Bardoni A, Felisari G, Sironi M, Comi G, Lai M, Robotti M, Bresolin N (2000) Loss of Dp140 regulatory sequences is associated with cognitive impairment in dystrophinopathies. Neuromuscul Disord 10:194–199
Barnard AM, Lott DJ, Batra A, Triplett WT, Forbes SC, Riehl SL, Willcocks RJ, Smith BK, Vandenborne K, Walter GA (2019) Imaging respiratory muscle quality and function in Duchenne muscular dystrophy. J Neurol 266:2752–2763
Battini R, Lenzi S, Lucibello S, Chieffo D, Moriconi F, Cristofani P, Bulgheroni S, Cumbo F, Pane M, Baranello G, Alfieri P, Astrea G, Cioni G, Vicari S, Mercuri E (2021) Longitudinal data of neuropsychological profile in a cohort of Duchenne muscular dystrophy boys without cognitive impairment. Neuromuscul Disord 31:319–327
Bello L, Morgenroth LP, Gordish-Dressman H, Hoffman EP, McDonald CM, Cirak S, CINRG investigators (2016) DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology 87:401–409
Bello L, Pegoraro E (2019) The “usual suspects”: genes for inflammation, fibrosis, regeneration, and muscle strength modify Duchenne muscular dystrophy. J Clin Med 8:649
Benarroch L, Bonne G, Rivier F, Hamroun D (2019) The 2020 version of the gene table of neuromuscular disorders. Neuromuscul Disord 29:980–1018
Bernert G, Hahn A, Köhler C, Meyer S, Schara U, Schlachter K, Trollmann R, Walter MC (2021) Expert recommendation: treatment of nonambulatory patients with Duchenne muscular dystrophy. Nervenarzt 92:359–366
Bhosle RC, Michele DE, Campbell KP, Li Z, Robson RM (2006) Interactions of intermediate filament protein synemin with dystrophin and utrophin. Biochem Biophys Res Commun 346:768–777
Biressi S, Filareto A, Rando TA (2020) Stem cell therapy for muscular dystrophies. J Clin Invest 130:5652–5664
Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, Case LE, Cripe L, Hadjiyannakis S, Olson AK, Sheehan DW, Bolen J, Weber DR, Ward LM, DMD Care Considerations Working Group (2018) Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 17:347–361
Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, Pandya S, Street N, Tomezsko J, Wagner KR, Ward LM, Weber DR, DMD Care Considerations Working Group (2018) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 17:251–267
Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Colvin MK, Cripe L, Herron AR, Kennedy A, Kinnett K, Naprawa J, Noritz G, Poysky J, Street N, Trout CJ, Weber DR, Ward LM, DMD Care Considerations Working Group (2018) Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol 17:445–455
Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barišić N, Brabec P, Lahdetie J, Walter MC, Schreiber-Katz O, Karcagi V, Garami M, Viswanathan V, Bayat F, Buccella F, Kimura E, Koeks Z, van den Bergen JC, Rodrigues M, Roxburgh R, Lusakowska A, Kostera-Pruszczyk A, Zimowski J, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Jeannet PY, Joncourt F, Díaz-Manera J, Gallardo E, Karaduman AA, Topaloğlu H, El Sherif R, Stringer A, Shatillo AV, Martin AS, Peay HL, Bellgard MI, Kirschner J, Flanigan KM, Straub V, Bushby K, Verschuuren J, Aartsma-Rus A, Béroud C, Lochmüller H (2015) The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 36:395–402
Böhm SV, Roberts RG (2009) Expression of members of the dystrophin, dystrobrevin, and dystrotelin superfamily. Crit Rev Eukaryot Gene Expr 19:89–108
Bonilla E, Samitt CE, Miranda AF, Hays AP, Salviati G, DiMauro S, Kunkel LM, Hoffman EP, Rowland LP (1988) Duchenne muscular dystrophy: deficiency of dystrophin at the muscle cell surface. Cell 54:447–452
Bonomo AC, Pinto-Mariz F, Riederer I, Benjamim CF, Butler-Browne G, Mouly V, Savino W (2020) Crosstalk between innate and T cell adaptive immunity with(in) the muscle. Front Physiol 11:573347
Boussaïd G, Stalens C, Devaux C, Segovia-Kueny S, Lofaso F, Reveillere C (2020) Impact of mechanical ventilation methods on the life perception of subjects with Duchenne muscular dystrophy: French cross-sectional survey. Respir Care 65:1712–1720
Braat E, Hoste L, De Waele L, Gheysens O, Vermeersch P, Goffin K, Pottel H, Goemans N, Levtchenko E (2015) Renal function in children and adolescents with Duchenne muscular dystrophy. Neuromuscul Disord 25:381–387
Brashear SE, Wohlgemuth RP, Gonzalez G, Smith LR (2021) Passive stiffness of fibrotic skeletal muscle in mdx mice relates to collagen architecture. J Physiol 599:943–962
Brinkmeier H, Ohlendieck K (2014) Chaperoning heat shock proteins: proteomic analysis and relevance for normal and dystrophin-deficient muscle. Proteomics Clin Appl 8:875–895
Camps J, Breuls N, Sifrim A, Giarratana N, Corvelyn M, Danti L, Grosemans H, Vanuytven S, Thiry I, Belicchi M, Meregalli M, Platko K, MacDonald ME, Austin RC, Gijsbers R, Cossu G, Torrente Y, Voet T, Sampaolesi M (2020) Interstitial cell remodeling promotes aberrant adipogenesis in dystrophic muscles. Cell Rep 31:107597
Capitanio D, Moriggi M, Gelfi C (2017) Mapping the human skeletal muscle proteome: progress and potential. Expert Rev Proteomics 14:825–839
Capitanio D, Moriggi M, Torretta E, Barbacini P, De Palma S, ViganòA LH, Muntoni F, Ferlini A, Mora M, Gelfi C (2020) Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: changes contributing to preserve muscle function in Becker muscular dystrophy patients. J Cachexia Sarcopenia Muscle 11:547–563
Capote J, DiFranco M, Vergara JL (2010) Excitation-contraction coupling alterations in mdx and utrophin/dystrophin double knockout mice: a comparative study. Am J Physiol Cell Physiol 298:C1077–C1086
Cappella M, Elouej S, Biferi MG (2021) The potential of induced pluripotent stem cells to test gene therapy approaches for neuromuscular and motor neuron disorders. Front Cell Dev Biol 9:662837
Carberry S, Zweyer M, Swandulla D, Ohlendieck K (2012) Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in the aged mdx diaphragm model of Duchenne muscular dystrophy. Int J Mol Med 30:229–234
Carr SJ, Zahedi RP, Lochmüller H, Roos A (2018) Mass spectrometry-based protein analysis to unravel the tissue pathophysiology in Duchenne muscular dystrophy. Proteomics Clin Appl 12(2) https://doi.org/10.1002/prca.201700071
Catapano F, Domingos J, Perry M, Ricotti V, Phillips L, Servais L, Seferian A, Groot I, Krom YD, Niks EH, Verschuuren JJ, Straub V, Voit T, Morgan J, Muntoni F (2018) Downregulation of miRNA-29, -23 and -21 in urine of Duchenne muscular dystrophy patients. Epigenomics 10:875–889
Chal J, Pourquié O (2017) Making muscle: skeletal myogenesis in vivo and in vitro. Development 144:2104–2122
Chang NC, Chevalier FP, Rudnicki MA (2016) Satellite cells in muscular dystrophy - lost in polarity. Trends Mol Med 22:479–496
Chang WJ, Iannaccone ST, Lau KS, Masters BS, McCabe TJ, McMillan K, Padre RC, Spencer MJ, Tidball JG, Stull JT (1996) Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci USA 93:9142–9147
Choi YA, Chun SM, Kim Y, Shin HI (2018) Lower extremity joint contracture according to ambulatory status in children with Duchenne muscular dystrophy. BMC Musculoskelet Disord 19:287
Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S (2013) Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 45:2191–2199
Clemens PR, Rao VK, Connolly AM, Harper AD, Mah JK, Smith EC, McDonald CM, Zaidman CM, Morgenroth LP, Osaki H, Satou Y, Yamashita T, Hoffman EP, Investigators CINRGDNHS (2020) Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol 77:982–991
Cohen S, Nathan JA, Goldberg AL (2015) Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14:58–74
Constantin B (2014) Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta 1838:635–642
Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifirò G (2020) Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis 15:141
Culligan K, Banville N, Dowling P, Ohlendieck K (2002) Drastic reduction of calsequestrin-like proteins and impaired calcium binding in dystrophic mdx muscle. J Appl Physiol 92:435–445
Culligan K, Glover L, Dowling P, Ohlendieck K (2001) Brain dystrophin-glycoprotein complex: persistent expression of beta-dystroglycan, impaired oligomerization of Dp71 and up-regulation of utrophins in animal models of muscular dystrophy. BMC Cell Biol 2:2
Culligan K, Ohlendieck K (2002) Diversity of the brain dystrophin-glycoprotein complex. J Biomed Biotechnol 2:31–36
Cutroneo G, Lentini S, Favaloro A, Anastasi G, Di Mauro D (2012) Costameric proteins: from benchside to future translational cardiovascular research. Ann Cardiol Angeiol (Paris) 61:55–60
Cyrulnik SE, Fee RJ, Batchelder A, Kiefel J, Goldstein E, Hinton VJ (2008) Cognitive and adaptive deficits in young children with Duchenne muscular dystrophy (DMD). J Int Neuropsychol Soc 14:853–861
Cyrulnik SE, Fee RJ, De Vivo DC, Goldstein E, Hinton VJ (2007) Delayed developmental language milestones in children with Duchenne’s muscular dystrophy. J Pediatr 150:474–478
Cyrulnik SE, Hinton VJ (2008) Duchenne muscular dystrophy: a cerebellar disorder? Neurosci Biobehav Rev 32:486–496
Dabaj I, Ferey J, Marguet F, Gilard V, Basset C, Bahri Y, Brehin AC, Vanhulle C, Leturcq F, Marret S, Laquerrière A, Schmitz-Afonso I, Afonso C, Bekri S, Tebani A (2021) Muscle metabolic remodelling patterns in Duchenne muscular dystrophy revealed by ultra-high-resolution mass spectrometry imaging. Sci Rep 11:1906
Darmahkasih AJ, Rybalsky I, Tian C, Shellenbarger KC, Horn PS, Lambert JT, Wong BL (2020) Neurodevelopmental, behavioral, and emotional symptoms common in Duchenne muscular dystrophy. Muscle Nerve 61:466–474
Davidson ZE, Truby H (2009) A review of nutrition in Duchenne muscular dystrophy. J Hum Nutr Diet 22:383–393
Demer JL (2015) Compartmentalization of extraocular muscle function. Eye (Lond) 29:157–162
Desguerre I, Mayer M, Leturcq F, Barbet JP, Gherardi RK, Christov C (2009) Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J Neuropathol Exp Neurol 68:762–773
Deshmukh AS, Murgia M, Nagaraj N, Treebak JT, Cox J, Mann M (2015) Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol Cell Proteomics 14:841–853
Deshmukh AS, Steenberg DE, Hostrup M, Birk JB, Larsen JK, Santos A, Kjøbsted R, Hingst JR, Schéele CC, Murgia M, Kiens B, Richter EA, Mann M, Wojtaszewski JFP (2021) Deep muscle-proteomic analysis of freeze-dried human muscle biopsies reveals fiber type-specific adaptations to exercise training. Nat Commun 12:304
Dietz AR, Connolly A, Dori A, Zaidman CM (2020) Intramuscular blood flow in Duchenne and Becker muscular dystrophy: quantitative power Doppler sonography relates to disease severity. Clin Neurophysiol 131:1–5
Doglio L, Pavan E, Pernigotti I, Petralia P, Frigo C, Minetti C (2011) Early signs of gait deviation in Duchenne muscular dystrophy. Eur J Phys Rehabil Med 47:587–594
Doorenweerd N, Mahfouz A, van Putten M, Kaliyaperumal R, T’ Hoen PAC, Hendriksen JGM, Aartsma-Rus AM, Verschuuren JJGM, Niks EH, Reinders MJT, Kan HE, Lelieveldt BPF (2017) Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep 7:12575
Dowling JJ, Gonorazky H, Cohn RD, Campbell C (2018) Treating pediatric neuromuscular disorders: the future is now. Am J Med Genet A 176:804–841
Dowling JJ, Weihl CC, Spencer MJ (2021) Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat Rev Mol Cell Biol. (in press)https://doi.org/10.1038/s41580-021-00389-z
Doran P, Dowling P, Lohan J, McDonnell K, Poetsch S, Ohlendieck K (2004) Subproteomics analysis of Ca2+-binding proteins demonstrates decreased calsequestrin expression in dystrophic mouse skeletal muscle. Eur J Biochem 271:3943–3952
Doran P, Martin G, Dowling P, Jockusch H, Ohlendieck K (2006) Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 6:4610–4621
Dowling P, Doran P, Ohlendieck K (2004) Drastic reduction of sarcalumenin in Dp427 (dystrophin of 427 kDa)-deficient fibres indicates that abnormal calcium handling plays a key role in muscular dystrophy. Biochem J 379:479–488
Dowling P, Gargan S, Murphy S, Zweyer M, Sabir H, Swandulla D, Ohlendieck K (2021) The dystrophin node as integrator of cytoskeletal organization, lateral force transmission, fiber stability and cellular signaling in skeletal muscle. Proteomes 9:9
Dowling P, Gargan S, Zweyer M, Henry M, Meleady P, Swandulla D, Ohlendieck K (2020) Proteome-wide changes in the mdx-4cv spleen due to pathophysiological cross talk with dystrophin-deficient skeletal muscle. iScience 23:101500
Dowling P, Gargan S, Zweyer M, Sabir H, Henry M, Meleady P, Swandulla D, Ohlendieck K (2021) Proteomic profiling of the interface between the stomach wall and the pancreas in dystrophinopathy. Eur J Transl Myol 31:9627
Dowling P, Gargan S, Zweyer M, Swandulla D, Ohlendieck K (2020) Proteomic profiling of fatty acid binding proteins in muscular dystrophy. Expert Rev Proteomics 17:137–148
Dowling P, Holland A, Ohlendieck K (2014) Mass spectrometry-based identification of muscle-associated and muscle-derived proteomic biomarkers of dystrophinopathies. J Neuromuscul Dis 1:15–40
Dowling P, Lohan J, Ohlendieck K (2003) Comparative analysis of Dp427-deficient mdx tissues shows that the milder dystrophic phenotype of extraocular and toe muscle fibres is associated with a persistent expression of beta-dystroglycan. Eur J Cell Biol 82:222–230
Dowling P, Murphy S, Ohlendieck K (2016) Proteomic profiling of muscle fibre type shifting in neuromuscular diseases. Expert Rev Proteomics 13:783–799
Dowling P, Murphy S, Zweyer M, Raucamp M, Swandulla D, Ohlendieck K (2019) Emerging proteomic biomarkers of X-linked muscular dystrophy. Expert Rev Mol Diagn 19:739–755
Dowling P, Zweyer M, Raucamp M, Henry M, Meleady P, Swandulla D, Ohlendieck K (2020) Proteomic and cell biological profiling of the renal phenotype of the mdx-4cv mouse model of Duchenne muscular dystrophy. Eur J Cell Biol 99:151059
Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A (2021) Duchenne muscular dystrophy. Nat Rev Dis Primers 7:13
Duchenne de Boulogne GBA (1968) Studies on pseudohypertrophic muscular paralysis or myosclerotic paralysis. Arch Neurol 19:629–636
Durbeej M, Larsson E, Ibraghimov-Beskrovnaya O, Roberds SL, Campbell KP, Ekblom P (1995) Non-muscle alpha-dystroglycan is involved in epithelial development. J Cell Biol 130:79–91
Egan B, Zierath JR (2013) Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17:162–184
Emery AE (2002) The muscular dystrophies. Lancet 359:687–695
Ervasti JM, Campbell KP (1991) Membrane organization of the dystrophin-glycoprotein complex. Cell 66:1121–1131
Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP (1990) Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345:315–319
Even PC, Decrouy A, Chinet A (1994) Defective regulation of energy metabolism in mdx-mouse skeletal muscles. Biochem J 304:649–654
Fadic R, Mezzano V, Alvarez K, Cabrera D, Holmgren J, Brandan E (2006) Increase in decorin and biglycan in Duchenne muscular dystrophy: role of fibroblasts as cell source of these proteoglycans in the disease. J Cell Mol Med 10:758–769
Fayssoil A, Chaffaut C, Ogna A, Stojkovic T, Lamothe L, Mompoint D, Meng P, Prigent H, Clair B, Behin A, Laforet P, Bassez G, Carlier R, Orlikowski D, Amthor H, Quijano Roy S, Crenn P, Chevret S, Eymard B, Lofaso F, Annane D (2019) Echographic assessment of diaphragmatic function in Duchenne muscular dystrophy from childhood to adulthood. J Neuromuscul Dis 6:55–64
Fayssoil A, Chaffaut C, Prigent H, Laforet P, Clair B, Orlikowski D, Ogna A, Chevret S, Meng P, Annane D, Lofaso F, Crenn P (2021) Nutritional status, swallowing disorders, and respiratory prognosis in adult Duchenne muscular dystrophy patients. Pediatr Pulmonol 56:2146–2154
Felisari G, Martinelli Boneschi F, Bardoni A, Sironi M, Comi GP, Robotti M, Turconi AC, Lai M, Corrao G, Bresolin N (2000) Loss of Dp140 dystrophin isoform and intellectual impairment in Duchenne dystrophy. Neurology 55:559–564
Finn DM, Ohlendieck K (1997) Rabbit brain and muscle isoforms containing the carboxy-terminal domain of 427 kDa skeletal muscle dystrophin exhibit similar biochemical properties. Neurosci Lett 222:25–28
Finn DM, Ohlendieck K (1998) Oligomerization of beta-dystroglycan in rabbit diaphragm and brain as revealed by chemical crosslinking. Biochim Biophys Acta 1370:325–336
Flanigan KM (2014) Duchenne and Becker muscular dystrophies. Neurol Clin 32:671–688
Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM, Pestronk A, Florence JM, Connolly AM, Mathews KD, Stephan CM, Laubenthal KS, Wong BL, Morehart PJ, Meyer A, Finkel RS, Bonnemann CG, Medne L, Day JW, Dalton JC, Margolis MK, Hinton VJ, United Dystrophinopathy Project Consortium, Weiss RB (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 30:1657–66
Formicola L, Marazzi G, Sassoon DA (2014) The extraocular muscle stem cell niche is resistant to ageing and disease. Front Aging Neurosci 6:328
Fortunato F, Rossi R, Falzarano MS, Ferlini A (2021) Innovative therapeutic approaches for duchenne muscular dystrophy. J Clin Med 10:820
Frank DE, Schnell FJ, Akana C, El-Husayni SH, Desjardins CA, Morgan J, Charleston JS, Sardone V, Domingos J, Dickson G, Straub V, Guglieri M, Mercuri E, Servais L, Muntoni F, SKIP-NMD Study Group (2020) Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 94:e2270–e2282
Friedrich O, von Wegner F, Chamberlain JS, Fink RH, Rohrbach P (2008) L-type Ca2+ channel function is linked to dystrophin expression in mammalian muscle. PLoS One 3:e1762
Fröhlich T, Kemter E, Flenkenthaler F, Klymiuk N, Otte KA, Blutke A, Krause S, Walter MC, Wanke R, Wolf E, Arnold GJ (2016) Progressive muscle proteome changes in a clinically relevant pig model of Duchenne muscular dystrophy. Sci Rep 6:33362
Garg S (2016) Management of scoliosis in patients with Duchenne muscular dystrophy and spinal muscular atrophy: a literature review. J Pediatr Rehabil Med 9:23–29
Gargan S, Dowling P, Zweyer M, Reimann J, Henry M, Meleady P, Swandulla D, Ohlendieck K (2021) Mass spectrometric profiling of extraocular muscle and proteomic adaptations in the mdx-4cv model of Duchenne muscular dystrophy. Life (Basel) 11:595
Gargan S, Dowling P, Zweyer M, Swandulla D, Ohlendieck K (2020) Identification of marker proteins of muscular dystrophy in the urine proteome from the mdx-4cv model of dystrophinopathy. Mol Omics 16:268–278
Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, Lamon S, Russell AP, Davies KE, Febbraio MA, Lynch GS (2012) Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 484:394–398
Gillies AR, Lieber RL (2011) Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 44:318–331
Giuliani G, Rosina M, Reggio A (2021) Signaling pathways regulating the fate of fibro/adipogenic progenitors (FAPs) in skeletal muscle regeneration and disease. FEBS J (in press) https://doi.org/10.1111/febs.16080
Goemans N, Buyse G (2014) Current treatment and management of dystrophinopathies. Curr Treat Options Neurol 16:287
Goudriaan M, Van den Hauwe M, Dekeerle J, Verhelst L, Molenaers G, Goemans N, Desloovere K (2018) Gait deviations in Duchenne muscular dystrophy-Part 1. A systematic review. Gait Posture 62:247–261
Goudriaan M, Van den Hauwe M, Simon-Martinez C, Huenaerts C, Molenaers G, Goemans N, Desloovere K (2018) Gait deviations in Duchenne muscular dystrophy-Part 2. Statistical non-parametric mapping to analyze gait deviations in children with Duchenne muscular dystrophy. Gait Posture 63:159–164
Gowran A, Brioschi M, Rovina D, Chiesa M, Piacentini L, Mallia S, Banfi C, Pompilio G, Santoro R (2021) Multiomic approaches to uncover the complexities of dystrophin-associated cardiomyopathy. Int J Mol Sci 22:8954
Gramolini AO, Wu J, Jasmin BJ (2000) Regulation and functional significance of utrophin expression at the mammalian neuromuscular synapse. Microsc Res Tech 49:90–100
Guevel L, Lavoie JR, Perez-Iratxeta C, Rouger K, Dubreil L, Feron M, Talon S, Brand M, Megeney LA (2011) Quantitative proteomic analysis of dystrophic dog muscle. J Proteome Res 10:2465–2478
Guiraud S, Aartsma-Rus A, Vieira NM, Davies KE, van Ommen GJ, Kunkel LM (2015) The pathogenesis and therapy of muscular dystrophies. Annu Rev Genomics Hum Genet 16:281–308
Gusel’nikova V, Antimonova O, Fedorova E, Shavlovsky M, Krutikov A, Mikhailova E, Gudkova A, Mikhailov V, Korzhevskii D (2018) Fluorescent characterization of amyloid deposits in the kidneys of mdx mice. Eur J Histochem 62:2870
Haenggi T, Schaub MC, Fritschy JM (2005) Molecular heterogeneity of the dystrophin-associated protein complex in the mouse kidney nephron: differential alterations in the absence of utrophin and dystrophin. Cell Tissue Res 319:299–313
Halayko AJ, Stelmack GL (2005) The association of caveolae, actin, and the dystrophin-glycoprotein complex: a role in smooth muscle phenotype and function? Can J Physiol Pharmacol 83:877–891
Halbert CL, Allen JM, Chamberlain JS (2018) AAV6 vector production and purification for muscle gene therapy. Methods Mol Bio 1687:257–266
Hardee JP, Martins KJB, Miotto PM, Ryall JG, Gehrig SM, Reljic B, Naim T, Chung JD, Trieu J, Swiderski K, Philp AM, Philp A, Watt MJ, Stroud DA, Koopman R, Steinberg GR, Lynch GS (2021) Metabolic remodeling of dystrophic skeletal muscle reveals biological roles for dystrophin and utrophin in adaptation and plasticity. Mol Metab 45:101157
Hathout Y, Brody E, Clemens PR, Cripe L, DeLisle RK, Furlong P, Gordish-Dressman H, Hache L, Henricson E, Hoffman EP, Kobayashi YM, Lorts A, Mah JK, McDonald C, Mehler B, Nelson S, Nikrad M, Singer B, Steele F, Sterling D, Sweeney HL, Williams S, Gold L (2015) Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci USA 112:7153–7158
Hathout Y, Seol H, Han MH, Zhang A, Brown KJ, Hoffman EP (2016) Clinical utility of serum biomarkers in Duchenne muscular dystrophy. Clin Proteomics 13:9
Hendriksen RG, Hoogland G, Schipper S, Hendriksen JG, Vles JS, Aalbers MW (2015) A possible role of dystrophin in neuronal excitability: a review of the current literature. Neurosci Biobehav Rev 51:255–262
Heydemann A (2018) Skeletal muscle metabolism in Duchenne and Becker muscular dystrophy-implications for therapies. Nutrients 10:796
Himič V, Davies KE (2021) Evaluating the potential of novel genetic approaches for the treatment of Duchenne muscular dystrophy. Eur J Hum Genet (in press). https://doi.org/10.1038/s41431-021-00811-2
Hinton VJ, Cyrulnik SE, Fee RJ, Batchelder A, Kiefel JM, Goldstein EM, Kaufmann P, De Vivo DC (2009) Association of autistic spectrum disorders with dystrophinopathies. Pediatr Neurol 41:339–346
Hinton VJ, Nereo NE, Fee RJ, Cyrulnik SE (2006) Social behavior problems in boys with Duchenne muscular dystrophy. J Dev Behav Pediatr 27:470–476
Hoffman EP (2020) The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J 287:3879–3887
Hoffman EP (2020) Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies. Acta Myol 39:179–186
Hoffman EP (2020) Pharmacotherapy of Duchenne muscular dystrophy. Handb Exp Pharmacol 261:25–37
Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928
Hoffman EP, Hudecki MS, Rosenberg PA, Pollina CM, Kunkel LM (1988) Cell and fiber-type distribution of dystrophin. Neuron 1:411–420
Hoh JFY (2021) Myosin heavy chains in extraocular muscle fibres: distribution, regulation and function. Acta Physiol (Oxf) 231:e13535
Hol EM, Pekny M (2015) Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol 32:121–130
Holland A, Carberry S, Ohlendieck K (2013) Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr Protein Pept Sci 14:680–697
Holland A, Dowling P, Meleady P, Henry M, Zweyer M, Mundegar RR, Swandulla D, Ohlendieck K (2015) Label-free mass spectrometric analysis of the mdx-4cv diaphragm identifies the matricellular protein periostin as a potential factor involved in dystrophinopathy-related fibrosis. Proteomics 15:2318–2331
Holland A, Dowling P, Zweyer M, Swandulla D, Henry M, Clynes M, Ohlendieck K (2013) Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 13:2312–2323
Holland A, Henry M, Meleady P, Winkler CK, Krautwald M, Brinkmeier H, Ohlendieck K (2015) Comparative label-free mass spectrometric analysis of mildly versus severely affected mdx mouse skeletal muscles identifies annexin, lamin, and vimentin as universal dystrophic markers. Molecules 20:11317–11344
Holland A, Murphy S, Dowling P, Ohlendieck K (2016) Pathoproteomic profiling of the skeletal muscle matrisome in dystrophinopathy associated myofibrosis. Proteomics 16:345–366
Holland A, Ohlendieck K (2014) Proteomic profiling of the dystrophin-deficient mdx phenocopy of dystrophinopathy-associated cardiomyopathy. Biomed Res Int 2014:246195
Holzmann-Littig C, Renders L, Steubl D (2021) Uromodulin - a new marker of kidney function? Clin Nephrol 95:347–349
Hopf FW, Turner PR, Steinhardt RA (2007) Calcium misregulation and the pathogenesis of muscular dystrophy. Subcell Biochem 45:429–464
Hotamisligil GS, Bernlohr DA (2015) Metabolic functions of FABPs–mechanisms and therapeutic implications. Nat Rev Endocrinol 11:592–605
Hrach HC, Mangone M (2019) miRNA profiling for early detection and treatment of duchenne muscular dystrophy. Int J Mol Sci 20:4638
Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP (1992) Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355:696–702
Ieronimakis N, Hays AL, Janebodin K, Mahoney WM Jr, Duffield JS, Majesky MW, Reyes M (2013) Coronary adventitial cells are linked to perivascular cardiac fibrosis via TGFβ1 signaling in the mdx mouse model of Duchenne muscular dystrophy. J Mol Cell Cardiol 63:122–134
Iskandar K, Dwianingsih EK, Pratiwi L, Kalim AS, Mardhiah H, Putranti AH, Nurputra DK, Triono A, Herini ES, Malueka RG, Gunadi LPS, Sunartini (2019) The analysis of DMD gene deletions by multiplex PCR in Indonesian DMD/BMD patients: the era of personalized medicine. BMC Res Notes 12:704
Itoh K, Jinnai K, Tada K, Hara K, Itoh H, Takahashi H (1999) Multifocal glial nodules in a case of Duchenne muscular dystrophy with severe mental retardation. Neuropathology 19:322–327
Jaka O, Casas-Fraile L, López de Munain A, Sáenz A (2015) Costamere proteins and their involvement in myopathic processes. Expert Rev Mol Med 17:e12
Johnson EK, Zhang L, Adams ME, Phillips A, Freitas MA, Froehner SC, Green-Church KB, Montanaro F (2012) Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS One 7:e43515
Jung S, Kim J (2020) Biomarker discovery and beyond for diagnosis of bladder diseases. Bladder (San Franc) 7:e40
Kaprielian RR, Stevenson S, Rothery SM, Cullen MJ, Severs NJ (2000) Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation 101:2586–2594
Kawamura K, Fukumura S, Nikaido K, Tachi N, Kozuka N, Seino T, Hatakeyama K, Mori M, Ito YM, Takami A, Hinotsu S, Kuno A, Kawasaki Y, Horio Y, Tsutsumi H (2020) Resveratrol improves motor function in patients with muscular dystrophies: an open-label, single-arm, phase IIa study. Sci Rep 10:20585
Ke Q, Zhao ZY, Mendell JR, Baker M, Wiley V, Kwon JM, Alfano LN, Connolly AM, Jay C, Polari H, Ciafaloni E, Qi M, Griggs RC, Gatheridge MA (2019) Progress in treatment and newborn screening for Duchenne muscular dystrophy and spinal muscular atrophy. World J Pediatr 15:219–225
Kennedy RA, Carroll K, McGinley JL, Paterson KL (2020) Walking and weakness in children: a narrative review of gait and functional ambulation in paediatric neuromuscular disease. J Foot Ankle Res 13:10
Kharraz Y, Guerra J, Pessina P, Serrano AL, Muñoz-Cánoves P (2014) Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed Res Int 2014:965631
Kim TW, Wu K, Xu JL, Black IB (1992) Detection of dystrophin in the postsynaptic density of rat brain and deficiency in a mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA 89:11642–11644
Kinali M, Arechavala-Gomeza V, Cirak S, Glover A, Guglieri M, Feng L, Hollingsworth KG, Hunt D, Jungbluth H, Roper HP, Quinlivan RM, Gosalakkal JA, Jayawant S, Nadeau A, Hughes-Carre L, Manzur AY, Mercuri E, Morgan JE, Straub V, Bushby K, Sewry C, Rutherford M, Muntoni F (2011) Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology 76:346–353
Kipke J, Birnkrant DJ, Jin JB, Aneja A, Bahler RC (2021) A systematic review of pharmacologic therapies for the cardiomyopathy of Duchenne muscular dystrophy. Pediatr Pulmonol 56:782–795
Klietsch R, Ervasti JM, Arnold W, Campbell KP, Jorgensen AO (1993) Dystrophin-glycoprotein complex and laminin colocalize to the sarcolemma and transverse tubules of cardiac muscle. Circ Res 72:349–360
Klingler W, Jurkat-Rott K, Lehmann-Horn F, Schleip R (2012) The role of fibrosis in Duchenne muscular dystrophy. Acta Myol 31:184–195
Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM (1987) Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50:509–517
Koenig M, Kunkel LM (1990) Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem 265:4560–4566
Koenig M, Monaco AP, Kunkel LM (1988) The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 53:219–228
Kononets O, Karaiev T, Tkachenko O, Lichman L (2020) Renal, hepatic and immune function indices in patients with duchenne muscular dystrophy. Georgian Med News 309:64–71
Koutsoulidou A, Phylactou LA (2020) Circulating biomarkers in muscular dystrophies: disease and therapy monitoring. Mol Ther Methods Clin Dev 18:230–239
Kueh SL, Dempster J, Head SI, Morley JW (2011) Reduced postsynaptic GABAA receptor number and enhanced gaboxadol induced change in holding currents in Purkinje cells of the dystrophin-deficient mdx mouse. Neurobiol Dis 43:558–564
Kunkel LM, Hejtmancik JF, Caskey CT, Speer A, Monaco AP, Middlesworth W, Colletti CA, Bertelson C, Müller U, Bresnan M, Shapiro F, Tantravahi U, Speer J, Latt SA, Bartlett R, Pericak-Vance MA, Roses AD, Thompson MW, Ray PN, Worton RG, Fischbeck KH, Gallano P, Coulon M, Duros C, Boue J, Junien C, Chelly J, Hamard G, Jeanpierre M, Lambert M, Kaplan JC, Emery A, Dorkins H, McGlade S, Davies KE, Boehm C, Arveiler B, Lemaire C, Morgan GJ, Denton MJ, Amos J, Bobrow M, Benham F, Boswinkel E, Cole C, Dubowitz V, Hart K, Hodgson S, Johnson L, Walker A, Roncuzzi L, Ferlini A, Nobile C, Romeo G, Wilcox DE, Affara NA, Ferguson-Smith MA, Lindolf M, Kaariainen H, de la Chapelle A, Ionasescu V, Searby C, Ionasescu R, Bakker E, van Ommen GJ, Pearson PL, Greenberg CR, Hamerton JL, Wrogemann K, Doherty RA, Polakowska R, Hyser C, Quirk S, Thomas N, Harper JF, Darras BT, Francke U (1986) Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature 322:73–77
Kupatt C, Windisch A, Moretti A, Wolf E, Wurst W, Walter MC (2021) Genome editing for Duchenne muscular dystrophy: a glimpse of the future? Gene Ther. (in press)https://doi.org/10.1038/s41434-021-00222-4
Kutluk MG, Doğan ÇS (2021) Kidney involvement and associated risk factors in children with Duchenne muscular dystrophy. Pediatr Nephrol 35:1953–1958
Lambert JT, Darmahkasih AJ, Horn PS, Rybalsky I, Shellenbarger KC, Tian C, Wong BL (2020) Neurodevelopmental, behavioral, and emotional symptoms in Becker muscular dystrophy. Muscle Nerve 61:156–162
Landfeldt E, Thompson R, Sejersen T, McMillan HJ, Kirschner J, Lochmüller H (2020) Life expectancy at birth in Duchenne muscular dystrophy: a systematic review and meta-analysis. Eur J Epidemiol 35:643–653
Law ML, Cohen H, Martin AA, Angulski ABB, Metzger JM (2020) Dysregulation of calcium handling in duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. J Clin Med 9:520
Le Borgne F, Guyot S, Logerot M, Beney L, Gervais P, Demarquoy J (2012) Exploration of lipid metabolism in relation with plasma membrane properties of Duchenne muscular dystrophy cells: influence of L-carnitine. PLoS One 7:e49346
Lee JW, Oh HJ, Choi WA, Kim DJ, Kang SW (2020) Relationship between eating and digestive symptoms and respiratory function in advanced duchenne muscular dystrophy patients. J Neuromuscul Dis 7:101–107
Lee-McMullen B, Chrzanowski SM, Vohra R, Forbes SC, Vandenborne K, Edison AS, Walter GA (2019) Age-dependent changes in metabolite profile and lipid saturation in dystrophic mice. NMR Biomed 32:e4075
Leyva-Leyva M, Sandoval A, Felix R, González-Ramírez R (2018) Biochemical and functional interplay between ion channels and the components of the dystrophin-associated glycoprotein complex. J Membr Biol 251:535–550
Li D, Mastaglia FL, Fletcher S, Wilton SD (2018) Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol Sci 39(11):982–994
Lidov HG, Byers TJ, Kunkel LM (1993) The distribution of dystrophin in the murine central nervous system: an immunocytochemical study. Neuroscience 54:167–187
Lidov HG, Kunkel LM (1998) Dystrophin and Dp140 in the adult rodent kidney. Lab Invest 78:1543–1551
Lidov HG, Selig S, Kunkel LM (1995) Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet 4:329–335
Lindsay A, Chamberlain CM, Witthuhn BA, Lowe DA, Ervasti JM (2019) Dystrophinopathy-associated dysfunction of Krebs cycle metabolism. Hum Mol Genet 28:942–951
Lionarons JM, Hoogland G, Hendriksen RGF, Faber CG, Hellebrekers DMJ, Van Koeveringe GA, Schipper S, Vles JSH (2019) Dystrophin is expressed in smooth muscle and afferent nerve fibers in the rat urinary bladder. Muscle Nerve 60:202–210
Lo Cascio CM, Goetze O, Latshang TD, Bluemel S, Frauenfelder T, Bloch KE (2016) Gastrointestinal dysfunction in patients with Duchenne muscular dystrophy. PLoS One 11:e0163779
Loh NY, Nebenius-Oosthuizen D, Blake DJ, Smith AJ, Davies KE (2001) Role of beta-dystrobrevin in nonmuscle dystrophin-associated protein complex-like complexes in kidney and liver. Mol Cell Biol 21:7442–7448
Loh NY, Newey SE, Davies KE, Blake DJ (2000) Assembly of multiple dystrobrevin-containing complexes in the kidney. J Cell Sci 113:2715–2724
Lohan J, Ohlendieck K (2004) Drastic reduction in the luminal Ca2+-binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. Biochim Biophys Acta 1689:252–258
Lombardo SD, Mazzon E, Mangano K, Basile MS, Cavalli E, Mammana S, Fagone P, Nicoletti F, Petralia MC (2019) Transcriptomic analysis reveals involvement of the macrophage migration inhibitory factor gene network in duchenne muscular dystrophy. Genes (Basel) 10:939
López-Hernández Y, Oropeza-Valdez JJ, Blanco-Sandate JO, Herrera-Van Oostdam AS, Zheng J, Chi Guo A, Lima-Rogel V, Rajabzadeh R, Salgado-Bustamante M, Adrian-Lopez J, Castillo CG, Robles Arguelles E, Monárrez-Espino J, Mandal R, Wishart DS (2020) The urinary metabolome of healthy newborns. Metabolites 10:165
Lorin C, Vögeli I, Niggli E (2015) Dystrophic cardiomyopathy: role of TRPV2 channels in stretch-induced cell damage. Cardiovasc Res 106:153–162
Loro E, Sengupta K, Bogdanovich S, Whig K, Schultz DC, Huryn DM, Khurana TS (2020) High-throughput identification of post-transcriptional utrophin up-regulators for Duchenne muscle dystrophy (DMD) therapy. Sci Rep 10:2132
Lourbakos A, Yau N, de Bruijn P, Hiller M, Kozaczynska K, Jean-Baptiste R, Reza M, Wolterbeek R, Koeks Z, Ayoglu B, de Klerk D, Campion G, Zaharieva I, Nadarajah VD, Nilsson P, Al-Khalili Szigyarto C, Muntoni F, Lochmüller H, Verschuuren JJ, Goemans N, Tulinius M, Niks EH, de Kimpe S, Aartsma-Rus A, ’t Hoen PAC, Spitali P, (2017) Evaluation of serum MMP-9 as predictive biomarker for antisense therapy in Duchenne. Sci Rep 7:17888
Lovering RM, Iyer SR, Edwards B, Davies KE (2020) Alterations of neuromuscular junctions in Duchenne muscular dystrophy. Neurosci Lett 737:135304
MacDonald EM, Cohn RD (2012) TGFβ signaling: its role in fibrosis formation and myopathies. Curr Opin Rheumatol 24:628–634
Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N (2014) A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 24:482–491
Mahyoob Rani AQ, Maeta K, Kawaguchi T, Awano H, Nagai M, Nishio H, Matsuo M (2019) Schwann cell-specific Dp116 is expressed in glioblastoma cells, revealing two novel DMD gene splicing patterns. Biochem Biophys Rep 20:100703
Malecova B, Gatto S, Etxaniz U, Passafaro M, Cortez A, Nicoletti C, Giordani L, Torcinaro A, De Bardi M, Bicciato S, De Santa F, Madaro L, Puri PL (2018) Dynamics of cellular states of fibro-adipogenic progenitors during myogenesis and muscular dystrophy. Nat Commun 9:3670
Manokaran RK, Aggarwala S, Kumar R, Gupta AK, Chakrabarty B, Jauhari P, Pandey RM, Gulati S (2020) Prevalence of smooth muscle dysfunction among children with Duchenne muscular dystrophy. Muscle Nerve 62:699–704
March JT, Golshirazi G, Cernisova V, Carr H, Leong Y, Lu-Nguyen N, Popplewell LJ (2018) Targeting TGFbeta signaling to address fibrosis using antisense oligonucleotides. Biomedicines 6:74
Mareedu S, Million ED, Duan D, Babu GJ (2021) Abnormal calcium handling in duchenne muscular dystrophy: mechanisms and potential therapies. Front Physiol 12:647010
Marini JF, Pons F, Leger J, Loffreda N, Anoal M, Chevallay M, Fardeau M, Leger JJ (1991) Expression of myosin heavy chain isoforms in Duchenne muscular dystrophy patients and carriers. Neuromuscul Disord 1:397–409
Marquis-Nicholson R, Lai D, Lan CC, Love JM, Love DR (2013) A streamlined protocol for molecular testing of the DMD gene within a diagnostic laboratory: a combination of array comparative genomic hybridization and bidirectional sequence analysis. ISRN Neurol 2013:908317
Matsuo M, Awano H, Maruyama N, Nishio H (2019) Titin fragment in urine: a noninvasive biomarker of muscle degradation. Adv Clin Chem 90:1–23
Matsuo M, Awano H, Matsumoto M, Nagai M, Kawaguchi T, Zhang Z, Nishio H (2017) Dystrophin Dp116: a yet to be investigated product of the Duchenne muscular dystrophy gene. Genes (Basel) 8:251
Matsumura T, Saito T, Fujimura H, Sakoda S (2012) Renal dysfunction is a frequent complication in patients with advanced stage of Duchenne muscular dystrophy]. Rinsho Shinkeigaku 52:211–217
Mázala DA, Novak JS, Hogarth MW, Nearing M, Adusumalli P, Tully CB, Habib NF, Gordish-Dressman H, Chen YW, Jaiswal JK, Partridge TA (2020) TGF-beta-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 5:e135703
McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, Clemens PR, Hoffman EP, Cnaan A, Gordish-Dressman H, Investigators CINRG (2018) Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 391:451–461
Mehler MF (2000) Brain dystrophin, neurogenetics and mental retardation. Brain Res Brain Res Rev 32:277–307
Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, Kaye EM, Mercuri E, Eteplirsen Study Group and Telethon Foundation DMD Italian Network (2016) Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 79:257–271
Mercado ML, Amenta AR, Hagiwara H, Rafii MS, Lechner BE, Owens RT, McQuillan DJ, Froehner SC, Fallon JR (2006) Biglycan regulates the expression and sarcolemmal localization of dystrobrevin, syntrophin, and nNOS. FASEB J 20:1724–1726
Mercuri E, Bönnemann CG, Muntoni F (2019) Muscular dystrophies. Lancet 394:2025–2038
Merlini L, Cecconi I, Parmeggiani A, Cordelli DM, Dormi A (2020) Quadriceps muscle strength in Duchenne muscular dystrophy and effect of corticosteroid treatment. Acta Myol 39:200–206
Messina S, Vita GL (2018) Clinical management of Duchenne muscular dystrophy: the state of the art. Neurol Sci 39:1837–1845
Meyers TA, Townsend D (2019) Cardiac pathophysiology and the future of cardiac therapies in Duchenne muscular dystrophy. Int J Mol Sci 20:4098
Micanovic R, LaFavers K, Garimella PS, Wu XR, El-Achkar TM (2020) Uromodulin (Tamm-Horsfall protein): guardian of urinary and systemic homeostasis. Nephrol Dial Transplant 35:33–43
Min YL, Bassel-Duby R, Olson EN (2019) CRISPR correction of Duchenne muscular dystrophy. Annu Rev Med 70:239–255
Mojumdar K, Giordano C, Lemaire C, Liang F, Divangahi M, Qureshi ST, Petrof BJ (2016) Divergent impact of Toll-like receptor 2 deficiency on repair mechanisms in healthy muscle versus Duchenne muscular dystrophy. J Pathol 239:10–22
Mojumdar K, Liang F, Giordano C, Lemaire C, Danialou G, Okazaki T, Bourdon J, Rafei M, Galipeau J, Divangahi M, Petrof BJ (2014) Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol Med 6:1476–1492
Morgenroth VH, Hache LP, Clemens PR (2012) Insights into bone health in Duchenne muscular dystrophy. Bonekey Rep 1:9
Morse CI, Higham K, Bostock EL, Jacques MF (2020) Urinary incontinence in men with Duchenne and Becker muscular dystrophy. PLoS One 15:e0233527
Motoki T, Shimizu-Motohashi Y, Saito I, Komaki H, Ishiyama A, Aibara K, Jogamoto T, Tezuka Y, Kawabe M, Makino A, Nagatani K, Tatara K, Kuwabara K, Kikuchi C, Fukuda M, Ishii E, Eguchi M (2020) Renal dysfunction can occur in advanced-stage Duchenne muscular dystrophy. Muscle Nerve 61:192–197
Mournetas V, Massouridès E, Dupont JB, Kornobis E, Polvèche H, Jarrige M, Dorval ARL, Gosselin MRF, Manousopoulou A, Garbis SD, Górecki DC, Pinset C (2021) Myogenesis modelled by human pluripotent stem cells: a multi-omic study of Duchenne myopathy early onset. J Cachexia Sarcopenia Muscle 12:209–232
Mueller AL, Bloch RJ (2020) Skeletal muscle cell transplantation: models and methods. J Muscle Res Cell Motil 41:297–311
Mukund K, Subramaniam S (2020) Skeletal muscle: a review of molecular structure and function, in health and disease. Wiley Interdiscip Rev Syst Biol Med 12:e1462
Muntoni F, Mateddu A, Serra G (1991) Passive avoidance behaviour deficit in the mdx mouse. Neuromuscul Disord 1:121–123
Muntoni F, Torelli S, Ferlini A (2003) Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2:731–740
Murgia M, Nagaraj N, Deshmukh AS, Zeiler M, Cancellara P, Moretti I, Reggiani C, Schiaffino S, Mann M (2015) Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep 16:387–395
Murphy S, Brinkmeier H, Krautwald M, Henry M, Meleady P, Ohlendieck K (2017) Proteomic profiling of the dystrophin complex and membrane fraction from dystrophic mdx muscle reveals decreases in the cytolinker desmoglein and increases in the extracellular matrix stabilizers biglycan and fibronectin. J Muscle Res Cell Motil 38:251–268
Murphy S, Dowling P, Zweyer M, Henry M, Meleady P, Mundegar RR, Swandulla D, Ohlendieck K (2017) Proteomic profiling of mdx-4cv serum reveals highly elevated levels of the inflammation-induced plasma marker haptoglobin in muscular dystrophy. Int J Mol Med 39:1357–1370
Murphy S, Dowling P, Zweyer M, Mundegar RR, Henry M, Meleady P, Swandulla D, Ohlendieck K (2016) Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J Proteomics 145:24–36
Murphy S, Dowling P, Zweyer M, Swandulla D, Ohlendieck K (2019) Proteomic profiling of giant skeletal muscle proteins. Expert Rev Proteomics 16:241–256
Murphy S, Ohlendieck K (2015) The biochemical and mass spectrometric profiling of the dystrophin complexome from skeletal muscle. Comput Struct Biotechnol J 14:20–27
Murphy S, Zweyer M, Henry M, Meleady P, Mundegar RR, Swandulla D, Ohlendieck K (2015) Label-free mass spectrometric analysis reveals complex changes in the brain proteome from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin Proteomics 12:27
Murphy S, Zweyer M, Henry M, Meleady P, Mundegar RR, Swandulla D, Ohlendieck K (2018) Proteomic profiling of liver tissue from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin Proteomics 15:34
Murphy S, Zweyer M, Mundegar RR, Swandulla D, Ohlendieck K (2018) Proteomic serum biomarkers for neuromuscular diseases. Expert Rev Proteomics 15:277–291
Murphy S, Zweyer M, Mundegar RR, Swandulla D, Ohlendieck K (2018) Proteomic identification of elevated saliva kallikrein levels in the mdx-4cv mouse model of Duchenne muscular dystrophy. Biochem Biophys Rep 18:100541
Murphy S, Zweyer M, Raucamp M, Henry M, Meleady P, Swandulla D, Ohlendieck K (2019) Proteomic profiling of the mouse diaphragm and refined mass spectrometric analysis of the dystrophic phenotype. J Muscle Res Cell Motil 40:9–28
Naidoo M, Anthony K (2020) Dystrophin Dp71 and the neuropathophysiology of duchenne muscular dystrophy. Mol Neurobiol 57:1748–1767
Nakamori M, Takahashi MP (2011) The role of alpha-dystrobrevin in striated muscle. Int J Mol Sci 12:1660–1671
Nance ME, Duan D (2015) Perspective on adeno-associated virus capsid modification for Duchenne muscular dystrophy gene therapy. Hum Gene Ther 26:786–800
Nerakh G, Ranganath P, Murugan S (2021) Next-generation sequencing in a cohort of asian indian patients with the Duchenne muscular dystrophy phenotype: diagnostic yield and mutation spectrum. J Pediatr Genet 10:23–28
Ng SY, Ljubicic V (2020) Recent insights into neuromuscular junction biology in Duchenne muscular dystrophy: impacts, challenges, and opportunities. EBioMedicine 61:103032
Nico B, Corsi P, Ria R, Crivellato E, Vacca A, Roccaro AM, Mangieri D, Ribatti D, Roncali L (2006) Increased matrix-metalloproteinase-2 and matrix-metalloproteinase-9 expression in the brain of dystrophic mdx mouse. Neuroscience 140:835–848
Nico B, Frigeri A, Nicchia GP, Corsi P, Ribatti D, Quondamatteo F, Herken R, Girolamo F, Marzullo A, Svelto M, Roncali L (2003) Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia 42:235–251
Nico B, Tamma R, Annese T, Mangieri D, De Luca A, Corsi P, Benagiano V, Longo V, Crivellato E, Salmaggi A, Ribatti D (2010) Glial dystrophin-associated proteins, laminin and agrin, are downregulated in the brain of mdx mouse. Lab Invest 90:1645–1660
Nix JS, Moore SA (2020) What every neuropathologist needs to know: the muscle biopsy. J Neuropathol Exp Neurol 79:719–733
Ohlendieck K (1996) Towards an understanding of the dystrophin-glycoprotein complex: linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur J Cell Biol 69:1–10
Ohlendieck K (2013) Proteomic identification of biomarkers of skeletal muscle disorders. Biomark Med 7:169–186
Ohlendieck K, Ervasti JM, Matsumura K, Kahl SD, Leveille CJ, Campbell KP (1991) Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron 7:499–508
Ohlendieck K, Ervasti JM, Snook JB, Campbell KP (1991) Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J Cell Biol 112:135–148
Ohlendieck K, Matsumura K, Ionasescu VV, Towbin JA, Bosch EP, Weinstein SL, Sernett SW, Campbell KP (1993) Duchenne muscular dystrophy: deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 43:795–800
Ohlendieck K, Swandulla D (2017) Molecular pathogenesis of Duchenne muscular dystrophy-related fibrosis. Pathologe 38:21–29
Okubo M, Minami N, Goto K, Goto Y, Noguchi S, Mitsuhashi S, Nishino I (2016) Genetic diagnosis of Duchenne/Becker muscular dystrophy using next-generation sequencing: validation analysis of DMD mutations. J Hum Genet 61:483–489
Ouisse LH, Remy S, Lafoux A, Larcher T, Tesson L, Chenouard V, Guillonneau C, Brusselle L, Vimond N, Rouger K, Péréon Y, Chenouard A, Gras-Le Guen C, Braudeau C, Josien R, Huchet C, Anegon I (2019) Immunophenotype of a rat model of Duchenne’s disease and demonstration of improved muscle strength after anti-CD45RC antibody treatment. Front Immunol 10:2131
Pane M, Lombardo ME, Alfieri P, D’Amico A, Bianco F, Vasco G, Piccini G, Mallardi M, Romeo DM, Ricotti V, Ferlini A, Gualandi F, Vicari S, Bertini E, Berardinelli A, Mercuri E (2012) Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype-genotype correlation. J Pediatr 161:705–709
Parisi L, Di Filippo T, Glorioso P, La Grutta S, Epifanio MS, Roccella M (2018) Autism spectrum disorders in children affected by Duchenne muscular dystrophy. Minerva Pediatr 70:233–239
Pedemonte M, Sandri C, Schiaffino S, Minetti C (1999) Early decrease of IIx myosin heavy chain transcripts in Duchenne muscular dystrophy. Biochem Biophys Res Commun 255:466–469
Perronnet C, Vaillend C (2010) Dystrophins, utrophins, and associated scaffolding complexes: role in mammalian brain and implications for therapeutic strategies. J Biomed Biotechnol 2010:849426
Petkova MV, Morales-Gonzales S, Relizani K, Gill E, Seifert F, Radke J, Stenzel W, Garcia L, Amthor H, Schuelke M (2016) Characterization of a Dmd (EGFP) reporter mouse as a tool to investigate dystrophin expression. Skelet Muscle 6:25
Pette D (2021) The significance of Gerta Vrbová’s low-frequency stimulation experiment. Eur J Transl Myol 31:9585
Pette D, Vrbová G (2017) The contribution of neuromuscular stimulation in elucidating muscle plasticity revisited. Eur J Transl Myol 27:6368
Politano L (2021) Read-through approach for stop mutations in Duchenne muscular dystrophy. An update Acta Myol 40:43–50
Preethish-Kumar V, Shah A, Kumar M, Ingalhalikar M, Polavarapu K, Afsar M, Rajeswaran J, Vengalil S, Nashi S, Thomas PT, Sadasivan A, Warrier M, Nalini A, Saini J (2020) In vivo evaluation of white matter abnormalities in children with Duchenne muscular dystrophy using DTI. AJNR Am J Neuroradiol 41:1271–1278
Prins KW, Humston JL, Mehta A, Tate V, Ralston E, Ervasti JM (2009) Dystrophin is a microtubule-associated protein. J Cell Biol 186:363–369
Quattrocelli M, Zelikovich AS, Salamone IM, Fischer JA, McNally EM (2021) Mechanisms and clinical applications of glucocorticoid steroids in muscular dystrophy. J Neuromuscul Dis 8:39–52
Rafii MS, Hagiwara H, Mercado ML, Seo NS, Xu T, Dugan T, Owens RT, Hook M, McQuillan DJ, Young MF, Fallon JR (2006) Biglycan binds to alpha- and gamma-sarcoglycan and regulates their expression during development. J Cell Physiol 209:439–447
Rayavarapu S, Coley W, Cakir E, Jahnke V, Takeda S, Aoki Y, Grodish-Dressman H, Jaiswal JK, Hoffman EP, Brown KJ, Hathout Y, Nagaraju K (2013) Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol Cell Proteomics 12:1061–1073
Reay DP, Bastacky SI, Wack KE, Stolz DB, Robbins PD, Clemens PR (2015) D-amino acid substitution of peptide-mediated NF-κB suppression in mdx mice preserves therapeutic benefit in skeletal muscle, but causes kidney toxicity. Mol Med 21:442–452
Rees ML, Lien CF, Górecki DC (2007) Dystrobrevins in muscle and non-muscle tissues. Neuromuscul Disord 17:123–134
Regensburger AP, Fonteyne LM, Jüngert J, Wagner AL, Gerhalter T, Nagel AM, Heiss R, Flenkenthaler F, Qurashi M, Neurath MF, Klymiuk N, Kemter E, Fröhlich T, Uder M, Woelfle J, Rascher W, Trollmann R, Wolf E, Waldner MJ, Knieling F (2019) Detection of collagens by multispectral optoacoustic tomography as an imaging biomarker for Duchenne muscular dystrophy. Nat Med 25:1905–1915
Reggiani C, Schiaffino S (2020) Muscle hypertrophy and muscle strength: dependent or independent variables? A provocative review. Eur J Transl Myol 30:9311
Rezniczek GA, Konieczny P, Nikolic B, Reipert S, Schneller D, Abrahamsberg C, Davies KE, Winder SJ, Wiche G (2007) Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J Cell Biol 176:965–977
Ricotti V, Mandy WP, Scoto M, Pane M, Deconinck N, Messina S, Mercuri E, Skuse DH, Muntoni F (2016) Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol 58:77–84
Ricotti V, Selby V, Ridout D, Domingos J, Decostre V, Mayhew A, Eagle M, Butler J, Guglieri M, Van der Holst M, Jansen M, Verschuuren JJGM, de Groot IJM, Niks EH, Servais L, Straub V, Voit T, Hogrel JY, Muntoni F (2019) Respiratory and upper limb function as outcome measures in ambulant and non-ambulant subjects with Duchenne muscular dystrophy: a prospective multicentre study. Neuromuscul Disord 29:261–268
Rizzo G, Di Maggio R, Benedetti A, Morroni J, Bouche M, Lozanoska-Ochser B (2020) Splenic Ly6Chi monocytes are critical players in dystrophic muscle injury and repair. JCI Insight 5:130807
Romo-Yáñez J, Rodríguez-Martínez G, Aragón J, Siqueiros-Márquez L, Herrera-Salazar A, Velasco I, Montanez C (2020) Characterization of the expression of dystrophins and dystrophin-associated proteins during embryonic neural stem/progenitor cell differentiation. Neurosci Lett 736:135247
Roos A, Thompson R, Horvath R, Lochmüller H, Sickmann A (2018) Intersection of proteomics and genomics to "solve the unsolved" in rare disorders such as neurodegenerative and neuromuscular diseases. Proteomics Clin Appl 12(2) https://doi.org/10.1002/prca.201700073
Rosenberg AS, Puig M, Nagaraju K, Hoffman EP, Villalta SA, Rao VA, Wakefield LM, Woodcock J (2015) Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med 7:299rv4
Ross LF, Clarke AJ (2017) A historical and current review of newborn screening for neuromuscular disorders from around the world: lessons for the United States. Pediatr Neurol 77:12–22
Rouillon J, Lefebvre T, Denard J, Puy V, Daher R, Ausseil J, Zocevic A, Fogel P, Peoc’h K, Wong B, Servais L, Voit T, Puy H, Karim Z, Svinartchouk F (2018) High urinary ferritin reflects myoglobin iron evacuation in DMD patients. Neuromuscul Disord 28:564–571
Rouillon J, Zocevic A, Leger T, Garcia C, Camadro JM, Udd B, Wong B, Servais L, Voit T, Svinartchouk F (2014) Proteomics profiling of urine reveals specific titin fragments as biomarkers of Duchenne muscular dystrophy. Neuromuscul Disord 24:563–573
Roy B, Griggs R (2021) Advances in treatments in muscular dystrophies and motor neuron disorders. Neurol Clin 39:87–112
Rufo A, Del Fattore A, Capulli M, Carvello F, De Pasquale L, Ferrari S, Pierroz D, Morandi L, De Simone M, Rucci N, Bertini E, Bianchi ML, De Benedetti F, Teti A (2011) Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J Bone Miner Res 26:1891–1903
Rupert JE, Jengelley DHA, Zimmers TA (2020) In vitro, in vivo, and in silico methods for assessment of muscle size and muscle growth regulation. Shock 53:605–615
Rybakova IN, Ervasti JM (1997) Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J Biol Chem 272:28771–28778
Rybalka E, Timpani CA, Stathis CG, Hayes A, Cooke MB (2015) Metabogenic and nutriceutical approaches to address energy dysregulation and skeletal muscle wasting in Duchenne muscular dystrophy. Nutrients 7:9734–9767
Ryder S, Leadley RM, Armstrong N, Westwood M, de Kock S, Butt T, Jain M, Kleijnen J (2017) The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis 12:79
Sabourin J, Lamiche C, Vandebrouck A, Magaud C, Rivet J, Cognard C, Bourmeyster N, Constantin B (2009) Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J Biol Chem 284:36248–36261
Saito T, Kawai M, Kimura E, Ogata K, Takahashi T, Kobayashi M, Takada H, Kuru S, Mikata T, Matsumura T, Yonemoto N, Fujimura H, Sakoda S (2017) Study of Duchenne muscular dystrophy long-term survivors aged 40 years and older living in specialized institutions in Japan. Neuromuscul Disord 27:107–114
Sansović I, Barišić I, Dumić K (2013) Improved detection of deletions and duplications in the DMD gene using the multiplex ligation-dependent probe amplification (MLPA) method. Biochem Genet 51:189–201
Santos AC, Olivera DM, Bertassoli BM, Viana DC, Vasconcelos BG, Assis Neto AC (2013) Morphologic features from mdx mice spleens, used for Duchenne muscular dystrophy studies. J Morphol Sci 30:21–27
Saure C, Caminiti C, Weglinski J, de Castro PF, Monges S (2018) Energy expenditure, body composition, and prevalence of metabolic disorders in patients with Duchenne muscular dystrophy. Diabetes Metab Syndr 12:81–85
Serrano AL, Muñoz-Cánoves P (2017) Fibrosis development in early-onset muscular dystrophies: mechanisms and translational implications. Semin Cell Dev Biol 64:181–190
Seto JT, Ramos JN, Muir L, Chamberlain JS, Odom GL (2012) Gene replacement therapies for duchenne muscular dystrophy using adeno-associated viral vectors. Curr Gene Ther 12:139–151
Sheikh O, Yokota T (2020) Advances in genetic characterization and genotype-phenotype correlation of Duchenne and Becker muscular dystrophy in the personalized medicine era. J Pers Med 10:111
Sheikh O, Yokota T (2021) Developing DMD therapeutics: a review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies. Expert Opin Investig Drugs 30:167–176
Shimizu-Motohashi Y, Komaki H, Motohashi N, Takeda S, Yokota T, Aoki Y (2019) Restoring dystrophin expression in Duchenne muscular dystrophy: current status of therapeutic approaches. J Pers Med 9:1
Shin J, Tajrishi MM, Ogura Y, Kumar A (2013) Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol 45:2266–2279
Shirley M (2021) Casimersen: first approval. Drugs 81:875–879
Shirokova N, Niggli E (2013) Cardiac phenotype of Duchenne muscular dystrophy: insights from cellular studies. J Mol Cell Cardiol 58:217–224
Shumyatcher Y, Shah TA, Noritz GH, Brouhard BH, Spirnak JP, Birnkrant DJ (2008) Symptomatic nephrolithiasis in prolonged survivors of Duchenne muscular dystrophy. Neuromuscul Disord 18:561–564
Siemionow M, Szilagyi E, Cwykiel J, Domaszewska-Szostek A, Heydemann A, Garcia-Martinez J, Siemionow K (2021) Transplantation of dystrophin expressing chimeric human cells of myoblast/mesenchymal stem cell origin improves function in Duchenne muscular dystrophy model. Stem Cells Dev 30:190–202
Signorelli M, Ayoglu B, Johansson C, Lochmüller H, Straub V, Muntoni F, Niks E, Tsonaka R, Persson A, Aartsma-Rus A, Nilsson P, Al-Khalili Szigyarto C, Spitali P (2020) Longitudinal serum biomarker screening identifies malate dehydrogenase 2 as candidate prognostic biomarker for Duchenne muscular dystrophy. J Cachexia Sarcopenia Muscle 11:505–517
Signorelli M, Ebrahimpoor M, Veth O, Hettne K, Verwey N, García-Rodríguez R, Tanganyika-deWinter CL, Lopez Hernandez LB, Escobar Cedillo R, Gómez Díaz B, Magnusson OT, Mei H, Tsonaka R, Aartsma-Rus A, Spitali P (2021) Peripheral blood transcriptome profiling enables monitoring disease progression in dystrophic mice and patients. EMBO Mol Med 13:e13328
Singer RH (2020) Distance meeting: farewell to professor David Yaffe - a pillar of the myogenesis field. Eur J Transl Myol 30(3):9327
Singh M, Jacobs IB, Spirnak JP (2007) Nephrolithiasis in patients with duchenne muscular dystrophy. Urology 70:643–645
Smith LR, Barton ER (2018) Regulation of fibrosis in muscular dystrophy. Matrix Biol 68:602–615
Smith LR, Hammers DW, Sweeney HL, Barton ER (2016) Increased collagen cross-linking is a signature of dystrophin-deficient muscle. Muscle Nerve 54:71–78
Snow WM, Anderson JE, Jakobson LS (2013) Neuropsychological and neurobehavioral functioning in Duchenne muscular dystrophy: a review. Neurosci Biobehav Rev 37:743–752
Soblechero-Martín P, López-Martínez A, de la Puente-Ovejero L, Vallejo-Illarramendi A, Arechavala-Gomeza V (2021) Utrophin modulator drugs as potential therapies for Duchenne and Becker muscular dystrophies. Neuropathol Appl Neurobiol (in press)https://doi.org/10.1111/nan.12735
Soeters PB, Shenkin A, Sobotka L, Soeters MR, de Leeuw PW, Wolfe RR (2021) The anabolic role of the Warburg, Cori-cycle and Crabtree effects in health and disease. Clin Nutr 40:2988–2998
Spencer MJ, Montecino-Rodriguez E, Dorshkind K, Tidball JG (2001) Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol 98:235–243
Spencer MJ, Tidball JG (2001) Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul Disord 11:556–564
Spitali P, Hettne K, Tsonaka R, Charrout M, van den Bergen J, Koeks Z, Kan HE, Hooijmans MT, Roos A, Straub V, Muntoni F, Al-Khalili-Szigyarto C, Koel-Simmelink MJA, Teunissen CE, Lochmüller H, Niks EH, Aartsma-Rus A (2018) Tracking disease progression non-invasively in Duchenne and Becker muscular dystrophies. J Cachexia Sarcopenia Muscle 9:715–726
Srivastava NK, Yadav R, Mukherjee S, Pal L, Sinha N (2017) Abnormal lipid metabolism in skeletal muscle tissue of patients with muscular dystrophy: in vitro, high-resolution NMR spectroscopy based observation in early phase of the disease. Magn Reson Imaging 38:163–173
Srivastava NK, Yadav R, Mukherjee S, Sinha N (2018) Perturbation of muscle metabolism in patients with muscular dystrophy in early or acute phase of disease: In vitro, high resolution NMR spectroscopy based analysis. Clin Chim Acta 478:171–181
Starosta A, Konieczny P (2021) Therapeutic aspects of cell signaling and communication in Duchenne muscular dystrophy. Cell Mol Life Sci 78:4867–4891
Stay TL, Miterko LN, Arancillo M, Lin T, Sillitoe RV (2019) In vivo cerebellar circuit function is disrupted in an mdx mouse model of Duchenne muscular dystrophy. Dis Model Mech 13 dmm040840
Steinberger M, Föller M, Vogelgesang S, Krautwald M, Landsberger M, Winkler CK, Kasch J, Füchtbauer EM, Kuhl D, Voelkl J, Lang F, Brinkmeier H (2015) Lack of the serum- and glucocorticoid-inducible kinase SGK1 improves muscle force characteristics and attenuates fibrosis in dystrophic mdx mouse muscle. Pflugers Arch 467:1965–1974
Strakova J, Kamdar F, Kulhanek D, Razzoli M, Garry DJ, Ervasti JM, Bartolomucci A, Townsend D (2018) Integrative effects of dystrophin loss on metabolic function of the mdx mouse. Sci Rep 8:13624
Strandberg K, Ayoglu B, Roos A, Reza M, Niks E, Signorelli M, Fasterius E, Pontén F, Lochmüller H, Domingos J, Ala P, Muntoni F, Aartsma-Rus A, Spitali P, Nilsson P, Szigyarto CA (2020) Blood-derived biomarkers correlate with clinical progression in Duchenne muscular dystrophy. J Neuromuscul Dis 7:231–246
Strehle EM, Straub V (2015) Recent advances in the management of Duchenne muscular dystrophy. Arch Dis Child 100:1173–1177
Sun C, Serra C, Lee G, Wagner KR (2020) Stem cell-based therapies for Duchenne muscular dystrophy. Exp Neurol 323:113086
Sun C, Shen L, Zhang Z, Xie X (2020) Therapeutic strategies for Duchenne muscular dystrophy: an update. Genes (Basel) 11:837
Suthar R, Reddy BVC, Malviya M, Sirari T, Attri SV, Patial A, Tageja M, Didwal G, Khandelwal NK, Saini AG, Saini L, Sahu JK, Dayal D, Sankhyan N (2021) Bone density and bone health alteration in boys with Duchenne muscular dystrophy: a prospective observational study. J Pediatr Endocrinol Metab 34:573–581
Szigyarto CA, Spitali P (2018) Biomarkers of Duchenne muscular dystrophy: current findings. Degener Neurol Neuromuscul Dis 8:1–13
Tadayoni R, Rendon A, Soria-Jasso LE, Cisneros B (2012) Dystrophin Dp71: the smallest but multifunctional product of the Duchenne muscular dystrophy gene. Mol Neurobiol 45:43–60
Takeshita E, Komaki H, Tachimori H, Miyoshi K, Yamamiya I, Shimizu-Motohashi Y, Ishiyama A, Saito T, Nakagawa E, Sugai K, Sasaki M (2018) Urinary prostaglandin metabolites as Duchenne muscular dystrophy progression markers. Brain Dev 40:918–925
Tamiyakul H, Kemter E, Kösters M, Ebner S, Blutke A, Klymiuk N, Flenkenthaler F, Wolf E, Arnold GJ, Fröhlich T (2020) Progressive proteome changes in the myocardium of a pig model for Duchenne muscular dystrophy. iScience 23:101516
Tennyson CN, Klamut HJ, Worton RG (1995) The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet 9:184–190
Terrill JR, Al-Mshhdani BA, Duong MN, Wingate CD, Abbas Z, Baustista AP, Bettis AK, Balog-Alvarez CJ, Kornegay JN, Nghiem PP, Grounds MD, Arthur PG (2020) Oxidative damage to urinary proteins from the GRMD dog and mdx mouse as biomarkers of dystropathology in Duchenne muscular dystrophy. PLoS One 15:e0240317
Thakur SS, Swiderski K, Ryall JG, Lynch GS (2018) Therapeutic potential of heat shock protein induction for muscular dystrophy and other muscle wasting conditions. Philos Trans R Soc Lond B Biol Sci 373:20160528
Thangarajh M (2019) The dystrophinopathies. Continuum (Minneap Minn) 25:1619–1639
Thangarajh M, Elfring GL, Trifillis P (2020) Longitudinal evaluation of working memory in Duchenne muscular dystrophy. J Clin Med 9:2940
Thangarajh M, Hendriksen J, McDermott MP, Martens W, Hart KA, Griggs RC, Muscle Study Group and TREAT-NMD (2019) Relationships between DMD mutations and neurodevelopment in dystrophinopathy. Neurology 93:e1597–e1604
Theret M, Rossi FMV, Contreras O (2021) Evolving roles of muscle-resident fibro-adipogenic progenitors in health, regeneration, neuromuscular disorders, and aging. Front Physiol 12:673404
Thomas GD (2013) Functional muscle ischemia in Duchenne and Becker muscular dystrophy. Front Physiol 4:381
Thompson R, Spendiff S, Roos A, Bourque PR, Warman Chardon J, Kirschner J, Horvath R, Lochmüller H (2020) Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet Neurol 19:522–532
Tidball JG, Welc SS, Wehling-Henricks M (2018) Immunobiology of inherited muscular dystrophies. Compr Physiol 8:1313–1356
Tinsley JM, Blake DJ, Roche A, Fairbrother U, Riss J, Byth BC, Knight AE, Kendrick-Jones J, Suthers GK, Love DR, Edwards YH, Davies KE (1992) Primary structure of dystrophin-related protein. Nature 360:591–593
Tracey I, Dunn JF, Radda GK (1996) Brain metabolism is abnormal in the mdx model of Duchenne muscular dystrophy. Brain 119:1039–1044
Trensz F, Haroun S, Cloutier A, Richter MV, Grenier G (2010) A muscle resident cell population promotes fibrosis in hindlimb skeletal muscles of mdx mice through the Wnt canonical pathway. Am J Physiol Cell Physiol 299:C939–C947
Tsonaka R, Signorelli M, Sabir E, Seyer A, Hettne K, Aartsma-Rus A, Spitali P (2020) Longitudinal metabolomic analysis of plasma enables modeling disease progression in Duchenne muscular dystrophy mouse models. Hum Mol Genet 29:745–755
Tuffery-Giraud S, Miro J, Koenig M, Claustres M (2017) Normal and altered pre-mRNA processing in the DMD gene. Hum Genet 136:1155–1172
Tyagi R, Aggarwal P, Mohanty M, Dutt V, Anand A (2020) Computational cognitive modeling and validation of Dp140 induced alteration of working memory in Duchenne Muscular Dystrophy. Sci Rep 10:11989
Uchino M, Hara A, Mizuno Y, Fujiki M, Nakamura T, Tokunaga M, Hirano T, Yamashita T, Uyama E, Ando Y, Mita S, Ando M (1996) Distribution of dystrophin and dystrophin-associated protein 43DAG (beta-dystroglycan) in the central nervous system of normal controls and patients with Duchenne muscular dystrophy. Intern Med 35:189–194
Ueda H, Baba T, Ohno S (2000) Current knowledge of dystrophin and dystrophin-associated proteins in the retina. Histol Histopathol 15:753–760
Ursitti JA, Lee PC, Resneck WG, McNally MM, Bowman AL, O’Neill A, Stone MR, Bloch RJ (2004) Cloning and characterization of cytokeratins 8 and 19 in adult rat striated muscle. Interaction with the dystrophin glycoprotein complex. J Biol Chem 279:41830–41838
Vaillend C, Billard JM, Laroche S (2004) Impaired long-term spatial and recognition memory and enhanced CA1 hippocampal LTP in the dystrophin-deficient Dmd(mdx) mouse. Neurobiol Dis 17:10–20
Van Pelt DW, Kharaz YA, Sarver DC, Eckhardt LR, Dzierzawski JT, Disser NP, Piacentini AN, Comerford E, McDonagh B, Mendias CL (2021) Multiomics analysis of the mdx/mTR mouse model of Duchenne muscular dystrophy. Connect Tissue Res 62:24–39
van Westering TLE, Johansson HJ, Hanson B, Coenen-Stass AML, Lomonosova Y, Tanihata J, Motohashi N, Yokota T, Takeda S, Lehtiö J, Wood MJA, El Andaloussi S, Aoki Y, Roberts TC (2020) Mutation-independent proteomic signatures of pathological progression in murine models of Duchenne muscular dystrophy. Mol Cell Proteomics 19:2047–2068
Verhaart IEC, Aartsma-Rus A (2019) Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol 15:373–386
Verma M, Fitzpatrick K, McLoon LK (2017) Extraocular muscle repair and regeneration. Curr Ophthalmol Rep 5:207–215
Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, Baeza-Raja B, Martínez de Lagrán M, Lafuste P, Ruiz-Bonilla V, Jardí M, Gherardi R, Christov C, Dierssen M, Carmeliet P, Degen JL, Dewerchin M, Muñoz-Cánoves P (2008) Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev 22:1747–1752
Villa CR, Kaddourah A, Mathew J, Ryan TD, Wong BL, Goldstein SL, Jefferies JL (2016) Identifying evidence of cardio-renal syndrome in patients with Duchenne muscular dystrophy using cystatin C. Neuromuscul Disord 26:637–642
Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG (2009) Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet 18:482–496
Villalta SA, Rinaldi C, Deng B, Liu G, Fedor B, Tidball JG (2011) Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum Mol Genet 20:790–805
Villanova M, Kazibwe S (2017) New survival target for Duchenne muscular dystrophy. Am J Phys Med Rehabil 96:e28–e30
Viollet L, Gailey S, Thornton DJ, Friedman NR, Flanigan KM, Mahan JD, Mendell JR (2009) Utility of cystatin C to monitor renal function in Duchenne muscular dystrophy. Muscle Nerve 40:438–442
Vuorinen A, Wilkinson IVL, Chatzopoulou M, Edwards B, Squire SE, Fairclough RJ, Bazan NA, Milner JA, Conole D, Donald JR, Shah N, Willis NJ, Martínez RF, Wilson FX, Wynne GM, Davies SG, Davies KE, Russell AJ (2021) Discovery and mechanism of action studies of 4,6-diphenylpyrimidine-2-carbohydrazides as utrophin modulators for the treatment of Duchenne muscular dystrophy. Eur J Med Chem 220:113431
Wada E, Hamano T, Matsui I, Yoshida M, Hayashi YK, Matsuda R (2019) Renal involvement in the pathogenesis of mineral and bone disorder in dystrophin-deficient mdx mouse. J Physiol Sci 69:661–671
Waite A, Brown SC, Blake DJ (2012) The dystrophin-glycoprotein complex in brain development and disease. Trends Neurosci 35:487–496
Ward LM, Hadjiyannakis S, McMillan HJ, Noritz G, Weber DR (2018) Bone health and osteoporosis management of the patient with Duchenne muscular dystrophy. Pediatrics 142:S34–S42
Watkins SC, Hoffman EP, Slayter HS, Kunkel LM (1988) Immunoelectron microscopic localization of dystrophin in myofibres. Nature 333:863–866
Webster C, Silberstein L, Hays AP, Blau HM (1988) Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 52:503–513
Wei X, Dai Y, Yu P, Qu N, Lan Z, Hong X, Sun Y, Yang G, Xie S, Shi Q, Zhou H, Zhu Q, Chu Y, Yao F, Wang J, He J, Yang Y, Liang Y, Yang Y, Qi M, Yang L, Wang W, Wu H, Duan J, Shen C, Wang J, Cui L, Yi X (2014) Targeted next-generation sequencing as a comprehensive test for patients with and female carriers of DMD/BMD: a multi-population diagnostic study. Eur J Hum Genet 22:110–118
Wei J, Gao Y (2021) Early disease biomarkers can be found using animal models urine proteomics. Expert Rev Proteomics 18:363–378
Wen Y, Parikh CR (2021) Current concepts and advances in biomarkers of acute kidney injury. Crit Rev Clin Lab Sci 8:1–24
Willcocks RJ, Arpan IA, Forbes SC, Lott DJ, Senesac CR, Senesac E, Deol J, Triplett WT, Baligand C, Daniels MJ, Sweeney HL, Walter GA, Vandenborne K (2014) Longitudinal measurements of MRI-T2 in boys with Duchenne muscular dystrophy: effects of age and disease progression. Neuromuscul Disord 24:393–401
Wilson J, Putt W, Jimenez C, Edwards YH (1999) Up71 and up140, two novel transcripts of utrophin that are homologues of short forms of dystrophin. Hum Mol Genet 8:1271–1278
Xu Y, Xie Y, Shao X, Ni Z, Mou S (2015) L-FABP: A novel biomarker of kidney disease. Clin Chim Acta 445:85–90
Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP (1995) SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem 270:11711–11714
Yuan C, Arora A, Garofalo AM, Grange RW (2021) Potential cross-talk between muscle and tendon in Duchenne muscular dystrophy. Connect Tissue Res 62:40–52
Zanotti S, Gibertini S, Mora M (2010) Altered production of extra-cellular matrix components by muscle-derived Duchenne muscular dystrophy fibroblasts before and after TGF-beta1 treatment. Cell Tissue Res 339:397–410
Zanotti S, Saredi S, Ruggieri A, Fabbri M, Blasevich F, Romaggi S, Morandi L, Mora M (2007) Altered extracellular matrix transcript expression and protein modulation in primary Duchenne muscular dystrophy myotubes. Matrix Biol 26:615–624
Zeiger U, Mitchell CH, Khurana TS (2010) Superior calcium homeostasis of extraocular muscles. Exp Eye Res 91:613–622
Zhang W, Liu Y, Zhang H (2021) Extracellular matrix: an important regulator of cell functions and skeletal muscle development. Cell Biosci 11:65
Zhang WR, Parikh CR (2019) Biomarkers of acute and chronic kidney disease. Annu Rev Physiol 81:309–333
Zhang A, Uaesoontrachoon K, Shaughnessy C, Das JR, Rayavarapu S, Brown KJ, Ray PE, Nagaraju K, van den Anker JN, Hoffman EP, Hathout Y (2015) The use of urinary and kidney SILAM proteomics to monitor kidney response to high dose morpholino oligonucleotides in the mdx mouse. Toxicol Rep 2:838–849
Zhao M, Li M, Yang Y, Guo Z, Sun Y, Shao C, Li M, Sun W, Gao Y (2017) A comprehensive analysis and annotation of human normal urinary proteome. Sci Rep 7:3024
Ziemba M, Barkhouse M, Uaesoontrachoon K, Giri M, Hathout Y, Dang UJ, Gordish-Dressman H, Nagaraju K, Hoffman EP (2021) Biomarker-focused multi-drug combination therapy and repurposing trial in mdx mice. PLoS One 16:e0246507
Zou X, Ouyang H, Pang D, Han R, Tang X (2021) Pathological alterations in the gastrointestinal tract of a porcine model of DMD. Cell Biosci 11:131
Zubrzycka-Gaarn EE, Bulman DE, Karpati G, Burghes AH, Belfall B, Klamut HJ, Talbot J, Hodges RS, Ray PN, Worton RG (1988) The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature 333:466–469
Acknowledgements
We acknowledge the Open Access funding enabled and organised by the project DEAL, and would like to thank our colleagues for their continuous support of our research into muscular dystrophy, especially Drs. Paul Dowling (Maynooth University) and Paula Meleady (Dublin City University), as well as Drs. Hemmen Sabir, Stephan Baader and Jens Reimann (University of Bonn). We are grateful to Diplom-Biologin Margit Zweyer (University of Bonn) for providing microscopical images.
Funding
Open Access funding provided by the IReL Consortium. Funding was provided by the Kathleen Lonsdale Institute for Human Health Research, Maynooth University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ohlendieck, K., Swandulla, D. Complexity of skeletal muscle degeneration: multi-systems pathophysiology and organ crosstalk in dystrophinopathy. Pflugers Arch - Eur J Physiol 473, 1813–1839 (2021). https://doi.org/10.1007/s00424-021-02623-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-021-02623-1