
Abstract
Erythropoietin (EPO) is a haematopoietic hormone that regulates erythropoiesis, but the EPO-receptor (EpoR) is also expressed in non-haematopoietic tissues. Stimulation of the EpoR in cardiac and skeletal muscle provides protection from various forms of pathological stress, but its relevance for normal muscle physiology remains unclear. We aimed to determine the contribution of the tissue-specific EpoR to exercise-induced remodelling of cardiac and skeletal muscle. Baseline phenotyping was performed on left ventricle and m. gastrocnemius of mice that only express the EpoR in haematopoietic tissues (EpoR-tKO). Subsequently, mice were caged in the presence or absence of a running wheel for 4 weeks and exercise performance, cardiac function and histological and molecular markers for physiological adaptation were assessed. While gross morphology of both muscles was normal in EpoR-tKO mice, mitochondrial content in skeletal muscle was decreased by 50%, associated with similar reductions in mitochondrial biogenesis, while mitophagy was unaltered. When subjected to exercise, EpoR-tKO mice ran slower and covered less distance than wild-type (WT) mice (5.5 ± 0.6 vs. 8.0 ± 0.4 km/day, p < 0.01). The impaired exercise performance was paralleled by reductions in myocyte growth and angiogenesis in both muscle types. Our findings indicate that the endogenous EPO-EpoR system controls mitochondrial biogenesis in skeletal muscle. The reductions in mitochondrial content were associated with reduced exercise capacity in response to voluntary exercise, supporting a critical role for the extra-haematopoietic EpoR in exercise performance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Erythropoietin (EPO) is a hormone that is secreted by the adult kidneys and the foetal liver and regulates erythropoiesis in the haematopoietic tissues [46, 61]. EPO stimulates erythropoiesis by binding to a homodimeric EPO-receptor (EpoR) on erythroid progenitor cells, which promotes their survival, proliferation and differentiation into erythrocytes [46, 61]. In certain pathological conditions such as chronic renal failure, EPO production becomes insufficient and anaemia ensues [46, 54, 61]. Treatment with recombinant human EPO (rhEPO) does not only increase haematocrit in these patients, but also improves quality of life and promotes exercise capacity [9, 31]. rhEPO is also used to enhance exercise performance by various endurance athletes without anaemia [3, 4].
The mechanisms underlying the effects of rhEPO on exercise are generally thought to result from the associated increase in haematocrit, under the assumption that this augments oxygen delivery to aerobic tissues [4, 25,26,27]. Although this may partially explain the beneficial effects in patients with severe anaemia, extra-haematopoietic effects may also contribute to the improvements in quality of life and exercise performance [14, 25]. For instance, studies have shown that rhEPO activates mitochondrial biogenesis and subsequently promotes exercise capacity, prior to an elevation in haematocrit [7]. Also, enhancement of mitochondrial function was observed in skeletal muscle fibres of athletes after rhEPO administration [39]. Indeed, most of the effects of rhEPO on exercise performance in endurance athletes are not exclusively explained by changes in haematocrit and the accessory effects could therefore result from non-haematopoietic effects [2].
There are several lines of evidence supporting the concept that EPO augments exercise performance by activating an EpoR subtype in non-haematopoietic tissues [36]. This EpoR has been discovered in a variety of non-haematopoietic tissues, including cardiac and skeletal muscle, endothelial cells, neurones and kidneys [34, 36, 46]. Evidence for its role in tissue physiology is based upon studies where rhEPO protected these tissues from various forms of pathological stress [10, 12, 24, 55, 56], even when administered in doses that are too low to stimulate erythropoiesis [23]. More direct evidence for the significance of the extra-haematopoietic EPO-EpoR system comes from studies with a mouse model that only expresses the EpoR in haematopoietic cells, but lack EpoR expression in all other tissues (EpoR-tissue knock-out, EpoR-tKO mice) [47]. While these EpoR-tKO mice display a normal phenotype with preserved erythropoiesis, their non-haematopoietic tissues are more vulnerable to pathological stress [10, 50]. For instance, cardiac and skeletal muscle of EpoR-tKO mice are more susceptible to ischemic or toxic injury and also develop more severe heart failure when subjected to pressure overload [16, 33, 48].
While this evidence indicates that the EpoR expressed in muscle is critical for the adaptation to pathological stress, its relevance for muscle physiology is not well described. We hypothesised that the EpoR expressed in cardiac and skeletal muscle is critical for the physiological adaptation to exercise. To test this hypothesis, we subjected EpoR-tKO mice to a model of voluntary wheel running.
Methods
Ethical approval
The experimental protocol was accepted by The Animal Ethical Committee of the University of Groningen (DEC4585, IvD 1583–02-011, 1583–02-014). In addition, all animal experiments followed the protocols from Directive 2010/63/EU of the European Parliament.
Mouse model
We employed the well-described EpoR-/-rescued mouse model in which EpoR-tissue knock-out mice have been rescued from lethality by a transgene that drives EpoR expression exclusively in haematopoietic tissues [47]. These mice (EpoR-tissue knock-out; EpoR-tKO) develop normally and are fertile without a clear (cardiac) phenotype, despite the absence of the EPO receptor in all non-haematopoietic tissues (Fig. 1) [47]. EpoR-tKO mice originated from the Riken BioResource Research Center (Riken BRC) repository in Japan. Thereafter, the EpoR-tKO mice were bred and housed at the central animal facility of the University of Groningen and back-crossed into our local C57Bl/6 J genetic background. Age and gender-matched wild-type (WT) mice were used as controls. Genotyping of EpoR-tKO mice was performed as described previously [47].

Baseline muscle phenotype of tissue-specific erythropoietin receptor deficient mice. Phenotyping of tissue-specific erythropoietin receptor deficient (EpoR-tKO) mice. Shown are (a) conceptual illustration of the mouse model, in which the erythropoietin receptor (EpoR) is absent in non-haematopoietic tissues of EpoR-tKO mice, (b) left ventricular (LV) mRNA expression of the EpoR, (n = 2–5/group), (c) heart weight to body weight ratio in milligrams per gram (mg/g), (d) cardiomyocyte cross-sectional area in micrometres squared (μm2) and typical examples, and (e) skeletal myocyte cross-sectional area (μm2) including typical examples, (n = 11–13/group). WT wild-type mice, EpoR-tKO EpoR-tissue knock-out mice. Data are presented as mean value ± standard error of the mean (SEM). The illustration in panel a contains images from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 unported license
Experimental model
At 8–12 weeks of age, WT and EpoR-tKO male mice were randomised to the presence or absence of a running wheel present in their individual cage. The voluntary running wheel (VWR) was present for the whole duration of the experiment, which was a period of 4 weeks (referred to as WT Run or EpoR-tKO Run). Control littermates were housed similarly but without a running wheel and therefore remained sedentary for this period (referred to as WT Sed or EpoR-tKO Sed). An additional group of male mice was selected for phenotyping of basal mitochondrial parameters. In total, 89 male mice were used for this study. All mice had ad libitum access to food and water.
Exercise performance
A cyclometer was attached to the running wheel, which measured daily exercise parameters, including speed, distance and time on the running wheel, as described previously [1].
Cardiac function parameters and haemodynamics
Following completion of the 4-week experiment and 2 days prior to sacrifice, cardiac function was determined by echocardiography (GE Vivid 7 Dimension, using a 13-MHz probe) as described before [29]. Subsequently, on the day of sacrifice, haemodynamic parameters were determined with a microtip pressure–volume transducer as described previously (Millar Instr. Inc., Houston, TX, USA) [37]. All cardiac function assessments were performed under general anaesthesia (isoflurane 2%).
Organ, body and blood measurements
After haemodynamic measurements, blood was drawn and hearts plus skeletal muscle were collected. After weighing the hearts, the left ventricle (LV) and the gastrocnemius skeletal muscle were fixated for immunohistochemistry or snap frozen for molecular analysis.
Histology
After isolation of LV and m. gastrocnemius, cardiac muscle was cut at the mid-papillary level, and skeletal muscle was cut at the mid-belly level. Sections were either fixated in 4% paraformaldehyde and embedded in paraffin after dehydration steps (Leica TP1020, Germany) or fixated in Tissue-Tec and subsequently slowly frozen in liquid nitrogen. Following fixation and embedding processes, sections were cut using a microtome and were thereafter processed onto slides.
(Cardio) Myocyte size
To determine (cardio) myocyte cross sectional area, deparaffinised 3-μm thick sections were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, USA) and wheat germ agglutinin (WGA) (Sigma-Aldrich, USA), as described before [60]. Stained transverse sections were visualised with fluorescence microscopy to generate images for analysis (Zeiss KS400, Germany). Quantification fields were chosen at random at × 40 magnification; five fields per sample were counted and averaged.
Capillary density
To measure capillary density as a measure of adaptive angiogenesis, endothelial cells were incubated with the endothelial cell antigen CD31 (purified rat anti-mouse CD31 antibody #550,274, BD Biosciences, USA). Prior to incubation of antibodies, cryosections were fixated in acetone, air-dried and blocked for endogenous peroxidases. After primary, secondary (unconjugated rabbit anti-rat IgG antibody, #AI-4001, Vector Laboratories, USA) and tertiary (anti-rabbit polymer-HRP, #K4008, EnVision kit, Denmark) antibody incubation, AEC + solution was applied and counterstained with Mayer’s hematoxylin. Capillaries were quantified from at least eight randomly chosen fields at × 40 magnification (Zeiss KS400, Germany), and adaptive angiogenesis was presented as capillary to (cardio)myocyte ratio [5].
Electron microscopy
To assess basal mitochondrial quantity, large-scale electron microscopy (EM), known as nanotomy (for nano-anatomy), was performed. Samples from skeletal muscle were cut into pieces of approximately 1 mm2 and were fixated in 2% glutaraldehyde and 2% paraformaldehyde solution in 0.1 M sodium cacodylate directly after sacrifice. Following fixation, samples were further processed with 1% osmium tetroxide and 1.5% potassium ferrocyanide and, finally, after dehydration embedded in EPON epoxy resin. Sections of 80 nm were cut using an ultramicrotome, which was followed by contrast staining with 4% neodymium acetate for 30 min [19]. Finally, sections were observed using scanning and transmission electron microscopy (STEM) (Zeiss Supra55, Oberkochen Germany), as described before [15, 18, 45]. This microscope was connected to an external scan generator ATLAS5 (Fibics, Canada) to produce many high-resolution images that are stitched to generate a large Nanotomy ‘map’. TIFF files were converted to html links and uploaded to the website www.nanotomy.org. Of each nanotomy file, at least six high-resolution images, selected randomly in the map, were saved for analysis of mitochondrial quantity. Analysis of images was performed in ImageJ (Fiji- ImageJ version of Java 6, USA).
Quantitative real-time polymerase chain reaction
To assess molecular analysis at mRNA level, qRT-PCR was performed as described before [58]. Total RNA from LV and m. gastrocnemius was isolated using the Trizol isolation method (Invitrogen Corporation, USA). Quantification of RNA concentrations was performed using Nanodrop (ThermoFisher, USA). cDNA synthesis was performed with equal RNA concentrations (500 ng of total RNA) using the Quantitect Reverse Transcription kit (Qiagen, the Netherlands). qRT-PCR was performed using SYBR Green dye super mix and the running protocol included 3 min at 95 °C, followed by 35 cycles of 15 s at 95 °C, 30 s at 60 °C, followed by dissociation and melting steps (Bio-Rad CFX384, USA). Data were processed according to the ddCT method, and were normalised for housekeeping gene 36B4 and control group WT sedentary. The Online Resource with Supplementary Table S1 contains the primer sequences.
Mitochondrial DNA to nuclear DNA ratio (mitochondrial DNA copy number)
To quantify basal mitochondrial DNA copy number as a measure for mitochondrial biogenesis, qRT-PCR was performed for genes encoding mitochondrial proteins. DNA was isolated from LV and m. gastrocnemius using the Nucleospin Tissue XS kit (Macherey–Nagel, Germany); DNA concentrations were quantified using Nanodrop (Thermofisher, USA). A total of 10 μg DNA was used for qRT-PCR, using the standard running protocol as described above. To determine mitochondrial DNA copy number, nuclear DNA-encoded mitochondrial protein hexokinase 2 (HK2) and mitochondrial DNA (mtDNA)-encoded protein NADH dehydrogenase (ND1) were detected [41]. mtDNA copy number was calculated using the ddCT method. HK2 served as reference gene and the WT sedentary group served as control. Supplementary Table S1 contains the primer sequences.
Western blot
To detect molecular changes at protein level, Western blot was performed [59], specifically to assess levels of the key regulators of mitochondrial biogenesis and mitophagy; nuclear respiratory factor 2 (NRF2), outer mitochondrial membrane protein TOM20 (TOM20), ubiquitin protein 62 (p62) and light chain 3B (LC3B). Total protein was extracted with RIPA buffer and fresh sodium vanadate, phosphatases and proteases buffers (Sigma-Aldrich, USA). Quantification of protein concentrations was performed using the BCA protein assay (Pierce. No. 232250, ThermoFisher, USA). Equal concentrations of protein were boiled and loaded on SDS-PAGE. Semi-dry blotting transferred the proteins onto PVDF membranes. Membranes were incubated with the following primary antibodies: polyclonal anti-NRF2 (ab31163, Abcam, UK), monoclonal anti-TOM20 (#42,406, Cell Signalling, USA), monoclonal anti-sqstm1/p62 antibody (ab56416, Abcam, UK), monoclonal anti-LC3B antibody (#2775, Cell Signalling, USA) and monoclonal anti-GAPDH antibody as a loading control (10R-G109A, Fitzgerald, USA). This was followed by secondary antibody incubation, and proteins were detected with enhanced chemiluminescence (NEL120E001EA, Western lightening ECL Pro, Perkin Elmer, USA) using ImageQuant imager. Western blots were quantified in ImageJ and values were normalised for loading control GAPDH expressions as well as for the control WT sedentary group.
Statistical analyses
Results are presented as mean value ± standard error of the mean (SEM). Statistical comparisons between two groups with normally distributed data were performed with a student’s t test; if data was not normally distributed, the non-parametric Mann Whitney U test was performed. Statistical comparisons among more than two groups were performed by one-way ANOVA with Bonferroni post hoc test, if distributed normally. Otherwise, the non-parametric Kruskal–Wallis test was used followed by the Mann–Whitney U test. Statistical significance was stated if the p value was < 0.05, of which all p values were two-tailed. Statistical analysis was performed in STATA v11SE software (College Station, TX) and Graph Pad Prism software (Version 7, USA).
Results
Phenotyping of basal cardiac and skeletal muscle in tissue-specific erythropoietin receptor-deficient (EpoR-tKO) mice
A conceptual illustration of the mouse model is presented in Fig. 1a. In accordance with this concept, EpoR mRNA expression was undetectable in cardiac muscle from EpoR-tKO mice (Fig. 1b), while haematocrit levels were unaltered, and thus, erythropoiesis was preserved (Supplementary Fig. S1). Cardiac mass and cardiomyocyte cross-sectional area were comparable between EpoR-tKO and WT mice (Fig. 1c–d), as was the mRNA expression of natriuretic peptides and muscle isoforms (Supplementary Fig. S1). Skeletal myocyte size in m. gastrocnemius was also comparable between EpoR-tKO and WT mice (Fig. 1e).
Basal mitochondrial biogenesis is disrupted in skeletal but not in cardiac muscle of EpoR-tKO mice
The generation of new mitochondria, and thereby increasing the mitochondrial content, is known as a process called mitochondrial biogenesis and is regulated by multiple signalling pathways and transcription factors [11, 40]. Interestingly, rhEPO has been previously shown to enhance exercise capacity and stimulate mitochondrial biogenesis through regulation of markers including eNOS and PGC-1α [7]. In our model, mitochondrial content expressed as the mitochondrial to nuclear DNA ratio was unaltered in cardiac muscle as was the mRNA expression of molecular markers for mitochondrial biogenesis (Fig. 2a–b). In skeletal muscle, however, mitochondrial DNA content was reduced by 50% in EpoR-tKO mice compared to WT mice (Fig. 2c). Similarly, protein levels of the outer mitochondrial membrane protein (TOM20) were also significantly lower in skeletal muscle of EpoR-tKO mice (Fig. 2e–f, Supplementary Fig. S2). These reductions in mitochondrial content were associated with similar reductions in the expression of key regulators of mitochondrial biogenesis, eNOS, SIRT1, PGC-1α and NRF2 (Fig. 2d). In addition, protein levels of NRF2, a key regulator of mitochondrial biogenesis, were also decreased in EpoR deficient mice (Fig. 2e–f, Supplementary Fig. S2). To corroborate whether the differences in mitochondrial DNA content and TOM20 protein levels paralleled with similar reductions in the actual number of mitochondria, we next performed electron microscopy (EM) of m. gastrocnemius muscle. Strikingly, quantification of the number of mitochondria in our EM datasets revealed that the number of interfibrillar mitochondria was reduced in skeletal muscle of EpoR-tKO mice (Fig. 2g, available for complete datasets). This was accompanied by a trend towards a decrease in mRNA expression of oxidative muscle fibre types, which may also reflect a reduction in mitochondrial abundance (Supplementary Fig. S3a) [40].

Mitochondrial biogenesis in basal cardiac and skeletal muscle. Cardiac and skeletal muscle mitochondrial biogenesis parameters at baseline are depicted (n = 11–13/group). Shown are (a) left ventricular (LV) mitochondrial to nuclear DNA ratio (mtDNA/nDNA), (b) LV mRNA expression of markers for mitochondrial biogenesis, (c) skeletal muscle (SM) mitochondrial to nuclear DNA ratio (mtDNA/nDNA), (d) SM mRNA expression of markers for mitochondrial biogenesis, (e) typical examples of SM protein levels of NRF2 and TOM20 (n = 3/group), (f) quantification of full Western blots of NRF2 and TOM20 (n = 4–5/group), and (g) typical electron microscopy examples of m. gastrocnemius (white arrows indicating mitochondria) and quantification of mitochondrial (mito) number per field of 55μm2; six high power fields were analysed per animal, (n = 2/group). eNOS endothelial nitric oxide synthase, SIRT1 sirtuin 1, PGC-1α peroxisome proliferator-activated receptor gamma coactivator 1-alpha, NRF2 nuclear respiratory factor 2, TOM20 outer mitochondrial membrane protein. WT wild-type mice, EpoR-tKO EpoR-tissue knock-out mice. Data are presented as mean value ± standard error of the mean (SEM). WT vs. EpoR-tKO: *p < 0.05, **p < 0.01. Raw data with zoomable files at high resolution are accessible at www.nanotomy.org
Next, we aimed to determine whether the reductions in mitochondrial content and biogenesis were also associated with enhanced elimination of mitochondria, through a process called mitophagy [11]. We therefore assessed p62, LC3B I and LC3B II at protein level, which are markers for mitophagy. We did not observe significant differences in protein levels of mitophagy markers in skeletal muscle of EpoR-tKO mice compared to WT mice (Supplementary Fig. S4a–b). Finally, to assess whether mitochondrial performance was altered in EpoR-tKO mice, oxygen consumption in isolated mitochondria was assessed. Surprisingly, the changes in mitochondrial content were not accompanied by changes in mitochondrial respiratory capacity (Supplementary Fig. S3b) [57]. Together, these data indicate that the EpoR expressed in skeletal muscle regulates mitochondrial biogenesis, without affecting mitochondrial oxidative capacity or mitophagy.
Exercise performance is impaired in EpoR-tKO mice despite normal haematocrit levels
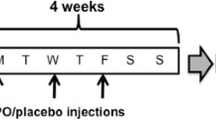
We next sought to determine whether the reductions in mitochondrial content in skeletal muscle affected exercise performance. For this purpose, WT and EpoR-tKO mice were housed individually in the presence or absence of a running wheel in their cage (Fig. 3a). With preserved erythropoiesis (Supplementary Table S2), EpoR-tKO running mice had a lower average speed compared to WT mice, spent less time on the running wheel and ran significantly fewer daily and kilometres (Fig. 3b–d). Consequently, mice in the EpoR-tKO Run group covered less total distance compared to mice in the WT Run group (Fig. 3e).

Exercise performance in wild-type and EpoR-tissue knock-out mice. Exercise parameters during 4-week period of voluntary wheel running (n = 11–12/group). Shown are (a) experimental design including voluntary wheel running protocol, (b) average daily running speed in kilometres per hour (km/h), (c) average running time in minutes per day (min/day), (d) average daily running distance in kilometres per day (km/day) and (e) total running distance in kilometres (km) that mice achieved in the 4-week voluntary wheel running experiment. WT wild-type mice, EpoR-tKO EpoR-tissue knock-out mice, Run running. Data are presented as mean value ± standard error of the mean (SEM). WT vs. EpoR-tKO: **p < 0.01. Part of the illustration in panel a contains images from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 unported license
Cardiac adaptation to exercise is impaired in EpoR-tKO mice
Cardiac function
To determine if loss of EpoR signalling in the cardiac muscle affected the adaptive changes in cardiac performance induced by exercise, intracardiac pressure measurements and echocardiography were performed. Voluntary exercise resulted in a significant improvement in both contractility and relaxation of the left ventricle (dP/dt max, dP/dt min, respectively), as well as a comparable effect in fractional shortening among both groups (Supplementary Table S3). However, left ventricular end-diastolic pressure (LVEDP), mean arterial pressure (MAP) and systolic blood pressure (SBP) were considerably increased in EpoR-tKO mice compared to WT mice after exercise (Fig. 4a, Supplementary Table S3).

Functional, structural and angiogenic changes in cardiomyocytes: immunohistochemistry analysis. Functional, structural and angiogenic changes in cardiomyocytes after 4 weeks of voluntary wheel running (n = 10–13/group). Shown are (a) intra cardiac measurement of left ventricular end-diastolic pressure (LVEDP) in millimetre of mercury (mmHg) and (b) heart weight (HW) in milligrams (mg). Panel c displays cardiomyocyte cross-sectional area in micrometres-squared (μm2) and (d) typical examples of cardiomyocyte size. In panel e, the capillary-to-cardiomyocyte ratio is shown, together with typical examples of capillary density in panel f. WT wild-type mice, EpoR-tKO EpoR-tissue knock-out mice, Sed sedentary, Run running. Data are presented as mean value ± standard error of the mean (SEM). Sed vs. Run: *p < 0.05, ***p < 0.001, Sed vs. Sed and Run vs. Run: # p < 0.05, ## p < 0.01
Cardiac hypertrophy
Exercise resulted in a significant increase in cardiac weight in WT mice (WT Sed 155 ± 3.6 vs. WT Run 195 ± 6.6 mg, p < 0.05) (Fig. 4b). In contrast, exercise did not significantly increase cardiac mass in EpoR-tKO mice (Fig. 4b). Body weight was comparable among the experimental groups (Supplementary Table S2). Accordingly, when cardiac weight was adjusted for body weight, a similar trend was observed (Supplementary Table S2).
Analogous to pathological stress, the cardiac physiological response to sustained exercise includes increasing cardiomyocyte size [44]. Exercise resulted in a 26% increase in cardiomyocyte cross-sectional area (CSA) in WT mice (Fig. 4c–d). In contrast, exercise did not result in a considerable increase in CSA in EpoR-tKO mice (Fig. 4c–d). Similarly, the increase in the mRNA expression of alpha-actinin 1 (ACTA-1), a molecular marker of muscle growth, was more pronounced in WT Run than in the EpoR-tKO Run group (Supplementary Table S4). Furthermore, atrial natriuretic peptide (ANP) expression was increased and a shift towards β-myosin heavy chain (β-MHC) isoforms was observed in the LV of EpoR-tKO Run mice, suggesting that exercise induced subtle maladaptive remodelling in EpoR-tKO mice [32] (Supplementary Table S4).
The angiogenic response in cardiac muscle
In response to exercise, cardiomyocyte hypertrophy is paralleled by a corresponding expansion of the capillary network [44]. Cardiac capillary-to-myocyte ratio was slightly, but significantly reduced in sedentary EpoR-tKO mice compared to sedentary WT mice (p < 0.01) (Fig. 4e–f). Furthermore, the normal increase in capillary density in response to exercise that was observed in WT Run mice was abrogated in the EpoR-tKO Run group (Fig. 4e–f). Similarly, the mRNA expression of vascular endothelial growth factor α (VEGF-α), a critical regulator of angiogenesis, was substantially increased in response to exercise in WT mice, but not altered by exercise in EpoR-tKO mice (Supplementary Table S4).
The adaptive response of skeletal muscle to exercise is impaired in EpoR-tKO mice
Myocyte hypertrophy
Similar to cardiac muscle, skeletal muscle fibres also develop myocyte hypertrophy in response to exercise [40]. Exercise resulted in a 32% increase in myocyte CSA in the WT Run group, but this increase upon exercise was absent in EpoR-tKO mice (Fig. 5a–b). This pattern was similar for the mRNA expression of ACTA-1, and can be observed in Supplementary Table S4.

Structural and angiogenic changes in skeletal myocytes: immunohistochemistry analysis. Structural and angiogenic changes in skeletal myocytes after 4 weeks of voluntary wheel running (n = 10–13/group). Shown are (a) skeletal myocyte (SM) cross-sectional area (CSA) in micrometres-squared (μm2) and (b) typical examples of SM size. In panel c, the skeletal capillary-to-myocyte ratio is shown, together with typical examples of capillary density in panel d. WT wild-type mice, EpoR-tKO EpoR-tissue knock-out mice, Sed sedentary, Run running. Data are presented as mean value ± standard error of the mean (SEM). Sed vs. Run: *p < 0.05, **p < 0.01
The angiogenic response in the skeletal muscle
Physiological myocyte hypertrophy in skeletal muscle is also paralleled by expansion of the capillary network [40]. To determine whether the EpoR is also critical for adaptive angiogenesis in skeletal muscle, capillary density was evaluated in the m. gastrocnemius. In this skeletal muscle, an abrogated response in capillary/myocyte ratio and VEGF-α mRNA expression was also observed in EpoR-tKO mice (Fig. 5c–d, Supplementary Table S4).
Discussion
The role of the non-haematopoietic EpoR in the adaptive response to physiological stress remains unexplored. In this study, we aimed to determine the role of the EpoR in cardiac and skeletal muscle in the physiological adaptation to exercise. While EpoR-tKO mice were phenotypically normal, mitochondrial content in skeletal muscle of EpoR-tKO mice was significantly reduced, and mitochondrial biogenesis was impaired compared to wild-type mice. EpoR-tKO mice also displayed reduced exercise performance in response to voluntary wheel running, associated with reductions in myocyte hypertrophy and adaptive angiogenesis in both cardiac and skeletal muscle. Taken together, our findings indicate that the endogenous EPO-EpoR system is critical for basal mitochondrial biogenesis in skeletal muscle. We also show that the associated reductions in mitochondrial content are accompanied by impaired exercise performance, indicating that the extra-haematopoietic EpoR is critical for the physiological adaptation to exercise.
Cardiac and skeletal muscle require considerable plasticity to adapt to continuous variations in demand. The response to endurance exercise training is remarkably similar in cardiac and skeletal muscle, including an increase in myocyte size, expansion of the capillary network, changes in muscle fibre isoforms and an increase in mitochondrial density [40, 43]. While several key regulators of physiological hypertrophy have been identified [32], the role of the endogenous EPO-EpoR system in cardiac and skeletal muscle has not been well described. The tissue-specific EpoR has however been widely studied in pathological conditions and findings from these studies suggest that the EpoR exerts protective effects in response to pathological stress [16, 33, 48]. Current knowledge therefore mainly focusses on the effects of the tissue-specific EpoR in disease but knowledge about its physiological role is limited. The sparse evidence indicating a critical role for this system in the response to exercise was often confounded by the concomitant changes in haematocrit. For instance, mutated mice with systemic EPO-deficiency have reduced exercise performance, associated with oxidative and metabolic stress in skeletal muscle and reduced expression of fast oxidative muscle fibres; yet, these observations were done in the presence of severe anaemia [30]. rhEPO also improves exercise performance and induces mitochondrial biogenesis in cardiac and skeletal muscle of healthy mice through activation of key regulators of physiological hypertrophy, including AKT, eNOS and PGC-1α [7]. Importantly, these mitochondrial effects preceded any changes in haematocrit, suggesting that they were regulated by a tissue-specific receptor. Mitochondrial respiration was also increased in permeabilised skeletal muscle fibres obtained from athletes after prolonged treatment with rhEPO [39]. Overexpression of EPO in mice also activated PGC-1α and increased the proportion of oxidative muscle fibres, whereas the opposite was observed in our mouse model [51]. While these studies clearly indicate that EPO stimulates oxidative metabolism in muscle cells, it is impossible to exclude the possibility that these results were confounded by off target effects of EPO. Our study suggests that the tissue-specific EpoR is a crucial regulator of basal mitochondrial biogenesis as well as structural and metabolic adaptations to exercise. Regarding the difference in capillary density, it should be noted that we cannot reach definitive conclusions about cause and effect. The insufficient angiogenic response could result from loss of EpoR signalling, but could also be a secondary effect related to reduced oxidative muscle fibres.
The first indication suggesting that a functional EPO receptor was present in the cardiac and skeletal muscle dates back almost two decades [6]. While the presence of a functional extra-haematopoietic EPO-receptor was scrutinised at first, studies using comprehensive analysis have firmly established the presence of a functional EPO receptor in muscle cells [35, 42, 52]. The extra-haematopoietic EpoR comprises a distinct isoform of the EpoR, with a distinct pattern of regulation, and heterodimerises with other receptors such as the beta common receptor (βcR) to compose the innate repair receptor (IRR) [8, 10]. The IRR potentially plays a role in repair mechanisms in response to injury and inflammation; however, some controversy has been described regarding the mechanisms of interaction between these receptor isoforms [8, 10]. Yet, these differences between the tissue and haematopoietic EPO receptor suggest that the tissue receptor can be targeted specifically and several compounds are currently under investigation [8, 10]. This would offer the possibility to exploit the beneficial effects of rhEPO for exercise performance without the untoward effects on haematocrit. Whether these isoforms are capable of regulating exercise performance and exercise-induced muscle growth remains unknown.
Literature describes some degree of discrepancy regarding the role of the skeletal muscle EpoR. Some studies observe little to no effects of the EpoR in skeletal muscle whereas other studies suggest a beneficial role for an EpoR in skeletal muscle [20,21,22, 51]. For instance, EPO administration shows activation of AKT signalling, but this was accompanied by limited effects on protein synthesis in skeletal muscle after exercise [21]. Also, the role of the EpoR in myogenesis was found to be negligible [22]. However, findings that describe the limited role for the EpoR in skeletal muscle adaptations were based on a single EPO injection or in vitro EPO treatment and do not specifically target the EpoR as described above. Contrarily to the above, our results are in line with previous reports that have indeed demonstrated the role of the EpoR in basal mitochondrial muscle metabolism in EpoR-tKO mice [51]. These mechanisms have been attributed to the regulation of AMPK and PGC-1α, which in turn modulate oxidative muscle fibre reprogramming [51]. With our study, we now provide the evidence that the EpoR in skeletal muscle is indeed critical for basal mitochondrial biogenesis. Our data suggests that these changes are associated with reduced exercise capacity and impaired adaptation of cardiac and skeletal muscle to voluntary exercise. Future investigations should be focussed on unravelling the possible underlying mechanisms of these adaptations, such as the regulation of AMPK. Taken together with our findings, the EpoR in skeletal muscle could be a novel target to improve skeletal muscle quality in patients with muscle fatigue, mitochondrial myopathies or heart failure-associated exercise intolerance [13, 38, 53]. Of note, ageing EpoR-tKO mice, mostly pronounced in females, also develop severe obesity and insulin resistance, suggesting that targeting the tissue-specific EPO receptor might also be an effective therapy for diabetes [49].
The mechanistic pathways underlying the cardiac effects as a result of the absent EpoR in non-haematopoietic tissues remain unexplored. In our study, we have observed a rise in left ventricular end diastolic pressure, together with similar effects in mean arterial pressure as well as systolic blood pressure in response to exercise. These functional parameters were accompanied by increments of ANP expression and a shift towards β-myosin heavy chain isoforms. Together, these effects describe a phenotype suiting pathological rather than physiological adaptation. We may argue that these effects are primarily due the loss of the EpoR in cardiac tissue; however, these effects may also be related to secondary effects exerted upon the heart. Our findings suggest the primary role for EpoR in response to physiological stress; unfortunately, it remains questionable whether these effects are in fact primary. Therefore, future studies are required in order to determine the specific underlying mechanisms of the impaired physiological response.
Several limitations of our analysis need to be acknowledged. For instance, this is a model of voluntary wheel running in which the workload on cardiac and skeletal muscle is relatively subtle and the amount of exercise varied between the groups, whereas forced or exhaustive exercise may cause more rigorous and equal effects on cardiac and skeletal muscle. Nevertheless, exercise-induced hypertrophy in our WT mice was substantial and similar studies also demonstrated substantial muscle growth induced by voluntary wheel running models [1, 17, 28]. Nonetheless, the role of the EpoR in the adaptation to more strenuous forms of exercise may also be different. The varied amount of exercise performed, however, can be attributed to our findings of decreased mitochondrial content in skeletal muscle of EpoR-tKO mice, resulting in impaired exercise performance and physiological adaptation. Next to this limitation, it should also be acknowledged that we compared mitochondrial content and molecular markers for mitochondrial biogenesis in our study. From this data, we cannot conclude definitely that the actual generation of new mitochondria was perturbed. Finally, this is a mouse model in which mice were rescued from lethality by erythroid lineage-restricted expression of the EpoR. Therefore, the outcomes cannot be interpreted as for cardiac and skeletal myocyte-specific KO mice.
In conclusion, this study provides evidence that the endogenous EPO-EpoR system controls mitochondrial biogenesis in skeletal muscle. We also provide evidence that reduced mitochondrial biogenesis in EpoR-deficient mice was associated with impaired exercise performance, suggesting that the EpoR expressed in cardiac and skeletal muscle is critical for physiological exercise and its adaptation.
Data availability
The data from the current study are available from the corresponding author on reasonable request.
References
Allen DL, Harrison BC, Maass A, Bell ML, Byrnes WC, Leinwand LA (2001) Cardiac and skeletal muscle adaptations to voluntary wheel running in the mouse. J Appl Physiol 90:1900–1908. https://doi.org/10.1152/jappl.2001.90.5.1900
Annaheim S, Jacob M, Krafft A, Breymann C, Rehm M, Boutellier U (2016) RhEPO improves time to exhaustion by non-hematopoietic factors in humans. Eur J Appl Physiol 116:623–633. https://doi.org/10.1007/s00421-015-3322-6
Atkinson TS, Kahn MJ (2020) Blood doping: then and now. A narrative review of the history, science and efficacy of blood doping in elite sport. Blood Rev 39:100632
Birzniece V (2015) Doping in sport: effects, harm and misconceptions. Intern Med J 45:239–248. https://doi.org/10.1111/imj.12629
de Boer RA, Pinto YM, Suurmeijer AJH, Pokharel S, Scholtens E, Humler M, Saavedra JM, Boomsma F, van Gilst WH, van Veldhuisen DJ (2003) Increased expression of cardiac angiotensin II type 1 (AT(1)) receptors decreases myocardial microvessel density after experimental myocardial infarction. Cardiovasc Res 57:434–442
Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, Salio M, Cerami A, Brines M (2003) Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sci U S A 100:4802–4806. https://doi.org/10.1073/pnas.0630444100
Carraway MS, Suliman HB, Jones WS, Chen C-W, Babiker A, Piantadosi CA (2010) Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ Res 106:1722–1730. https://doi.org/10.1161/CIRCRESAHA.109.214353
Cheung Tung Shing KS, Broughton SE, Nero TL, Gillinder K, Ilsley MD, Ramshaw H, Lopez AF, Griffin MDW, Parker MW, Perkins AC, Dhagat U (2018) EPO does not promote interaction between the erythropoietin and beta-common receptors. Sci Rep 8:1–16. https://doi.org/10.1038/s41598-018-29865-x
Cohen RS, Karlin P, Yushak M, Mancini D, Maurer MS (2009) The effect of erythropoietin on exercise capacity, left ventricular remodeling, pressure-volume relationships, and quality of life in older patients with anemia and heart failure with preserved ejection fraction. Congest Hear Fail 16:96–103. https://doi.org/10.1111/j.1751-7133.2009.00135.x
Collino M, Thiemermann C, Cerami A, Brines M (2015) Flipping the molecular switch for innate protection and repair of tissues: long-lasting effects of a non-erythropoietic small peptide engineered from erythropoietin. Pharmacol Ther 151:32–40. https://doi.org/10.1016/j.pharmthera.2015.02.005
Dorn GW, Vega RB, Kelly DP (2015) Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 29:1981–1991
Ghezzi P, Bernaudin M, Bernaudinb M, Bianchi R, Bianchic R, Blomgren K, Brines M, Campana W, Cavaletti G, Cerami A, Chopp M, Coleman T, Digicaylioglu M, Ehrenreich H, Erbayraktar S, Erbayraktar Z, Gassmann M, Genc S, Gokmen N, Grasso G, Juul S, Lipton SA, Hand CC, Latini R, Lauria G, Leist M, Newton SS, Petit E, Probert L, Sfacteria A, Siren A-L, Talan M, Thiemermann C, Westenbrink D, Yaqoob M, Zhu C (2010) Erythropoietin: not just about erythropoiesis. Lancet (London, England) 375:2142. https://doi.org/10.1016/S0140-6736(10)60992-0
Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, LeBrasseur NK, Yan Z, Spiegelman BM (2007) Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J Biol Chem 282:30014–30021. https://doi.org/10.1074/jbc.M704817200
Hardeman M, Alexy T, Brouwer B, Connes P, Jung F, Kuipers H, Baskurt OK (2014) EPO or PlacEPO? Science versus practical experience: panel discussion on efficacy of erythropoetin in improving performance. Biorheology 51:83–90. https://doi.org/10.3233/BIR-140655
Hoes MF, Grote Beverborg N, Kijlstra JD, Kuipers J, Swinkels DW, Giepmans BNG, Rodenburg RJ, van Veldhuisen DJ, de Boer RA, van der Meer P (2018) Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail 20:910–919. https://doi.org/10.1002/ejhf.1154
Jia Y, Suzuki N, Yamamoto M, Gassmann M, Noguchi CT (2012) Endogenous erythropoietin signaling facilitates skeletal muscle repair and recovery following pharmacologically induced damage. FASEB J 26:2847–2858. https://doi.org/10.1096/fj.11-196618
Konhilas JP, Widegren U, Allen DL, Paul AC, Cleary A, Leinwand LA (2005) Loaded wheel running and muscle adaptation in the mouse. Am J Physiol - Hear Circ Physiol 289:H455-65. https://doi.org/10.1152/ajpheart.00085.2005
Kuipers J, de Boer P, Giepmans BNG (2015) Scanning EM of non-heavy metal stained biosamples: large-field of view, high contrast and highly efficient immunolabeling. Exp Cell Res 337:202–207. https://doi.org/10.1016/j.yexcr.2015.07.012
Kuipers J, Giepmans BNG (2020) Neodymium as an alternative contrast for uranium in electron microscopy. Histochem Cell Biol 153:271–277. https://doi.org/10.1007/s00418-020-01846-0
Lamon S, Russell AP (2013) The role and regulation of erythropoietin (EPO) and its receptor in skeletal muscle: how much do we really know? Front Physiol 4:176. https://doi.org/10.3389/fphys.2013.00176
Lamon S, Zacharewicz E, Arentson-Lantz E, Della GPA, Ghobrial L, Gerlinger-Romero F, Garnham A, Paddon-Jones D, Russell AP (2016) Erythropoietin does not enhance skeletal muscle protein synthesis following exercise in young and older adults. Front Physiol 7:292. https://doi.org/10.3389/fphys.2016.00292
Lamon S, Zacharewicz E, Stephens AN, Russell AP (2014) EPO-receptor is present in mouse C2C12 and human primary skeletal muscle cells but EPO does not influence myogenesis. Physiol Rep 2:e00256. https://doi.org/10.1002/phy2.256
Lipsic E, Westenbrink BD, van der Meer P, van der Harst P, Voors AA, van Veldhuisen DJ, Schoemaker RG, van Gilst WH (2008) Low-dose erythropoietin improves cardiac function in experimental heart failure without increasing haematocrit. Eur J Heart Fail 10:22–29. https://doi.org/10.1016/j.ejheart.2007.10.008
Lu J, Dai Q, ming, Ma G shan, Zhu Y hong, Chen B, Li B, Yao Y yu, (2017) Erythropoietin attenuates cardiac dysfunction in rats by inhibiting endoplasmic reticulum stress-induced diabetic cardiomyopathy. Cardiovasc Drugs Ther 31:367–379. https://doi.org/10.1007/s10557-017-6742-1
Lundby C, Olsen NV (2011) Effects of recombinant human erythropoietin in normal humans. J Physiol 589:1265–1271. https://doi.org/10.1113/jphysiol.2010.195917
Lundby C, Thomsen JJ, Boushel R, Koskolou M, Warberg J, Calbet JAL, Robach P (2007) Erythropoietin treatment elevates haemoglobin concentration by increasing red cell volume and depressing plasma volume. J Physiol 578:309–314. https://doi.org/10.1113/jphysiol.2006.122689
Mancini DM, Katz SD, Lang CC, LaManca J, Hudaihed A, Androne AS (2003) Effect of erythropoietin on exercise capacity in patients with moderate to severe chronic heart failure. Circulation 107:294–299. https://doi.org/10.1161/01.CIR.0000044914.42696.6A
Manzanares G, Brito-Da-Silva G, Gandra PG (2019) Voluntary wheel running: patterns and physiological effects in mice. Brazilian J Med Biol Res 52https://doi.org/10.1590/1414-431X20187830
Meems LMG, Cannon MV, Mahmud H, Voors AA, van Gilst WH, Silljé HHW, Ruifrok WPT, de Boer RA (2012) The vitamin D receptor activator paricalcitol prevents fibrosis and diastolic dysfunction in a murine model of pressure overload. J Steroid Biochem Mol Biol 132:282–289. https://doi.org/10.1016/j.jsbmb.2012.06.004
Mille-Hamard L, Billat VL, Henry E, Bonnamy B, Joly F, Benech P, Barrey E (2012) Skeletal muscle alterations and exercise performance decrease in erythropoietin-deficient mice: a comparative study. BMC Med Genomics 5:29. https://doi.org/10.1186/1755-8794-5-29
Muirhead N, Bargman J, Burgess E, Jindal KK, Levin A, Nolin L, Parfrey P (1995) Evidence-based recommendations for the clinical use of recombinant human erythropoietin. Am J Kidney Dis 26:S1-24
Nakamura M, Sadoshima J (2018) Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 15:387–407. https://doi.org/10.1038/s41569-018-0007-y
Nakano M, Satoh K, Fukumoto Y, Ito Y, Kagaya Y, Ishii N, Sugamura K, Shimokawa H (2007) Important role of erythropoietin receptor to promote VEGF expression and angiogenesis in peripheral ischemia in mice. Circ Res 100:662–669. https://doi.org/10.1161/01.RES.0000260179.43672.fe
Noguchi CT, Wang L, Rogers HM, Teng R, Jia Y (2008) Survival and proliferative roles of erythropoietin beyond the erythroid lineage. Expert Rev Mol Med 10:e36. https://doi.org/10.1017/S1462399408000860
Ogilvie M, Yu X, Nicolas-Metral V, Pulido SM, Liu C, Ruegg UT, Noguchi CT (2000) Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. J Biol Chem 275:39754–39761. https://doi.org/10.1074/jbc.M004999200
Ostrowski D, Heinrich R (2018) Alternative erythropoietin receptors in the nervous system. J Clin Med 7:24. https://doi.org/10.3390/jcm7020024
Pacher P, Nagayama T, Mukhopadhyay P, Bátkai S, Kass DA (2008) Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc 3:1422–1434. https://doi.org/10.1038/nprot.2008.138
Pin F, Busquets S, Toledo M, Camperi A, Lopez-Soriano FJ, Costelli P, Argilés JM, Penna F (2015) Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 6:43202–43215. https://doi.org/10.18632/oncotarget.6439
Plenge U, Belhage B, Guadalupe-Grau A, Andersen PR, Lundby C, Dela F, Stride N, Pott FC, Helge JW, Boushel R (2012) Erythropoietin treatment enhances muscle mitochondrial capacity in humans. Front Physiol 3:50. https://doi.org/10.3389/fphys.2012.00050
Qaisar R, Bhaskaran S, Van Remmen H (2016) Muscle fiber type diversification during exercise and regeneration. Free Radic Biol Med 98:56–67. https://doi.org/10.1016/j.freeradbiomed.2016.03.025
Quiros PM, Goyal A, Jha P, Auwerx J (2017) Analysis of mtDNA/nDNA ratio in mice. Curr Protoc Mouse Biol 7:47–54. https://doi.org/10.1002/cpmo.21
Rundqvist H, Rullman E, Sundberg CJ, Fischer H, Eisleitner K, Ståhlberg M, Sundblad P, Jansson E, Gustafsson T (2009) Activation of the erythropoietin receptor in human skeletal muscle. Eur J Endocrinol 161:427–434. https://doi.org/10.1530/EJE-09-0342
Seo DY, Kwak HB, Kim AH, Park SH, Heo JW, Kim HK, Ko JR, Lee SJ, Bang HS, Sim JW, Kim M, Han J (2019) Cardiac adaptation to exercise training in health and disease. Pflugers Arch Eur. J. Physiol 472:155–168
Shimizu I, Minamino T (2016) Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol 97:245–262. https://doi.org/10.1016/j.yjmcc.2016.06.001
Sokol E, Kramer D, Diercks GFH, Kuipers J, Jonkman MF, Pas HH, Giepmans BNG (2015) Large-scale electron microscopy maps of patient skin and mucosa provide insight into pathogenesis of blistering diseases. J Invest Dermatol 135:1763–1770. https://doi.org/10.1038/jid.2015.109
Suresh S, Rajvanshi PK, Noguchi CT (2020) The Many facets of erythropoietin physiologic and metabolic response. Front Physiol 10:1534
Suzuki N, Ohneda O, Takahashi S, Higuchi M, Mukai HY, Nakahata T, Imagawa S, Yamamoto M (2002) Erythroid-specific expression of the erythropoietin receptor rescued its null mutant mice from lethality. Blood 100:2279–2288. https://doi.org/10.1182/blood-2002-01-0124
Tada H, Kagaya Y, Takeda M, Ohta J, Asaumi Y, Satoh K, Ito K, Karibe A, Shirato K, Minegishi N, Shimokawa H (2006) Endogenous erythropoietin system in non-hematopoietic lineage cells plays a protective role in myocardial ischemia/reperfusion. Cardiovasc Res 71:466–477. https://doi.org/10.1016/j.cardiores.2006.05.010
Teng R, Gavrilova O, Suzuki N, Chanturiya T, Schimel D, Hugendubler L, Mammen S, Yver DR, Cushman SW, Mueller E, Yamamoto M, Hsu LL, Noguchi CT (2011) Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nat Commun 2:520. https://doi.org/10.1038/ncomms1526
Wang L, Di L, Noguchi CT (2014) Erythropoietin, a novel versatile player regulating energy metabolism beyond the erythroid system. Int J Biol Sci 10:921–939. https://doi.org/10.7150/ijbs.9518
Wang L, Jia Y, Rogers H, Suzuki N, Gassmann M, Wang Q, McPherron AC, Kopp JB, Yamamoto M, Noguchi CT (2013) Erythropoietin contributes to slow oxidative muscle fiber specification via PGC-1α and AMPK activation. Int J Biochem Cell Biol 45:1155–1164. https://doi.org/10.1016/j.biocel.2013.03.007
Wang L, Jia Y, Rogers H, Wu YP, Huang S, Noguchi CT (2012) GATA-binding protein 4 (GATA-4) and T-cell acute leukemia 1 (TAL1) regulate myogenic differentiation and erythropoietin response via cross-talk with Sirtuin1 (Sirt1). J Biol Chem 287:30157–30169. https://doi.org/10.1074/jbc.M112.376640
Weiss K, Schär M, Panjrath GS, Zhang Y, Sharma K, Bottomley PA, Golozar A, Steinberg A, Gerstenblith G, Russell SD, Weiss RG (2017) Fatigability, exercise intolerance, and abnormal skeletal muscle energetics in heart failure. Circ Heart Fail 10:e004129. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004129
Westenbrink BD, De Boer RA, Voors AA, Van Gilst WH, Van Veldhuisen DJ (2008) Anemia in chronic heart failure: etiology and treatment options. Curr Opin Cardiol 23:141–147
Westenbrink BD, Oeseburg H, Kleijn L, van der Harst P, Belonje AMS, Voors AA, Schoemaker RG, de Boer RA, van Veldhuisen DJ, van Gilst WH (2008) Erythropoietin stimulates normal endothelial progenitor cell-mediated endothelial turnover, but attributes to neovascularization only in the presence of local ischemia. Cardiovasc drugs Ther 22:265–274. https://doi.org/10.1007/s10557-008-6094-y
Westenbrink BD, Ruifrok W-PT, Voors AA, Tilton RG, van Veldhuisen DJ, Schoemaker RG, van Gilst WH, de Boer RA (2010) Vascular endothelial growth factor is crucial for erythropoietin-induced improvement of cardiac function in heart failure. Cardiovasc Res 87:30–39. https://doi.org/10.1093/cvr/cvq041
Yu H, Tigchelaar W, Koonen DPY, Patel HH, de Boer RA, van Gilst WH, Westenbrink BD, Silljé HHW (2013) AKIP1 expression modulates mitochondrial function in rat neonatal cardiomyocytes. PLoS ONE 8:e80815. https://doi.org/10.1371/journal.pone.0080815
Yu H, Tigchelaar W, Lu B, van Gilst W, de Boer R, Westenbrink B, Silljé H (2013) AKIP1, a cardiac hypertrophy induced protein that stimulates cardiomyocyte growth via the Akt pathway. Int J Mol Sci 14:21378–21393. https://doi.org/10.3390/ijms141121378
Yurista SR, Matsuura TR, Silljé HHW, Nijholt KT, McDaid KS, Shewale SV, Leone TC, Newman JC, Verdin E, van Veldhuisen DJ, de Boer RA, Kelly DP, Westenbrink BD (2020) Ketone ester treatment improves cardiac function and reduces pathologic remodeling in preclinical models of heart failure. Circ Hear Fail. https://doi.org/10.1161/circheartfailure.120.007684
Yurista SR, Silljé HHW, Nijholt KT, Dokter MM, van Veldhuisen DJ, de Boer RA, Westenbrink BD (2020) Factor Xa inhibition with apixaban does not influence cardiac remodelling in rats with heart failure after myocardial infarction. Cardiovasc Drugs Ther. https://doi.org/10.1007/s10557-020-06999-7
Zhang Y, Wang L, Dey S, Alnaeeli M, Suresh S, Rogers H, Teng R, Noguchi CT (2014) Erythropoietin action in stress response, tissue maintenance and metabolism. Int J Mol Sci 15:10296–10333
Acknowledgements
We thank Ben N.G. Giepmans (UMCG) for advice on nanotomy. Animal and/or laboratory techniques were performed with the help of Bianca Meijeringh, Linda van Genne, Inge Vreeswijk-Baudoin, Silke Oberdorf-Maass, Janny Takens, Martin Dokter and Marloes Schouten.
Funding
This study was funded in part by grants from the Netherlands Organization for Research (VENI 016–106-117) and Netherlands Heart Foundation (2007T046, 2019T064). Part of the work has been performed in the UMCG Microscopy and Imaging Centre (UMIC), sponsored by ZonMW 91111.006.
Author information
Authors and Affiliations
Contributions
WPTR, AHM, RAdB and BDW were involved in conception and design of experiment. KTN, LMGM, WPTR, SRY, MGPG, BM and AHGW performed data collection, analysis and interpretation. KTN, LMGM, WPTR, AHM, SRY, MGPG, DJvV, WHvG, HHWS, RAdB and BDW were responsible for the article drafting and revising. All authors approved the final version of the manuscript, all authors met the criteria for authorship and all who qualify for authorship are listed.
Corresponding author
Ethics declarations
Ethics approval
The experimental protocol was accepted by The Animal Ethical Committee of the University of Groningen (DEC4585/IvD 1583–02-011, 1583–02-014). In addition, all animal experiments followed the protocols from Directive 2010/63/EU of the European Parliament.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The UMCG, which employs several of the authors, has received research grants and/or fees from AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk and Roche. Dr. de Boer received speaker fees from Abbott, AstraZeneca, Novartis and Roche. The other authors do not have any conflict of interest to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
• Ablation of the erythropoietin receptor (EpoR) in skeletal muscle results in defective mitochondrial biogenesis.
• EpoR deficiency in cardiac and skeletal muscle reduces exercise performance.
• EpoR deficiency disrupts physiological myocyte hypertrophy and adaptive angiogenesis in cardiac and skeletal muscle.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nijholt, K.T., Meems, L.M.G., Ruifrok, W.P.T. et al. The erythropoietin receptor expressed in skeletal muscle is essential for mitochondrial biogenesis and physiological exercise. Pflugers Arch - Eur J Physiol 473, 1301–1313 (2021). https://doi.org/10.1007/s00424-021-02577-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-021-02577-4