
Abstract
Vesicle fusion is elementary for intracellular trafficking and release of signal molecules, thus providing the basis for diverse forms of intercellular communication like hormonal regulation or synaptic transmission. A detailed characterization of the mechanisms underlying exocytosis is key to understand how the nervous system integrates information and generates appropriate responses to stimuli. The machinery for vesicular release employs common molecular players in different model systems including neuronal and neuroendocrine cells, in particular members of the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) protein family, Sec1/Munc18-like proteins, and other accessory factors. To achieve temporal precision and speed, excitable cells utilize specialized regulatory proteins like synaptotagmin and complexin, whose interplay putatively synchronizes vesicle fusion and enhances stimulus-secretion coupling. In this review, we aim to highlight recent progress and emerging views on the molecular mechanisms, by which constitutively forming SNAREpins are organized in functional, tightly regulated units for synchronized release. Specifically, we will focus on the role of vesicle associated membrane proteins, also referred to as vesicular SNAREs, in fusion and rapid cargo discharge. We will further discuss the functions of SNARE regulators during exocytosis and focus on chromaffin cell as a model system of choice that allows for detailed structure-function analyses and direct measurements of vesicle fusion under precise control of intracellular [Ca]i.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exocytosis is a fundamental and ubiquitous cellular process, by which secretory organelles merge with the plasma membrane in order to release various cargo molecules (e.g., transmitters, hormones, enzymes, or chemokines). A wealth of experimental evidence has firmly established that members of the SNARE protein family form the minimal molecular machine for membrane fusion [13, 136]. In mammals, the SNARE superfamily contains more than 30 members that exhibit distinct subcellular localization [21, 56] and putatively mediate different intracellular trafficking events [6]. As a common structural feature all SNARE proteins contain a homologous 60–70 amino acid-long domain, also referred to as SNARE motif [144, 150]. SNARE motifs form amphiphatic helices with periodic heptad repeats of hydrophobic residues. The four SNARE helices can assemble into a stable complex [55, 103], as first shown by Rothman and colleagues [127, 128]. In case of exocytosis, the three cognate SNARE proteins synaptobrevin (syb, also known as vesicle associated membrane protein (VAMP)), syntaxin (stx), and SNAP-25 assemble into a functional complex, with syb and stx each contributing one SNARE motif, while SNAP-25 inserts two motifs into the assembly. Although singular SNARE motifs are largely unstructured in solution, the three SNARE motifs of syb, stx, and SNAP-25 spontaneously assemble into a stable complex that is exceptionally resistant to heat denaturation [31] and SDS treatment [49]. As shown in crystallization studies of the Brunger group [138], the four α-helices within the fully assembled SNARE complex form a rod-like coiled-coil bundle. This 12-nm-long core complex is stabilized by nonpolar side-chain interactions of conserved residues in all four helices, thereby forming 15 “hydrophobic layers” numbered from − 7 to + 8 [58, 138]. Intriguingly, a central “0” layer of the complex is composed of conserved polar amino acids: one arginine (R) and three glutamine (Q) residues [32, 159]. This feature is highly conserved throughout the SNARE superfamily [3, 62, 109] and led to the classification of SNAREs into R- and Q-SNAREs, which is used in parallel to the functional categorization in vesicular (v)-SNAREs and target (t)-SNAREs in the context of exocytosis.
While the importance of SNARE proteins for exocytosis has been established beyond any doubt by the action of clostridial neurotoxins (for a detailed review see [105]), by genetic mutants affecting SNARE protein function [20, 33, 157], or by knockout models [8, 12, 26, 121, 129, 130, 148], our knowledge of the exact molecular mechanism of Ca2+-regulated exocytosis is still far from being complete. In particular, the SNARE-lipid interactions and mechanistic intermediates leading to membrane merger and fusion pore formation are still elusive. Reduced in vitro models could demonstrate that membrane-anchored SNARE proteins enable fusion of liposomes, albeit at a slow time scale [102, 149]. Yet, the challenge remains to unravel how specific kinetic properties and Ca2+ dependency of physiological secretion are conferred to the SNARE machinery. Neuroendocrine cells of the adrenal medulla have been instrumental in elucidating critical mechanistic aspects of Ca2+-dependent exocytosis. Here, we will discuss recent advances in our understanding of SNARE function, especially highlighting studies that employed adrenal chromaffin cell as a model system.
Studying exocytosis in chromaffin cells
Chromaffin cells of the adrenal medulla are specialized neuroendocrine cells belonging to the sympathetic nervous system. Apart from several peptide hormones and bioactive peptides, these cells release either adrenaline (80%) or noradrenaline (20%) from large-dense core vesicles into the bloodstream [36]. Forming a sympathetic paraganglion, chromaffin cells receive excitatory cholinergic innervation from preganglionic splanchnic fibers. Released acetylcholine depolarizes chromaffin cells and elicits action potentials that induce Ca2+ influx via voltage-dependent Ca2+ channels, which in turn triggers the exocytosis of secretory granules [16, 82, 106] (for detailed review, see [107]). While the specific physiological role of secretion from the adrenal medulla is reviewed in detail in a sister article (Smith and Eiden, this issue), we discuss here the specific properties that make chromaffin cells a superior model system for studying exocytosis.
The greatest benefit of chromaffin cells is given by the accessibility of granule secretion through multiple experimental read-outs: The spherical shape of chromaffin cells makes them well suited for voltage clamping experiments using the patch clamp technique [44]. The compact cellular structure facilitates high-resolution membrane capacitance measurements [99], which can be used to follow exo- and endocytosis based on changes in plasma membrane area. In their seminal work, Neher and Marty observed discrete capacitance steps in the range of 0.4–80 fF using the whole-cell configuration, which is consistent with the fusion of single or multiple vesicles upon stimulation. To induce secretion in membrane capacitance recordings, chromaffin cells can be loaded via the patch pipette with intracellular solutions containing elevated Ca2+ levels (nM-μM) [96, 97, 126] or with caged-Ca2+ compounds that can be photolyzed by transient UV illumination, granting full control over the intracellular Ca2+ concentration [100]. Indeed, Ca2+ uncaging experiments in chromaffin cells have been crucial in determining secretion kinetics as well as in delineating vesicle pools with distinct release readiness (the so-called readily releasable pool (RRP) and slowly releasable pool (SRP)), which seem to be associated with different “primed” states of secretory vesicles [145]. The high time resolution of these experiments also allowed to assay the latency of secretion and the Ca2+ dependency of release rates, delivering reliable information on ‘stimulation-secretion coupling’ (for detailed review, see [113]). Since chromaffin granules release oxidizable cargo molecules (catecholamines), transmitter discharge from single vesicles can be directly recorded as oxidation currents by a polarized carbon fiber electrode [71]. Such amperometric recordings provide a high temporal resolution, monitoring discrete phases of transmitter discharge from a single chromaffin granule [23, 154]. Apart from electrophysiological methods, chromaffin cells have also been one of the first model systems, in which the fusion of single vesicles with the plasma membrane could actually be visualized. In particular, vesicle movement near the plasma membrane can be easily studied by “total internal fluorescence” microscopy [134, 135]. These experiments exploited the fact that spherical chromaffin cells flatly attach to a glass surface forming a “foot print” and that chromaffin granules can be readily marked and tracked by expression of vesicular cargo (e.g., neuropeptide Y) carrying a fluorophore tag.
The molecular players responsible for biogenesis, targeting, docking, and fusion of secretory vesicles have been well characterized in chromaffin cells [4, 34, 51, 119, 129, 139] and largely match the components in the exocytotic machinery of neurons [14]. This similarity has facilitated the use of chromaffin cells as a model for neuronal exocytosis, covering many mechanistic aspects of synaptic transmission. That being said, chromaffin cells lack morphologically distinct sites for vesicle fusion similar to presynaptic “active zones” in neurons. Still, hot-spots for Ca2+ entry and granule secretion have been described in cultured chromaffin cells [95, 117]. Owing to these benefits, it is no surprise that chromaffin cells have been extensively used as the model system of choice for studying proteins, lipids, and second messenger systems controlling magnitude and kinetics of vesicle fusion as well as release rate of cargo molecules from single vesicles. In the following part of this review, we will discuss the mechanisms underlying SNARE-mediated, Ca2+-triggered membrane merger, giving special attention to studies that have utilized chromaffin cells to delineate the SNARE action—from priming of vesicles to fusion pore opening and its final expansion.
The SNARE hypothesis: complex assembly and force transduction
How exactly SNARE complexes induce membrane fusion is key to our understanding of the exocytosis mechanism. In the absence of SNARE proteins, spontaneous membrane fusion rarely occurs, as the negative charges of phospholipid headgroups and their associated hydration shells keep biological membranes at a distance of at least 1–2 nm [77, 115]. Energy-consuming steps like the approximation of membranes against repulsive forces, shedding of the hydration shells, and reorganization of the lipid bilayers put an energetic toll on the fusion reaction, which has been estimated to exceed 40 κ B T (where κ B is Boltzmann’s constant and T is temperature) from theoretical studies [24, 53, 84, 151]. Notably, a recent study utilizing liposome fusion assay approximated the energy cost to be ~ 30 κ B T for membrane merger [35]. While it is widely accepted that formation of the thermodynamically stable SNARE complex delivers the free energy for membrane fusion [73] (for review, see [116]), different energy estimates prevented a definite answer to the question whether the energy of a single SNARE complexes is sufficient to drive membrane merger [73, 80, 81, 152]. However, recent work suggested that one SNARE complex or a low number of SNARE complexes are formed during the fusion of vesicles with the plasma membrane [91, 93, 114, 124, 142].
Based on structural and biochemical data, it has been envisioned that complex assembly starts at the N-termini of the four aligned SNARE motifs and subsequently progresses toward their C-terminal ends [47, 48, 74, 110, 138]. Functional evidence for progressive zippering was delivered by Xu et al. by testing the effect of a monoclonal antibody that recognizes an N-terminal epitope within SNAP-25 (amino acid 20–40) [157]. Characterizing secretion properties in chromaffin cells with membrane capacitance measurements and Ca2+ uncaging, they could show that a loose N-terminal SNARE assembly results in a decreased rate of sustained release, suggesting that SNARE complex initialization is hindered. In addition, a selective loss of the fast burst component indicated that the bound antibody somehow delayed the transition of partially zippered to fully assembled complexes, thus determining the balance between SRP and RRP. Another insight into SNARE assembly came from experiments, in which N- or C- terminal protein fragments of the stx-1A SNARE motif were infused in permeabilized PC12 cells. Intriguingly, only a N-terminal but not of a C-terminal fragment inhibited Ca2+-triggered release of norepinephrine, which again emphasizes the functional polarization of SNARE motifs [86]. While it would be also possible that SNARE assembly occurs in one step after the Ca2+ triggering [143], most evidence points to the occurrence of a distinctive partially assembled fusion intermediate (Fig. 1). Sorensen and colleagues directly tested the idea of progressive SNARE zippering in mouse chromaffin cells and systematically analyzed the phenotypical properties of mutations that interfere with the formation of different hydrophobic SNARE layers [131, 147]. Expressing either SNAP-25 or syb-2 mutants in corresponding null-background chromaffin cells, the Sorensen group performed combined membrane capacitance measurements and Ca2+-uncaging experiments in order to analyze vesicle pools and transition kinetics. By this method, they showed that mutations in the membrane proximal C-terminus of the SNARE complex preferentially compromised Ca2+-induced fusion triggering, whereas mutations in the middle or in the N-terminus of the complex selectively impeded vesicle priming without detectable changes in triggering rate. These findings support the notion of sequential assembly, starting with zippering at the N-terminus during vesicle priming and ending with C-terminal assembly of the SNARE complex that coincides with membrane fusion.

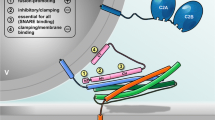
Model of SNARE-mediated Ca2+-triggered exocytosis. The core fusion machinery comprises the three SNARE proteins syb-2 (blue), stx-1A (orange; Habc domain not shown), and SNAP-25 (green; depicted without linker for clarity) as well as the accessory factors syt (dark blue) and cplx (pink) (topmost panel). PM, plasma membrane; v, vesicle. (1) Trans-SNARE complexes are nucleated during initial tethering and docking steps, and progressive N- to C-terminal zippering of the SNARE complex results in formation of a primed partially assembled intermediate. This primed state is supported by syt and cplx, the latter putatively preventing premature fusion. Primed vesicle pools can be estimated from the size of the initial membrane capacitance increase (inset). (2) Fusion triggering involves Ca2+ binding to syt and correlated conformational changes of both, syt and cplx, thereby lifting the fusion clamp and allowing for C-terminal SNARE assembly. Molecular straining of membranes and SNARE TMD dynamics putatively initiate lipid stalk formation. Fusion rate can be estimated by kinetic analysis of membrane capacitance recordings (inset). (3) Merger of the proximal leaflets results in hemifusion, as suggested by imaging of the fluorescently labeled cytosolic leaflet (F PH) of the plasma membrane versus the fluid phase marker (F 655) in chromaffin cells [163], (kindly provided by L.G. Wu). (4) Rearrangement of the distal leaflets forms the nascent fusion pore, through which cargo molecules diffuse out of the vesicle. Transmitter discharge from single granule, as measured by amperometry (inset), is limited initially by the diameter of the fusion pore neck. TMD conformational properties efficiently regulate the rate of fusion pore expansion, likely by modulating membrane curvature [27]
For regulated exocytosis the SNARE zippering process should be discontinuous allowing for a partially assembled intermediate state, wherein the SNARE complex rests, before Ca2+ stimulus induces final membrane fusion. SNARE folding experiments with optical tweezers recently delivered evidence for the existence of such a metastable assembly state [72, 165], which is possibly stabilized by SNARE regulators like complexins [65], as will be discussed in a later chapter. The transition from a partly unstructured, flexible SNARE configuration to a fully assembled rod-like structure should result in mechanical force transfer from the SNARE complex onto the adjoining membrane segments until the relative positions of the transmembrane anchors of stx-1A and syb-2 have converged in the process. Interestingly, an X-ray crystallographic study [133] that analyzed neuronal SNARE complexes containing full-length stx-1A and syb-2 suggested that helical assembly may extend beyond the SNARE motifs, encompassing JMRs and TMDs of both transmembrane proteins. The resulting SNARE force may not only be used to pull the secretory organelle toward the plasma membrane but might also locally strain the lipid bilayers, thereby directly contributing to membrane merger. Based on these considerations, it has been posited that the length of the linker peptide that connects the SNARE motif with the transmembrane domain (TMD) should be critical for fusion efficiency [88]. In case of the v-SNARE syb-2, insertion of a 12 or 24 amino acid-long flexible linker between the SNARE motif and TMD abolished synaptic vesicle exocytosis [25]. Experiments in syb-2/cellubrevin (ceb) double null chromaffin cells expressing syb-2 variants with increasing linker length (insertion of 2–22 amino acids) gradually reduced the primed pool of vesicles, attenuated early fusion pore jitter, and slowed the kinetics of cargo discharge from single vesicle in a linker length-dependent fashion [60]. These results suggested that continuous molecular straining of SNAREs on membranes drives vesicles from priming to membrane merger. A similar requirement for a tight coupling between SNARE motif and TMD has been shown in bovine chromaffin cells [10] and in syb-2−/− hippocampal neurons [43]. In the neuronal system, the “degree” of coupling between syb-2 TMD and SNARE motif affected the synchronized release component of synaptic transmission as well as the waveform of quantal events, again pointing to deficits in vesicle fusion and transmitter discharge. In the same line, insertion of 3 or 7 amino acid-long linkers between the stx-1A TMD and its SNARE motif strongly inhibited evoked synaptic transmission, reiterating the importance of tight mechanical coupling of both elements [164].
v-SNARE action in protein-lipid interplay and fusion pore opening
The formation of the SNARE complex putatively forces vesicle and plasma membrane into close apposition, which may facilitate the establishment of first hydrophobic contacts, serving as a starting point for significant rearrangements between the merging membranes [149]. In silico analyses have suggested that hydrophobic nucleation events, wherein tails of lipids belonging to opposite membranes interconnect the adjacent leaflets (lipid splay), are highly energy demanding [57, 115, 125]. Therefore, it is possible that the SNARE machinery may not only employ inter-membranous force (by protein-protein interactions) but also direct protein-lipid interactions to facilitate membrane merger (Fig. 1). The juxtamembrane regions (JMR) and TMDs of the SNARE proteins are prime candidates for such SNARE-lipid interactions. Since several interesting ideas about SNARE-lipid recently arose from work with syb-2, we will focus the discussion on v-SNARE-mediated membrane interactions.
Lipid interactions of the juxtamembrane domain of syb-2
Tryptophan (Trp) residues are frequently found at water-lipid interfaces of transmembrane proteins due to their amphiphatic character [61]. The JMR of syb-2 contains two Trp residues (W89, W90), which are conserved throughout the animal kingdom and could possibly contribute to critical SNARE-lipid interactions during the fusion mechanism. Experiments in reconstitution assays have demonstrated that the two Trp residues in the JMR indeed interact with the phospholipid bilayer [66, 112] and may even define the orientation of the TMD within the vesicular membrane [9]. Interestingly, a site-directed spin-labeling EPR study proposed that substitution of the Trp residues by hydrophilic serine residues accelerated the rate of SNARE complex formation [67], which suggests that JMR-membrane interactions serve an inhibitory role by restricting access to C-terminal portions of the SNARE motif. However, later reports could not confirm this observation, showing that neither kinetics nor efficacy of SNARE complex formation was altered, when Trp residues were mutated [50, 123]. While deletion or substitution of Trps by serine residues did not change SNARE-mediated liposome fusion [123], secretion of human growth hormone in PC12 cells expressing a syb-2 variant containing a W89A/W90A double mutation was substantially reduced [111]. Furthermore, expression of the W89A/W90A double mutant in murine cortical syb-2−/− neurons caused a severe reduction in evoked release and a substantial enhancement of spontaneous release, a phenotype that is reminiscent of the secretion phenotype resulting from the knockdown of the accessory protein complexin (cplx) [87]. Although the W89A/W90A mutation does not alter cplx-SNARE complex interaction, the phenotypic similarities of cplx knockdown and Trp mutations led the authors to conclude that the Trp residues in the JMR are involved in syb-2-cplx interplay [87] or unmask a secondary Ca2+ sensor [160] (see also [98]). Fang et al. [30] reported that the W89A/W90A double mutant in v-SNARE-deficient chromaffin cells enhanced the frequency of fusion events upon subtle depolarization of chromaffin cells with 5 mM KCl, increased the quantal size, and reduced secretion in comparison to controls upon stronger stimulation by 100 mM KCl. Contradicting the view of a JMR-based fusion clamp, Borisovska et al. [7] found that substitution of the Trp residues did not change the rate of v-SNARE-dependent exocytosis at submicromolar [Ca]i, but strongly reduced the capacitance increase in response to a stepwise increase in [Ca]I (15–25 μM). Furthermore, no changes in quantal signaling with respect charge, amplitude or kinetics could be detected using carbon fiber amperometry. Additional molecular dynamics simulations of C-terminal syb-2 fragments containing the JMR and the membrane-embedded TMD rather suggested that Trp residues within the JMR of syb-2 play a crucial role in positioning the neighboring lysine and arginine residues at the membrane-water interface, which creates a positive electrostatic potential at the site of action. This positive membrane surface potential could reduce repulsive forces between the negatively charged vesicle and plasma membrane, thereby stabilizing the primed state. Regardless of the exact molecular mechanism, the available data indicate that protein-lipid interplay between the syb-2 JMR and vesicular membrane are important for the maintenance of the primed, release-ready state of the vesicle, either by energetically stabilizing it or by preventing premature fusion.
Function of the transmembrane domain of syb-2
Previous in vitro analyses suggested that SNARE TMDs in isolation (in the absence of the cytoplasmic SNARE domain) actively promote liposome fusion [69, 70]. Furthermore, experiments in reduced model systems have shown that replacing the SNARE TMDs with lipidic anchors is detrimental for fusion between liposomes [89], cell-cell fusion with ectopically expressed “flipped” SNAREs [41], and lipid nanodisc fusion with liposome [5, 122] and for yeast vacuole fusion [42, 108, 118]. Evidently, SNARE TMDs may actively contribute to membrane merger rather than serving as passive membrane anchors. In sharp contrast, the role of SNARE TMDs in neurotransmitter release from live cells has been controversially discussed. The substitution of the syb-2 TMD with an acylated, lipid-anchoring motif of CSP-α was reported to allow for an substantial rescue of evoked synaptic transmission in murine cortical neurons and also supported spontaneous release of miniature currents similar to wild-type protein [164]. However, the same syb-2/CSP-α chimera was shown to be inefficient in supporting exocytosis in neurons as well as mouse chromaffin cells [17]. Moreover, high-resolution membrane capacitance measurements and carbon fiber amperometry have indicated that CSP-α anchored syb-2 proteins failed to promote efficient exocytosis as well as transmitter discharge from chromaffin granules [27]. These results provided strong evidence for an important role of SNARE TMDs in Ca2+-triggered release.
Generally, the fusion pore has been conceived as a purely lipidic structure that results from progressive membrane rearrangements starting with the establishment of a hemifused intermediate (for detailed review, see [68]), Fig. 1). The existence of such a hemifused state, in which only the outer leaflets of two bilayers merge, while the inner leaflets remain intact, was first demonstrated for membrane fusion of influenza hemagglutinin viruses. In this viral system, mutant hemagglutinin without TMD arrested fusion at an intermediated state allowing for lipid mixing but not for content exchange [59, 90]. Later, hemifused intermediates were also shown in SNARE-dependent liposome fusion [158, 161], in cell-cell fusion [41], in lipid nanodisc-liposome fusion [122], and even in Ca2+-triggered neurotransmitter release from chromaffin cells [163]. To actively support membrane fusion in this scenario, TMDs-lipid interactions may either facilitate the formation of the initial hemifused state or support the transition to the opened fusion pore. While several recent studies have presented clues, suggesting that v-SNARE TMDs indeed serve important functions in these particular steps, little consent about the underlying mechanism has been reached. Liposome fusion experiments and structural analysis with EPR have pointed out that lipid interactions may switch the conformation of v-SNARE TMD dimers from an open to a closed scissor-like configuration, which could help to modify membrane curvature and thus facilitate fusion [140]. Moreover, results from a fusion assay between v-SNARE-containing lipid nanodiscs and t-SNARE liposomes suggested that a critical number of native v-SNARE TMDs is required to open and maintain the fusion pore [122], implying that the TMD facilitates the transition from lipid stalk to open fusion pore. Extending the idea of v-SNARE-mediated mechanical straining of the membrane, the group of Manfred Lindau investigated the possibility that the SNARE TMDs could be actually pulled through the vesicular membrane and thereby induce reorganization and merger of affected membrane segments. In support of this notion, Ngatchou et al. have shown that addition of polar residues (lysine or glutamate) at the luminal end of syb-2 strongly suppressed exocytosis [101]. Since the inhibition of granule fusion was clearly determined by the polarity of the replacing amino acids, this observation might indicate that this TMD region is shifting its position into the membrane or even transit through it [101]. However, a lot of molecular details with respect to the fate of the syb-2 TMD are still unclear. We performed a systematic structure-function analysis in chromaffin cells and neurons to provide evidence for yet another role of the syb-2 TMD that is tied to the high incidence of helix-destabilizing ß-branched amino acids (isoleucine/valine) within its sequence [27]. Intriguingly, our data showed that substitution of the native TMD core by a leucine-containing peptide that is strictly helical significantly reduced Ca2+-triggered release and also slowed down the kinetics of catecholamine discharge from single granules in chromaffin cells. In reverse, TMDs mutants containing an increased number of ß-branched amino acids could again fully support secretion but accelerated the rate of fusion pore expansion over that in wild-type controls [27]. Comparing these functional results with conformational data from molecular dynamic simulations of membrane-embedded syb-2 TMD mutants, we found a direct correlation between the conformational flexibility of the TMD and its ability to facilitate pore expansion kinetics. Overall, these data have suggested that structural flexibility of v-SNARE TMDs might play a role at different steps of the fusion mechanism (Fig. 1). In the initial phase of membrane merger, structural properties of the TMD might promote lipid splay in the neighboring membrane area [115] and thereby facilitate fusion. Similarly, conformational dynamics of the TMD seem to facilitate of fusion pore expansion, most likely by affecting the negative curvature of the narrow fusion pore neck.
Still, alternative hypotheses on fusion pore structure have been brought forward in the literature. For example, SNARE TMDs might be organized in a barrel-like arrangement to directly form a proteinaceous fusion pore that in principle would resemble an ion channel [54, 75, 76]. The idea of a channel-like fusion pore first arose from membrane capacitance measurements in mast cells, where the initial fusion pore conductance was similar to that of a large ion channel, and fusion pores showed a “flickering” behavior just like ion channels [94, 132]. More direct support of this view was then provided by a systematic mutational analysis, in which single amino acids in the stx TMD were substituted by bulky residues, in particular Trp [46]. Performing amperometric recordings in PC12 cells, the authors demonstrated that mutations reducing pore conductance resided on the same face of the TMD helix, thus putatively lining the pore. This analysis was recently also extended to the syb-2 TMD and identified not one but two mutation-sensitive surfaces on the TMD pointing in opposite directions, which would suggest a deviating structural organization of the hemi-channel formed by syb-2 TMDs compared to its counterpart on the plasma membrane [19]. These results have been interpreted as evidence for a fusion pore made up from multiple SNARE TMDs organized in a gap junction-like arrangement in SNARE-bridged membranes [18, 45, 141]. However, the hypothesis of a proteinaceous fusion is difficult to reconcile with the observation that membrane-anchored syb-2 lacking the TMD was still able to support some chromaffin granule exocytosis above the level of v-SNARE null background [17, 27].
In this context, it is important to note that the lipid composition of the adjoining membranes is also a critical determinant of membrane fusion. Previous studies have provided evidence that various species of membrane phospholipids can affect bilayer merger by virtue of their shape or charge (for a detailed review, see [2]). Since putative fusion intermediates require phospholipids to arrange in membrane structures with high curvature [22], it stands to reason that lipids that can support the curvature of the fusion pore neck (negatively curved cytosolic side and positively curved luminal side) also promote fusion. Several studies have demonstrated that manipulation of the lipid composition in neuroendocrine cells alters granule secretion [1, 162]. In particular, application of the curvature perturbing lysophosphatidyl choline (LPC), which induces positive membrane curvature due to its large head-group area relative to the carbonyl chain, has been demonstrated to either reduce or enhance exocytosis depending on the side of action (intra- or extracellularly). In reverse, phospholipids with a negative curvature like oleic acid exerted opposite effects. Still, the challenge remains to elucidate the functional interplay between protein-lipid interfaces and lipid microdomains in exocytosis research.
Regulatory brakes on SNARE assembly en route to membrane fusion
Importance of release-ready vesicles
Since SNARE proteins are constitutively active in membrane fusion [149], the fusion process requires additional regulatory factors that allow the exquisite kinetic control and reliability, which are hallmarks of Ca2+-regulated exocytosis. As a result, secretory organelles transit through different mechanistic steps in the fusion process, from “docking” (attachment of the vesicles to the plasma membrane) to priming (molecular maturation that render vesicles release competent), and finally Ca2+-dependent triggering [155]. As mentioned above, advanced biophysical analyses of single SNARE assembly have shown that cognate v- and t-SNAREs have the propensity to zipper in a discontinuous fashion allowing for metastable intermediates [37, 165]. In the last decade, significant efforts have been made to identify potential SNARE-interacting proteins that may stabilize a fusion intermediate and thus could function as a “fusion clamp” impeding premature fusion. While a detailed review of SNARE regulatory proteins and their mechanistic functions in determining release-ready vesicle pools is beyond the scope of this review, we still want to briefly outline the role of cplx and synaptotagmin (syt), two accessory proteins, whose interplay seems to be intimately tied to establishment and relief of the fusion clamp (Fig. 1). These factors are believed to act in tandem to establish a state of vesicle maturation, from which secretion can progress in a highly synchronized fashion (for detailed review, see [92]).
Actions of Cplx and Syt in imparting synchronicity to the release apparatus
Genetic deletion of cplx-2 in chromaffin cells strongly diminishes the exocytotic burst (EB) in chromaffin cells, which is usually observed in response to a stepwise increase in intracellular [Ca]i [15, 28]. Similarly, a loss of syt-1 nearly abolishes the readily releasable pool (RRP) component of the EB [146] and slows down the kinetics of the secretion from the slowly releasable pool (SRP) [28]. Furthermore, studies analyzing synaptic transmission at the neuromuscular junction of D. melanogaster and C. elegans cplx-null mutants as well as work on secretion in cplx-2 knockout chromaffin cells reported a dramatic increase in spontaneous or tonic release, which implies an additional inhibitory effects of cplx on neurotransmission [28, 52, 85]. A similar increase in spontaneous release has also been seen with cplx-1/-2 double knockdown of murine cortical neurons [87]. In contrast, genetic ablation of the putative calcium sensor syt-1 elevated the rate of spontaneous fusion in some [11, 29, 63, 78, 104, 137, 156] but not in other systems [28, 38, 79]. These disparate results may in part be due to substituting syt isoforms, improperly replacing the Ca2+ sensor [63, 137] or to secondary effects of the syt-1 knockout phenotype impairing also the inhibitory impact of GABAergic interneurons on spontaneous glutamatergic signaling [153]. In any case, these results indicate that cplx and syt do not always have mechanistically overlapping functions in fusion clamping.
In the quest for the particular mechanisms, by which cplx clamps release, experiments in reduced systems demonstrated that cplx can inhibit fusion between liposomes carrying only v- and t-SNAREs [120] or between cells expressing such flipped SNARE variants [39]. In close correlation, experiments in mouse chromaffin cells showed that the clamp action of cplx-2 persisted even in the common absence of syt-1 and syt-7—the two major Ca2+ sensors present in chromaffin cells [28]. Therefore, both in vitro and in vivo experiments support a model, wherein cplx directly hinders SNARE activity, arresting premature vesicle fusion.
If cplx acts as a “prefusion clamp” that stabilizes half-zippered SNARE complexes, it is mandatory that this molecular clamp can be rapidly lifted upon a Ca2+ stimulus to enable a synchronous secretory response with short delay. Experimental clues from in vitro studies [40, 64, 83, 120] have suggested that, upon Ca2+ binding, syt likely reverses the “clamped” state by affecting the conformation of cplx bound to SNARE complex, which leads to completion of SNARE zippering and membrane merger. Direct evidence for this putative cplx/syt antagonism recently came from experiments in mouse chromaffin cells [28]. Removal of syt-1 in chromaffin cells resulted in slower expansion of the initial narrow fusion pore, as analyzed by carbon fiber amperometry, while additional loss of cplx-2 (cplx-2−/−; syt-1−/−) reversed this fusion pore phenotype. Furthermore, overexpression of cplx-2 reproduced the syt-1 ko phenotype, indicating competitive actions of both SNARE regulators. Moreover, in the presence of syt-1, cplx-2 deficiency had no effect on the fusion pore expansion rate at high [Ca]i, but increasingly sped up the rate of pore expansion with lowering [Ca]i. These results can be explained well in the context of a syt-1/cplx-2 antagonism, wherein cpx-2 exerts a decelerating (clamping) effect on fusion pore expansion, which is overcome by syt-1 in a Ca2+-dependent fashion. Collectively, these results clearly illustrate the antagonistic actions of both SNARE regulators in controlling the kinetics of the early fusion pore.
Concluding remarks
Based on the available experimental data, it is apparent that SNARE proteins are the major “work horses” driving membrane fusion, while SNARE regulators provide the temporal speed and precision that are essential for fast Ca2+-triggered exocytosis. Intriguingly, experimental results from chromaffin cells have pinpointed intermediate steps of membrane fusion (priming, fusion triggering, and slow/rapid pore expansion) that are affected by SNARE force, SNARE TMD-mediated protein-lipid interactions, and SNARE regulators like cplx and syt. Evidently, these results underline the necessity that different molecular players orchestrate and converge on the same cellular process to guide the vesicles from priming to fusion pore opening and its expansion, ensuring rapid cargo discharge with a sub-millisecond precision.
Abbreviations
- SNAREs:
-
N-Ethylmaleimide-sensitive factor (NSF) attachment protein receptors
- v-SNARE:
-
Vesicular SNARE
- t-SNARE:
-
Target SNARE
- SNAP25:
-
Synaptosomal-associated protein, 25 kDa
- syb-2:
-
Synaptobrevin II
- stxIa:
-
Syntaxin Ia
- syt:
-
Synaptotagmin
- cplx:
-
Complexin
- Ceb:
-
Cellubrevin
- VAMP:
-
Vesicular associated membrane protein
- PC12:
-
Pheochromocytoma cell line
- [Ca]i:
-
Intracellular calcium
- NMJ:
-
Neuromuscular junction
References
Amatore C, Arbault S, Bouret Y, Guille M, Lemaitre F, Verchier Y (2006) Regulation of exocytosis in chromaffin cells by trans-insertion of lysophosphatidylcholine and arachidonic acid into the outer leaflet of the cell membrane. Chembiochem: Eur J Chem Biol 7:1998–2003
Ammar MR, Kassas N, Chasserot-Golaz S, Bader MF, Vitale N (2013) Lipids in regulated exocytosis: what are they doing? Front Endocrinol 4:125
Antonin W, Fasshauer D, Becker S, Jahn R, Schneider TR (2002) Crystal structure of the endosomal SNARE complex reveals common structural principles of all SNAREs. Nat Struct Biol 9:107–111
Baltazar G, Tome A, Carvalho AP, Duarte EP (2000) Differential contribution of syntaxin 1 and SNAP-25 to secretion in noradrenergic and adrenergic chromaffin cells. Eur J Cell Biol 79:883–891
Bao H, Goldschen-Ohm M, Jeggle P, Chanda B, Edwardson JM, Chapman ER (2016) Exocytotic fusion pores are composed of both lipids and proteins. Nat Struct Mol Biol 23:67–73
Bonifacino JS, Glick BS (2004) The mechanisms of vesicle budding and fusion. Cell 116:153–166
Borisovska M, Schwarz YN, Dhara M, Yarzagaray A, Hugo S, Narzi D, Siu SW, Kesavan J, Mohrmann R, Bockmann RA, Bruns D (2012) Membrane-proximal tryptophans of synaptobrevin II stabilize priming of secretory vesicles. J Neurosci: Off J Soc Neurosci 32:15983–15997
Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Sudhof T, Bruns D (2005) v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J 24:2114–2126
Bowen M, Brunger AT (2006) Conformation of the synaptobrevin transmembrane domain. Proc Natl Acad Sci U S A 103:8378–8383
Bretou M, Anne C, Darchen F (2008) A fast mode of membrane fusion dependent on tight SNARE zippering. J Neurosci: Off J Soc Neurosci 28:8470–8476
Broadie K, Bellen HJ, DiAntonio A, Littleton JT, Schwarz TL (1994) Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proc Natl Acad Sci U S A 91:10727–10731
Broadie K, Prokop A, Bellen HJ, O'Kane CJ, Schulze KL, Sweeney ST (1995) Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron 15:663–673
Bruns D, Jahn R (2002) Molecular determinants of exocytosis. Pflugers Arch: Eur J Physiol 443:333–338
Burgoyne RD (1995) Mechanisms of catecholamine secretion from adrenal chromaffin cells. J Physiol Pharmacol: Off J Pol Physiol Soc 46:273–283
Cai H, Reim K, Varoqueaux F, Tapechum S, Hill K, Sorensen JB, Brose N, Chow RH (2008) Complexin II plays a positive role in Ca2+-triggered exocytosis by facilitating vesicle priming. Proc Natl Acad Sci U S A 105:19538–19543
Cena V, Nicolas GP, Sanchez-Garcia P, Kirpekar SM, Garcia AG (1983) Pharmacological dissection of receptor-associated and voltage-sensitive ionic channels involved in catecholamine release. Neuroscience 10:1455–1462
Chang CW, Chiang CW, Gaffaney JD, Chapman ER, Jackson MB (2016) Lipid-anchored synaptobrevin provides little or no support for exocytosis or liposome fusion. J Biol Chem 291:2848–2857
Chang CW, Chiang CW, Jackson MB (2017) Fusion pores and their control of neurotransmitter and hormone release. J Gen Physiol 149:301–322
Chang CW, Jackson MB (2015) Synaptobrevin transmembrane domain influences exocytosis by perturbing vesicle membrane curvature. Biophys J 109:76–84
Chen YA, Scales SJ, Patel SM, Doung YC, Scheller RH (1999) SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell 97:165–174
Chen YA, Scheller RH (2001) SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol 2:98–106
Chernomordik LV, Zimmerberg J, Kozlov MM (2006) Membranes of the world unite! J Cell Biol 175:201–207
Chow R, Von Rüden L (1995) Electrochemical detection of secretion from single cells. In: Sakmann B, Neher E (eds) Single-channel recording. Springer, US, pp 245–275
Cohen FS, Melikyan GB (2004) The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J Membr Biol 199:1–14
Deak F, Shin OH, Kavalali ET, Sudhof TC (2006) Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci: Off J Soc Neurosci 26:6668–6676
Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL (1998) Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci: Off J Neurosci 18:2028–2039
Dhara M, Yarzagaray A, Makke M, Schindeldecker B, Schwarz Y, Shaaban A, Sharma S, Bockmann RA, Lindau M, Mohrmann R, and Bruns D (2016) v-SNARE transmembrane domains function as catalysts for vesicle fusion. eLife 5:e17571. https://doi.org/10.7554/eLife.17571
Dhara M, Yarzagaray A, Schwarz Y, Dutta S, Grabner C, Moghadam PK, Bost A, Schirra C, Rettig J, Reim K, Brose N, Mohrmann R, Bruns D (2014) Complexin synchronizes primed vesicle exocytosis and regulates fusion pore dynamics. J Cell Biol 204:1123–1140
DiAntonio A, Parfitt KD, Schwarz TL (1993) Synaptic transmission persists in synaptotagmin mutants of Drosophila. Cell 73:1281–1290
Fang Q, Zhao Y, Lindau M (2013) Juxtamembrane tryptophans of synaptobrevin 2 control the process of membrane fusion. FEBS Lett 587:67–72
Fasshauer D, Otto H, Eliason WK, Jahn R, Brunger AT (1997) Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J Biol Chem 272:28036–28041
Fasshauer D, Sutton RB, Brunger AT, Jahn R (1998) Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci U S A 95:15781–15786
Fergestad T, Wu MN, Schulze KL, Lloyd TE, Bellen HJ, Broadie K (2001) Targeted mutations in the syntaxin H3 domain specifically disrupt SNARE complex function in synaptic transmission. J Neurosci: Off J Soc Neurosci 21:9142–9150
Fisher RJ, Pevsner J, Burgoyne RD (2001) Control of fusion pore dynamics during exocytosis by Munc18. Science 291:875–878
Francois-Martin C, Rothman JE, Pincet F (2017) Low energy cost for optimal speed and control of membrane fusion. Proc Natl Acad Sci U S A 114:1238–1241
Ganong WF (2005) Review of medical physiology. McGraw-Hill Medical, New York p. 928 p
Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, Zhang Y (2012) Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337:1340–1343
Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79:717–727
Giraudo CG, Eng WS, Melia TJ, Rothman JE (2006) A clamping mechanism involved in SNARE-dependent exocytosis. Science 313:676–680
Giraudo CG, Garcia-Diaz A, Eng WS, Yamamoto A, Melia TJ, Rothman JE (2008) Distinct domains of complexins bind SNARE complexes and clamp fusion in vitro. J Biol Chem 283:21211–21219
Giraudo CG, Hu C, You D, Slovic AM, Mosharov EV, Sulzer D, Melia TJ, Rothman JE (2005) SNAREs can promote complete fusion and hemifusion as alternative outcomes. J Cell Biol 170:249–260
Grote E, Baba M, Ohsumi Y, Novick PJ (2000) Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J Cell Biol 151:453–466
Guzman RE, Schwarz YN, Rettig J, Bruns D (2010) SNARE force synchronizes synaptic vesicle fusion and controls the kinetics of quantal synaptic transmission. J Neurosci: Off J Soc Neurosci 30:10272–10281
Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch: Eur J Physiol 391:85–100
Han X, Jackson MB (2005) Electrostatic interactions between the syntaxin membrane anchor and neurotransmitter passing through the fusion pore. Biophys J 88:L20–L22
Han X, Wang CT, Bai J, Chapman ER, Jackson MB (2004) Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304:289–292
Hanson PI, Heuser JE, Jahn R (1997) Neurotransmitter release—four years of SNARE complexes. Curr Opin Neurobiol 7:310–315
Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE (1997) Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 90:523–535
Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Sudhof TC, Niemann H (1994) Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J 13:5051–5061
Hernandez JM, Stein A, Behrmann E, Riedel D, Cypionka A, Farsi Z, Walla PJ, Raunser S, Jahn R (2012) Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336:1581–1584
Hohne-Zell B, Gratzl M (1996) Adrenal chromaffin cells contain functionally different SNAP-25 monomers and SNAP-25/syntaxin heterodimers. FEBS Lett 394:109–116
Huntwork S, Littleton JT (2007) A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci 10:1235–1237
Jackson MB (2009) Minimum membrane bending energies of fusion pores. J Membr Biol 231:101–115
Jackson MB (2010) SNARE complex zipping as a driving force in the dilation of proteinaceous fusion pores. J Membr Biol 235:89–100
Jahn R, Lang T, Sudhof TC (2003) Membrane fusion. Cell 112:519–533
Jahn R, Sudhof TC (1999) Membrane fusion and exocytosis. Annu Rev Biochem 68:863–911
Kasson PM, Lindahl E, Pande VS (2010) Atomic-resolution simulations predict a transition state for vesicle fusion defined by contact of a few lipid tails. PLoS Comput Biol 6:e1000829
Katz L, Hanson PI, Heuser JE, Brennwald P (1998) Genetic and morphological analyses reveal a critical interaction between the C-termini of two SNARE proteins and a parallel four helical arrangement for the exocytic SNARE complex. EMBO J 17:6200–6209
Kemble GW, Danieli T, White JM (1994) Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 76:383–391
Kesavan J, Borisovska M, Bruns D (2007) v-SNARE actions during Ca(2+)-triggered exocytosis. Cell 131:351–363
Killian JA, von Heijne G (2000) How proteins adapt to a membrane-water interface. Trends Biochem Sci 25:429–434
Kloepper TH, Kienle CN, Fasshauer D (2007) An elaborate classification of SNARE proteins sheds light on the conservation of the eukaryotic endomembrane system. Mol Biol Cell 18:3463–3471
Kochubey O, Schneggenburger R (2011) Synaptotagmin increases the dynamic range of synapses by driving Ca(2)+-evoked release and by clamping a near-linear remaining Ca(2)+ sensor. Neuron 69:736–748
Krishnakumar SS, Radoff DT, Kummel D, Giraudo CG, Li F, Khandan L, Baguley SW, Coleman J, Reinisch KM, Pincet F, Rothman JE (2011) A conformational switch in complexin is required for synaptotagmin to trigger synaptic fusion. Nat Struct Mol Biol 18:934–940
Kummel D, Krishnakumar SS, Radoff DT, Li F, Giraudo CG, Pincet F, Rothman JE, Reinisch KM (2011) Complexin cross-links prefusion SNAREs into a zigzag array. Nat Struct Mol Biol 18:927–933
Kweon DH, Kim CS, Shin YK (2003) Insertion of the membrane-proximal region of the neuronal SNARE coiled coil into the membrane. J Biol Chem 278:12367–12373
Kweon DH, Kim CS, Shin YK (2003) Regulation of neuronal SNARE assembly by the membrane. Nat Struct Biol 10:440–447
Kweon DH, Kong B, Shin YK (2017) Hemifusion in synaptic vesicle cycle. Front Mol Neurosci 10:65
Langosch D, Crane JM, Brosig B, Hellwig A, Tamm LK, Reed J (2001) Peptide mimics of SNARE transmembrane segments drive membrane fusion depending on their conformational plasticity. J Mol Biol 311:709–721
Langosch D, Hofmann M, Ungermann C (2007) The role of transmembrane domains in membrane fusion. Cell Mol Life Sci: CMLS 64:850–864
Leszczyszyn DJ, Jankowski JA, Viveros OH, Diliberto EJ Jr, Near JA, Wightman RM (1990) Nicotinic receptor-mediated catecholamine secretion from individual chromaffin cells. Chemical evidence for exocytosis. J Biol Chem 265:14736–14737
Li F, Kummel D, Coleman J, Reinisch KM, Rothman JE, Pincet F (2014) A half-zippered SNARE complex represents a functional intermediate in membrane fusion. J Am Chem Soc 136:3456–3464
Li F, Pincet F, Perez E, Eng WS, Melia TJ, Rothman JE, Tareste D (2007) Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat Struct Mol Biol 14:890–896
Lin RC, Scheller RH (1997) Structural organization of the synaptic exocytosis core complex. Neuron 19:1087–1094
Lindau M, Almers W (1995) Structure and function of fusion pores in exocytosis and ectoplasmic membrane fusion. Curr Opin Cell Biol 7:509–517
Lindau M, Alvarez de Toledo G (2003) The fusion pore. Biochim Biophys Acta 1641:167–173
Lipowsky R (1995) The morphology of lipid membranes. Curr Opin Struct Biol 5:531–540
Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ (1993) Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell 74:1125–1134
Liu H, Dean C, Arthur CP, Dong M, Chapman ER (2009) Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci: Off J Soc Neurosci 29:7395–7403
Liu W, Montana V, Parpura V, Mohideen U (2009) Single molecule measurements of interaction free energies between the proteins within binary and ternary SNARE complexes. J Nanoneurosci 1:120–129
Liu W, Parpura V (2009) Single molecule probing of SNARE proteins by atomic force microscopy. Ann N Y Acad Sci 1152:113–120
Livett BG (1993) Chromaffin cells: roles for vesicle proteins and Ca2+ in hormone secretion and exocytosis. Trends Pharmacol Sci 14:345–348
Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Sollner TH (2012) Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J 31:3270–3281
Markin VS, Albanesi JP (2002) Membrane fusion: stalk model revisited. Biophys J 82:693–712
Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS (2011) Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol: CB 21:97–105
Matos MF, Mukherjee K, Chen X, Rizo J, Sudhof TC (2003) Evidence for SNARE zippering during Ca2+-triggered exocytosis in PC12 cells. Neuropharmacology 45:777–786
Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC (2009) Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323:516–521
McNew JA, Weber T, Engelman DM, Sollner TH, Rothman JE (1999) The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol Cell 4:415–421
McNew JA, Weber T, Parlati F, Johnston RJ, Melia TJ, Sollner TH, Rothman JE (2000) Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J Cell Biol 150:105–117
Melikyan GB, White JM, Cohen FS (1995) GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J Cell Biol 131:679–691
Mohrmann R, de Wit H, Verhage M, Neher E, Sorensen JB (2010) Fast vesicle fusion in living cells requires at least three SNARE complexes. Science 330:502–505
Mohrmann R, Dhara M, and Bruns D (2015) Complexins: small but capable. Cellular and molecular life sciences: CMLS
Mohrmann R, Sorensen JB (2012) SNARE requirements en route to exocytosis: from many to few. J Mol Neurosci: MN 48:387–394
Monck JR, Fernandez JM (1996) The fusion pore and mechanisms of biological membrane fusion. Curr Opin Cell Biol 8:524–533
Monck JR, Robinson IM, Escobar AL, Vergara JL, Fernandez JM (1994) Pulsed laser imaging of rapid Ca2+ gradients in excitable cells. Biophys J 67:505–514
Moser T, Neher E (1997) Estimation of mean exocytic vesicle capacitance in mouse adrenal chromaffin cells. Proc Natl Acad Sci U S A 94:6735–6740
Moser T, Neher E (1997) Rapid exocytosis in single chromaffin cells recorded from mouse adrenal slices. J Neurosci: Off J Soc Neurosci 17:2314–2323
Neher E (2010) Complexin: does it deserve its name? Neuron 68:803–806
Neher E, Marty A (1982) Discrete changes of cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proc Natl Acad Sci U S A 79:6712–6716
Neher E, Zucker RS (1993) Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron 10:21–30
Ngatchou AN, Kisler K, Fang Q, Walter AM, Zhao Y, Bruns D, Sorensen JB, Lindau M (2010) Role of the synaptobrevin C terminus in fusion pore formation. Proc Natl Acad Sci U S A 107:18463–18468
Nickel W, Weber T, McNew JA, Parlati F, Sollner TH, Rothman JE (1999) Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci U S A 96:12571–12576
Otto H, Hanson PI, Jahn R (1997) Assembly and disassembly of a ternary complex of synaptobrevin, syntaxin, and SNAP-25 in the membrane of synaptic vesicles. Proc Natl Acad Sci U S A 94:6197–6201
Pang ZP, Sun J, Rizo J, Maximov A, Sudhof TC (2006) Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J 25:2039–2050
Pantano S, Montecucco C (2014) The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell Mol Life Sci: CMLS 71:793–811
Penner R, Neher E (1988) The role of calcium in stimulus-secretion coupling in excitable and non-excitable cells. J Exp Biol 139:329–345
Perlman RL, Chalfie M (1977) Catecholamine release from the adrenal medulla. Clin Endocrinol Metab 6:551–576
Pieren M, Desfougeres Y, Michaillat L, Schmidt A, Mayer A (2015) Vacuolar SNARE protein transmembrane domains serve as nonspecific membrane anchors with unequal roles in lipid mixing. J Biol Chem 290:12821–12832
Pobbati AV, Razeto A, Boddener M, Becker S, Fasshauer D (2004) Structural basis for the inhibitory role of tomosyn in exocytosis. J Biol Chem 279:47192–47200
Pobbati AV, Stein A, Fasshauer D (2006) N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science 313:673–676
Quetglas S, Iborra C, Sasakawa N, De Haro L, Kumakura K, Sato K, Leveque C, Seagar M (2002) Calmodulin and lipid binding to synaptobrevin regulates calcium-dependent exocytosis. EMBO J 21:3970–3979
Quetglas S, Leveque C, Miquelis R, Sato K, Seagar M (2000) Ca2+-dependent regulation of synaptic SNARE complex assembly via a calmodulin- and phospholipid-binding domain of synaptobrevin. Proc Natl Acad Sci U S A 97:9695–9700
Rettig J, Neher E (2002) Emerging roles of presynaptic proteins in Ca++-triggered exocytosis. Science 298:781–785
Rickman C, Hu K, Carroll J, Davletov B (2005) Self-assembly of SNARE fusion proteins into star-shaped oligomers. Biochem J 388:75–79
Risselada HJ, Kutzner C, Grubmuller H (2011) Caught in the act: visualization of SNARE-mediated fusion events in molecular detail. Chembiochem : Eur J Chem Biol 12:1049–1055
Rizo J, Rosenmund C (2008) Synaptic vesicle fusion. Nat Struct Mol Biol 15:665–674
Robinson IM, Finnegan JM, Monck JR, Wightman RM, Fernandez JM (1995) Colocalization of calcium entry and exocytotic release sites in adrenal chromaffin cells. Proc Natl Acad Sci U S A 92:2474–2478
Rohde J, Dietrich L, Langosch D, Ungermann C (2003) The transmembrane domain of Vam3 affects the composition of cis- and trans-SNARE complexes to promote homotypic vacuole fusion. J Biol Chem 278:1656–1662
Roth D, Burgoyne RD (1994) SNAP-25 is present in a SNARE complex in adrenal chromaffin cells. FEBS Lett 351:207–210
Schaub JR, Lu X, Doneske B, Shin YK, McNew JA (2006) Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat Struct Mol Biol 13:748–750
Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294:1117–1122
Shi L, Shen QT, Kiel A, Wang J, Wang HW, Melia TJ, Rothman JE, Pincet F (2012) SNARE proteins: one to fuse and three to keep the nascent fusion pore open. Science 335:1355–1359
Siddiqui TJ, Vites O, Stein A, Heintzmann R, Jahn R, Fasshauer D (2007) Determinants of synaptobrevin regulation in membranes. Mol Biol Cell 18:2037–2046
Sinha R, Ahmed S, Jahn R, Klingauf J (2011) Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc Natl Acad Sci U S A 108:14318–14323
Smirnova YG, Marrink SJ, Lipowsky R, Knecht V (2010) Solvent-exposed tails as prestalk transition states for membrane fusion at low hydration. J Am Chem Soc 132:6710–6718
Smith CB, Betz WJ (1996) Simultaneous independent measurement of endocytosis and exocytosis. Nature 380:531–534
Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75:409–418
Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362:318–324
Sorensen JB, Matti U, Wei SH, Nehring RB, Voets T, Ashery U, Binz T, Neher E, Rettig J (2002) The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc Natl Acad Sci U S A 99:1627–1632
Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003) Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114:75–86
Sorensen JB, Wiederhold K, Muller EM, Milosevic I, Nagy G, de Groot BL, Grubmuller H, Fasshauer D (2006) Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J 25:955–966
Spruce AE, Breckenridge LJ, Lee AK, Almers W (1990) Properties of the fusion pore that forms during exocytosis of a mast cell secretory vesicle. Neuron 4:643–654
Stein A, Weber G, Wahl MC, Jahn R (2009) Helical extension of the neuronal SNARE complex into the membrane. Nature 460:525–528
Steyer JA, Almers W (1999) Tracking single secretory granules in live chromaffin cells by evanescent-field fluorescence microscopy. Biophys J 76:2262–2271
Steyer JA, Horstmann H, Almers W (1997) Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature 388:474–478
Sudhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–477
Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Sudhof TC (2007) A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450:676–682
Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395:347–353
Tagaya M, Toyonaga S, Takahashi M, Yamamoto A, Fujiwara T, Akagawa K, Moriyama Y, Mizushima S (1995) Syntaxin 1 (HPC-1) is associated with chromaffin granules. J Biol Chem 270:15930–15933
Tong J, Borbat PP, Freed JH, Shin YK (2009) A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc Natl Acad Sci U S A 106:5141–5146
Ungermann C, Langosch D (2005) Functions of SNAREs in intracellular membrane fusion and lipid bilayer mixing. J Cell Sci 118:3819–3828
van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R (2010) One SNARE complex is sufficient for membrane fusion. Nat Struct Mol Biol 17:358–364
van den Bogaart G, Thutupalli S, Risselada JH, Meyenberg K, Holt M, Riedel D, Diederichsen U, Herminghaus S, Grubmuller H, Jahn R (2011) Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nat Struct Mol Biol 18:805–812
Vites O, Florin EL, Jahn R (2008) Docking of liposomes to planar surfaces mediated by trans-SNARE complexes. Biophys J 95:1295–1302
Voets T (2000) Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron 28:537–545
Voets T, Moser T, Lund PE, Chow RH, Geppert M, Sudhof TC, Neher E (2001) Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci U S A 98:11680–11685
Walter AM, Wiederhold K, Bruns D, Fasshauer D, Sorensen JB (2010) Synaptobrevin N-terminally bound to syntaxin-SNAP-25 defines the primed vesicle state in regulated exocytosis. J Cell Biol 188:401–413
Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5:19–26
Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92:759–772
Weimbs T, Low SH, Chapin SJ, Mostov KE, Bucher P, Hofmann K (1997) A conserved domain is present in different families of vesicular fusion proteins: a new superfamily. Proc Natl Acad Sci U S A 94:3046–3051
Weis WI, Scheller RH (1998) Membrane fusion. SNARE the rod, coil the complex. Nature 395:328–329
Wiederhold K, Fasshauer D (2009) Is assembly of the SNARE complex enough to fuel membrane fusion? J Biol Chem 284:13143–13152
Wierda KD, Sorensen JB (2014) Innervation by a GABAergic neuron depresses spontaneous release in glutamatergic neurons and unveils the clamping phenotype of synaptotagmin-1. J Neurosci: Off J Soc Neurosci 34:2100–2110
Wightman RM, Jankowski JA, Kennedy RT, Kawagoe KT, Schroeder TJ, Leszczyszyn DJ, Near JA, Diliberto EJ Jr, Viveros OH (1991) Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc Natl Acad Sci U S A 88:10754–10758
Wojcik SM, Brose N (2007) Regulation of membrane fusion in synaptic excitation-secretion coupling: speed and accuracy matter. Neuron 55:11–24
Xu J, Pang ZP, Shin OH, Sudhof TC (2009) Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci 12:759–766
Xu T, Rammner B, Margittai M, Artalejo AR, Neher E, Jahn R (1999) Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell 99:713–722
Xu Y, Zhang F, Su Z, McNew JA, Shin YK (2005) Hemifusion in SNARE-mediated membrane fusion. Nat Struct Mol Biol 12:417–422
Yang B, Gonzalez L Jr, Prekeris R, Steegmaier M, Advani RJ, Scheller RH (1999) SNARE interactions are not selective. Implications for membrane fusion specificity. J Biol Chem 274:5649–5653
Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Sudhof TC (2010) Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory alpha helix. Neuron 68:907–920
Yoon TY, Okumus B, Zhang F, Shin YK, Ha T (2006) Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci U S A 103:19731–19736
Zhang Z, Jackson MB (2010) Membrane bending energy and fusion pore kinetics in Ca(2+)-triggered exocytosis. Biophys J 98:2524–2534
Zhao WD, Hamid E, Shin W, Wen PJ, Krystofiak ES, Villarreal SA, Chiang HC, Kachar B, Wu LG (2016) Hemi-fused structure mediates and controls fusion and fission in live cells. Nature 534:548–552
Zhou P, Bacaj T, Yang X, Pang ZP, Sudhof TC (2013) Lipid-anchored SNAREs lacking transmembrane regions fully support membrane fusion during neurotransmitter release. Neuron 80:470–483
Zorman S, Rebane AA, Ma L, Yang G, Molski MA, Coleman J, Pincet F, Rothman JE, Zhang Y (2014) Common intermediates and kinetics, but different energetics, in the assembly of SNARE proteins. elife 3:e03348
Acknowledgments
The work was supported by grants from the DFG (SFB894 and SFB1027) to DB and RM, and HOMFOR (MD).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the special issue on Chromaffin Cells in Pflügers Archiv – European Journal of Physiology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dhara, M., Mohrmann, R. & Bruns, D. v-SNARE function in chromaffin cells. Pflugers Arch - Eur J Physiol 470, 169–180 (2018). https://doi.org/10.1007/s00424-017-2066-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-017-2066-z