
Abstract
The clinically established proarrhythmic effect of bradycardia and antiarrhythmic effect of lidocaine (10 μM) were reproduced in hypokalaemic (3.0 mM K+) Langendorff-perfused murine hearts paced over a range (80–180 ms) of baseline cycle lengths (BCLs). Action potential durations (at 90% repolarization, APD90s), transmural conduction times and ventricular effective refractory periods (VERPs) were then determined from monophasic action potential records obtained during a programmed electrical stimulation procedure in which extrasystolic stimuli were interposed following regular stimuli at successively decreasing coupling intervals. A novel graphical analysis of epicardial and endocardial, local and transmural relationships between APD90, corrected for transmural conduction time where appropriate, and VERP yielded predictions in precise agreement with the arrhythmogenic findings obtained over the entire range of BCLs studied. Thus, in normokalaemic (5.2 mM K+) hearts a statistical analysis confirmed that all four relationships were described by straight lines of gradients not significantly (P > 0.05) different from unity that passed through the origin and thus subtended constant critical angles, θ with the abscissa (45.8° ± 0.9°, 46.6° ± 0.5°, 47.6° ± 0.5° and 44.9° ± 0.8°, respectively). Hypokalaemia shifted all points to the left of these reference lines, significantly (P < 0.05) increasing θ at BCLs of 80–120 ms where arrhythmic activity was not observed (∼63°, ∼54°, ∼55° and ∼58°, respectively) and further significantly (P < 0.05) increasing θ at BCLs of 140–180 ms where arrhythmic activity was observed (∼68°, ∼60°, ∼61° and ∼65°, respectively). In contrast, the antiarrhythmic effect of lidocaine treatment was accompanied by a significant (P < 0.05) disruption of this linear relationship and decreases in θ in both normokalaemic (∼40°, ∼33°, ∼39° and ∼41°, respectively) and hypokalaemic (∼40°, ∼44°, ∼50° and ∼48°, respectively) hearts. This extended a previous approach that had correlated alterations in transmural repolarization gradients with arrhythmogenicity in murine models of the congenital long QT syndrome type 3 and hypokalaemia at a single BCL. Thus, the analysis in terms of APD90 and VERP provided a more sensitive indication of the effect of lidocaine than one only considering transmural repolarization gradients and may be particularly applicable in physiological and pharmacological situations in which these parameters diverge.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A dependence of arrhythmogenicity upon heart rate, frequently quantified in terms of baseline cycle length (BCL), is well established in a range of clinical and experimental situations. Thus, in hereditary arrhythmic syndromes including the congenital long QT syndrome type 3 (LQT3) [43, 55], the Brugada syndrome [1] and catecholaminergic polymorphic ventricular tachycardia [33, 49] bradycardia is associated with the initiation of arrhythmic activity, while rapid pacing is antiarrhythmic. In LQT3 this relationship is further associated with the exacerbation of an already existent QT prolongation [30], reflecting increased action potential duration [50]. Such clinical observations have been replicated in animal models of LQT3, which have additionally demonstrated abnormal differences between epicardial and endocardial action potential durations [6, 12, 51].
This relationship between arrhythmogenicity, BCL and QT interval has also been observed in situations of acquired electrocardiographic QT prolongation resulting from treatment with drugs including antiarrhythmics [9, 36, 56, 57] and macrolide antibiotics [26, 28], cardiac hypertrophy [50] and metabolic abnormalities including hypokalaemia [10, 45]. Such phenomena have frequently been associated with alterations in repolarizing K+ currents influencing the time course of action potential recovery [17, 54, 58]. In such situations arrhythmic activity is suppressed by the commonly used class 1b antiarrhythmic agent lidocaine [10, 45].
The present study begins by demonstrating an increase in the incidence of arrhythmic activity as BCL is increased over a physiological range for the first time in hypokalaemic hearts of any species, directly reflecting clinical observations [10, 45]. This is also the first time that such a finding has been reported in murine hearts under any conditions. It then uses an analysis of arrhythmogenicity involving local and transmural relationships between the recovery of membrane voltage and of excitability following depolarization developed in a previous paper [37] as a platform for the investigation of this phenomenon. This represents the first exploration of the effect of BCL on such relationships in any experimental system, whether murine or otherwise.
Furthermore, this study introduces novel graphical plots representing local and transmural relationships between action potential duration (quantified at 90% repolarization, APD90) and ventricular effective refractory period (VERP) able to display data obtained over a range of BCLs. These are used to demonstrate for the first time that in the normokalaemic case APD90 and VERP, and local and transmural relationships between them, alter with BCL in a manner that prevents arrhythmogenesis. In sharp contrast this relationship is shown to break down in the hypokalaemic case, with APD90 exceeding VERP at long BCLs, in association with arrhythmogenesis. Furthermore, treatment with lidocaine is demonstrated to result in the breakdown of this relationship with VERP exceeding APD90, in association with an anti-arrhythmic effect. Finally, this analysis is compared with a generalization of a previous analysis that related arrhythmogenicity to changes in relationships between the recovery of membrane voltage in the epicardium and in the endocardium [17, 18, 27, 51, 52]. This analysis is extended from a single BCLs to a range of BCLs and displayed as a novel plot.
Materials and methods
Mice were housed at 21 ± 1°C with 12 h light/dark cycles and were fed sterile chow (RM3 Maintenance Diet, SDS, Witham, Essex, UK) with free access to water. Wild-type 129 Sv mice aged 3–6 months were used in procedures, all of which complied with UK Home Office regulations (Animals (Scientific Procedures) Act 1986). All solutions were based on bicarbonate-buffered Krebs–Henseleit solution (mM: NaCl 119, NaHCO3 25, KCl 4, KH2PO4 1.2, MgCl2 1, CaCl2 1.8, glucose 10 and Na-pyruvate 2; pH adjusted to 7.4) bubbled with 95% O2/5% CO2 (British Oxygen Company, Manchester, U.K). Hypokalaemic (3.0 mM K+) solutions were prepared by reduction of KCl content. Lidocaine-containing normokalaemic and hypokalaemic solutions were prepared by adding lidocaine (Sigma-Aldrich, Poole, UK) to a final concentration of 10 μM.
A Langendorff perfusion protocol previously adapted for murine hearts [2] was used. In brief, mice were killed by cervical dislocation (Schedule 1: UK Animals [Scientific Procedures] Act 1986). Hearts were then quickly excised and placed in ice-cold bicarbonate-buffered Krebs–Henseleit solution. A short section of aorta was cannulated under the surface of the solution and attached to a custom-made 21-gauge cannula filled with the same solution using an aneurysm clip (Harvard Apparatus, Edenbridge, Kent, UK). Fresh Krebs–Henseleit solution was then passed through 200- and 5-μm filters (Millipore, Watford, UK), warmed to 37°C using a water jacket and circulator (Techne model C-85A, Cambridge, UK) and used for constant-flow retrograde perfusion at 2–2.5 ml/min via a peristaltic pump (Watson–Marlow Bredel model 505S, Falmouth, Cornwall, UK). Hearts were regarded as suitable for experimentation if, on rewarming, they regained a healthy pink colour and began to contract spontaneously.
Epicardial monophasic action potential (MAP) electrodes (Hugo Sachs, Harvard Apparatus, UK) were placed against the basal left ventricular epicardium. In addition, small windows were created in the septum between the left and right ventricles to allow access to the left ventricular endocardium [4]. Custom-made endocardial MAP electrodes comprising two twisted strands of high-purity Teflon-coated 0.25 mm diameter silver wire (Advent Research Materials, UK) were constructed. The Telflon coats were removed from the distal 1 mm of the electrodes, which were then galvanically chlorided to eliminate DC offset, inserted and placed against the septal endocardial surface. MAPs were then amplified, band-pass filtered (0.5 Hz to 1 kHz: Gould 2400S, Gould-Nicolet Technologies, Ilford, Essex, UK) and digitised at a sampling frequency of 5 kHz (micro1401, Cambridge Electronic Design, Cambridge, UK). Analysis of MAPs was performed using Spike II software (Cambridge Electronic Design, Cambridge, UK).
A bipolar platinum stimulating electrode (1 mm inter-pole spacing) was placed against the basal right ventricular epicardium. Square-wave stimuli (Grass S48 stimulator, Grass-Telefactor, Slough, UK) of 2-ms duration and with amplitudes of twice the diastolic threshold were initially applied to hearts at a constant baseline cycle length of 125 ms for at least 10 min and until MAPs showed stable baselines, rapid upstroke phases that reached consistent amplitudes and smooth repolarization phases [11, 19]. Hearts were then exposed to test solutions for 20 min, during which time stimulation was continued, before subsequent recordings were made.
Action potential duration (at 90% repolarization, APD90) was quantified during regular stimulation at baseline cycle lengths (BCLs) varied in 5-ms increments between 80 and 180 ms. Hearts were subjected to an adapted form of an extrasystolic stimulation procedure previously used to assess arrhythmogenicity and refractoriness in both human [42] and murine [15] studies of long QT syndrome, described in detail later. This procedure was used to assess arrhythmogenicity and determine ventricular effective refractory periods (VERPs) at BCLs varied in 10-ms increments between 80 and 180 ms. The possibility that events evoked by extrasystolic stimuli rather represented motion artefacts was excluded by their being reproducible between hearts and appearing identical in both electrodes.
All data are presented as means ± standard errors of the means (SEM) and include both the number of repetitions and the number of hearts. Comparisons were made using analysis of variance other than where stated. A significance threshold of P ≤ 0.05 was applied in all cases.
Results
The initial experiments quantitatively assessed the effect of steady-state baseline cycle length (BCL) on the inducibility of arrhythmic activity in hearts exposed to normokalaemic (5.2 mM K+), hypokalaemic (3.0 mM K+), normokalaemic lidocaine-containing (10 μM) and hypokalaemic lidocaine-containing (10 μM) test solutions. Hearts were first exposed to the test solutions for 20 min, while stimulated at a BCL of 125 ms to allow a steady state to be reached. Arrhythmogenicity was then assessed during 1-h periods of regular stimulation at BCLs of 80 and 180 ms. In addition, arrhythmogenicity was studied using an extrasystolic stimulation procedure previously established in human long QT syndrome (LQTS) [42] and adapted for use in murine models [15]. This involved a standardised stimulation protocol consisting of regular (S1) stimulation at BCLs of 80 and 180 ms, interrupted by extrasystolic (S2) stimuli imposed following every eighth S1 stimulus. The S1S2 interval was decremented in 1-ms steps with each successive stimulus cycle from the BCL. This was continued until the S2 stimulus either appeared to initiate arrhythmic activity, confirmed by the persistence of irregular waveforms on cessation of stimulation, or failed to initiate a MAP, suggesting that the ventricular effective refractory period (VERP) had been reached.
Arrhythmic activity occurs in hypokalaemic hearts paced at long but not short baseline cycle lengths
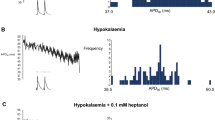
While studies under normokalaemic conditions confirmed an absence of arrhythmic activity at either BCL (0 out of five hearts subjected to regular stimulation, 0 out of seven hearts subjected to S2 stimulation), studies under hypokalaemic conditions (Fig. 1) revealed arrhythmogenicity at the longest (A, five out of five hearts subjected to regular stimulation, seven out of seven hearts subjected to S2 stimulation) but not the shortest (B, 0 out of five hearts subjected to regular stimulation, one out of nine hearts subjected to S2 stimulation) BCL studied. These results correlated with clinical observations associating hypokalaemia with arrhythmogenicity.

Arrhythmic activity in hypokalemic hearts. Epicardial monophasic action potential recordings obtained during regular stimulation (left) and following the application of extrasystolic (S2) stimuli at S1S2 intervals just greater than the ventricular effective refractory period (right) in hypokalemic (3.0 mM K+) hearts paced at baseline cycle lengths of 180 ms (a) and 80 ms (b). Single vertical lines indicate the timing of S1 stimuli and double lines indicate the timing of S2 stimuli
Incidence of arrhythmic activity in hypokalaemic hearts decreases with decreasing baseline cycle length
Arrhythmogenicity was then studied over a range of BCLs using the S2 stimulation protocol. Table 1 demonstrates a graded dependence of the incidence of arrhythmic activity upon BCL through the entire range of BCLs studied. The experiments then went on to confirm the clinically established antiarrhythmic effect of lidocaine under these experimental conditions [10, 34, 45] over all BCLs. Thus, S2 stimulation failed to initiate arrhythmic activity at any BCL in either normokalaemic or hypokalaemic hearts exposed to lidocaine (seven and six hearts at each BCL, respectively).
Action potential duration and refractory period vary with baseline cycle length
The subsequent experiments made systematic measurements of action potential shapes, as represented in their durations at 90% repolarization (APD90s), and VERPs through the entire range of BCLs, thereby extending previous experiments restricted to a single BCL [37]. Figure 2 displays the resulting findings, showing APD90s (open symbols) and VERPs (filled symbols) obtained from both the epicardium (circles) and the endocardium (squares).

Effect of baseline cycle length on action potential duration and ventricular effective refractory period. Relationship between epicardial (circles) and endocardial (squares) action potential duration (at 90% repolarization, APD90, open symbols), ventricular effective refractory period (VERP, filled symbols) and cycle length in hearts exposed to normokalaemic (5.2 mM K+; a, b) and hypokalaemic (3.0 mM K+; c, d) test solutions, and normokalaemic (e, f) and hypokalaemic (g, h) test solutions containing lidocaine (10 μM)
Thus Fig. 2a and b show that in normokalaemic hearts both epicardial and endocardial APD90 varied with BCL, decreasing from 51.2 ± 1.7 and 60.6 ± 6.0 ms, respectively, at a BCL of 180 ms to 31.7 ± 2.0 and 31.7 ± 1.8 ms, respectively, at a BCL of 80 ms (n = 12; seven hearts). VERP showed a similar trend, decreasing from 53 ± 6 and 57 ± 1 ms, respectively at a BCL of 180 ms to 32 ± 2 and 26 ± 1 ms, respectively, at a BCL of 80 ms (n = 9; seven hearts).
In hypokalaemic hearts, both epicardial and endocardial APD90 again varied with BCL, decreasing from 71.4 ± 6.2 and 60.1 ± 6.3 ms, respectively, at a BCL of 180 ms to 37.3 ± 3.5 and 36.0 ± 3.5 ms, respectively, at a BCL of 80 ms (n = 11; seven hearts; Fig. 2c,d). Compared to normokalaemic hearts, VERP was significantly decreased at all BCLs (P < 0.01). However, VERP did not vary significantly (P > 0.05) with BCL (n = 8; seven hearts).
In normokalaemic hearts treated with lidocaine both epicardial and endocardial APD90 continued to vary with BCL, decreasing from 59.5 ± 4.2 and 56.1 ± 8.1 ms, respectively, at a BCL of 180 ms to 32.4 ± 2.2 and 28.7 ± 2.4 ms, respectively, at a BCL of 80 ms (n = 12; seven hearts; Fig. 2e,f). As compared to normokalaemic hearts VERP was significantly increased at all BCLs (P < 0.01). However, VERP did not vary significantly (P > 0.05) with BCL (n = 12; seven hearts).
In hypokalaemic hearts treated with lidocaine both epicardial and endocardial APD90 again varied with BCL, decreasing from 59.2 ± 3.4 and 87.7 ± 2.6 ms, respectively, at a BCL of 180 ms to 32.2 ± 2.3 and 29.6 ± 2.1 ms, respectively, at a BCL of 80 ms (n = 9; six hearts; Fig. 2g,h). Compared to normokalaemic hearts, VERP was significantly increased at all BCLs (P < 0.05). VERP also decreased with decreasing BCL from 65 ± 4 and 62 ± 8 ms, respectively, at a BCL of 180 ms to 45 ± 2 and 42 ± 5 ms, respectively, at a BCL of 80 ms (n = 8; seven hearts).
Graphical analysis of arrhythmogenic tendency
The above data permitted an extension of a previous analysis of arrhythmogenicity obtained in murine hearts studied under similar conditions of hypokalaemia and exposure to lidocaine but then confined to a single BCL [37]. That earlier study successfully analysed arrhythmic propensity in terms of possible reexcitation dependent on the relative time courses of the recoveries of membrane voltage and of excitability from total refractoriness to a finite threshold for excitation following action potential depolarization. This could involve either local myocardial regions or, as suggested for congenital long QT syndrome type 3, transmural interactions through the thickness of the myocardial wall [25, 27, 44, 51].
Figures 3 and 4 thus represent equivalent parameters of epicardial (circles) and endocardial (squares) APD90 plotted against VERP in a graphical form for all BCLs studied for all four test solutions (a–h, respectively). These provide possible criteria for local (Fig. 3) and transmural (Fig. 4) reexcitation as in the previous study. In Fig. 4, APD90s are corrected for the time taken for depolarization to propagate from endocardium to epicardium, given by the difference in the latencies between stimulation and endocardial depolarization and between stimulation and epicardial depolarization (Δlatency). Thus, Figs. 3a,b and 4a,b demonstrate that normokalaemic hearts showed similar local and transmural relationships whether obtained from the epicardium or endocardium.

Local relationships between epicardial and endocardial action potential duration and ventricular effective refractory period obtained over a range of baseline cycle lengths. Local relationships between epicardial (circles) and endocardial (squares) action potential duration (at 90% repolarization, APD90) and ventricular effective refractory period (VERP) in hearts exposed to normokalaemic (5.2 mM K+; a, b) and hypokalaemic (3.0 mM K+; c, d) test solutions, and normokalaemic (e, f) and hypokalaemic (g, h) test solutions containing lidocaine (10 μM) and stimulated at baseline cycle lengths of between 80 ms and 180 ms. Filled symbols indicate conditions resulting in arrhythmic activity in >50% of hearts

Transmural relationships between action potential duration and ventricular effective refractory period allowing for transmural conduction time obtained over a range of baseline cycle lengths. Transmural relationships between epicardial (circles) and endocardial (squares) action potential duration (at 90% repolarization, APD90) plus transmural conduction time (Δlatency) and ventricular effective refractory period (VERP) in hearts exposed to normokalaemic (5.2 mM K+; a, b) and hypokalaemic (3.0 mM K+; c, d) test solutions, and normokalaemic (e, f) and hypokalaemic (g, h) test solutions containing lidocaine (10 μM) and stimulated at baseline cycle lengths of between 80 ms and 180 ms. Filled symbols indicate conditions resulting in arrhythmic activity in >50% of hearts
All these above relationships were linear. Thus, fits to the linear function y = mx + c, where m denotes the constant gradient and c the intercept with the ordinate, gave r2 values of 0.98, 0.99, 0.99 and 0.98, respectively (P << 0.01 in all cases) and corresponding reduced χ2 values of 8.13, 5.14, 5.22 and 1.15, respectively. However, the values of c were statistically indistinguishable from zero (P > 0.05) in all cases (4.79 ± 4.31, 4.54 ± 1.32, 2.85 ± 1.32 and 9.87 ± 3.56, respectively) and exclusion of this constant term did not result in a significantly worse fit. Thus, repeating a regression analysis this time fitting the line y = mx gave indistinguishable regression values (with P<<0.01) and gave a second set of reduced χ2 values (10.2, 6.3, 6.5 and 1.3, respectively). This permitted the derivation of an F statistic for the appropriate, N + 1 = 6 data points given by \( F = N{\left( {\frac{{\chi ^{2}_{1} - \chi ^{2}_{2} }}{{\chi ^{2}_{2} }}} \right)} \) in which the subscripts on the right hand side of the equation denote the number of parameters involved [3]. This yielded P values of > 0.25 in all cases, suggesting that the two fits were statistically indistinguishable and justifying adoption of the simpler function.
Taken together, statistical analysis of these data therefore confirmed a linear relationship uniquely involving the parameters of APD90 (corrected if appropriate) and VERP, generalising the analysis presented by Sabir et al. [37] for a single BCL to all BCLs. Furthermore, the resulting values of m were matched to and indistinguishable from each other and from unity in all, epicardial and endocardial, local and transmural, cases (P > 0.05). This permitted description of the relationship between APD90 and VERP in terms of m, or in terms of a critical angle, θ obtained by fitting the line \( y = x \cdot \tan \theta \) to the data points. Thus, in the latter analysis in normokalaemic (5.2 mM K+) hearts θ took statistically indistinguishable (P > 0.05) values of 45.8 ± 0.9°, 46.6 ± 0.5°, 47.6 ± 0.5° and 44.9 ± 0.8° in the four cases, respectively.
The proarrhythmic effect of hypokalemia corresponds to increases in the critical angle, θ, while the antiarrhythmic effect of lidocaine-exposure corresponds to decreases in θ
Data points obtained through each experimental condition could then be compared with these normokalaemic reference lines. Hypokalaemic situations where arrhythmogenesis was observed corresponded to points falling to the left of these lines (Figs. 3 and 4). In contrast where arrhythmogenesis was prevented by the addition of lidocaine points fell to the right of the reference lines. These geometrical relationships were quantified using a simple approach that obtained the angle subtended by each data point and the abscissa at the origin. This provided a graphical expression of a simple quotient:
correcting for Δlatency where appropriate. This value was then compared with the corresponding angle shown by the respective reference lines. This approach additionally reflected the requirement that any function relating the necessarily positive values of APD90 to VERP would traverse the origin: a zero APD90 i.e. no action potential would correspond to a zero VERP. Thus, values of θ greater or less than that of the reference lines would suggest pro- and antiarrhythmic situations, respectively. Furthermore, the extent to which θ remains constant as BCL varies would provide an indication of the extent to which the relationship between APD90 and VERP is maintained under any given experimental condition. The analysis that follows explores the extent to which such an analysis provides results that correlate with observed arrhythmogenicity.
Figures 3c,d and 4c,d show that exposure of hearts to hypokalaemic test solution indeed resulted in significant increases in local and transmural θ values (P > 0.05) in relation to the reference values at all BCLs studied. All hypokalaemic (3.0 mM) hearts gave points that lay to the left of the reference line, corresponding to θ values of greater than 45°. Those that did not demonstrate arrhythmic activity (during pacing at BCLs between 80 and 120 ms, open symbols) gave points that lay closer to the reference line than those that were arrhythmogenic (paced at BCLs of between 120 and 180 ms, filled symbols). In the first case, the mean local θ values were ∼63° in epicardium (Fig. 3c) and ∼54° in the endocardium (Fig. 3d), while the corresponding transmural values were ∼55° (Fig. 4c) and ∼58° (Fig. 4d), respectively. In the second case the corresponding values were ∼68° (Fig. 3c), ∼60° (Fig. 3d), ∼61° (Fig. 4c) and ∼65° (Fig. 4d).
Exposure of normokalaemic hearts to lidocaine significantly reduced (P < 0.05) mean local and transmural θ values in relation to the reference values at all BCLs studied. Thus, mean local θ values were ∼40° in the epicardium (Fig. 3e) and ∼33° in the endocardium (Fig. 3f), while the corresponding transmural values were ∼39° (Fig. 4e) and ∼41° (Fig. 4f), respectively. Exposure of hypokalaemic hearts to lidocaine again significantly reduced (P < 0.05) mean local and transmural θ values in relation to the reference values at all BCLs, accompanying its antiarrhythmic effect as described above. Thus, local mean local θ values were ∼40° in the epicardium (Fig. 3g) and ∼44° in the endocardium (Fig. 3h), while corresponding transmural values were ∼50° (Fig. 4g) and ∼48° (Fig. 4h), respectively.
Hypokalaemia alters the relationship between epicardial and endocardial action potential durations
These findings were then compared with the results of an extension of a previous analysis [17, 52], which involved the comparison of endocardial and epicardial action potential durations. This had proved successful in the assessment of arrhythmogenicity in a murine model of the congenital long QT syndrome type 3 (LQT3) [52], a situation in which APD90 and VERP might be expected always to vary concordantly, and in a murine model of hypokalemia [17] at a single BCL. The present study extends use of this analysis from a single BCL to a range of BCLs, plotting endocardial APD90 on the abscissa and epicardial APD90 on the ordinate over the range of BCLs under the four above conditions (Fig. 5a–d). Filled symbols indicate conditions under which arrhythmic activity was observed in more than 50% of hearts.

Relationships between epicardial and endocardial action potential durations (at 90% repolarization, APD90s) obtained over a range of baseline cycle lengths. Hearts exposed to normokalaemic (5.2 mM K+; a) and hypokalaemic (3.0 mM K+; b) test solutions, and normokalaemic (c) and hypokalaemic (d) test solutions containing lidocaine (10 μM) and stimulated at baseline cycle lengths of between 80 ms and 180 ms. Filled symbols indicate conditions resulting in arrhythmic activity in >50% of hearts
In contrast to the previous analysis, the normokalaemic plot (Fig. 5a) did not give a linear relationship between endocardial and epicardial APD90s, but rather was fitted by a quadratic function. Thus, the curve \( y = ax^{2} + bx + c \), where a = −10.7 ± 12.6, b = 1.8 ± 0.5 and c = −0.01 ± 0.0 gave a reduced χ 2 of 3.3, while fitting the line y = mx + c gave a reduced χ 2 of 4.23, permitting calculation of an F statistic for the appropriate, N + 1 = 20 data points. This gave a probability of exceeding F of <0.05, indicating that inclusion of the quadratic term was statistically justified.
Hypokalaemia and lidocaine exposure resulted in loss of the proportional relationship between (corrected) APD90 and VERP in all cases (Fig. 5b–d). While the departure of points to the left of the reference curve under hypokalaemic conditions were less marked than in the previous analysis, it was nonetheless demonstrable (Fig. 5b). Exposure of both normokalaemic (Fig. 5c) and hypokalaemic (Fig. 5d) hearts to lidocaine gave points that were shifted towards more positive endocardial APD90 values than seen in Fig. 5b. Thus, in hypokalaemic hearts lidocaine displaced points to the right, towards the reference curve (Fig. 5d), while in normokalaemic hearts lidocaine displaced points to the left, away from the reference curve (Fig. 5c). These observations may be attributable to the disproportionate effect of lidocaine on VERP as opposed to APD90.
Taken together these findings demonstrate that the analysis of arrhythmogenicity in terms of the relative time courses of recovery of membrane voltage and recovery of excitability following depolarization presented by Sabir et al. [37] for hypokalaemic conditions can be extended from a single BCL to a range of BCLs. While a previous analysis of arrhythmogenicity in terms of the relationship between endocardial and epicardial action potential durations proposed for LQT3 [52] and hypokalemia [17] successfully predicted arrhythmogenicity, analyses incorporating VERP proved more sensitive.
Discussion
The proarrhythmic effects of bradycardia and contrasting antiarrhythmic effects of rapid pacing have been described in a number of clinical situations [1, 33, 49]. These associations are especially marked in the presence both of congenital [43, 55] and of acquired (review in [28, 50]) electrocardiographic QT prolongation, of which clinical hypokalaemia constitutes a common example. In the latter situation, arrhythmogenesis is suppressed by treatment with the class 1b antiarrhythmic agent lidocaine [10, 45]. The present study sought to investigate the basis for these phenomena using the intact murine heart as an experimental model for arrhythmogenicity, as established on previous occasions [2, 11, 12, 15, 17, 37, 47, 51, 52].
The murine heart is well established as a model for human cardiac disease and importantly allows scope for the introduction of genetic modifications [24]. Hence, it has previously yielded important findings concerning mechanisms underlying arrhythmogenicity in conditions including the congenital long QT syndromes ([11, 12; see review in [39, 47, 51, 52]), the Brugada syndrome [32, 46], catecholaminergic polymorphic ventricular tachycardia [5, 23], cardiomyopathy [21] and hypokalemia [17, 37, 38]. Structurally, the sinoatrial node, atrioventricular node and His-Purkinje system are similar in murine hearts and in the hearts of larger mammals including humans [35], while there are notable differences in size and in basal rate [53]. Nonetheless, murine and human ventricular action potentials share rapid depolarization phases attributable to Na+ currents [14], making the murine heart useful for studying the effects of Na+ channel blocking drugs such as lidocaine. While differences in kinetics of the main currents responsible for the repolarization phase (the transient outward current, I to, in mice and the delayed rectifier and slowly inactivating delayed rectifier currents, I Kr and I Ks, in humans) result in the absence of a plateau phase and a shorter ventricular action potential duration in mice [8]; all such currents are K+-sensitive [17, 31, 58], making the murine heart suitable for the study of clinical hypokalaemia. Furthermore, murine and human hearts exhibit similar spatial gradients in action potential duration [19], similar relationships between action potential duration and refractory period [11, 20] and near identical transmural conduction velocities [16, 22], making murine hearts at the very least suitable for the investigation of fundamental mechanisms underlying arrhythmogenicity.
Thus, arrhythmogenicity was studied in hearts subjected to a regular stimulation protocol and to an extrasystolic stimulation protocol, previously used to assess arrhythmogenicity and refractoriness in both human [42] and murine [15] studies of the congenital long QT syndromes, at a range of baseline cycle lengths (BCLs). This represents the first experimental exploration of the effect of BCL on arrhythmogenicity under hypokalaemic conditions in any species and of the effect of BCL on arrhythmogenicity in the murine heart under any conditions. Under normokalaemic conditions, hearts gave monophasic action potentials (MAPs) that consistently demonstrated stable waveforms and rhythms during both protocols. We report for the first time that hypokalaemic hearts demonstrated periods of both transient and sustained arrhythmic activity when these protocols were applied at long, but not short, BCLs. Furthermore, arrhythmic activity was never observed during treatment with lidocaine under either condition during either protocol. Taken together, these findings thus parallel the clinical observations.
The experiments accordingly extended the analysis of arrhythmic propensity established by Sabir et al. [37] from single BCL to a range of BCLs. As in that previous study, the analysis that followed explored the tendency towards reexcitation and arrhythmogenesis in terms of relative changes in the time courses of the recovery of membrane voltage and the recovery of excitability after depolarization. It went on to describe propensity to reexcitation of both the epicardium and the endocardium either arising locally or from a transmural source using novel graphical representations. These plotted action potential durations (at 90% repolarization, APD90s) obtained through the range of BCLs explored against the corresponding ventricular effective refractory periods (VERPs). In transmural plots, APD90s were corrected for the time taken for depolarization to propagate from endocardium to epicardium, given by the difference in the latencies between stimulation and endocardial depolarization and between stimulation and epicardial depolarization (Δlatency).
A statistical analysis suggested that the relationships between APD90 and VERP varied along linear functions in normokalaemic hearts. Furthermore, the functions consistently passed through the origin, excluding any necessity for additional constant terms, and did so with gradients of near unity. Together these findings extend previous results from murine [19], canine [13], and human [20] studies. The various relationships between APD90 and VERP could then be described by critical angles, θ unique to each case for all BCLs. We demonstrate for the first time that hypokalaemia disrupted these simple linear relationships and additionally significantly increased all values of θ at all BCLs. Furthermore, the largest values of θ were observed in hearts paced at the longest BCLs (140–180 ms), which were in turn associated with the highest incidences of arrhythmic activity. Treatment of both normokalaemic and hypokalaemic hearts with lidocaine resulted in significantly lower θ values than were obtained in the absence of the drug, again at all BCLs. This paralleled its antiarrhythmic effect in hypokalaemic hearts. However, under normokalaemic conditions, lidocaine abolished the directly proportional relationship between APD90 and VERP and could thereby exert a rate-limiting effect in vivo at short BCLs.
In summary, using an experimental model we successfully replicated the clinical effects of BCL on arrhythmogenicity and in particular the association between bradycardia and arrhythmogenesis [1, 10, 28, 33, 43, 45, 49, 50, 55]. This led to the development of criteria predictive of such arrhythmogenicity that compared local and transmural, epicardial and endocardial relationships between APD90 and VERP. Such an approach agrees with and extends previous criteria for arrhythmogenicity invoking transmural repolarization gradients expressed through the relationship between epicardial and endocardial APD90 values [17, 51]. Such criteria have been shown to predict arrhythmogenicity in murine models of the congenital long QT syndrome type 3 (LQT3) [51] and of hypokalemia [17, 38]. However, these studies were restricted to a single BCL. Furthermore, LQT3 results from a Na+ channel mutation and therefore produces proportionate prolongations of APD90 and VERP [51]. Hence, in LQT3 the transmural relationships between APD90 and VERP would be expected to be in direct agreement with the transmural differences in action potential repolarization time. This contrasts with situations illustrated in the present paper where APD90 and VERP sharply diverge, particularly in the presence of lidocaine.
Thus, the analyses presented in this study represent a generalization of previous criteria, which is applicable over a range of BCL and in situations where changes in APD90 and VERP occur independently. Thus, it would predict that action potential prolongation should prove especially pro-arrhythmic where APD90 is disproportionately prolonged as compared to VERP. Such an interpretation would be applicable in explaining the contrasting pro- or anti-arrhythmic effects of various agents sharing apparently similar effects on action potential waveforms. Thus, the proarrhythmic effects of the anti-psychotic agents haloperidol [41] and sulpiride [48] and of the antibiotic agent sparfloxacin [40] in canine models, which demonstrate action potential characteristics almost identical to those in humans, are associated with disproportionate increases action potential duration as compared to VERP. Conversely, the anti-arrhythmic effects of various class 1 agents including lidocaine are associated with disproportionate increases in VERP compared to action potential duration [7, 29], as reflected in this study. It would then directly follow that procedures used to predict the proarrhythmic potential of QT prolonging drugs and arrhythmic propensity in patients exhibiting QT prolongation should incorporate assessment of VERP and should be conducted at a range of heart rates.
References
Antzelevitch C, Brugada P, Brugada J, Brugada R, Towbin JA, Nademanee K (2003) Brugada syndrome: 1992–2002: a historical perspective. J Am Coll Cardiol 41:1665–1671
Balasubramaniam R, Grace AA, Saumarez RC, Vandenberg JI, Huang CL (2003) Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J Physiol 552:535–546
Bevington PR (1969) Data reduction and error analysis for the physical sciences. McGraw-Hill, New York
Casimiro MC, Knollmann BC, Ebert SN, Vary JC Jr, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K (2001) Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc Natl Acad Sci USA 98:2526–2531
Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG (2005) Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res 96:e77–e82
Chinushi M, Kasai H, Tagawa M, Washizuka T, Hosaka Y, Chinushi Y, Aizawa Y (2002) Triggers of ventricular tachyarrhythmias and therapeutic effects of nicorandil in canine models of LQT2 and LQT3 syndromes. J Am Coll Cardiol 40:555–562
Costard-Jaeckle A, Liem LB, Franz MR (1989) Frequency-dependent effect of quinidine, mexiletine, and their combination on postrepolarization refractoriness in vivo. J Cardiovasc Pharmacol 14:810–817
Danik S, Cabo C, Chiello C, Kang S, Wit AL, Coromilas J (2002) Correlation of repolarization of ventricular monophasic action potential with ECG in the murine heart. Am J Physiol Heart Circ Physiol 283:H372–H381
Davy JM, Weissenburger J, Ertzbischoff O, Lainee P, Chezalviel F, Poirier JM, Cheymol G, Motte G (1988) Sotalol-induced torsades de pointe in the conscious dog with atrioventricular block. Role of hypokalemia. Arch Mal Coeur Vaiss 81:1117–1124
el-Sherif N, Zeiler RH, Craelius W, Gough WB, Henkin R (1988) QTU prolongation and polymorphic ventricular tachyarrhythmias due to bradycardia-dependent early afterdepolarizations. Afterdepolarizations and ventricular arrhythmias. Circ Res 63:286–305
Fabritz L, Kirchhof P, Franz MR, Eckardt L, Monnig G, Milberg P, Breithardt G, Haverkamp W (2003) Prolonged action potential durations, increased dispersion of repolarization, and polymorphic ventricular tachycardia in a mouse model of proarrhythmia. Basic Res Cardiol 98:25–32
Fabritz L, Kirchhof P, Franz MR, Nuyens D, Rossenbacker T, Ottenhof A, Haverkamp W, Breithardt G, Carmeliet E, Carmeliet P (2003) Effect of pacing and mexiletine on dispersion of repolarisation and arrhythmias in ΔKPQ SCN5A (long QT3) mice. Cardiovasc Res 57:1085–1093
Franz MR, Chin MC, Sharkey HR, Griffin JC, Scheinman MM (1990) A new single catheter technique for simultaneous measurement of action potential duration and refractory period in vivo. J Am Coll Cardiol 16:878–886
Guo W, Xu H, London B, Nerbonne JM (1999) Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J Physiol 521:587–599
Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, Grace AA, Huang CL (2005) Paced electrogram fractionation analysis of arrhythmogenic tendency in ΔKPQ SCN5A mice. J Cardiovasc Electrophysiol 16:1329–1340
Higuchi T, Nakaya Y (1984) T wave polarity related to the repolarization process of epicardial and endocardial ventricular surfaces. Am Heart J 108:290–295
Killeen M, Thomas G, Gurung I, Goddard C, Fraser J, Mahaut-Smith M, Colledge H, Grace A, Huang C (2006) Arrhythmogenic mechanisms in the isolated perfused hypokalemic murine heart. Acta Physiologica 189:33–46
Kirchhof P, Degen H, Franz MR, Eckardt L, Fabritz L, Milberg P, Laer S, Neumann J, Breithardt G, Haverkamp W (2003) Amiodarone-induced postrepolarization refractoriness suppresses induction of ventricular fibrillation. J Pharmacol Exp Ther 305:257–263
Knollmann BC, Katchman AN, Franz MR (2001) Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol 12:1286–1294
Koller BS, Karasik PE, Solomon AJ, Franz MR (1995) Relation between repolarization and refractoriness during programmed electrical stimulation in the human right ventricle. Implications for ventricular tachycardia induction. Circulation 91:2378–2384
Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, Demetris AJ, Feldman AM (1997) Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-α. Circ Res 81:627–635
Liu G, Iden JB, Kovithavongs K, Gulamhusein R, Duff HJ, Kavanagh KM (2004) In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J Physiol 555:267–279
Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG (2006) Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res 99:292–298
London B (2001) Cardiac arrhythmias: from (transgenic) mice to men. J Cardiovasc Electrophysiol 12:1089–1091
Lubinski A, Lewicka-Nowak E, Kempa M, Baczynska AM, Romanowska I, Swiatecka G (1998) New insight into repolarization abnormalities in patients with congenital long QT syndrome: the increased transmural dispersion of repolarization. Pacing Clin Electrophysiol 21:172–175
Milberg P, Eckardt L, Bruns HJ, Biertz J, Ramtin S, Reinsch N, Fleischer D, Kirchhof P, Fabritz L, Breithardt G, Haverkamp W (2002) Divergent proarrhythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J Pharmacol Exp Ther 303:218–225
Milberg P, Reinsch N, Wasmer K, Monnig G, Stypmann J, Osada N, Breithardt G, Haverkamp W, Eckardt L (2005) Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc Res 65:397–404
Morissette P, Hreiche R, Turgeon J (2005) Drug-induced long QT syndrome and torsade de pointes. Can J Cardiol 21:857–864
Nakaya Y, Nii H, Nomura M, Fujino K, Mori H (1989) Effects of lidocaine and quinidine on post-repolarization refractoriness after the basic and premature action potentials: consideration of aim of antiarrhythmic drug therapy. Am Heart J 118:907–912
Nemec J, Buncova M, Bulkova V, Hejlik J, Winter B, Shen WK, Ackerman MJ (2004) Heart rate dependence of the QT interval duration: differences among congenital long QT syndrome subtypes. J Cardiovasc Electrophysiol 15:550–556
Nerbonne JM (2000) Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J Physiol 525:285–298
Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA (2002) Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA 99:6210–6215
Postma AV, Denjoy I, Kamblock J, Alders M, Lupoglazoff JM, Vaksmann G, Dubosq-Bidot L, Sebillon P, Mannens MM, Guicheney P, Wilde AA (2005) Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet 42:863–870
Rehnqvist N, Ericsson CG, Eriksson S, Olsson G, Svensson G (1984) Comparative investigation of the antiarrhythmic effect of propafenone (Rytmonorm) and lidocaine in patients with ventricular arrhythmias during acute myocardial infarction. Acta Med Scand 216:525–530
Rentschler S, Vaidya DM, Tamaddon H, Degenhardt K, Sassoon D, Morley GE, Jalife J, Fishman GI (2001) Visualization and functional characterization of the developing murine cardiac conduction system. Development 128:1785–1792
Roden DM, Hoffman BF (1985) Action potential prolongation and induction of abnormal automaticity by low quinidine concentrations in canine Purkinje fibers. Relationship to potassium and cycle length. Circ Res 56:857–867
Sabir IN, Fraser JA, Killeen MJ, Grace AA, Huang C (2007) The contribution of refractoriness to arrhythmic substrate in hypokalemic Langendorff-perfused murine hearts. Pflugers Arch PMID: 17295037 (Electronic publication ahead of print)
Sabir IN, Killeen MJ, Goddard CA, Thomas G, Gray S, Grace AA, Huang CL (2007) Transient alterations in transmural repolarization gradients following premature ventricular depolarizations in arrhythmogenic hypokalemic Langendorff-perfused murine hearts. J Physiol PMID:17331992 (Electronic publication ahead of print)
Salama G, London B (2007) Mouse models of long QT syndrome. J Physiol 578:43–53
Satoh Y, Sugiyama A, Chiba K, Tamura K, Hashimoto K (2000) QT-prolonging effects of sparfloxacin, a fluoroquinolone antibiotic, assessed in the in vivo canine model with monophasic action potential monitoring. J Cardiovasc Pharmacol 36:510–515
Satoh Y, Sugiyama A, Tamura K, Hashimoto K (2000) Effect of magnesium sulfate on the haloperidol-induced QT prolongation assessed in the canine in vivo model under the monitoring of monophasic action potential. Jpn Circ J 64:445–451
Saumarez RC, Pytkowski M, Sterlinski M, Hauer RN, Derksen R, Lowe MD, Szwed H, Huang CL, Ward DE, Camm AJ, Grace AA (2006) Delayed paced ventricular activation in the long QT syndrome is associated with ventricular fibrillation. Heart Rhythm 3:771–778
Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R (2001) Genotype–phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103:89–95
Shimizu W, Antzelevitch C (1997) Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation 96:2038–2047
Shimizu W, Tanaka K, Suenaga K, Wakamoto A (1991) Bradycardia-dependent early afterdepolarizations in a patient with QTU prolongation and torsade de pointes in association with marked bradycardia and hypokalemia. Pacing Clin Electrophysiol 14:1105–1111
Stokoe KS, Balasubramaniam R, Goddard CA, Colledge WH, Grace AA, Huang CL (2007) Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/− murine hearts. J Physiol PMID:17303635 (Electronic publication ahead of print
Stokoe KS, Thomas G, Goddard CA, Colledge WH, Grace AA, Huang CL (2007) Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/Δ murine hearts modelling long QT syndrome 3. J Physiol 578:69–84
Sugiyama A, Satoh Y, Shiina H, Takeda S, Hashimoto K (2002) Torsadegenic action of the antipsychotic drug sulpiride assessed using in vivo canine models. J Cardiovasc Pharmacol 40:235–245
Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, Saito A, Kurosaki K, Jouo K, Koujiro M, Konishi S, Matsuoka S, Oono T, Hayakawa S, Miura M, Ushinohama H, Shibata T, Niimura I (2003) Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 89:66–70
Swynghedauw B, Baillard C, Milliez P (2003) The long QT interval is not only inherited but is also linked to cardiac hypertrophy. J Mol Med 81:336–345
Thomas G, Gurung IS, Killeen MJ, Hakim P, Goddard CA, Mahaut-Smith MP, Colledge WH (2006) Effects of L-type Ca2+ channel antagonism on ventricular arrhythmogenesis in ΔKPQ Scn5a (long QT 3) murine hearts. J Physiol 578:85–97
Thomas G, Killeen MJ, Gurung IS, Hakim P, Balasubramaniam RN, Goddard CA, Grace AA, Huang CL (2006) Mechanisms of ventricular arrhythmogenesis in mice following targeted disruption of KCNE1. J Physiol 578:97–114
Vaidya D, Morley GE, Samie FH, Jalife J (1999) Reentry and fibrillation in the mouse heart. A challenge to the critical mass hypothesis. Circ Res 85:174–181
Vandenberg CA (1987) Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc Natl Acad Sci USA 84:2560–2564
Viskin S (2000) Cardiac pacing in the long QT syndrome: review of available data and practical recommendations. J Cardiovasc Electrophysiol 11:593–600
Voigt L, Coromilas J, Saul BI, Kassotis J (2003) Amiodarone-induced torsade de pointes during bladder irrigation: an unusual presentation—a case report. Angiology 54:229–231
Vos MA, Verduyn SC, Gorgels AP, Lipcsei GC, Wellens HJ (1995) Reproducible induction of early afterdepolarizations and torsade de pointes arrhythmias by d-sotalol and pacing in dogs with chronic atrioventricular block. Circulation 91:864–872
Yang T, Roden DM (1996) Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation 93:407–411
Acknowledgements
We thank the James Baird Fund, the Frank Elmore Fund, the Medical Research Council, the Engineering and Physical Sciences Research Council, the Wellcome Trust and the British Heart Foundation for their generous support. JAF holds a Research Fellowship at Gonville & Caius College, Cambridge.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sabir, I.N., Fraser, J.A., Cass, T.R. et al. A quantitative analysis of the effect of cycle length on arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch - Eur J Physiol 454, 925–936 (2007). https://doi.org/10.1007/s00424-007-0255-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-007-0255-x