
Abstract
Background
Mutations in the RPGR gene predominantly cause rod photoreceptor disorders with a large variability in clinical course. In this report, we describe two families with mutations in this gene and cone involvement.
Methods
We investigated an X-linked cone dystrophy family (1) with 25 affected males, 25 female carriers, and 21 non-carriers, as well as a small family (2) with one affected and one unaffected male. The RPGR gene was analyzed by direct sequencing. All medical records were evaluated, and all available data on visual acuity, color vision testing, ophthalmoscopy, fundus photography, fundus autofluorescence, Goldmann perimetry, SD-OCT, dark adaptation, and full-field electroretinography (ERG) were registered. Cumulative risks of visual loss were studied with Kaplan–Meier product-limit survival analysis.
Results
Both families had a frameshift mutation in ORF15 of the RPGR gene; family 1 had p.Ser1107ValfsX4, and family 2 had p.His1100GlnfsX10. Mean follow up was 13 years (SD 10). Virtually all affected males showed reduced photopic and normal scotopic responses on ERG. Fifty percent of the patients had a visual acuity of <0.5 at age 35 years (SE 2.2), and 75% of the patients was legally blind at age 60 years (SE 2.3). Female carriers showed no signs of ocular involvement.
Conclusions
This report describes the clinical course and visual prognosis in two families with cone dystrophy due to RPGR mutations in the 3’ terminal region of ORF15. Remarkable features were the consistent, late-onset phenotype, the severe visual outcome, and the non-expression in female carriers. Expression of RPGR mutations in this particular region appears to be relatively homogeneous and predisposed to cones.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cone dystrophy (CD) is a progressive disease in which the cone photoreceptors are primarily affected. Patients experience increasing photophobia, and develop loss of visual acuity, color vision disturbances, and central visual field defects. An important clinical hallmark for diagnosis is reduction of cone responses with preservation of rod responses on full-field electroretinogram (ERG) [1, 2]. All three inheritance patterns occur in CD; 15% is autosomal dominant, 80% autosomal recessive, and 5% X-linked. Revelation of the genetic background and genotype-phenotype correlation in CD is currently ongoing, improving clinical counseling and prediction of visual decline.
A common cause of X-linked retinitis pigmentosa (RP) are mutations in the retinitis pigmentosa GTPase regulator gene (RPGR; Xp21.1) [3–8] The gene has also been reported in X-linked cone-rod dystrophy (CRD) [9–13] or even in pure CD [3, 14, 15]. Although the clinical course of patients with RPGR mutations and RP has been evaluated [16], how the disease progresses in CD patients is currently unknown.
In this report, we present two families with CD due to mutations at the 3’ terminal end of ORF15. We carefully studied the course of disease in 26 affected males and 25 female carriers during a long period of follow-up, and established a model for visual outcome that can be used for clinical counseling.
Patients and methods
Study population
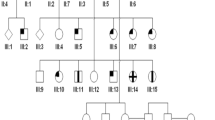
We studied a pedigree of 71 relatives (25 affected males; 25 female carriers; 21 non-carriers) of one Dutch family (family 1), as well as one affected male and one unaffected sibling of the second family (family 2) (Fig. 1). All affected males of family 1 had been diagnosed with CD. We were not able to contact five patients and 11 female carriers. All other patients were ascertained at three medical centers in the Netherlands: the Netherlands Institute for Neuroscience (NIN), Erasmus Medical Center Rotterdam, and the St. Oosterschelde Hospital in Goes. The study was approved by the Medical Ethics Committee of Erasmus Medical Center and adhered to the tenets of the Declaration of Helsinki. All participants provided signed, informed consent for participation in the study, retrieval of medical records, and use of blood and DNA for research.

a Pedigree of family 1 with X-linked cone dystrophy (CD) showing segregation of the c.3317dup (p.Ser1107ValfsX4) mutation in ORF15 of the RPGR gene. b Pedigree of family 2 with CD showing segregation of the c.3300_3301del (p.His1100GlnfsX10) mutation in ORF15 of the RPGR gene. Open square and circle, unaffected males and females; black square, affected males; open circle with a dot, an obligate female carrier; dashed symbols denote deceased individuals
Clinical examination
Medical charts were retrieved from the patients’ ophthalmologists, and all available data on Snellen visual acuity, color vision (Hardy-Rand-Rittler color-vision test, Ishihara pseudoisochromatic plates, or Farnsworth Panel D15/D28 test), Goldmann perimetry, slit-lamp and fundus examination, and ERGs were recorded. Patients with incomplete or no available data from the previous 5 years were invited for a work-up examination at one of the clinics. All patients underwent a full-field ERG with contact lens electrodes, following the recommendations of the International Society for Clinical Electrophysiology of Vision (ISCEV) [17]. Subsequently, we performed a Goldmann Weeker’s dark adaptometry (DA), spectral domain-ocular coherence tomography (SD-OCT), fundus photography, and fundus autofluorescence (FAF) in a subset of carriers and patients.
Molecular genetic analysis
Blood samples were obtained from all participating family members of family 1 (n = 87), and family 2 (n = 3). DNA was isolated from peripheral blood lymphocytes by standard procedures. Molecular analyses were carried out as described [18]. In short, the coding region and intron/exon boundaries of the RPGR ORF15 region were amplified by polymerase chain reaction (PCR) using M13 tail sequences at the 5’ ends of the target-specific primers. Sanger sequencing reactions were performed using these M13 tail sequences. Primer sequences and reaction conditions are available upon request.
Statistical analysis
Proportions of categorical variables between two groups were compared using the Chi-square test. Cumulative risks of visual loss were studied with Kaplan–Meier product-limit survival analysis, with low vision (visual acuity < 0.5) or legal blindness (visual acuity ≤0.1) as outcomes. Participants older than 60 were pooled to maintain unbiased estimates.
Results
Mutation analysis
Pedigrees of both families are provided in Fig. 1. In 25 affected males and in 25 female carriers of family 1, the 1-bp insertion c.3317dup (p.Ser1107ValfsX4) in RPGR ORF15 (NM_001034853.1) was detected as the causative mutation (Fig. 2a). This earlier described frameshift mutation [11] is located at the C-terminus of the ORF15 protein and results in premature truncation of the protein. Full co-segregation of the mutation and the disease was observed in the pedigree; no mutation was detected in 10 unaffected males and 11 unaffected females.

a Sequence chromatograms of the RPGR ORF15 gene for family 1 with the c.3317dup (p.Ser1107ValfsX4) mutation. The top chromatogram shows a hemizygous mutated allele (duplication of an adenine nucleotide in the A base stretch); the middle chromatogram a heterozygous mutated sequence in a female carrier; the bottom chromatogram shows the normal allele. b Sequence chromatograms of the RPGR ORF15 gene for family 2 with the c.3300_3301del (p.His1100GlnfsX10) mutation. The top chromatogram shows the hemizygous deleted allele; the middle chromatogram shows the heterozygous mutated allele in a female carrier; the bottom chromatogram the normal allele. The displayed chromatograms in a and b are reverse sequences causing the typical disturbed sequence patterns in such a heterozygous carrier chromatogram to the left
In the proband of family 2, a novel 2-bp deletion c.3300_3301del (p.His1100GlnfsX10) in RPGR ORF15 was identified (Fig. 2b). This mutation also resulted in a frameshift mutation, leading to a premature truncation of the protein at amino acid 1110 of the C-terminus. This mutation was also present in his unaffected daughter, and was not identified in his unaffected brother.
Clinical findings and risk of visual decline
For family 1, the clinical findings of all affected males (n = 25), female carriers (n = 25), and non-carriers (n = 21) are summarized in Table 1. Data on multiple visits were available with a mean follow-up of 13 years (SD 10). The mean age at onset was 36.2 (SD 3.1) years, and all patients showed progression of the disease. Hemeralopia was a first symptom in 8/25 (32%), preceding visual loss. Nystagmus was not observed in any patient. High myopia, refractive error of −6 D or worse, was common (19/25, 76%) in male patients, significantly more common than in female carriers (2/25, 8%; p< 0.001). High myopia was not present in non-carriers. All three color axes were severely disturbed in all patients, except in one. This patient was relatively young (21 years), had normal visual acuity, only mild color vision disturbances, and a normal macula.
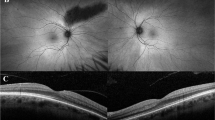
Macular appearance deteriorated progressively in most patients, and ranged from no abnormalities to a Bull’s eye maculopathy or atrophy (Fig. 3). Goldmann perimetry was abnormal in all patients, and showed a reduced central sensitivity (n = 3) or a (relative) central scotoma (n = 22). ERG demonstrated reduced or absent photopic responses with normal scotopic responses in 24/25 patients. In these patients, the latter remained normal during follow-up. In the remaining patient (age 72 years), the ERG did show slightly reduced rod responses, however, this occurred only after 22 years of disease duration. All carriers (and non-carriers) had a normal visual acuity, normal macular appearance, and normal ERG. Figure 4 illustrates the clinical findings of patient V-7 and carrier VI-4 of family 1, and shows a fundus photograph, FAF, SD-OCT, ERG, and DA-curve.

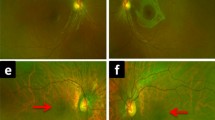
a Composite fundus photograph of the left eye of patient V-4, of family 1 with a c.3317dup (p.Ser1107ValfsX4) mutation in the RPGR ORF15 gene, performed at age 27 years. No macular abnormalities are apparent yet. b Composite fundus photograph of the left eye of patient IV-19 of family 1 with the c.3317dup (p.Ser1107ValfsX4) mutation in RPGR ORF15, performed at age 71 years. The retina shows retinal pigment epithelium atrophy in the macular region and peripapillary atrophy. The patient has high myopia

Fundus photographs, fundus autofluorescence images (FAF), spectral domain-OCTs (SD-OCT), full-field electroretinograms (ERG), and Goldman-Weekers dark adaptation curves (DA) of the left eye of patient V-7, performed at age 50, and carrier VI-6, performed at age 24, both of family 1. The ERGs were performed according to the standard International Society for Clinical Electrophysiology of Vision (ISCEV) protocol. Best-corrected visual acuity of the patient and carrier was 0.10 and 1.0, respectively. a Fundus photograph of patient V-7, showing peripapillary atrophy and pigment clumping with atrophic lesions in the macula. The patient has high myopia (S-9.0 D). The arrow denotes the position of the SD-OCT. The OCT signifies thinning of the fovea (158 μm), pigment clumping, and atrophy of the RPE. The FAF shows a high-density autofluorescent peri-macular ring. The ERG of this patient is shown at the left with traces of an age-related control person at right for purposes of comparison. Replications of the responses are shown as thin traces, the average as solid. Note the significantly reduced cone-specific responses with preserved rod responses. b Fundus photograph of carrier VI-4, showing a normal optic nerve and macula. The arrow denotes the position of the SD-OCT image. The OCT denotes a slightly reduced foveal thickness (232 μm), but an otherwise intact photoreceptor and RPE cell layer. FAF shows no abnormalities. The ERG is shown at the left with normal control traces at right. Replications of the responses are shown as thin traces, the average as solid. No abnormalities were present. c The DA-curves of patient V-7 (diamonds), carrier VI-4 (squares) and a normal control person (grey dots) with time (min) on the X-axis and threshold (dB) on the Y-axis. The cone plateau for the patient is elevated by ~1 log-unit. The S2 phase, i.e., the straight lines in the initial rod adaptation phase, has the same slope as the normal control although the level is 1 log-unit apart. The latter can be explained by the difficulty of the patient to observe the fixation mark due to his low vision. The threshold for the ERG response in the extended ISCEV ERG protocol was not elevated. The DA for the carrier did not differ from normal
Figure 5 shows the cumulative risk of visual decline as a function of age in all affected males of family 1. Low vision was present in 50% of patients at age 35 years (SE 2.2); and in 75% of patients at 40 years (SE 1.1). Legal blindness was present in 50% at 47 years (SE 3.4), and in 75% at 60 years (SE 2.3).

a Kaplan–Meier survival curves showing the cumulative incidence of low vision (visual acuity <0.50) as a function of age. b Kaplan–Meier survival curves showing the cumulative incidence of legal blindness (visual acuity ≤0.10) as a function of age
With respect to family 2, the proband developed color vision disturbances at age 16 years, and a progressively deteriorating visual acuity since age 43 years to counting fingers at age 75 years. He had high myopia, severely disturbed color vision in all three axes, and atrophic lesions in the macula. Goldmann perimetry showed a central scotoma with a normal periphery, and ERG revealed absent photopic responses with normal rod responses. His brother had a normal visual acuity with no color vision disturbances and a normal macular appearance.
Discussion
In this report, we describe two families with RPGR mutations in the 3’ terminal end of ORF15 and cone pathology. The onset in these families was relatively late, i.e., third and fourth decades, and visual acuity declined to legal blindness within two decades thereafter. The onset and clinical course were rather homogeneous in all affected individuals. It was remarkable that rod function remained unaffected in almost all patients. Only in one patient with a long duration of disease, rod responses were slightly diminished. This coincided with a normal peripheral visual field and no nyctalopia, indicating that the rod decay in this person was not clinically significant.
The RPGR gene is known to express different isoforms caused by alternative splicing [3]. Two of these protein isoforms are localized in the connecting cilium of cone and rod photoreceptors. The first is encoded by the most commonly expressed transcript, RPGR-ORF15, containing exons 1 through ORF15 (a large exon consisting of exon 15 extending into a part of intron 15). The other, RPGR-ex1-19, contains exons 1 through 19 but lacks exons 14 and 15 [3]. Exons 1–14 contain six RCC1-like domains; their main function is to facilitate interaction with other proteins [19–22]. Mutations within these domains lead to reduced or abolished protein interactions [19]. The ORF15 region encodes for a repetitive glycine and glutamic acid rich protein domain of unknown function [3, 4]. It has been demonstrated in the mouse that reduction of this repetitive region can preserve a normal protein function [21]. Most reported mutations in the first 14 exons are nonsense or frameshift mutations which can lead to nonsense-mediated decay of the mRNA (NMD), and low or absent levels of the transcript. In contrast, nonsense or frameshift mutations in ORF15 are less likely to lead to NMD since this is the last exon of the transcript [23], and a series of truncated proteins of varying length can be found [7].
Until now, 300 pathogenic mutations have been discovered in the RPGR gene. Most lead to RP (n = 287), only seven mutations have been associated with CRD (Fig. 6) [24]. Systemic involvement like respiratory tract infections or hearing loss has been described in only a few patients [25]. One paper described atrophic macular degeneration [26] and three earlier publications reported a phenotype of pure CD [3, 14, 15]. More than half (164/287; 57%) of the mutations leading to RP occur in the ORF15 region; and all mutations that cause CRD and CD have been found at this site. No mutations have been detected in exon 16–19 (Fig. 6).

Localization of all known mutations in the RPGR ORF15 isoform (n = 300) stratified per phenotype and per exon. *: Retinitis pigmentosa; n = 287 (large asterisk symbolizes 50 different mutations). +: Cone-rod dystrophy; n = 7. $: Cone dystrophy; n = 4. #: Retinitis pigmentosa with systemic ciliary dysfunction; n = 2
Literature on the clinical course in CD patients with mutations in RPGR is lacking. In this report, we describe the onset and course of visual acuity in 25 patients of a family with a known duplication (c.3317dup), and in a proband with a newly identified deletion (c.3300_3301del) in ORF15. Both are frameshift mutations located in the terminal 3’ region of this exon, causing premature termination of the protein. Both mutations resulted in a loss of only ~40-50 amino acids at the C-terminus. We propose that this could explain the late onset of the disease with legal blindness occurring after a mean age of 60 years. In comparison, the mean age of legal blindness for mutations in the first 14 exons and in the 5’ region of ORF15 was 45 years in RP patients [16].
Why do some ORF15 mutations affect rods, and others cause predominantly cone involvement? [27] Several speculations can be made. Mutant mice with different mutations exhibit mis-localization of rod and/or cone (rhod)opsins. Which opsin is mis-localized most depends on the type of mutation [28]. In humans, there is also a genotype-phenotype relation: certain mutations predispose to rod cell loss and others affect mostly cones. The RPGR protein is expressed in the basal body of the connecting cilium of both rod and cone photoreceptors. The connecting cilium serves as a conduit for bidirectional transport of protein complexes, and is important for the outer segment disc morphogenesis and renewal [29]. For execution of its transport function, the RPGR protein interacts with other ciliary proteins like RPGRIP1, CEP290, NPHP5, and NPHP6 [30]. These protein interactions are known to differ among rods and cones, and certain mutations may have more effect on cone interactions than those of rods and vice versa.
The duplication of family 1 (c.3317dup) had been reported earlier by Demirci et al., who observed this mutation in an individual with CRD [11]. In our family, the rods were spared in virtually all patients. Variation in phenotype in patients with identical mutations may be due to post-translational effects by modifying genes or environmental factors [31]. However, the homogeneity of the phenotype in family 1 signifies that these effects are probably small.
The female carriers in both our families did not have any ocular involvement. In many reports of RP families with ORF15 mutations, carriers have mild or even severe ophthalmologic abnormalities, ranging from pigmentary changes in the periphery to severe visual loss [5, 32, 33]. Mutations causing a widespread photoreceptor decay may be more difficult to compensate for the non-mutated X-allele than mutations causing a more restricted dysfunction of the photoreceptors [5].
A large proportion of the CD patients in our families had high myopia of more than 6 D (17/25;73%). Photoreceptor disorders commonly have high myopia, independent of genetic etiology or inheritance mode [34–36]. The reasons for this phenomenon remain unclear. One explanation may be that scleral growth is stimulated by signals from dysfunctional photoreceptors; another is that high myopia and primary photoreceptor disorders are part of the same syndrome.
In conclusion, this is a comprehensive clinical report of two families with pure cone involvement due to RPGR ORF15 mutations. Our survival analyses can be useful to counsel such patients. This report adds to the growing evidence that RPGR, an established gene for rod cell loss, can be restricted to cone cell loss.
References
Michaelides M, Hardcastle AJ, Hunt DM, Moore AT (2006) Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv Ophthalmol 51:232–258
Michaelides M, Hunt DM, Moore AT (2004) The cone dysfunction syndromes. Br J Ophthalmol 88:291–297
Vervoort R, Lennon A, Bird AC, Tulloch B, Axton R, Miano MG, Meindl A, Meitinger T, Ciccodicola A, Wright AF (2000) Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet 25:462–466
Vervoort R, Wright AF (2002) Mutations of RPGR in X-linked retinitis pigmentosa (RP3). Hum Mutat 19:486–500
Neidhardt J, Glaus E, Lorenz B, Netzer C, Li Y, Schambeck M, Wittmer M, Feil S, Kirschner-Schwabe R, Rosenberg T, Cremers FP, Bergen AA, Barthelmes D, Baraki H, Schmid F, Tanner G, Fleischhauer J, Orth U, Becker C, Wegscheider E, Nürnberg G, Nürnberg P, Bolz HJ, Gal A, Berger W (2008) Identification of novel mutations in X-linked retinitis pigmentosa families and implications for diagnostic testing. Mol Vis 14:1081–1093
Bader I, Brandau O, Achatz H, Apfelstedt-Sylla E, Hergersberg M, Lorenz B, Wissinger B, Wittwer B, Rudolph G, Meindl A, Meitinger T (2003) X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Invest Ophthalmol Vis Sci 44:1458–1463
Shu X, Black GC, Rice JM, Hart-Holden N, Jones A, O’Grady A, Ramsden S, Wright AF (2007) RPGR mutation analysis and disease: an update. Hum Mutat 28:322–328
Jin ZB, Gu F, Ma X, Nao-i N (2007) Identification of a novel RPGR exon ORF15 mutation in a family with X-linked retinitis pigmentosa. Arch Ophthalmol 125:1407–1412
Bergen AAB, Pinckers AJLG (1997) Localization of a novel X-linked progressive cone dystrophy gene to Xq27: evidence for genetic heterogeneity. Am J Hum Genet 60:1468–1473
Mears AJ, Hiriyanna S, Vervoort R, Yashar B, Gieser L, Fahrner S, Daiger SP, Heckenlively JR, Sieving PA, Wright AF, Swaroop A (2000) Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am J Hum Genet 67:1000–1003
Demirci FY, Rigatti BW, Wen G, Radak AL, Mah TS, Baic CL, Traboulsi EI, Alitalo T, Ramser J, Gorin MB (2002) X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet 70:1049–1053
Jalkanen R, Demirci FY, Tyynismaa H, Bech-Hansen T, Meindl A, Peippo M, Mäntyjärvi M, Gorin MB, Alitalo T (2003) A new genetic locus for X linked progressive cone-rod dystrophy. J Med Genet 40:418–423
Ebenezer ND, Michaelides M, Jenkins SA, Audo I, Webster AR, Cheetham ME, Stockman A, Maher ER, Ainsworth JR, Yates JR, Bradshaw K, Holder GE, Moore AT, Hardcastle AJ (2005) Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci 46:1891–1898
Yang Z, Peachey NS, Moshfeghi DM, Thirumalaichary S, Chorich L, Shugart YY, Fan K, Zhang K (2002) Mutations in the RPGR gene cause X-linked cone dystrophy. Hum Mol Genet 11:605–611
Robson AG, Michaelides M, Luong VA, Holder GE, Bird AC, Webster AR, Moore AT, Fitzke FW (2008) Functional correlates of fundus autofluorescence abnormalities in patients with RPGR or RIMS1 mutations causing cone or cone rod dystrophy. Br J Ophthalmol 92:95–102
Sandberg MA, Rosner B, Weigel-DiFranco C, Dryja TP, Berson EL (2007) Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Invest Ophthalmol Vis Sci 48:1298–1304
Marmor MF (1989) An international standard for electroretinography. Doc Ophthalmol 73:299–302
Booij JC, Bakker A, Kulumbetova J, Moutaoukil Y, Smeets B, Verheij J, Kroes HY, Klaver CC, van Schooneveld M, Bergen AA, Florijn RJ (2011) Simultaneous mutation detection in 90 retinal disease genes in multiple patients using a custom-designed 300-kb retinal resequencing chip. Ophthalmology 118:160–167
Roepman R, van Duijnhoven G, Rosenberg T, Pinckers AJ, Bleeker-Wagemakers LM, Bergen AA, Post J, Beck A, Reinhardt R, Ropers HH, Cremers FP, Berger W (1996) Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum Mol Genet 5:1035–1041
Roepman R, Bernoud-Hubac N, Schick DE, Maugeri A, Berger W, Ropers HH, Cremers FP, Ferreira PA (2000) The retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of rod photoreceptors. Hum Mol Genet 9:2095–2105
Hong DH, Pawlyk BS, Adamian M, Li T (2004) Dominant, gain-of-function mutant produced by truncation of RPGR. Invest Ophthalmol Vis Sci 45:36–41
He S, Parapuram SK, Hurd TW, Behnam B, Margolis B, Swaroop A, Khanna H (2008) Retinitis Pigmentosa GTPase Regulator (RPGR) protein isoforms in mammalian retina: insights into X-linked retinitis pigmentosa and associated ciliopathies. Vision Res 48:366–376
Nagy E, Maquat LE (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 23:198–199
Fokkema IFAC, Den Dunnen JT, Taschner PEM (2005) LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-Box” approach. Hum Mutat 26:63–68
Zito I, Downes SM, Patel RJ, Cheetham ME, Ebenezer ND, Jenkins SA, Bhattacharya SS, Webster AR, Holder GE, Bird AC, Bamiou DE, Hardcastle AJ (2003) RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J Med Genet 40:609–615
Ayyagari R, Demirci FY, Liu J, Bingham EL, Stringham H, Kakuk LE, Boehnke M, Gorin MB, Richards JE, Sieving PA (2002) X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics 80:166–171
Demirci FY, Gupta N, Radak AL, Rigatti BW, Mah TS, Milam AH, Gorin MB (2005) Histopathologic study of X-linked cone-rod dystrophy (CORDX1) caused by a mutation in the RPGR exon ORF15. Am J Ophthalmol 139:386–388
Hong DH, Pawlyk BS, Shang J, Sandberg MA, Berson EL, Li T (2000) A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc Natl Acad Sci USA 97:3649–3654
Zhao Y, Hong DH, Pawlyk B, Yue G, Adamian M, Grynberg M, Godzik A, Li T (2003) The retinitis pigmentosa GTPase regulator (RPGR)-interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci USA 100:3965–3970
Roepman R, Wolfrum U (2007) Protein networks and complexes in photoreceptor cilia. Subcell Biochem 43:209–235
Walia S, Fishman GA, Swaroop A, Branham KE, Lindeman M, Othman M, Weleber RG (2008) Discordant phenotypes in fraternal twins having an identical mutation in exon ORF15 of the RPGR gene. Arch Ophthalmol 126:379–384
Adamian M, Pawlyk BS, Hong DH, Berson EL (2006) Rod and cone opsin mislocalization in an autopsy eye from a carrier of X-linked retinitis pigmentosa with a Gly436Asp mutation in the RPGR gene. Am J Ophthalmol 142:515–518
Amzallag T, Puech B, Hache JC, François P (1990) Progressive cone dystrophy: electrophysiological changes in female carriers. J Fr Ophtalmol 13:421–428
Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL (2003) RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet 73:1131–1146
Pras E, Abu A, Rotenstreich Y, Avni I, Reish O, Morad Y, Reznik-Wolf H, Pras E (2009) Cone-rod dystrophy and a frameshift mutation in the PROM1 gene. Mol Vis 15:1709–1716
Smith M, Whittock N, Searle A, Croft M, Brewer C, Cole M (2007) Phenotype of autosomal dominant cone-rod dystrophy due to the R838C mutation of the GUCY2D gene encoding retinal guanylate cyclase-1. Eye (Lond) 21:1220–1225
Acknowledgements
This study was supported by Prof. Dr. Henkes Stichting, Nijmeegse Oogonderzoek Stichting, Stichting Wetenschappelijk Onderzoek Oogziekenhuis (SWOO), The Rotterdam Eye Hospital, Macula Degeneratie Fonds (MD Fonds), Algemene Nederlandse Vereniging ter Voorkoming van Blindheid (ANVVB), Dr. F.P. Fischer Stichting, Gelderse Blinden Stichting, Landelijke Stichting voor Blinden en Slechtzienden (LSBS), Stichting Blindenhulp, Stichting Blinden-penning, Stichting Nederlands Oogheelkundig Onderzoek (SNOO), Stichting Ondersteuning Oogheelkunde’s-Gravenhage (OOG), Stichting ter Verbetering van het Lot der Blinden. We thank Gabriëlle Buitendijk for her help with the composite fundus photographs of Fig. 3, and Jacoline ten Brink with her help on the pedigree and the molecular work in family 1.
Financial disclosures
None.
Conflict of interest statement
None declared.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
C.C.W. Klaver and A.A.H.J. Thiadens had full access to all the data in the study. They take responsibility for the integrity of the data and the accuracy of the data analysis and agree to allow Graefes Archive for Clinical and Experimental Ophthalmology to review our data upon request.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Thiadens, A.A.H.J., Soerjoesing, G.G., Florijn, R.J. et al. Clinical course of cone dystrophy caused by mutations in the RPGR gene. Graefes Arch Clin Exp Ophthalmol 249, 1527–1535 (2011). https://doi.org/10.1007/s00417-011-1789-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-011-1789-3