
Abstract
Dentatorubral–pallidoluysian atrophy (DRPLA) is a rare neurodegenerative disorder caused by CAG repeat expansions in the atrophin-1 gene and is inherited in an autosomal dominant fashion. There are currently no disease-modifying treatments available. The broad development of therapies for DRPLA, as well as other similar rare diseases, has hit a roadblock due to the rarity of the condition and the wide global distribution of patients and families, consequently inhibiting biomarker development and therapeutic research. Considering the shifting focus towards diverse populations, widespread genetic testing, rapid advancements in the development of clinical and wet biomarkers for Huntington’s disease (HD), and the ongoing clinical trials for antisense oligonucleotide (ASO) therapies, the prospect of developing effective treatments in rare disorders has completely changed. The awareness of the HD ASO program has prompted global collaboration for rare disorders in natural history studies and the development of biomarkers, with the eventual goal of undergoing treatment trials. Here, we discuss DRPLA, which shares similarities with HD, and how in this and other repeat expansion disorders, neurogenetics groups like ours at UCL are gearing up for forthcoming natural history studies to accelerate future ASO treatment trials to hopefully emulate the progress seen in HD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Current understanding of DRPLA
Dentatorubral–pallidoluysian atrophy (DRPLA) is a rare autosomal dominant neurodegenerative disorder, characterized by progressive cerebellar ataxia, myoclonus, epilepsy, dementia, choreoathetosis, and psychiatric symptoms [90]. The condition was first described by Titica and van Boegard in 1946, whereby two cases in a family with progressive choreoathetosis, ataxia, and dementia were reported [88]. The term “hereditary DRPLA” was later coined by Naito and Oyanagi in 1982 [61]. DRPLA is classified within the spinocerebellar ataxia (SCA) group, which represents a heterogeneous group of > 40 autosomal dominantly inherited diseases [44]. DRPLA is caused by a CAG-polyglutamine (polyQ) repeat expansion. Nine such polyQ diseases have currently been identified in humans, including Huntington’s disease (HD), spinal and bulbar muscular atrophy (SBMA), SCA 1, 2, 3, 6, 7, and 17 [82].
DRPLA is the result of an unstable CAG repeat expansion in exon 5 of the atrophin-1 (ATN1) gene [46, 60]. The number of repeats in normal individual chromosomes ranges typically between 6 and 35. Full penetrance occurs at ≥ 48 CAG repeats, whilst alleles of 35–47 repeats are incompletely penetrant and are usually associated with a milder clinical phenotype [13, 37, 38, 46, 55, 60]. Characterized by genetic anticipation, with paternal transmission resulting in more prominent anticipation than maternal transmission, DRPLA symptoms present more severely and earlier in each subsequent generation [56, 90]. The CAG repeat load is also associated with the phenotype, whereby the longer the size of expanded CAG repeats, the earlier the age of onset and death, the more severe the symptoms and long-term disability, and the poorer the prognosis [34, 37, 55]. Figure 1 illustrates the currently known features of DRPLA.

Main genetic, clinical, neuroimaging and neuropathological features of DRPLA. a Illustration of location of the trinucleotide repeat within the gene—In DRPLA, the CAG trinucleotide repeat expansion occurs in coding exon 5 of the atrophin-1 (ATN1) gene, which is located on chromosome 12p13.31. Normal alleles in the ATN1 gene have 6–35 CAG repeats. ≥ 20 repeats are considered normal mutable alleles that expand on transmission and result in symptoms in the next generation, and ≥ 48 repeats demonstrate fully penetrant clinical phenotype. The unstable CAG repeat sequence causes a polyglutamine (polyQ) expansion in the atrophin-1 protein [13, 25, 44, 55, 60, 68, 94]. b Clinical features—The primary clinical features of DRPLA are ataxia and cognitive impairment, however, the age of onset affects the clinical presentation, with different symptoms observed between adult-onset and juvenile-onset DRPLA (Wardle et al. [98]; [34, 48, 55]. c Neuroimaging findings—[47, 106]. d Neuropathological findings—[36, 84, 106, 107]
Due to the heterogeneity in clinical presentation, based on the prominent genetic anticipation and age of onset, diagnosing DRPLA can often be challenging, with symptoms associated with a broad differential diagnosis. Whilst epileptic seizures are common in juvenile-onset patients (onset prior to the age of 20), the frequency of seizures is reduced after the age of 20, and rare in patients with an onset after the age of 40. Patients with an onset after the age of 20 tend to present with cerebellar ataxia, choreoathetosis and dementia, often making the disease difficult to differentiate from clinical mimics including HD and other hereditary SCAs [13, 61]. Further, brain MRI findings in DRPLA are variable, with case reports of early-stage patients often presenting with only mild changes, whilst late stages of the disease are associated with non-specific changes such as atrophy of the cerebellum and brainstem, complicating the differentiation of the disease from other neurological disorders [35, 45, 77, 83, 92].
Global burden
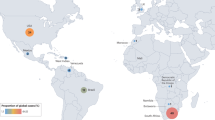
Defining global burden through natural history studies is important to understand the impact of condition and to identify disease biomarkers in the preparation for therapeutic trials. DRPLA is most commonly recognised in populations of Japanese ancestry and has an estimated incidence in Japan of 2–7 per million [26, 71]. DRPLA is considered to be the third most common autosomal dominant ataxia in the Japanese population, accounting for approximately 7.3–20% of autosomal dominant SCA [54, 87, 91]. Whilst it is believed to be rare in non-Asian populations, there are no accurate reports on the worldwide prevalence of DRPLA, with current estimates based on the evaluation of cohorts diagnosed with SCA, suggesting that the prevalence of DRPLA is likely to be underestimated [6, 91, 96]. In Singapore, Korea and China, the frequencies of DRPLA have been found to be 3.4%, 3.4%, and 1%, respectively [42, 50, 111]. In South America, the DRPLA frequency has found to be 0.14% and 3.1% in SCA cohorts from Brazil and Venezuela, respectively [8, 65]. In Europe, findings have been variable, with reports of the frequency in Portuguese cohorts with autosomal dominant ataxias of DRPLA ranging from 4 to 11.2%, whilst in Spain, the frequency was reported as 3.3% [18, 39, 93]. In South Wales, France, and Italy, the frequency amongst cohorts with SCA has been reported as 5%, 0.25%, and 0.45–1%, respectively [11, 27, 49, 97]. Figure 2 illustrates the estimated number of cases around the world.

Estimated number of reported cases of DRPLA based on a literature search. a World map with dots corresponding to the number of reported cases of DRPLA, as per studies shown in (b). b Table of DRPLA families/cases found outside of Japan as reported in literature. DRPLA literature search for studies written in English published between January 1945 and July 2020 was performed on PubMed and Scopus databases, using the following key words: DRPLA; dentatorubral–pallidoluysian atrophy; Naito-Oyanagi disease; Haw-River syndrome; or ataxia. Cases for Thailand and Germany based on personal communication
Developing a treatment approach for DRPLA and other repeat expansion disorders: drawing upon insights observed for HD
There are currently no treatments to prevent or stop the disease progression in DRPLA [90]. Whilst the exact pathophysiology of DRPLA is unclear, the literature overall points towards the idea that the expanded polyQ stretch leads to a “gain-of-toxic” function of the mutant protein on neuronal cells [89]. To downregulate the levels of the pathological polyQ proteins, RNA-targeting therapies may hold promise in the treatment of DRPLA, in particular, antisense oligonucleotides (ASO) therapy [16]. Therapeutic ASOs are single-stranded synthetic DNA molecules that work by binding to complementary target mRNA through Watson and Crick hybridization to interfere with normal gene expression and protein synthesis. ASOs affect gene expression through three mechanisms: RNase H-mediated degradation of mRNA, blocking ribosomes from binding to mRNA and preventing protein translation, or by modulating splicing of pre-mRNA [70, 102]. Figure 3 shows the normal steps of gene expression and the mechanisms by which therapeutic ASOs can influence this process. The scope of ASO therapeutics has expanded considerably in recent years, with an emphasis particularly placed on rare untreatable conditions, which cannot be easily addressed with small molecule drugs. ASO therapeutics have shown promise in several neurological disorders. For example, Nusinersen and Eteplisren are FDA-approved ASO treatment options for spinal muscular atrophy (SMA) and Duchenne muscular dystrophy (DMD), respectively, whilst clinical trials are ongoing for ASO treatments for amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), and HD [75, 104]. Figure 4 highlights the progress of therapeutic ASO development for repeat expansion neurological disorders.

Normal process of protein synthesis and common mechanisms of ASOs on target RNA. Normal process of protein synthesis (left): DNA is transcribed to pre-messenger RNA (pre-mRNA), which contains coding (exon) and non-coding (intron) regions between 5′ and 3′ untranslated regions (UTR). Pre-mRNA undergoes post-transcriptional modifications into mature mRNA, including 5′ capping, removal of introns (splicing), and polyadenylation (poly-A tail). The mature mRNA undergoes ribosome-dependent protein synthesis. Mechanisms of ASOs on target RNA (right): Splicing modulation—ASO binds to pre-mRNA intron/exon junctions and modulate splicing to include or skip the target exon, resulting in the synthesis of a modified protein. Translation inhibition—ASO binds to the mRNA and sterically blocks and prevents the binding of ribosomes to the mRNA, inhibiting translation and resulting in reduced protein synthesis. RNase H-mediated degradation of mRNA—ASO binds to the mRNA to form an RNA–DNA hybrid, allowing the recruitment of RNase H nuclease, inducing degradation of the target mRNA, resulting in reduced protein synthesis [22, 70, 73, 78, 102]. Diagram created on biorender.com

Progress in the development of ASO therapies for repeat expansions associated with neurological disorders. Table shows stages in the research towards developing ASO therapies for neurological repeat expansion disorders. Information. Chr: chromosome; HD; Huntington’s disease; ALS: amyotrophic lateral sclerosis; SCA: spinocerebellar ataxia; DM1: myotonic dystrophy; SBMA: spinobulbar muscular atrophy
HD is the most widely studied CAG repeat expansion disorder and has gained significant attention for ASO therapeutics. Based on the success seen in preclinical studies of non-human animals, ASO clinical trials are ongoing for HD [78]. HTTRX is an ASO that targets the mutant and wild-type alleles with the purpose of reducing levels of the mutant Huntingtin protein (mHTT). Through phase 1–2a clinical trial in early-stage HD patients, it was found that CSF mHTT levels showed dose-dependent decrease by up to 40%. No significant safety concerns were reported, though levels of CSF neurofilament light chain (NfL), a marker of neuroaxonal damage, were shown to be increased in the final study visit [86]. Preclinical studies have also been conducted for SCA. In early manifest transgenic SCA3 mice, ATXN3-targeting ASO resulted in sustained reduction of polyQ-expanded ATXN3, accompanied by rescued motor impairment [57]. Further, in SCA2 mouse models, the delivery of ATXN2-targeting ASO led to the downregulation of the ATXN2 mRNA and protein, delayed onset of the SCA2 phenotype, with improved motor performance [74]. These findings indicate a promising proof-in-concept for ASO therapy as an approach for polyQ conditions.
The advances in HD are suggestive of the possibility of adopting similar methods to define biomarkers and treat DRPLA. Before clinically meaningful interventions can be discovered, a greater understanding of DRPLA disease progression and the identification of wet biomarkers must be pursued. Despite the significant advancements made for other neurodegenerative diseases, such as HD and AD, biomarkers in biological fluid, such as blood and CSF, have not been found for DRPLA [16]. Potential biomarkers including glial fibrillary acidic protein, DJ-1, and tau have been studied in SCA1, SCA2, and SCA6 patients, where only CSF tau was significantly higher in patients than controls, though levels did not correlate with CAG repeat size and disease severity [9]. CSF and plasma/serum NfL have been shown to be a notable biomarker in many neurodegenerative conditions, including HD, AD, ALS, and multiple sclerosis [12, 23, 30, 101]. In a small cohort of repeat-expansion SCA patients, serum NfL was found to be higher in patients than controls; however, the correlation with disease severity was not analysed [103]. Another potentially important biomarker for DRPLA disease progression and severity, which may also act as a potential therapeutic target, is repeat expansion somatic instability. This has been observed in HD mouse models and human brain tissue; it is worth exploring as a prominent biomarker for DRPLA and other repeat expansion disorders [3, 24, 85]. In addition to being clinically beneficial in regard to improving diagnostic accuracy and monitoring disease progression, biological biomarkers for DRPLA would also be influential in research. For example, they would provide greater accuracy in clinical trial recruitment, objective monitoring of disease-related biological changes, tracking adverse effects and response to treatment interventions [1]. Studies in larger cohorts are needed to gather data on the role of biological biomarkers DRPLA.
Insight into the natural progression of rare diseases is an essential step in facilitating the process of drug development [64]. To garner progress towards the discovery of disease-modifying treatments for DRPLA, emphasis must be placed on natural history studies to enhance our understanding of disease progression and to identify reproducible, validated biomarkers (Fig. 5). For rare diseases, this entails international collaboration to understand the longitudinal clinical progression in statistically large numbers of cases, ideally with n > 20 patients from multiple geographical areas, alongside matched controls. Initial investigative markers of disease progression would include clinical rating scales, imaging techniques and EEG, and objective, fluid-based biomarkers. In DRPLA, and other rare repeat expansion disorders, prominent analysis will comprise of DNA extracted from multiple fluids to investigate somatic instability, RNA extracted from blood and fibroblast cell lines, and extraction of serum/plasma and/or CSF for the examination of biomarkers such as NfL. The goal over the next 3 years will be to enhance our insight of DRPLA clinical features, imaging and fluid biomarkers, disease progression, and to uncover methods to monitor response to therapeutic intervention.

Facilitating DRPLA therapeutic development through understanding of natural history and discovery of biomarkers. Natural history studies follow the course of a disease from prior to inception, through the presymptomatic and clinical stages, to the point it ends (the patient is either cured, chronically disabled or dead, without external intervention) [20]. International, collaborative clinical studies are paramount to the DRPLA drug discovery process by identifying milestones of the disease progression and facilitating the discovery of longitudinal or cross-sectional biomarkers to objectively track disease-related biological changes. The discovery of biomarkers is, in turn, essential for clinical trials. The figure showcases the process by which a clinical and biobank resource for DRPLA can be uncovered, through collaborative efforts. Several methods are used for biomarker discovery; in particular, ‘omics’ technologies contribute towards the rapid discovery and validation of biomarkers. Genomics allows the identification of gene mutations or polymorphisms; transcriptomics can identify changes in RNA; epigenetics can identify modified epigenetic mechanisms; metabolomics and proteomics can identify small molecule metabolites and protein biomarkers in human biological fluid, respectively [1, 2, 28, 31, 41]. MS: multiple spectrometry; NfL: neurofilament light chain; NMR: nuclear magnetic resonance; MALDI-TOF MS: matrix-assisted laser desorption/ionization time of flight mass spectrometry; MRI: magnetic resonance imaging; EEG: electroencephalogram; EDTA: Ethylenediamine tetraacetic acid; CSF: cerebrospinal fluid
The next few years for DRPLA
The future for DRPLA and other rare disorders is one of momentous opportunity. The knowledge gained in the scientific community from previous successful (and many unsuccessful) trials for SMA, HD, and other similar diseases have defined the foundations required to understand disease progression and how to see the reversal. The current development of collaborative natural history and biomarker studies for DRPLA by our group at UCL alongside other institutions gives hope to DRPLA patients and families for advancements over the next few years. Whilst it is expected that many clinical, fluid or imaging markers of disease will overlap with other conditions, namely fluid NfL and MRI sequences, it can be postulated that DRPLA-specific markers may be discovered, for example, disease-associated protein levels such as ATN1, or somatic instability in the repeat expansion in biosamples. Though funding is challenging in rare disorders, natural history studies, in tandem with open-access data, imaging, wet biomarker and fibroblast repositories are essential. Ascertaining a wide resource for use by future researchers is crucial in the drive towards discoveries that may potentially benefit patient care.
Change history
21 June 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00415-021-10644-0
References
Ashizawa T, Öz G, Paulson HL (2018) Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol 14(10):590–605. https://doi.org/10.1038/s41582-018-0051-6
Augustine EF, Adams HR, Mink JW (2013) Clinical trials in rare disease: challenges and opportunities. J Child Neurol 28(9):1142–1150. https://doi.org/10.1177/0883073813495959
Aviolat H, Pinto RM, Godschall E et al (2019) Assessing average somatic CAG repeat instability at the protein level. Sci Rep 9:19152. https://doi.org/10.1038/s41598-019-55202-x
Becher MW, Rubinsztein DC, Leggo J, Wagster MV, Stine OC, Ranen NG, Barron L (1997) Dentatorubral and pallidoluysian atrophy (DRPLA) Clinical and neuropathological findings in genetically confirmed north American and European pedigrees. Movement Disord 12(4):519–530. https://doi.org/10.1002/mds.870120408
Bhanushali AA, Venkatesan R, Das BR (2020) Spinocerebellar Ataxias in India: three-year molecular data from a central reference laboratory. Neurol India 68:86–91. https://www.neurologyindia.com/text.asp?2020/68/1/86/279666
Bhowmik A, Rangaswamiaih S et al (2015) Molecular genetic analysis of trinucleotide repeat disorders (TRDs) in Indian population and application of repeat primed PCR. Eur J Med Genet 58:160–167. https://doi.org/10.1016/j.ejmg.2014.12.010
Bidollari E, Rotundo G, Altieri F, Amicucci M, Wiquel D, Ferrari D, Goldoni M, Bernardini L, Consoli F, De Luca A, Fanelli S, Lamorte G, D'Agruma L, Vescovi AL, Squitieri F, Rosati J (2019) Generation of induced pluripotent stem cell line CSSi008-A (4698) from a patient affected by advanced stage of Dentato-Rubral-Pallidoluysian atrophy (DRPLA). Stem Cell Res 40:101551. https://doi.org/10.1016/j.scr.2019.101551
Braga-Neto P, Pedroso JL, Furtado GV et al (2017) Dentatorubro-Pallidoluysian Atrophy (DRPLA) among 700 Families with Ataxia in Brazil. Cerebellum 16:812–816. https://doi.org/10.1007/s12311-017-0862-9
Brouillette A, Gulin O, Gomez C (2015) Cerebrospinal fluid biomarkers in spinocerebellar ataxia: a pilot study. Dis Markers. https://doi.org/10.1155/2015/413098
Brunetti-Pierri N, Wilfong AA, Hunter JV, Craigen WJ (2006) A severe case of dentatorubro-pallidoluysian atrophy (DRPLA) with microcephaly, very early onset of seizures, and cerebral white matter involvement. Neuropediatrics 37(5):308–311. https://doi.org/10.1055/s-2006-955967
Brusco A, Gellera C, Cagnoli C et al (2004) Molecular genetics of hereditary spinocerebellar ataxia: mutation analysis of spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian Families. Arch Neurol 61(5):727–733. https://doi.org/10.1001/archneur.61.5.727
Byrne LM, Rodrigues FB, Blennow K, Durr A, Leavitt BR, Roos R, Scahill RI, Tabrizi SJ, Zetterberg H, Langbehn D, Wild EJ (2017) Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol 16(8):601–609. https://doi.org/10.1016/S1474-4422(17)30124-2
Carroll LS, Massey TH, Wardle M, Peall KJ (2018) Dentatorubral-pallidoluysian atrophy: an update. Tremor Other Hyperkinetic Movements 8:577. https://doi.org/10.7916/D81N9HST
Casseron W, Azulay JP, Broglin D, Kaphan E, Genton P, Le Ber I, Gastaut JL (2004) Phenotype variability in a caucasian family with dentatorubral-pallidoluysian atrophy. Eur Neurol 52(3):175–176. https://doi.org/10.1159/000081859
Chen J, Zeng Z, Wu J et al (2013) The clinical and genetical characteristic of one dentatorubral-pallidoluysian atrophy pedigree with an onset of cognitive impairment. Chin J Neurol 46:962–969. https://doi.org/10.3760/cma.j.issn.1006-7876.2013.10.011
Coarelli G, Brice A, Durr A (2018) Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Research. 7: F1000. https://doi.org/10.12688/f1000research.15788.1
Connarty M, Dennis NR, Patch C et al (1996) Molecular re-investigation of patients with Huntington's disease in Wessex reveals a family with dentatorubral and pallidoluysian atrophy. Hum Genet 97:76–78. https://doi.org/10.1007/BF00218837
Coutinho P, Ruano L, Loureiro JL et al (2013) Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 70(6):746–755. https://doi.org/10.1001/jamaneurol.2013.1707
Cox H, Costin-Kelly NM, Ramani P, Whitehouse WP (2000) An established case of dentatorubral pallidoluysian atrophy (DRPLA) with unusual features on muscle biopsy. Eur J Paediat Neurol 4(3):119–123. https://doi.org/10.1053/ejpn.2000.0279
de Paz MP et al (2010) Rare diseases epidemiology research. In: Posada M, Groft S (eds) Rare diseases epidemiology. Advances in experimental medicine and biology, vol 686. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-9485-8_2
Destée A, Delalande I, Vuillaume I, Schraen-Maschke S, Defebvre L, Sablonnière B (2000) The first identified French family with dentatorubral-pallidoluysian atrophy. Movement Disord 15(5):996–999. https://doi.org/10.1002/1531-8257(200009)15:5<996:aid-mds1036>3.0.co;2-9
Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A, Bahal R (2020) Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med 9(6):2004. https://doi.org/10.3390/jcm9062004
Disanto G, Barro C, Benkert P, Naegelin Y, Schädelin S, Giardiello A, Zecca C, Blennow K, Zetterberg H, Leppert D, Kappos L, Gobbi C, Kuhle J (2017) Serum neurofilament light: a biomarker of neuronal damage in multiple sclerosis. Ann Neurol 81(6):857–870. https://doi.org/10.1002/ana.24954
Dragivela E, Henricks A, Teed A et al (2009) Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis 33:37–47. https://doi.org/10.1016/j.nbd.2008.09.014
Ellerby L (2019) Repeat expansion disorders: mechanisms and therapeutics. Neurotherapeutics 16:924–927. https://doi.org/10.1007/s13311-019-00823-3
Fanto M, Charroux B (2010) Atrophin-1. Encyclopedia Movement Disord 2010:100–103. https://doi.org/10.1016/B978-0-12-374105-9.00303-8
Filla A, Mariotti C, Caruso G, Coppola G, Cocozza S, Castaldo I, Calabrese O, Salvatore E, De Michele G, Riggio MC, Pareyson D, Gellera C, Di Donato S (2000) Relative frequencies of CAG expansions in spinocerebellar ataxia and dentatorubropallidoluysian atrophy in 116 Italian families. Eur Neurol 44(1):31–36. https://doi.org/10.1159/000008189
Fitzgerald G (2016) Measure for measure: biomarker standards and transparency. Sci Transl Med. 8:34310. https://doi.org/10.1126/scitranslmed.aaf8590
Friedrich J, Kordasiewicz HB, O'Callaghan B, Handler HP, Wagener C, Duvick L, Swayze EE, Rainwater O, Hofstra B, Benneyworth M, Nichols-Meade T, Yang P, Chen Z, Ortiz JP, Clark HB, Öz G, Larson S, Zoghbi HY, Henzler C, Orr HT (2018) Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI insight 3(21):e123193. https://doi.org/10.1172/jci.insight.123193
Gaiani A, Martinelli I, Bello L, Querin G, Puthenparampil M, Ruggero S, Toffanin E, Cagnin A, Briani C, Pegoraro E, Sorarù G (2017) Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: neurofilament light chain levels in definite subtypes of disease. JAMA Neurol 74(5):525–532. https://doi.org/10.1001/jamaneurol.2016.5398
Garcia-Gutierrez MS, Navarrete F, Sala F, Gasparyan A, Austrich-Olivares A, Manzanares J (2020) Biomarkers in psychiatry: concept, definition, types and relevance to the clinical reality. Front Psychiatry 11:432. https://doi.org/10.3389/fpsyt.2020.00432
Grimaldi S, Cupidi C, Smirne N, Bernardi L, Giacalone F, Piccione G, Basiricò S, Mangano GD, Nardello R, Orsi L, Grosso E, Laganà V, Mitolo M, Maletta RG, Bruni AC (2019) The largest caucasian kindred with dentatorubral-pallidoluysian atrophy: a founder mutation in Italy. Movement Disord 34(12):1919–1924. https://doi.org/10.1002/mds.27879
Hao Y, Gu W, Wang G et al (2010) Clinical and genetic study of one DRPLA case. Chin J Contemporary Neurol Neurosurg 10:637–641. https://doi.org/10.3969/j.issn.1672-6731.2010.06.012
Hasegawa A, Ikeuchi T, Koike R, Matsubara N, Tsuchiya M, Nozaki H, Homma A, Idezuka J, Nishizawa M, Onodera O (2010) Long-term disability and prognosis in dentatorubral-pallidoluysian atrophy: a correlation with CAG repeat length. Movement Disord 25(11):1694–1700. https://doi.org/10.1002/mds.23167
Hatano T, Okuma Y, Iijima M, Fujishima K, Goto K, Mizuno Y (2003) Cervical dystonia in dentatorubral-pallidoluysian atrophy. Acta Neurol Scand 108(4):287–289. https://doi.org/10.1034/j.1600-0404.2003.00150.x
Hayashi Y, Kakita A, Yamada M, Egawa S, Oyanagi S, Naito H, Takahashi H (1998) Hereditary dentatorubral-pallidoluysian atrophy: ubiquitinated filamentous inclusions in the cerebellar dentate nucleus neurons. Acta Neuropathol 95(5):479–482. https://doi.org/10.1007/s004010050828
Ikeuchi T, Koide R, Onodera O et al (1995) Dentatorubralpallidoluysian atrophy (DRPLA). Molecular basis for wide clinical features of DRPLA. Clin Neurosci 3:23–27
Ikeuchi T, Koide R, Tanaka H, Onodera O, Igarashi S, Takahashi H, Sato K (1995) Dentatorubral-pallidoluysian atrophy: clinical features are closely related to unstable expansions of trinucleotide (CAG) repeat. Ann Neurol 37(6):769–775. https://doi.org/10.1002/ana.410370610
Infante J, Combarros O, Volpini V, Corral J, Llorca J, Berciano J (2005) Autosomal dominant cerebellar ataxias in Spain: molecular and clinical correlations, prevalence estimation and survival analysis. Acta Neurol Scand 111(6):391–399. https://doi.org/10.1111/j.1600-0404.2005.00400.x
Jauvin D, Chrétien J, Pandey SK, Martineau L, Revillod L, Bassez G, Lachon A, MacLeod AR, Gourdon G, Wheeler TM, Thornton CA, Bennett CF, Puymirat J (2017) Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol Ther Nucleic Acids 7:465–474. https://doi.org/10.1016/j.omtn.2017.05.007
Jewell NP (2016) Natural history of diseases: statistical designs and issues. Clin Pharmacol Ther 100(4):353–361. https://doi.org/10.1002/cpt.423
Jin D, Oh M, Song S et al (1999) Frequency of spinocerebellar ataxia types 1, 2, 3, 6, 7 and dentatorubral pallidoluysian atrophy mutations in Korean patients with spinocerebellar ataxia. J Neurol 246:207–210. https://doi.org/10.1007/s004150050335
Kim H, Yun JY, Choi KG, Koo H, Han HJ (2018) Sleep related problems as a nonmotor symptom of dentatorubropallidoluysian atrophy. J Korean Med Sci 33(17):e130. https://doi.org/10.3346/jkms.2018.33.e130
Klockgether T, Mariotti C, Paulson HL (2019) Spinocerebellar ataxia. Nat Rev Dis Primers 5:24. https://doi.org/10.1038/s41572-019-0074-3
Kobayashi J, Nagao M, Kawata A, Matsubara S (2009) A case of late adult-onset dentatorubral-pallidoluysian atrophy mimicking central pontine myelinolysis. J Neurol 256(8):1369–1371. https://doi.org/10.1007/s00415-009-5111-9
Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Saito M (1994) Unstable expansion of CAG repeat in hereditary dentatorubral–pallidoluysian atrophy (DRPLA). Nat Genet 6(1):9–13. https://doi.org/10.1038/ng0194-9
Koide R, Onodera O, Ikeuchi T, Kondo R, Tanaka H, Tokiguchi S, Shimizu N (1997) Atrophy of the cerebellum and brainstem in dentatorubral pallidoluysian atrophy: influence of CAG repeat size on MRI findings. Neurology 49(6):1605–1612. https://doi.org/10.1212/WNL.49.6.1605
Latimer CS, Flanagan ME, Cimino PJ, Jayadev S, Davis M, Hoffer ZS, Montine TJ, Gonzalez-Cuyar LF, Bird TD, Keene CD (2017) Neuropathological comparison of adult onset and juvenile Huntington's disease with cerebellar atrophy: a report of a father and son. J Huntington's Dis 6(4):337–348. https://doi.org/10.3233/JHD-170261
Le Ber I, Camuzat A, Castelnovo G et al (2003) Prevalence of dentatorubral-pallidoluysian atrophy in a large series of white patients with cerebellar ataxia. Arch Neurol 60(8):1097–1099. https://doi.org/10.1001/archneur.60.8.1097
Lee IH, Soong BW, Lu YC, Chang YC (2001) Dentatorubropallidoluysian atrophy in Chinese. Arch Neurol 58(11):1905–1908. https://doi.org/10.1001/archneur.58.11.1905
Licht DJ, Lynch DR (2002) Juvenile dentatorubral-pallidoluysian atrophy: new clinical features. Pediatr Neurol 26(1):51–54. https://doi.org/10.1016/s0887-8994(01)00346-0
Liu H, Xie L, Su C et al (2017) Clinical features, electroneurophysiology, neuroimaging and gene analysis of one juvenile dentatorubral-pallidoluysian atrophy pedigree. Chin J Neurol 50:506–510. https://doi.org/10.3760/cma.j.issn.1006-7876.2017.07.006
Matsuzono K, Imamura K, Murakami N, Tsukita K, Yamamoto T, Izumi Y, Kaji R, Ohta Y, Yamashita T, Abe K, Inoue H (2017) Antisense oligonucleotides reduce RNA foci in spinocerebellar ataxia 36 patient iPSCs. Mol Ther Nucleic ACIDS 8:211–219. https://doi.org/10.1016/j.omtn.2017.06.017
Maruyama H, Izumi Y, Morino H et al (2002) Difference in disease-free survival curve and regional distribution according to subtype of spinocerebellar ataxia: A study of 1,286 Japanese patients. Int J Med Genetics 114:578–583. https://doi.org/10.1002/ajmg.10514
Maruyama S, Saito Y, Nakagawa E, Saito T, Komaki H, Sugai K, Sasaki M, Kumada S, Saito Y, Tanaka H, Minami N, Goto Y (2012) Importance of CAG repeat length in childhood-onset dentatorubral-pallidoluysian atrophy. J Neurol 259(11):2329–2334. https://doi.org/10.1007/s00415-012-6493-7
Mazzoni P, Jain S (2007) Myoclonus. Neurobiol Dis 30:305–317. https://doi.org/10.1016/B978-012088592-3/50032-3
McLoughlin H, Moore L, Chopra R et al (2018) Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3. Ann Neurol 84:64–77. https://doi.org/10.1002/ana.25264
Munhoz RP, Bergeron C, Lang AE (2004) Sporadic case of dentatorubral pallidoluysian atrophy with no CAG repeat expansion and no intranuclear inclusions. Movement Disord 19(5):580–583. https://doi.org/10.1002/mds.10674
Muñoz E, Milà M, Sánchez A et al (1999) Dentatorubropallidoluysian atrophy in a Spanish family: a clinical, radiological, pathological, and genetic study. J Neurol Neurosurg Psychiatry 67:811–814
Nagafuchi S, Yanagisawa H, Sato K et al (1994) Expansion of an unstable CAG trinucleotide on chromosome 12p in dentatorubral and pallidoluysian atrophy. Nat Genet 6:14–18
Naito H, Oyanagi S (1982) Familial myoclonus epilepsy and choreoathetosis: hereditary dentatorubral-pallidoluysian atrophy. Neurology 32(8):798–798. https://doi.org/10.1212/WNL.32.8.798
Niu C, Prakash TP, Kim A, Quach JL, Huryn LA, Yang Y, Lopez E, Jazayeri A, Hung G, Sopher BL, Brooks BP, Swayze EE, Bennett CF, La Spada AR (2018) Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci Transl Med 10(465):8677. https://doi.org/10.1126/scitranslmed.aap8677
Nørremølle A, Nielsen JE, Sørensen SA, Hasholt L (1995) Elongated CAG repeats of the B37 gene in a Danish family with dentato-rubro-pallido-luysian atrophy. Hum Genet 95(3):313–318
Gavin P (2015) The importance of natural histories for rare diseases. Expert Opin Orphan Drugs 3(8):855–857. https://doi.org/10.1517/21678707.2015.1063415
Paradisi I, Ikonomu V, Arias S (2016) Spinocerebellar ataxias in Venezuela: genetic epidemiology and their most likely ethnic descent. J Hum Genet 61:215–222. https://doi.org/10.1038/jhg.2015.131
Potter NT, Meyer MA, Zimmerman AW, Eisenstadt ML, Anderson IJ (1995) Molecular and clinical findings in a family with dentatorubral-pallidoluysian atrophy. Ann Neurol 37(2):273–277. https://doi.org/10.1002/ana.410370220
Pujana MA, Corral J, Gratacòs M, Combarros O, Berciano J, Genís D (1999) Spinocerebellar ataxias in Spanish patients: genetic analysis of familial and sporadic cases. Hum Genet 104(6):516–522
Rodriquez C, Todd P (2019) New pathologic mechanisms in nucleotide repeat expansion disorders. Neurobiol Dis 130:104515. https://doi.org/10.1016/j.nbd.2019.104515
Rodriguez CM, Wright SE, Kearse MG et al (2020) A native function for RAN translation and CGG repeats in regulating fragile X protein synthesis. Nat Neurosci 23:386–397. https://doi.org/10.1038/s41593-020-0590-1
Rossor A, Reilly M, Sleigh J (2018) Antisense oligonucleotides and other genetic therapies made simple. Pract Neurol 18:126–131. https://doi.org/10.1136/practneurol-2017-001764
Ruano L, Melo C, Silva MC, Coutinho P (2014) The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42(3):174–183. https://doi.org/10.1159/000358801
Sahashi K, Katsuno M, Hung G et al (2015) Silencing neuronal mutant androgen receptor in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 24:5985–5994. https://doi.org/10.1093/hmg/ddv300
Schoch K, Miller T (2017) Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 94:1056–1070. https://doi.org/10.1016/j.neuron.2017.04.010
Scoles D, Meera P, Schneider M et al (2017) Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 544:362–366. https://doi.org/10.1038/nature22044
Scoles DR, Minikel EV, Pulst SM (2019) Antisense oligonucleotides: a primer. Neurol Genetics 5(2):e323. https://doi.org/10.1212/NXG.0000000000000323
Sharma P, Shaikh R, Shamim U et al (2020) Genetically confirmed first Indian dentato-rubral-pallidoluysian atrophy kindred: a case report. Ann Mov Disord 3:60–64
Shiraishi H, Egawa K, Ito T, Kawano O, Asahina N, Kohsaka S (2017) Efficacy of perampanel for controlling seizures and improving neurological dysfunction in a patient with dentatorubral-pallidoluysian atrophy (DRPLA). Epilepsy Behav Case Rep 8:44–46. https://doi.org/10.1016/j.ebcr.2017.05.004
Silva A, Lobo D, Martins I et al (2020) Antisense oligonucleotide therapeutics in neurodegenerative diseases: the case of polyglutamine disorders. Brain 143:407–429. https://doi.org/10.1093/brain/awz328
Silver MR, Sethi KD, Mehta SH et al (2015) Case report of optic atrophy in Dentatorubropallidoluysian Atrophy (DRPLA). BMC Neurol 15:260. https://doi.org/10.1186/s12883-015-0520-0
Simpson M, Smith A, Kent H, Roxburgh R (2012) Neurological picture. Distinctive MRI abnormalities in a man with dentatorubral-pallidoluysian atrophy. J Neurol Neurosurg Psychiatry 83(5):529–530. https://doi.org/10.1136/jnnp-2011-301612
Smith J, Gonda V, Malamud N (1958) Unusual form of cerebellar ataxia: combined dentato-rubral and pallido-luysian degeneration. Neurology 8:205
Stoyas C, Spada A (2018) The CAG–polyglutamine repeat diseases: a clinical, molecular, genetic, and pathophysiologic nosology. Handbook Clin Neurol 147:143–170. https://doi.org/10.1016/B978-0-444-63233-3.00011-7
Sugiyama A, Sato N, Nakata Y et al (2018) Clinical and magnetic resonance imaging features of elderly onset dentatorubral–pallidoluysian atrophy. J Neurol 265:322–329. https://doi.org/10.1007/s00415-017-8705-7
Sunami Y, Koide R, Arai N, Yamada M, Mizutani T, Oyanagi K (2011) Radiologic and neuropathologic findings in patients in a family with dentatorubral-pallidoluysian atrophy. AJNR Am J Neuroradiol 32(1):109–114. https://doi.org/10.3174/ajnr.A2252
Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, Wheeler VC (2009) Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet 18(16):3039–3047. https://doi.org/10.1093/hmg/ddp242
Tabrizi S, Leavitt B, Landwehrmeyer B et al (2019) Targeting Huntingtin expression in patients with Huntington’s disease. NEJM 380:2307–2316. https://doi.org/10.1056/NEJMoa190090
Takano H, Cancel G, Ikeuchi T, Lorenzetti D, Mawad R, Stevanin G, Sasaki R (1998) Close associations between prevalences of dominantly inherited spinocerebellar ataxias with CAG-repeat expansions and frequencies of large normal CAG alleles in Japanese and Caucasian populations. Am J Hum Genet 63(4):1060–1066. https://doi.org/10.1086/302067
Titica J, van Bogaert LUDO (1946) Heredo-degenerative hemiballismus: a contribution to the question of primary atrophy of the corpus Luysii. Brain 69(4):251–263
Tsuji S (2000) [Dentatorubral-pallidoluysian atrophy (DRPLA)–discovery of the disease, DRPLA gene and the pathophysiology] Rinsho Shinkeigaku. Clin Neurol 40(12):1287–1289
Tsuji S (2012) Dentatorubral–pallidoluysian atrophy. Handbook Clin Neurol 103:587–594. https://doi.org/10.1016/B978-0-444-51892-7.00041-3
Tsuji S, Onodera O, Goto J et al (2008) Sporadic ataxias in Japan—a population-based epidemiological study. Cerebellum 7:189. https://doi.org/10.1007/s12311-008-0028-x
Tunc S, Tadic V, Zühlke C, Hellenbroich Y, Brüggemann N (2018) Pearls & Oy-sters: Family history of Huntington disease disguised a case of dentatorubral-pallidoluysian atrophy. Neurology 90(3):142–143. https://doi.org/10.1212/WNL.0000000000004833
Vale J, Bugalho P, Silveira I, Sequeiros J, Guimarães J, Coutinho P (2010) Autosomal dominant cerebellar ataxia: frequency analysis and clinical characterization of 45 families from Portugal. Eur J Neurol 17(1):124–128. https://doi.org/10.1111/j.1468-1331.2009.02757.x
Veneziano L, Mantuano E, Catalli C, Gellera C, Durr A, Romano S, Spadaro M, Frontali M, Novelletto A (2014) A shared haplotype for dentatorubropallidoluysian atrophy (DRPLA) in Italian families testifies of the recent introduction of the mutation. J Hum Genet 59(3):153–157. https://doi.org/10.1038/jhg.2013.137
Vinton A, Fahey M, O'Brien T et al (2005) Dentatorubral-pallidoluysian atrophy in three generations, with clinical courses from nearly asymptomatic elderly to severe juvenile, in an Australian family of Macedonian descent. AJMG 136A:201–204
Wardle M, Majounie E, Williams NM et al (2008) Dentatorubral pallidoluysian atrophy in South Wales. J Neurol Neurosurg Psychiatry 79:804–807. https://doi.org/10.1136/jnnp.2007.128074
Wardle M, Majounie E, Muzaimi MB, Williams NM, Morris HR, Robertson NP (2009) The genetic aetiology of late-onset chronic progressive cerebellar ataxia. A population-based study. J Neurol 256(3):343–348. https://doi.org/10.1007/s00415-009-0015-2
Wardle M, Majounie E, Muzaimi MB, Williams NM, Morris HR, Robertson NP (2009) The genetic aetiology of late-onset chronic progressive cerebellar ataxia A population-based study. J Neurol 256(3):343–348. https://doi.org/10.1007/s00415-009-0015-2
Wardle M, Morris HR, Robertson NP (2009) Clinical and genetic characteristics of non-Asian dentatorubral-pallidoluysian atrophy: a systematic review. Movement Disord 24(11):1636–1640. https://doi.org/10.1002/mds.22642
Warner TT, Williams LD, Walker RWH, Flinter F, Robb SA, Bundey SE, Harding AE (1995) A clinical and molecular genetic study of dentatorubropallidoluysian atrophy in four European families. Ann Neurol 37(4):452–459. https://doi.org/10.1002/ana.410370407
Weston P, Poole T, Ryan NS, Nair A, Liang Y, Macpherson K, Druyeh R, Malone IB, Ahsan RL, Pemberton H, Klimova J, Mead S, Blennow K, Rossor MN, Schott JM, Zetterberg H, Fox NC (2017) Serum neurofilament light in familial Alzheimer disease: a marker of early neurodegeneration. Neurology 89(21):2167–2175. https://doi.org/10.1212/WNL.0000000000004667
Wild EJ, Tabrizi SJ (2017) Therapies targeting DNA and RNA in Huntington's disease. Lancet Neurol 16(10):837–847. https://doi.org/10.1016/S1474-4422(17)30280-6
Wilke C, Bender F, Hayer SN, Brockmann K, Schöls L, Kuhle J, Synofzik M (2018) Serum neurofilament light is increased in multiple system atrophy of cerebellar type and in repeat-expansion spinocerebellar ataxias: a pilot study. J Neurol 265(7):1618–1624. https://doi.org/10.1007/s00415-018-8893-9
Wurster CD, Ludolph AC (2018) Antisense oligonucleotides in neurological disorders. Thera Adv Neurol Disord 11:1756286418776932. https://doi.org/10.1177/1756286418776932
Yam WK, Wu NS, Lo IF, Ko CH, Yeung WL, Lam ST (2004) Dentatorubral-pallidoluysian atrophy in two Chinese families in Hong Kong. Hong Kong Med J 10(1):53–56
Yamada M (2010) Dentatorubral-pallidoluysian atrophy (DRPLA): The 50th Anniversary of Japanese Society of Neuropathology. Neuropathology 30:453–457. https://doi.org/10.1111/j.1440-1789.2010.01120.x
Yamada M, Wood JD, Shimohata T, Hayashi S, Tsuji S, Ross CA, Takahashi H (2001) Widespread occurrence of intranuclear atrophin-1 accumulation in the central nervous system neurons of patients with dentatorubral-pallidoluysian atrophy. Ann Neurol 49(1):14–23. https://doi.org/10.1002/1531-8249(200101)49:1%3C14:AID-ANA5%3E3.0.CO;2-X
Yiş U, Dirik E, Gündoğdu-Eken A, Başak AN (2009) Dentatorubral pallidoluysian atrophy in a Turkish family. Turk J Pediatr 51(6):610–612
Zádori D, Tánczos T, Jakab K, Vécsei L, Klivényi P (2015) The first identified Central-Eastern European patient with genetically confirmed dentatorubral-pallidoluysian atrophy. Ideggyogyaszati szemle 68(1–2):68–71
Zhang Z, Hao Y (2013) Genetics and clinical study of Chinese kindreds with dentatorubral pallidoluysian atrophy. Chin J Med Genetics 30:31–35. https://doi.org/10.3760/cma.j.issn.1003-9406.2013.01.008
Zhao Y, Tan EK, Law HY, Yoon CS, Wong MC, Ng I (2002) Prevalence and ethnic differences of autosomal-dominant cerebellar ataxia in Singapore. Clin Genet 62:478–481. https://doi.org/10.1034/j.1399-0004.2002.620610.x
Acknowledgements
We thank all patients and families for their support, as well as clinical and laboratory collaborators who are essential to our work. We are grateful to Ataxia UK, CureDRPLA and The National Institute for Health Research University College London Hospitals Biomedical Research Centre for funding and supporting the current recruitment and forthcoming natural history study into DRPLA; for which we are enthusiastic to receive email contact (h.houlden@ucl.ac.uk) and enroll patients from around the world.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaudhry, A., Anthanasiou-Fragkouli, A. & Houlden, H. DRPLA: understanding the natural history and developing biomarkers to accelerate therapeutic trials in a globally rare repeat expansion disorder. J Neurol 268, 3031–3041 (2021). https://doi.org/10.1007/s00415-020-10218-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10218-6