
Abstract
Myasthenia gravis (MG) is the archetypic disorder of both the neuromuscular junction and autoantibody-mediated disease. In most patients, IgG1-dominant antibodies to acetylcholine receptors cause fatigable weakness of skeletal muscles. In the rest, a variable proportion possesses antibodies to muscle-specific tyrosine kinase while the remainder of seronegative MG is being explained through cell-based assays using a receptor-clustering technique and, to a lesser extent, proposed new antigenic targets. The incidence and prevalence of MG are increasing, particularly in the elderly. New treatments are being developed, and results from the randomised controlled trial of thymectomy in non-thymomatous MG, due for release in early 2016, will be of particular clinical value. To help navigate an evidence base of varying quality, practising clinicians may consult new MG guidelines in the fields of pregnancy, ocular and generalised MG (GMG). This review focuses on updates in epidemiology, immunology, therapeutic and clinical aspects of GMG in adults.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Myasthenia gravis (MG) represents the archetypic disorder of both the neuromuscular junction (NMJ) and autoantibody-mediated disease. In most patients, IgG1-dominant antibodies to acetylcholine receptors (AChRs) cause fatigable weakness of skeletal muscles with an ocular onset in up to 85 % [1]. A variable proportion of patients lacking AChR antibodies, termed seronegative MG (SNMG), possess antibodies to muscle-specific tyrosine kinase (MuSK) [2, 3] and intriguingly, these antibodies are principally IgG4 [3–5]. The remainder of SNMG is now rapidly being explained via cell-based assays (CBAs) using a receptor-clustering technique [6–8], and, to a lesser extent, proposed new antigenic targets [9].
The incidence and prevalence of MG are increasing, particularly in older individuals [10, 11]. However, MG remains a rare disease and there are well-documented impediments to clinical trials including low participant recruitment [12]. Indeed, the EPITOME trial [13] in ocular MG (OMG) had to close recently due to failure to recruit adequate numbers [14]. Nevertheless, rituximab appears to show promise in MuSK MG [15] and a much-anticipated randomised controlled trial (RCT) of thymectomy in non-thymomatous MG [16] is due to report in early 2016. These results will be of great value since thymectomy has been offered for many years in this setting, without incontrovertible evidence of benefit compared to purely medical management [17, 18].
Expert clinical guidelines have reviewed pregnancy in MG [19], and management guidelines have been published for OMG [20] and generalised MG (GMG) (with some comments on OMG) [21]. This review will focus on GMG, as recent updates on congenital myasthenia [22] and OMG [23] have already been published. However, in addition to the epidemiology, immunology, therapeutics and clinical management of GMG, ongoing efforts to define the risk of generalisation (ROG) from ocular to generalised MG will be described.
Epidemiology: the changing face of myasthenia gravis
Calculations of total MG incidence and prevalence, based on 55 studies spanning 1950–2007, have yielded a pooled incidence rate (IR) of 5.3 per million person-years and a prevalence rate (PR) of 77.7 cases per million of the population [10]. Marked heterogeneity and the varying quality of epidemiological studies, were, not surprisingly, notable factors influencing these estimations over so many years [10]. Nevertheless, it is well recognised that MG prevalence has been rising since the middle of the last century [24], with improved recognition and diagnosis, medical and intensive care advances and patient longevity all playing a role [1, 10, 24].
The yearly incidence has also risen in all studies performed more recently [24, 25], due to a pronounced increase among older males as well as females [25, 26]. It remains appreciable even after adjustment for life expectancy [11, 27–29] and is not paralleled in younger females or children [30]. Studies of late-onset MG (LOMG) are hampered by the lack of unanimously agreed age of onset, with suggested cut-off points ranging from 40 to 75 years [1, 26, 28, 31–34] (see Box 1). The different HLA haplotype association in LOMG patients has been recognised since the 1980s [35], but the increase in incidence could also be related to environmental aspects [36] and better case detection [28].
Described immunological changes that occur with ageing including diminished B and T cell repertoires and activation, but environmental factors are also implicated [36]. Although some investigators have reported a higher rate of thymomas in LOMG [28], thymic hyperplasia is less common in older individuals [31–33, 37] and thymectomy unusual unless thymoma is present, limiting samples available for study [1]. The advent of robotic and other minimally invasive operative techniques may alter this scenario, since data now suggest that the operation is safe in older individuals and potentially beneficial if hyperplasia is present [37, 38]. Whether the surgical fitness of these elderly Japanese cohorts can be extrapolated to other elderly populations requires consideration.
Currently, a UK multicentre trial to define immunological, phenotypic and clinical features, including optimal treatment, of LOMG is recruiting and at the time of writing is at 41 % of target [39]. This is important as, historically, there is evidence of misdiagnosis among older individuals, particularly with cerebrovascular disease [26] and new diagnoses have been made in those as old as 98 [40]. Older patients are less likely to enter complete stable remission [27, 31] and are more likely to suffer exacerbation with poorer outcomes, including death [32, 41]. Their management is also challenging because they are more likely to have co-morbidities [33] and are at higher risk of side-effects from acetylcholinesterase inhibition [42] and steroids [33].
The epidemiological story in MuSK
MuSK MG has a younger age of onset and female predominance [43–47]. Initially, MuSK antibodies were reported to be prevalent in 70 % of ‘seronegative’ sera [2]. Subsequent cohorts, probably including patients with less severe disease, detected MuSK positivity less frequently in SNMG, but with wide variations [48]. For example, 3.8 % of SNMG tested positive for MuSK in a Chinese cohort [49] whereas in Italy the rate reached 47.4 % [44]. Accordingly, incidence and prevalence rates in MuSK MG epidemiological studies have differed with higher rates in Greece [annual IR of 0.32 patients/million population per year and prevalence rates (PR) of 2.92 per million population] compared to The Netherlands (where these rates are 0.10 and 1.9, respectively) [50, 51]. The variation in incidence in these studies and others [48; A Vincent, unpublished observations] support an environmental factor, but since there is also an association with HLA DQ5 across Dutch, Italian and Turkish populations [52–54], it suggests this acts on patients with a genetic predisposition.
Immunological advances: new techniques, an emerging pathogenic player and proposed new antigenic targets
Cell-based assays using clustered acetylcholine receptors
Traditionally, radioimmunoassays (RIA) (where the antigen is present in solution) are used for antibody ascertainment in MG. However, AChRs expressed on a mammalian cell line (human embryonic kidney cells, HEK) more closely mirror physiological conditions and increase sensitivity and specificity compared to solid phase assays for many antigenic targets [55, 56]. For MG, a clustered AChR cell-based assay (CBA) was established, in which the AChR subunits are expressed together with rapsyn, a post-synaptic protein crucial for AChR clustering at the NMJ. Using this method, AChR antibodies could be identified in 66 % of previously SNMG sera. These antibodies were primarily of the IgG1 sub-type and, in agreement with their pathogenic potential, could activate complement [6] and transfer neuromuscular transmission defects to mice [7].
50 % of previously seronegative OMG patients also had IgG1 clustered AChR antibodies [7]. As in all similar studies, it is not always clear whether the cohorts used are representative of the spectrum of incident cases, but clustered CBA antibodies have been found helpful in the diagnosis of young or childhood MG with often mild or ocular disease, responding well to immunotherapies [8]. The phenotype may become better defined as more laboratories adopt the clustered CBA [57].
The use of CBAs, with clustered antigens if appropriate, should improve both diagnosis and management of previously seronegative patients. Preliminary results from Oxford suggest 8 % of patients without MuSK or other antibodies bound detectably to MuSK expressed on HEK cells [56]. An international study probing seronegative sera from 13 countries for binding to MuSK by CBA identified antibodies in 13 % of samples [58], but many of the antibodies detected were predominantly of the IgM type, which is of unknown relevance.
IgG4: a new pathogenic player
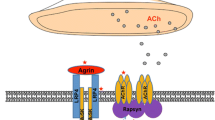
MuSK is a post-synaptic protein which is critical for the development and maintenance of the NMJ [reviewed in 59]. Agrin is released from the presynaptic nerve terminal and binds to low-density lipoprotein receptor 4 (Lrp4), which in turn binds to and activates MuSK. This leads to MuSK phosphorylation, ultimately resulting in the clustering of rapsyn and AChR on top of post-synaptic folds; this AChR clustering is essential for efficient transmission at the NMJ [60, 61].
MuSK antibodies are mainly IgG4 [3], in contrast to the IgG1 and IgG3 dominance of AChR antibodies. AChR antibodies principally act through complement activation, and by cross-linking and internalisation of receptors [1], both of which require antibody divalency. However, MuSK IgG4 antibodies are thought to be single chain rather than divalent, and do not activate complement or cause cross-linking of the MuSK molecules [4, 62]. Nevertheless, the pathogenicity of MuSK IgG4 antibodies has been demonstrated and overall the titres in a patient relate well to disease severity with reductions on remission [63, 64]. Injection of purified IgG4 into experimental animals leads to defects in neuromuscular transmission with AChR loss [65] as does injection of purified IgG [66].
MuSK IgG4 antibodies act through direct inhibition of MuSK-Lrp4 binding, without receptor dimerization or endocytosis, whereas this was not found with the remaining IgG1–3 antibodies [4, 5]. However, both these IgG1–3 antibodies and IgG4 antibodies could disperse AChR clusters in a mouse muscle cell line [4], implying that IgG1–3 could play a role in the disease; it is possible that by binding divalently, and activating phosphorylation, they cause desensitisation of MuSK leading to loss of function by that mechanism.
IgG4 antibodies were previously little known in autoimmune disease and thought to occur as a benign phenomenon in conjunction with resolution of allergic reactions [4, 62]. However, they are recognised in other diseases, such as forms of pemphigus, and represent a field of growing interest [62].
Continuing the search for new epitopes
Although antibodies detectable via clustered CBAs are likely to be diagnostic in the majority of currently ‘seronegative’ patients, the hunt continues for antibodies to other elements of the NMJ which may be disease causative in smaller sub-sets of patients.
LRP4
As the partner of MuSK, Lrp4 is similarly essential in development and for normal function of the adult NMJ, where it performs both anterograde and retrograde signalling roles [67]. These roles highlighted it as a putative antigen of interest, and LRP4 antibodies have been reported in Japanese [68] and European [69, 70] patients. The antibodies were of the complement-activating IgG1 type [68] and impeded agrin-induced clustering of AChRs [69, 70]. These antibodies have now been examined by a number of groups and overall their presence in seronegative sera has varied widely (Table 1).
These discrepant results are likely to emanate at least in part from the different assays used by separate research groups [9]. The potential of certain commercial secondary antibodies to detect non-specific binding of IgM antibodies to MuSK has been noted (S Huda, unpublished results). In addition to clarifying the specificities and sensitivities of these different LRP4 antibody assays, the relevance of the antibodies when found in association with other pathogenic myasthenia antibodies (see Table 1) needs exploration. Further definition of the clinical phenotype of LRP4 disease is also required although present information suggests this is a predominantly female cohort with mild symptoms, similar to AChR-antibody-positive patients [69, 71].
Agrin and ColQ
Antibodies to agrin have recently been identified in a small number of ‘triple negative’ MG sera (samples negative for AChR, MuSK and LRP4 antibodies) at proportions ranging from 15 to 50 % [9, 72]. These antibodies have sometimes been at low titres [72] and can be found with [9, 72] or only with AChR or MuSK antibodies [73]. This suggests technical difficulties and methodologies need to improve before the significance of agrin antibodies can be evaluated.
ColQ tethers MuSK within the synapse [74] and is thought to interact also with MuSK. ColQ antibodies were reported in 3–4 % of all MG patient sera tested and 1.2–5.5 % of the AChR/MuSK/LRP4 negative samples [9, 75] but again the specificities are unclear and further work is required to delineate their role [75].
A link with antibody-mediated demyelinating disorders?
The co-occurrence of MG and demyelinating disorders happens more than would be expected by chance [76]. Recently, a cohort of 16 patients with neuromyelitis optica (NMO) and MG was described. In such cases, the MG tended to be mild and 90 % presented with a prior history of NMO; however, Aquaporin-4 (AQP4) antibodies could pre-date clinically evident NMO by up to 16 years [76]. Of note, in other case series of MG presenting with demyelinating disorders described as MS or ADEM, AQP4 testing was not reported; some of these may have represented NMO [77–79] and in at least one case this was subsequently confirmed [78]. LRP4 antibodies have also been described in NMO patients, albeit without a known diagnosis of MG, but not other neurological disorders [70]. Why these two diseases should occur in this order, with AQP4 antibodies rising over time, is not understood, although it is speculated that thymectomy may play a triggering role. Clinicians should at least be vigilant to the possible co-existence of NMO with MG, particularly in young patients with AChR-antibody-positive disease and should test for AQP4 antibodies in MG patients who develop MS or other demyelinating disorders [76].
Risk of generalisation from OMG: setbacks and progress
The debate over the risk of generalisation from ocular to generalised MG continues. Retrospective patient data indicate that most OMG patients who develop GMG do so in the first 2 years [80–82]. However, unanimity has not been reached on other risk factors, or on whether early intervention with immunosuppressants, particularly corticosteroids, can delay or prevent the onset of GMG [23, 83]. This is of growing relevance in the era of expanding therapeutic and surgical options, and to avoid exposing patients to unnecessary adverse effects of immunotherapies.
Several retrospective cohorts have reported lower rates of generalisation in patients treated with corticosteroids compared to those treated with acetylcholinesterase inhibitors alone [81, 82, 84], but solid conclusions cannot be drawn from these non-randomised studies where confounding factors such as duration of OMG, serological status and differing steroid regimes are present. In some cases, patients received lengthy courses of steroids (up to 92 months) or were maintained on low-dose steroid regimes, either of which could have masked the development of GMG [82, 84]. Moreover, Grob’s survey of nearly 2000 patients between 1940 and 2000 found stable rates of generalisation from OMG [85], which might seem to contradict a disease-modifying effect of corticosteroid treatment.
The EPITOME trial [13], which was due to address these issues, would therefore have been of great clinical utility but unfortunately its closure was recently announced due to poor recruitment [14]. A UK initiative to develop a prognostic ‘ROG’ score is still in progress and reported preliminary results at the 2015 Association of British Neurologists conference. Using available case notes, investigators identified the three positive factors most predictive of secondary generalisation as being thymic hyperplasia, seropositivity and co-morbidity (including but not limited to other autoimmune disorders) [86]. Such a model should now be prospectively validated and could help identify high-risk patients for whom early immunosuppression would be beneficial.
The therapeutic landscape
Pyridostigmine and corticosteroids retain a central role in the management of GMG [87]. Use of azathioprine as a steroid-sparing agent is supported by an RCT [88] but limited high quality evidence underlies many other immunosuppressants [87]. However, recently, a single-blinded trial proposed methotrexate as an alternative to azathioprine [89]. While this trial was devised to validate methotrexate in a resource-limited setting, it may have applicability for azathioprine-intolerant individuals.
MuSK MG patients have traditionally represented a clinical challenge as they exhibit poor response to acetylcholinesterase inhibitors [43–45, 90]. Rituximab, an anti-CD20 monoclonal antibody, is emerging as a potential option in this cohort [15, 91–93]. Following rituximab treatment, some patients even revert to a seronegative status [15]. Of particular interest, specific monitoring of IgG sub-classes in five clinically improved rituximab-treated MuSK MG individuals demonstrated significantly reduced IgG4 titres in all five. On the other hand, both clinical and serologic impact was much less favourable in AChR antibody patients treated in the same study [92]. Indeed, rituximab appears to be a useful treatment in other IgG4-related diseases and to act by eliminating a population of B- or plasma cells responsible for the production of IgG4 antibodies [62, 92]. Rituximab’s effect on T cell response may also be relevant, and an increase in T-regulatory cells has been observed post-rituximab administration in a refractory MuSK, but not AChR-positive, patient [94]. Further work is required to determine the optimal timing and administration schedule of rituximab in MG [93].
Another monoclonal antibody being considered for MG is eculizumab, which targets the C5 protein of the complement cascade, and so might protect the NMJ from complement-mediated damage. In a small phase II trial, there was significant change on the quantitative myasthenia gravis score (QMGS) with eculizumab compared to placebo [95]. A phase III trial is now in progress, aiming to enrol 92 patients [96]. The weekly dosing schedule and high cost of this medication may limit its use.
Early stage agents in development include EN101/Monarsen, an antisense oligonucleotide to mRNA of a splicing variant of acetylcholinesterase which is elevated in mice with experimental autoimmune MG and in patients. It is currently unclear whether clinical effect is due to inhibition of the splicing variant, producing symptomatic relief, or anti-inflammatory and immunomodulatory actions via the NF-κB pathway [97, 98]. Another drug in phase II studies is Tirasemtiv, which enhances skeletal muscle’s response to calcium and may be of benefit in combination with acetylcholinesterase inhibitors [99].
Emergency treatments
A RCT of plasma exchange (PLEX) compared to IVIg in myasthenic crisis found equivalence between the two treatments [100, 101]. After 2 weeks, similar numbers improved in both groups, as measured on the QMGS. Although more patients (17.5 %) had worse 2-week QMGS scores in the IVIg group compared to those receiving PLEX (2 %), this was non-significant [100]. Recent hospital data show a trend to declining use of PLEX, which may be prompted by the invasive nature of this treatment modality [41] and its lack of availability in many centres. Nevertheless, it is important to note that in several cohorts, MuSK MG patients appear to respond less well to IVIg than PLEX [43, 45, 47, 90].
Thymectomy: towards a definitive answer
The announcement of the results of the MGTX RCT of thymectomy vs. medical treatment in AChR-antibody-positive GMG patients will take place in Oxford in early 2016 and is likely to be a milestone event for myasthenic treatment. The protocol of this multicentre (>40 centre) trial has previously been published and is a single-blind, double armed trial evaluating trans-sternal thymectomy versus no operation in patients on prednisolone [16]. Some initial reports of thymic pathology in trial participants have been published and revealed 25–40 % thymic hyperplasia depending on the immunostaining method used, and parenchymal changes comparable to the non-MG population [102].
Where a thymoma is present, thymectomy is indicated to treat the tumour but not the myasthenia; however, frail and elderly patients are sometimes treated medically. On the other hand, in MuSK MG patients, thymic pathology is relatively rare [45, 90, 103, 104] although cannot be precluded [46, 105]. Few MuSK patients appear to improve following the procedure [43, 44] or, similar to AChR patients, are maintained on corticosteroids [47] which cloud the interpretation of any operative effects. Until MGTX reports, the situation remains difficult with no high quality evidence available to support decision making [16, 17]. The American Academy of Neurology (AAN) has developed guidelines to assist in this scenario [17], advising thymectomy be viewed as an option to improve clinical status and remission rates.
Another conundrum is whether a trans-sternal, minimally invasive or robotic approach offers best results. The MGTX will not answer this question, but it is sensible first to establish clinical benefit of any operative intervention prior to probing competing techniques [106]. Comparable results of around 28–34 % complete stable remission (CSR) or CSR and pharmacological remission (PR) have been achieved in single-centre, non-randomised case series from different hospitals [106–110]. One single-centre review of patient records displayed superior rates of complete remission with robotic (39.25 %) compared to thoracoscopic surgery (20.3 %), but the dates of all thoracoscopic surgeries predated robotic procedures, introducing the possibility of confounding historical factors [111].
New best practice guidelines
New best practice guidelines have been released in the past 2 years which address ocular and generalised MG as well as pregnancy in MG [19–21]. Key points are summarised in Box 2. An important message for expectant mothers is that birth plans should aim for a hospital delivery as babies are at risk of transient neonatal MG irrespective of the mother’s disease status. Therefore, home births and midwife-led units are not advised [21].
Conclusions
This paper has reviewed a number of evolving areas in GMG. In particular, patient diagnosis and management will improve as the pool of ‘seronegative’ MG decreases. However, care should be taken to establish the pathogenicity of newly identified antibodies. It is likely the field of IgG4-mediated disease will continue to gain scientific momentum.
More work is required to understand the phenomenon of increasing incidence of LOMG, with elderly patients posing a diagnostic and therapeutic challenge. Within the next 12 months, the results of the MGTX trial may answer one of the longest-standing questions in MG, namely, the role of thymectomy in non-thymomatous disease.
References
Meriggioli MN, Sanders DB (2009) Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol 8(5):475–490
Hoch W, McConville J, Helms S et al (2001) Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 7(3):365–368
McConville J, Farrugia ME, Beeson D et al (2004) Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol 55(4):580–584
Koneczny I, Cossins J, Waters P et al (2013) MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1-3 can disperse preformed agrin-independent AChR clusters. PLoS One 8(11):e80695
Huijbers MG, Zhang W, Klooster R et al (2013) MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci USA 110(51):20783–20788
Leite MI, Jacob S, Viegas S et al (2008) IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 131(Pt 7):1940–1952
Jacob S, Viegas S, Leite MI et al (2012) Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 69(8):994–1001
Rodríguez Cruz PM, Al-Hajjar M, Huda S et al (2015) Clinical features and diagnostic usefulness of antibodies to clustered acetylcholine receptors in the diagnosis of seronegative myasthenia gravis. JAMA Neurol 72(6):642–649
Cossins J, Belaya K, Zoltowska K et al (2012) The search for new antigenic targets in myasthenia gravis. Ann N Y Acad Sci 1275:123–128
Carr AS, Cardwell CR, McCarron PO et al (2010) A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol 18(10):46
Pakzad Z, Aziz T, Oger J (2011) Increasing incidence of myasthenia gravis among elderly in British Columbia, Canada. Neurology 76(17):1526–1528
Benatar M, Sanders DB, Burns TM et al (2012) Recommendations for myasthenia gravis clinical trials. Muscle Nerve 45(6):909–917
Benatar M, Sanders DB, Wolfe GI et al (2012) Design of the efficacy of prednisone in the treatment of ocular myasthenia (EPITOME) trial. Ann N Y Acad Sci 1275:17–22
Clinical trials.gov. Efficacy of prednisone in the treatment of ocular myasthenia (EPITOME). ClinicalTrials.gov identifier NCT00995722 [on-line]. Web-page last updated 2015 Jan 6. https://clinicaltrials.gov/ct2/show/NCT00995722?term=EPITOME&rank=1. Accessed 13 Aug 2015
Keung B, Robeson KR, DiCapua DB et al (2013) Long-term benefit of rituximab in MuSK autoantibody myasthenia gravis patients. J Neurol Neurosurg Psychiatry 84(12):1407–1409
Wolfe GI, Kaminski HJ, Jaretzki A et al (2003) Development of a thymectomy trial in nonthymomatous myasthenia gravis patients receiving immunosuppressive therapy. Ann N Y Acad Sci 998:473–480
Gronseth GS, Barohn RJ (2000) Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): report of the quality standards subcommittee of the American academy of neurology. Neurology 55(1):7–15
Cea G, Benatar M, Verdugo RJ et al (2013) Thymectomy for non-thymomatous myasthenia gravis. Cochrane Database Syst Rev 10:CD008111
Norwood F, Dhanjal M, Hill M et al (2014) Myasthenia in pregnancy: best practice guidelines from a UK multispecialty working group. J Neurol Neurosurg Psychiatry 85(5):538–543
Kerty E, Elsais A, Argov Z et al (2014) EFNS/ENS Guidelines for the treatment of ocular myasthenia. Eur J Neurol 21(5):687–693
Sussman J, Farrugia ME, Maddison P et al (2015) Myasthenia gravis: association of British Neurologists’ management guidelines. Pract Neurol 15(3):199–206
Cruz PMR, Palace J, Beeson D (2014) Inherited disorders of the neuromuscular junction: an update. J Neurol. 261(11):2234–2243
Wong SH, Huda S, Vincent A et al (2014) Ocular myasthenia gravis: controversies and updates. Curr Neurol Neurosci Rep 14(1):421
Phillips LH, Torner JC (1996) Epidemiologic evidence for a changing natural history of myasthenia gravis. Neurology 47(5):1233–1238
Poulas K, Tisbri E, Kokla A et al (2001) Epidemiology of seropositive myasthenia gravis in Greece. J Neurol Neurosurg Psychiatry 71(3):352–356
Vincent A, Clover L, Buckley C et al (2003) Evidence of underdiagnosis of myasthenia gravis in older people. J Neurol Neurosurg Psychiatry 74(8):1105–1108
Matsui N, Nakane S, Nakagawa Y et al (2009) Increasing incidence of elderly onset patients with myasthenia gravis in a local area of Japan. J Neurol Neurosurg Psychiatry 80(10):1168–1171
Murai H, Yamashita N, Watanabe M et al (2011) Characteristics of myasthenia according to onset-age: Japanese nationwide survey. J Neurol Sci 305(1–2):97–102
Somnier FE (2005) Increasing incidence of late-onset anti-AChR antibody-seropositive myasthenia gravis. Neurology 65(6):928–930
Parr JR, Andrew MJ, Finnis M et al (2014) How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch Dis Child 99(6):539–542
Alkhawajah N, Oger J (2013) Late-onset myasthenia gravis: a review when incidence in older adults keeps increasing. Muscle Nerve 48(5):705–710
de Meel RH, Lipka AF, van Zwet EW et al (2015) Prognostic factors for exacerbations and emergency treatments in myasthenia gravis. J Neuroimmunol 15(282):123–125
Evoli A, Batocchi AP, Minisci C et al (2000) Clinical characteristics and prognosis of myasthenia gravis in older people. J Am Geriatr Soc 48(11):1442–1448
Živković SA, Clemens PR, Lacomis D (2012) Characteristics of late-onset myasthenia gravis. J Neurol 259(10):2167–2171
Compston DA, Vincent A, Newsom-Davis J et al (1980) Clinical, pathological, HLA antigen and immunological evidence for disease heterogeneity in myasthenia gravis. Brain 103(3):579–601
Linton PJ, Dorshkind K (2004) Age-related changes in lymphocyte development and function. Nat Immunol 5(2):133–139
Uzawa A, Kawaguchi N, Kanai T et al (2015) Two-year outcome of thymectomy in non-thymomatous late-onset myasthenia gravis. J Neurol 262(4):1019–1023
Tsuchida M, Yamoto Y, Souma T et al (1999) Efficacy and safety of extended thymectomy for elderly patients with myasthenia gravis. Ann Thorac Surg 67(6):1563–1567
UK clinical research network study portfolio. A prospective study of late-onset myasthenia gravis. UKCRN 13582 [on-line]. http://public.ukcrn.org.uk/search/StudyDetail.aspx?StudyID=13582. Accessed 13 Aug 2015
Phillips LH, Juel VC (1999) Myasthenia gravis in the tenth decade. Muscle Nerve 22(9):1297–1298
Alshekhlee A, Miles JD, Katirji B et al (2009) Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 72(18):1548–1554
Punga AR, Sawada M, Stalberg EV (2008) Electrophysiological signs and the prevalence of adverse effects of acetylcholinesterase inhibitors in patients with myasthenia gravis. Muscle Nerve 37(3):300–307
Sanders DB, El-Salem K, Massey JM et al (2003) Clinical aspects of MuSK antibody positive seronegative MG. Neurology 60(12):1978–1980
Evoli A, Tonali PA, Padua L et al (2003) Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain 126(Pt 10):2304–2311
Guptill JT, Sanders DB, Evoli A (2011) Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 44(1):36–40
Lavrnic D, Losen M, Vujic A et al (2005) The features of myasthenia gravis with autoantibodies to MuSK. J Neurol Neurosurg Psychiatry 76(8):1099–1102
Pasnoor M, Wolfe GI, Nations S et al (2010) Clinical findings in MuSK-antibody positive myasthenia gravis: a US experience. Muscle Nerve 41(3):370–374
Vincent A, Leite MI (2005) Neuromuscular junction autoimmune disease: muscle specific kinase antibodies and treatments for myasthenia gravis. Curr Opin Neurol 18(5):519–525
Yeh JH, Chen WH, Chiu HC et al (2004) Low frequency of MuSK antibody in generalized seronegative myasthenia gravis among Chinese. Neurology 62(11):2131–2132
Tsiamalos P, Kordas G, Kokla A et al (2009) Epidemiological and immunological profile of muscle-specific kinase myasthenia gravis in Greece. Eur J Neurol 16(8):925–930
Niks EH, Kuks JB, Verschuuren JJ (2007) Epidemiology of myasthenia gravis with anti-muscle specific kinase antibodies in the Netherlands. J Neurol Neurosurg Psychiatry 78(4):417–418
Niks EH, Kuks JB, Roep BO et al (2006) Strong association of MuSK antibody–positive myasthenia gravis and HLA-DR14-DQ5. Neurology 66(11):1772–1774
Bartoccioni E, Scuderi F, Augugliaro et al (2009) HLA class II allele analysis in MuSK-positive myasthenia gravis suggests a role for DQ5. Neurology 72(2):195–197
Alahgholi-Hajibehzad M, Yilmaz V, Gülsen-Parman Y et al (2013) Association of HLA-DRB*1, -DRB*16 and -DQB1*05 with MuSK-myasthenia gravis in patients from Turkey. Hum Immunol 74(12):1633–1635
Waters P, McKeon A, Leite MI et al (2012) Serologic diagnosis of NMO. A multicenter comparison of aquaporin-4-IgG assays. Neurology 78(9):665–671
Rodríguez Cruz PM, Huda S, López-Ruiz P et al (2015) Use of cell-based assays in myasthenia gravis and other antibody-mediated diseases. Exp Neurol 270:66–71
Devic P, Petiot P, Simonet T et al (2014) Antibodies to clustered acetylcholine receptor: expanding the phenotype. Eur J Neurol 21(1):130–134
Tsonis AI, Zisimopoulou P, Lazaridis K et al (2015) MuSK autoantibodies in myasthenia gravis detected by cell based assay—a multinational study. J Neuroimmunol 15(284):10–17
Koneczny I, Cossins J, Vincent A (2014) The role of muscle-specific tyrosine kinase (MuSK) and mystery of MuSK myasthenia gravis. J Anat 224(1):29–35
Kim N, Steigler AL, Cameron TO et al (2008) Lrp4 is a receptor for agrin and forms a complex with MuSK. Cell 135(2):334–342
Zhang B, Luo S, Wang Q et al (2008) LRP4 serves as a coreceptor of agrin. Neuron 60(2):285–297
Huijbers MG, Querol LA, Niks EH et al (2015) The expanding field of IgG4-mediated neurological autoimmune disorders. Eur J Neurol 22(8):1151–1161
Bartoccioni E, Scuderi F, Minicuci GM et al (2006) Anti-MuSK antibodies: correlation with myasthenia gravis severity. Neurology 67(3):505–507
Niks EH, van Leeuwen Y, Leite MI et al (2008) Clinical fluctuations in MuSK myasthenia gravis are related to antigen-specific IgG4 instead of IgG1. J Neuroimmunol 195(1–2):151–156
Klooster R, Plomp JJ, Huijbers MG et al (2012) Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 135(Pt 4):1081–1101
Viegas S, Jacobson L, Waters P et al (2012) Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol 234(2):506–512
Yumoto N, Kim N, Burden S (2012) Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature 489(7416):438–442
Higuchi O, Hamuro J, Motomura M et al (2011) Autoantibodies to low-density lipoprotein receptor–related protein 4 in Myasthenia Gravis. Ann Neurol 69(2):418–422
Pevzner A, Schoser B, Peters K et al (2012) Anti-LRP4 autoantibodies in AChR- and MuSK-antibody negative myasthenia gravis. J Neurol 259(3):427–435
Zhang B, Tzartos JS, Belimezi M et al (2012) Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 69(4):445–451
Zisimopoulou P, Evangelakou P, Tzartos J et al (2014) A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun 52:139–145
Zhang B, Shen C, Bealmear B et al (2014) Autoantibodies to agrin in myasthenia gravis patients. PLoS One 9(3):e91816
Gasperi C, Melms A, Schoser B et al (2014) Anti-agrin autoantibodies in myasthenia gravis. Neurology 82(22):1976–1983
Cartaud A, Strochlic L, Guerra M et al (2004) MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J Cell Biol 165(4):505–515
Katarzyna M, Belaya K, Leite M et al (2015) Collagen Q—a potential target for autoantibodies in myasthenia gravis. J Neurol Sci 348(1–2):241–244
Leite MI, Coutinho E, Lana-Peixoto M et al (2012) Myasthenia gravis and neuromyelitis optica spectrum disorder. A multicenter study of 16 patients. Neurology 78(20):1601–1607
Gotkine M, Fellig Y, Abramsky O (2006) Occurrence of CNS demyelinating disease in patients with myasthenia gravis. Neurology 67(5):881–883
Isbister CM, Mackenzie PJ, Anderson D (2003) Co-occurrence of multiple sclerosis and myasthenia gravis in British Columbia. Mult Scler 9(6):550–553
Spillane J, Christofi G, Sidle KC et al (2013) Myasthenia gravis and neuromyelitis optica: a causal link. Mult Scler Relat Disord 2(3):233–237
Bever CT, Aquino AV, Penn AS et al (1983) Prognosis of ocular myasthenia. Ann Neurol 14(5):516–519
Sommer N, Sigg B, Melms A et al (1997) Ocular myasthenia gravis: response to long term immunosuppressive treatment. J Neurol Neurosurg Psychiatry 62(2):156–162
Kupersmith MJ (2009) Ocular myasthenia gravis: treatment successes and failures in patients with long-term follow-up. J Neurol 256(8):1314–1320
Wong SH, Plant GT, Cornblath W (2015) Does treatment of ocular myasthenia gravis with early immunosuppressive therapy prevent secondarily generalization and should it be offered to all such patients? J Neuroophthalmol [Epub ahead of print]
Mee J, Paine M, Byrne E et al (2003) Immunotherapy of ocular myasthenia gravis reduces conversion to generalized myasthenia gravis. J Neuroophthalmol 23(4):251–255
Grob D, Brunner N, Namba T et al (2008) Lifetime course of myasthenia gravis. Muscle Nerve 37(2):141–149
Wong S, Petrie A, Plant G (2015) Ocular MG: towards a risk of generalisation (‘ROG’) score. In: Association of British Neurologists (ABN) annual meeting, final programme and abstract book, 2015 May 20-2; Harrogate, UK: ABN; 2015 p 48
Kumar V, Kaminski HJ (2011) Treatment of myasthenia gravis. Curr Neurol Neurosci Rep 11(1):89–96
Palace J, Newsom-Davis J, Lecky B et al (1998) A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology 50(6):1778–1783
Heckmann J, Rawoot A, Bateman K et al (2011) A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 5(11):97
Zhou L, McConville J, Chaudhry V et al (2004) Clinical comparison of muscle-specific tyrosine kinase (MuSK) antibody-positive and -negative myasthenic patients. Muscle Nerve 30(1):55–60
Maddison P, McConville J, Farrugia ME et al (2011) The use of rituximab in myasthenia gravis and Lambert Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry 82(6):671–673
Díaz-Manera J, Martínez-Hernández E, Querol L et al (2012) Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 78(3):189–193
Iorio R, Damato V, Aboini PE et al (2015) Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta-analysis. J Neurol 262(5):1115–1119
Catzola V, Battaglia A, Buzzonetti A et al (2013) Changes in regulatory T-cells after rituximab in two patients with refractory myasthenia gravis. J Neurol 260(8):2163–2165
Howard JF, Barohn RJ, Cutter GR et al (2013) A randomized, double-blind, placebo-controlled phase II trial of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve 48(1):76–84
Clinical trials.gov. Safety and efficacy of eculizumab in refractory generalized myasthenia gravis (REGAIN study). ClinicalTrials.gov identifier NCT01997229 [on-line]. Web-page last updated 2015 July 6. https://clinicaltrials.gov/ct2/show/NCT01997229. Accessed 13 Aug 2015
Argov Z, McKee D, Agus S et al (2007) Treatment of human myasthenia with oral antisense suppression of acetylcholinesterase. Neurology 69(7):699–700
Sussman J, Argov Z, Wirguin Y et al (2012) Further developments with antisense treatment for myasthenia gravis. Ann N Y Acad Sci 1275:13–16
Sanders DB, Rosenfeld J, Dimachkie MM et al (2015) A double-blinded, randomized, placebo-controlled trial to evaluate efficacy, safety, and tolerability of single doses of Tirasemtiv in patients with acetylcholine receptor-binding antibody-positive myasthenia gravis. Neurotherapeutics 12(2):455–460
Barth D, Nouri MN, Ng E et al (2011) Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 76(23):2017–2023
Bril V, Barnett-Tapia C, Barth D et al (2012) IVIg and PLEX in the treatment of myasthenia gravis. Ann N Y Acad Sci 1275:1–6
Marx A, Pfister F, Schalke B et al (2012) Thymus pathology observed in the MGTX trial. Ann N Y Acad Sci 1275:92–100
Lauriola L, Ranelletti F, Maggiano N et al (2005) Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology 64(3):536–538
Leite MI, Ströbel P, Jones M et al (2005) Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol 57(3):444–448
Saka E, Topcuoglu MA, Akkaya B et al (2005) Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology 65(5):782–783 (author reply 782–3)
Shrager JB (2010) Extended transcervical thymectomy: the ultimate minimally invasive approach. Ann Thorac Surg 89(6):S2128–S2134
Freeman RK, Ascioti AJ, van Woerkom JM et al (2011) Long-term follow-up after robotic thymectomy for nonthymomatous myasthenia gravis. Ann Thorac Surg 92(3):1018–1022 (discussion 1022–3)
Marulli G, Schiavon M, Perissinotto E et al (2013) Surgical and neurologic outcomes after robotic thymectomy in 100 consecutive patients with myasthenia gravis. J Thorac Cardiovasc Surg 145(3):730–735 (discussion 735–6)
Spillane J, Hayward M, Hirsch NP et al (2013) Thymectomy: role in the treatment of myasthenia gravis. J Neurol 260(7):1798–1801
Keijzers M, de Baets M, Hochstenbag M et al (2015) Robotic thymectomy in patients with myasthenia gravis: neurological and surgical outcomes. Eur J Cardiothorac Surg 48(1):40–45
Rückert JC, Swierzy M, Ismail M (2011) Comparison of robotic and nonrobotic thoracoscopic thymectomy: a cohort study. J Thorac Cardiovasc Surg 141(3):673–677
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Angela Vincent and University of Oxford holds a patent for MuSK antibody assays, licensed to Athena Diagnostics, and receives royalties from this.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Binks, S., Vincent, A. & Palace, J. Myasthenia gravis: a clinical-immunological update. J Neurol 263, 826–834 (2016). https://doi.org/10.1007/s00415-015-7963-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-015-7963-5