
Abstract
Neuromyelitis optica (NMO, Devic’s syndrome), long considered a clinical variant of multiple sclerosis, is now regarded as a distinct disease entity. Major progress has been made in the diagnosis and treatment of NMO since aquaporin-4 antibodies (AQP4-Ab; also termed NMO-IgG) were first described in 2004. In this review, the Neuromyelitis Optica Study Group (NEMOS) summarizes recently obtained knowledge on NMO and highlights new developments in its diagnosis and treatment, based on current guidelines, the published literature and expert discussion at regular NEMOS meetings. Testing of AQP4-Ab is essential and is the most important test in the diagnostic work-up of suspected NMO, and helps to distinguish NMO from other autoimmune diseases. Furthermore, AQP4-Ab testing has expanded our knowledge of the clinical presentation of NMO spectrum disorders (NMOSD). In addition, imaging techniques, particularly magnetic resonance imaging of the brain and spinal cord, are obligatory in the diagnostic workup. It is important to note that brain lesions in NMO and NMOSD are not uncommon, do not rule out the diagnosis, and show characteristic patterns. Other imaging modalities such as optical coherence tomography are proposed as useful tools in the assessment of retinal damage. Therapy of NMO should be initiated early. Azathioprine and rituximab are suggested as first-line treatments, the latter being increasingly regarded as an established therapy with long-term efficacy and an acceptable safety profile in NMO patients. Other immunosuppressive drugs, such as methotrexate, mycophenolate mofetil and mitoxantrone, are recommended as second-line treatments. Promising new therapies are emerging in the form of anti-IL6 receptor, anti-complement or anti-AQP4-Ab biologicals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
NEMOS
The Neuromyelitis Optica Study Group (NEMOS; see http://www.nemos-net.de) was initiated in 2008 by neurologists at 25 German university and academic teaching hospitals as an open-access network to improve the care of patients with neuromyelitis optica (NMO). Since then, the group has organized a number of national and international meetings and symposia on NMO, collected and analyzed data on epidemiological, clinical, and magnetic resonance imaging (MRI) characteristics of NMO in a large German cohort [1], and published recommendations on the diagnosis and treatment of NMO in Germany [2]. In the following report, these recommendations are updated to reflect the most recent literature in the field and current scientific knowledge. The 2010 guidelines of the European Federation of Neurological Societies (EFNS) on the diagnosis and management of NMO, guidelines published by an international expert group [3], and the evidence-based guidelines on clinical evaluation and treatment of transverse myelitis published by the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology can also be referred to for additional information [4, 5].
Introduction
Neuromyelitis optica is an immune-mediated chronic inflammatory disease of the central nervous system (CNS) [1, 6, 7]. NMO was first described in the 19th century and was long considered a clinical variant of multiple sclerosis (MS) [8–13]. Clinically, it presents with optic neuritis (ON) and myelitis, often characterized by poor or no recovery. Imaging typically shows longitudinally extensive lesions spanning three or more vertebral segments. Histopathologically, NMO is characterized by astrocytic damage, demyelination, neuronal loss, and often pronounced necrosis [14–16]. The discovery of perivascular antibody and complement deposition within active lesions and the subsequent discovery of specific autoantibodies (aquaporin-4 antibodies, AQP4-Ab; also termed NMO-IgG) in the serum of NMO patients indicated that humoral immunity is involved in the majority of cases. AQP4-Ab-positive NMO is now distinguished from MS as an independent disease entity [17–27]. Accordingly, serological identification of NMO-IgG has also been included as an additional criterion in all diagnostic criteria for NMO currently in use [2, 4, 28, 29].
Epidemiology
Solid data on the incidence and prevalence of NMO are lacking. Its prevalence is estimated to range from less than 1 to 4.4/100.000 in the Western world [31–33]. In the past, many patients (>20 %) with NMO were misdiagnosed with MS, especially before NMO-IgG testing became widely available [1]. Notably more women than men have NMO (ratio 9:1, compared with just 2:1 in MS) [1, 34]. The median age at onset, 39 years, is approximately 10 years higher than in MS [1, 30]. However, cases of onset during childhood and in the elderly have been described [1, 35–38]. NMO takes either a relapsing or a monophasic course, with the former predominating (approximately 80–90 % of cases) [1, 30]. Compared with MS, AQP4-Ab-positive NMO is more frequently associated with other autoimmune diseases such as myasthenia gravis, systemic lupus erythematosus, Sjögren’s syndrome, celiac disease, and sarcoidosis [1, 39–52]. In up to 20–30 % of cases, NMO attacks are preceded by infection or vaccination [1, 7]. Age at onset and genetic factors may influence the clinical outcome [53].
Only few reports on the influence of pregnancies in NMO exist. Two studies reported an increase in relapse rate in the first 3 or 6 months, respectively, post partum [54, 55].
Diagnostic criteria
According to the criteria proposed by Wingerchuk et al. [28] in 2006, a diagnosis of NMO can be made with high specificity if, in addition to a history of at least one episode of ON and one episode of myelitis, two of the following three supporting criteria are met:
-
1.
Contiguous spinal cord MRI lesion extending over three or more vertebral segments
-
2.
Brain MRI not meeting Paty’s diagnostic criteria for MSFootnote 1 [56] at disease onset.Footnote 2
-
3.
NMO-IgG seropositive statusFootnote 3
Of note, the sensitivity and specificity of these criteria [28] were defined using brain MRI at disease onset as first preference. If the first scan available was taken at a later time and was negative for MS, it was assumed that the onset scan would also have been negative. By contrast, the authors did not indicate whether the brain MRI criterion should be applied at all if the first available scan was taken at a later time and met MS criteria. However, we believe that the diagnostic criteria proposed by Wingerchuk et al. should, in general, not be applied to rule out NMO if any of the paraclinical procedures required to evaluate the three supporting criteria were not performed. Of course, a diagnosis of NMO can be made if the index events and any two of the three supporting criteria are met, even though information on the third supporting criterion is not available.
More broadly, those criteria should be primarily used to make, rather than to exclude, a diagnosis of NMO, because brain lesions and (far more rarely) short spinal cord lesions—individually or combined—may in fact be present in patients with otherwise typical NMO (as confirmed by AQP4-Ab seropositivity and/or occurrence of longitudinal extensive transverse myelitis (LETM) in the later disease course in these patients) [1].
“NMO-spectrum disorder”—abortive and atypical manifestations
AQP4-Ab have been demonstrated in patients with conditions other than classical NMO, including isolated LETM, as defined by lesions spanning over more than three segments, monophasic or recurrent isolated ON, and certain types of brainstem encephalitis (particularly if the diencephalon or the medulla oblongata is involved) [57–59]. Brainstem manifestations frequently include intractable hiccups or vomiting, symptomatic narcolepsy, and neuroendocrine dysfunctions [58–60], and may also precede ON or myelitis [1, 61–63]. It has been suggested that posterior reversible encephalopathy syndrome might also present in the context of NMO [64]. Recently, olfactory dysfunction has been described in patients with NMO [65]. Whether AQP4-Ab causes damage outside the CNS (e.g., placenta [1–3], stomach [4], muscle [5, 6], or inner ear [7]) is currently under investigation.
In children, an even broader spectrum of encephalitic manifestations has been described, in particular regarding seizures [36–38]. In a German cohort, 152 of 175 patients (87 %) did not present at disease onset with simultaneous myelitis and bilateral ON, but with isolated (mostly unilateral) ON, isolated myelitis, or brainstem encephalitis. Similarly, 89 of 106 patients (84 %) presented with abortive or atypical symptoms in a British-Japanese cohort [1, 53]. As most of these patients later developed NMO, various groups have suggested classifying these symptoms—if occurring in the context of AQP4-Ab seropositivity—as ‘high-risk syndromes for NMO’ (HRS) and referring to AQP4-Ab-positive classical NMO and AQP4-Ab-positive HRS as ‘NMO spectrum disorder’ (NMOSD) or ‘autoimmune AQP4 channelopathy’ [74–77]. The inconsistent use of the term ‘NMOSD’ has recently been criticized [8].
Clinical evaluation when NMO is suspected
Medical history and physical examination
A detailed medical history is essential. The neurological and physical examination should focus not only on the primary symptoms, but also on disease indicators that could suggest alternative diagnoses or concomitant autoimmune disorders, which are frequently present in patients with AQP4-Ab-positive NMO [1, 45, 47]. Special attention should be paid to brainstem symptoms, neuropathic pain, and painful tonic spasm [78], which have been shown to occur more frequently in NMO than in MS, and which have a demonstrated serious impact on quality of life [1, 58–63, 79, 80].
Basic laboratory tests
The following tests are recommended for exclusion of differential diagnoses or confirmation of NMO-associated diseases: differential blood count, coagulation, serum chemistry, blood sedimentation, blood glucose, vitamin B12 [81], folic acid, antibodies associated with connective disorders (ANA/ENA, anti-ds-DNA antibodies, lupus anticoagulant, antiphospholipid antibodies, ANCA, etc. [45]), urine analysis and sediment, Treponema pallidum hemagglutination assay, and paraneoplastic antibodies (in particular, anti-CV2/CRMP5 [82] and anti-Hu). Based on clinical presentation and cerebrospinal fluid (CSF) results, analysis for copper deficiency (to exclude it as a cause of myelopathy) and zinc poisoning (if suspected) should be performed [83]. Moreover, recently, antibodies to myelin oligodendrocyte glycoprotein (MOG) have been reported in a subset of both adult and pediatric patients with (mostly AQP4-Ab-negative) NMO [84–86]; however, the exact diagnostic and therapeutic relevance of this finding is currently investigated [87].
Detection of AQP4 antibodies
Several techniques are currently available to test for serum AQP4-Ab and can be categorized according to whether they are tissue-, cell-, or protein-based [18, 25, 88–97]. Using these serological tests, AQP4-Ab are detected in 60–90 % of patients who meet the clinical and radiologic criteria for NMO. The specificity of these assays varies between ~90 and 100 %. So-called cell-based assays using HEK293 cells transfected with recombinant, full-length human AQP4 have shown higher sensitivity and specificity than indirect immunofluorescence (IHC) [88, 90, 95, 98], enzyme-linked immunosorbent assays [95], and, in particular, radioimmunoprecipitation assays [93]. The prevalence of AQP4-Ab seems to be higher in female patients and in patients with relapsing disease [1, 99]. AQP4-Ab serum levels have been shown to be higher during relapse than during remission [88, 95, 96, 98, 100, 101]. However, levels during relapse vary considerably both inter- and intraindividually, with no apparent threshold for relapse induction [100, 102]. AQP4-Ab remain detectable in many cases during immunosuppressive treatment (with the exception of plasma exchange), as long as sufficiently sensitive assays are used [100]. Whenever possible, however, AQP4-Ab testing should be performed on samples taken prior to treatment commencement [100]. Re-testing initially seronegative patients during an acute attack or a treatment-free interval may be advisable [38]. Routine testing of AQP4-IgM is currently not recommended [103]. The diagnostic value of AQP4-Ab in the CSF remains controversial [104, 105]. AQP4-IgG are relatively stable over a period of at least a week at room temperature or 4 °C [106]; however, shipment on dry ice may be advisable for low-titer or CSF samples.
While AQP4-Ab are potentially of high diagnostic and therapeutic relevance, a critical need exists to challenge the current clinical practice of AQP4-Ab testing, for the following reasons: (1) Due to the low incidence of AQP4-IgG-positive NMO, the vast number of patients currently tested for AQP4-Ab [107], the limited specificity of some diagnostic assays, and the insufficient number of controls included in almost all past studies, the ratio of false-positive to true-positive test results might be higher than generally expected. This is even more problematic in patients presenting with a first episode of isolated ON or brainstem encephalitis, who are less frequently positive for AQP4-Ab. (2) Assays with insufficient sensitivity, such as IHC, have been broadly used in the past and are still partly in use. False-negative results may lead to treatment with interferon-beta or natalizumab for suspected MS; these two drugs are thought to cause disease exacerbation or to have no therapeutic benefit, respectively, in patients with NMO. On the other hand, false-positive results might prompt treatment with immunosuppressants with no established efficacy in MS and potentially serious side effects. Manufacturer-independent, multicenter comparative trials that include multiple assays as well as a sufficient number of adequate controls (≥1,000) are urgently required. Ideally, AQP4-Ab test results should therefore be confirmed using a second, methodologically independent assay with high sensitivity and specificity, and, in the case of conflicting results, a third assay. Moreover, repeat testing is recommended in equivocal cases.
Cerebrospinal fluid diagnostics
Examination of CSF includes cell count, cytology, protein, lactate, albumin CSF/serum ratio, IgG, IgA, and IgM CSF/serum ratios, oligoclonal bands (OCB), and the MRZ (measles, rubella, and varicella zoster virus) reaction. Moderate pleocytosis (mostly lymphomonocytic) is often a feature of NMO, and can be more prominent than in MS, but usually less than in infectious myelitis [7, 108–111]. On the other hand white cell counts are normal in around 40 % of CSF samples during acute relapses in patients with AQP4-Ab positive NMO [111]. Neutrophil (sometimes also eosinophil) granulocytes are frequently detected and, especially if present along with elevated lactate levels, may lead to the incorrect diagnosis of infectious myelitis in individual patients [111–113]. OCBs are positive in approximately 30 % of cases [111]. Repeating the CSF analysis can be useful for individual cases, since—unlike in MS—most CSF alterations in NMO mainly present during acute events and disappear during remission [111]. Moreover, an initial finding of OCB positivity followed by OCB negativity later in the disease course is indicative of NMO [104, 111, 114], but not MS. Testing for a positive MRZ reaction (defined as intrathecal IgG synthesis against at least two of the three pathogens) can be useful for differentiating between NMO and MS, as it is frequently positive in MS but not in NMO [1, 115]. More recently, concentrations of interleukin-6 (IL-6) and of the soluble IL-6 receptor (sIL-6R) were found to be higher in the CSF of NMO patients than of MS patients, and these may prove to be useful markers for differentiating NMO from other demyelinating diseases [116–118]. Whether measurements of glial fibrillary acidic protein (GFAP) in serum and/or CSF are of differential diagnostic value in NMO remains to be clarified [119–124].
Electrophysiology
Visual evoked potentials, median and tibial somatosensory evoked potentials, and motor evoked potentials should be performed. Visual evoked potentials are frequently altered in NMO [125, 126]. A recent study found prolonged P100 latencies in around 40 % and reduced amplitudes or missing potentials in around 25 % of patients [125].
Imaging
Magnetic resonance imaging
Magnetic resonance imaging is the most important imaging technique in the differential diagnosis of NMO. Imaging of the entire CNS (cranial and spinal cord MRI) should always be performed, regardless of the primary presenting clinical signs and symptoms. Contrast agents are obligatory, as are follow-up examinations. Predominantly central longitudinally spinal cord lesions, usually extending over three or more vertebral segments, are typical of NMO [127]. These often, but not always, show contrast enhancement for weeks up to months after the onset of symptoms. Enhancement can be patchy and inhomogeneous. Extensive, centrally located necrosis and cavitation have been reported [128]. However, treatment can induce a marked improvement and sometimes full recovery. The lesions can also resemble ischemic lesions in the anterior spinal artery territory [129] or local tumours [130]. Additional presence of cerebral lesions does not exclude a diagnosis of NMO. Cerebral T2-/FLAIR hyperintensities exist in up to 60 % of NMO patients, although they are often clinically silent, frequently not classically oval-shaped (as typically seen in MS), and typically not visible on T1-weighted images [131, 132]. In two recent studies, 58 % and 63 %, respectively, of patients with NMOSD showed brain lesions and, of these, 18 % and 27 %, respectively, were considered diagnostic of MS [1, 132]. Brain lesions are generally located close to the ventricles, in the diencephalon and hypothalamus. Two recent ultrahigh-field MRI studies showed that—as opposed to MS lesions—NMO lesions in the brain are not characterized by central veins and that cortical lesions were absent in NMO [133, 134]; however, extensive lesions and MS-like findings are possible [57, 135–142].
Optical coherence tomography
Optical coherence tomography (OCT) is a rapid and non-invasive technique for imaging unmyelinated CNS axons within the retina (the so-called retinal nerve fiber layer, RNFL). Recent technical advances have facilitated the high-resolution depiction of deeper retinal layers such as the ganglion cell layer. OCT is an increasingly popular tool in neuroimmunological research. Damage (thinning) to the RNFL in MS patients with and without a history of ON has been demonstrated by numerous groups. The suitability of OCT as a means of measuring disease progression and as a response marker for neuroprotective therapies in MS and other neurological conditions is currently being investigated [143–150].
A single acute attack of ON causes more severe damage to the RNFL in NMO than in MS, reflecting the poorer visual outcome in NMO-associated ON [7, 151, 152]. While MS patients experience progressive reduction of the RNFL over time compared with healthy controls, accrual of RNFL loss in NMO seems to be related to clinical attacks [153–159]. Whether OCT may contribute to NMO differential diagnosis is currently under investigation [160].
Therapy
A curative treatment for NMO does not exist to date. Instead, the main treatment goals are:
1. Remission and improvement of relapse-associated symptoms
2. Long-term stabilization of disease course by means of relapse prevention
3. Symptomatic therapy of residual symptoms
This review focuses on relapse therapy and intermittent long-term therapy. For symptomatic treatment recommendations, please see the reviews of MS treatment by de Sa et al. [161] and Samkoff and Goodmann [162], both published in 2011, as the symptomatic management of NMO is similar.
The rarity of NMO and its frequently severe disease course hamper the performance of prospective, randomized controlled trials evaluating treatment efficacy. The recommendations presented here are thus mainly based on case reports, retrospective case series, and a few prospective studies, all of which only meet evidence class III–IV. Accordingly, several areas of ambiguity exist. In the case of seronegative NMO, which more often takes a monophasic course [1], it remains unclear whether the treatment should be the same as that for seropositive NMO. Therefore, infectious, parainfectious, metabolic, or paraneoplastic causes must definitely be ruled out before considering immunosuppressive treatments for patients with seronegative NMO. Similarly, no treatment studies focusing on patients with limited or atypical forms of APQ4-Ab-positive NMO have yet been performed. Despite this, early initiation of long-term immunosuppressive therapy to delay a second relapse is recommended, because such patients have a high risk of relapse and conversion to typical NMO [53, 163]. In most recent case series and retrospective studies, the efficacy of the investigated therapies was found to be the same for patients with typical NMO and with AQP4-Ab-positive NMOSD. In light of this, relapse and intermittent treatment of APQ4-Ab-positive patients with limited forms of NMO should follow that of patients with typical NMO.
Treatment of acute disease attacks
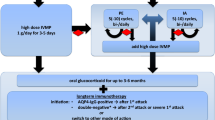
After standard neurological examination and the exclusion of infection, steroids are applied on five consecutive days with 1 g methylprednisolone (MP) per day i.v. in combination with a proton pump inhibitor and thrombosis prophylaxis [164]. In the case of a confirmed diagnosis of NMO, and depending on severity of the attack, an oral steroid tapering period should be considered.
If the patient’s condition does not sufficiently improve or the neurological symptoms worsen, therapeutic plasma exchange (TPE, five to seven cycles) can be performed [165–169]. Notably, TPE was effective both in seropositive and in seronegative patients with NMOSD in some studies [166, 170]. Early initiation of TPE might be associated with better clinical outcome [168, 171, 172]. In some cases, e.g., if contraindications for TPE exist, a second course of steroids can be applied at a higher dosage of up to five times 2 g MP [173, 174]. In a retrospective review of 10 patients treated with intravenous immunoglobulins (IVIg) for acute relapses because of lack of response to steroids with/without TPE, improvement was noted in about 50 % of patients [175].
If the patient is known to have responded well to TPE during earlier attacks and the present attack is severe, TPE can also be considered as a first measure. Immunoadsorption is an option for patients with contraindication for TPE, such as hypersensitivity reactions, or if TPE is not available [176]; however, whether the treatment has the same therapeutic efficacy as TPE has not been investigated to date.
Long-term treatment of NMO
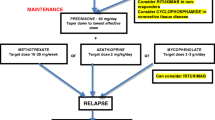
As NMO takes a relapsing course in most cases, with often incomplete recovery and rapid accumulation of neurological deficits, long-term immunosuppressive treatment should be initiated once the diagnosis of NMO has been confirmed. This also applies to APQ4-Ab-negative NMO patients with a severe first relapse and incomplete remission. However, as seronegative NMO more often follows a monophasic course, it may be justified to taper immunosuppressive therapy after some years of disease stability and after careful assessment of the risks and benefits in this group of patients.
Data on the long-term treatment (>5 years) of NMO are sparse, all retrospective, and mainly concern azathioprine (AZA) and rituximab (RX). Accordingly, AZA and RX are currently the most widely used first-line therapies in NMO. No studies comparing the efficacy of these two therapies have been published.
The following section discusses the currently most widely used therapy regimens and reports on new and emerging NMO therapies.
Azathioprine
Several studies, including a large retrospective review of 99 patients with NMO/NMOSD, have shown AZA to reduce relapse rate and ameliorate neurological disability in NMO [100, 177, 178]. A dosage regimen of 2.5–3 mg/kg body weight/day orally with monitoring of hematologic parameters and liver enzymes is recommended. The lymphocyte count should decrease to between 600 and 1,000/μl with AZA therapy and the mean erythrocyte volume should increase by about 5 % from baseline [177]. If the treatment response is lacking or side effects present, the dose should be adjusted or, if necessary, a different treatment should be applied. As the treatment may only take full effect after 3–6 months, it should initially be combined with oral steroid therapy (1 mg/kg body weight/day), as oral steroids have been shown to suppress disease activity in NMO [14, 179]. Blood cell count and liver enzyme monitoring are mandatory. Thiopurine methyltransferase enzyme activity (TMTP) testing can be performed before AZA therapy, if available, since patients with low activity may be at higher risk for severe side effects [180].
Rituximab
B cell depletion with RX has been demonstrated as effective in the treatment of NMO in several clinical case series and retrospective analyses [100, 102, 181–185]. Although the patients in these studies generally had already received one or more previous treatments, RX is now increasingly also used in treatment-naïve NMO patients with high disease activity. Thus, RX is another option for first-line treatment in NMO/NMOSD and for patients who have not responded to previous immunosuppressive therapy (e.g., AZA).
RX treatment can be initiated using one of two different regimens: either two 1 g infusions of RX at an interval of 2 weeks or four weekly 375 mg/m2 body surface area (BSA) applications. To prevent infusion-related side effects, premedication (1 g paracetamol, 100 mg prednisolone, 4 mg dimethindene maleate i.v.) should be dispensed. Additionally, the infusion should be administered at a sufficiently slow speed and monitored. Increasing evidence shows that incomplete B-cell depletion and/or B-cell repopulation is associated with relapse risk in NMO [100, 102, 183, 186]. Because most patients remain B-cell deficient for 6 months after RX treatment, re-dosing every 6 months is considered to be an adequate retreatment frequency [183]. CD19/20-positive B cells and/or CD27+ memory cells may be used as surrogate markers for treatment monitoring and re-dosing [100, 102, 183, 185]. Whether long-term RX treatment at lower doses does in fact suppress disease activity, as suggested by first patient therapy cohorts and recent investigations [185–187], requires further investigation. Individual patients with NMO have been treated up to eleven times with RX without major side effects and with an acceptable safety profile. Cases of progressive multifocal leukoencephalopathy (PML) have been reported in patients with cancer and rheumatological diseases treated with RX, mostly in combination with other immunosuppressive therapies. To date, no incidents of progressive PML have been reported in NMO patients during RX therapy. However, more data on the efficacy and safety of RX treatment in NMO are required.
Mycophenolate mofetil
In a retrospective analysis of 24 patients, treatment with mycophenolate mofetil (MMF) (median dose 2,000 mg/day, ranging between 750 and 3,000 mg) was associated with a reduction in relapse frequency and stable or reduced disability in patients with NMOSD. Half the patients in the study had previously been treated with AZA [188]. The treatment effect occurs more rapidly for MMF than for AZA. In patients experiencing side effects or poor response to AZA, MMF is recommended as an alternative treatment. PML has not yet been observed in NMO patients during treatment with MMF, but has been encountered in transplant recipients [189].
Immunoglobulins
Individual case reports and a 2012 case series have shown that high-dose IVIg are potentially beneficial [190–192]. For example, a case series of eight Spanish NMO patients showed positive results using bimonthly IVIg treatment (0.7 g/kg body weight/day for 3 days) for up to 2 years [192]. Thus, IVIg therapy is suggested as an alternative for patients with contraindication to one of the other treatments or, particularly, in children.
Mitoxantrone
Two recent observational studies [193, 194] have reported a 75–80 % reduction in relapse rate during treatment with mitoxantrone (treatment duration up to 22 months), underlining prior reports on the efficacy of mitoxantrone in NMO. A dose of 12 mg/m² BSA of mitoxantrone was administered i.v. monthly for 3–6 months, followed by infusions of 6–12 mg/m² every 3 months. The maximum dose of mitoxantrone was 100–120 mg/m² BSA. Whether other regimes (e.g., sole quarterly infusions, frequently used in MS) are as efficacious is not known. Due to the side effects (cardiotoxicity, therapy-related acute leukemia [195–197]) and the limited duration of the therapy, we recommend mitoxantrone as a second-line therapy when the treatments described above fail or cannot be applied. As for MS, we recommend that the maximum cumulative dose should not exceed 100 mg/m2 BSA. In individual cases, treatment with up to 140 mg/m2 BSA can be administered by a physician experienced in the therapy, but only if a stringent risk–benefit analysis is performed and cardiac function is monitored throughout the treatment.
Cyclophosphamide
While preliminary evidence from a number of case studies (AQP4-Ab-positive and -negative; systemic lupus erythematosus- and Sjögren’s syndrome-associated; daily oral dose in one, IV pulse in six, immunoablative in one; combination with steroids, IVIg, or AZA in all) suggested a possible treatment response to cyclophosphamide (CYC) [100, 198–201], a recent retrospective analysis of seven Brazilian NMO cases (pulsed IV CYC) failed to show such effect [202]. In another study, three of four patients treated with pulsed IV CYC had to be switched to methotrexate later due to treatment failure [203]. In light of this, CYC is only recommended when other immunosuppressive therapies fail or are not available. The treatment may be applied in immunoablative doses (2,000 mg/day for 4 days) or at a dose of 600 mg/m2 BSA per administration (together with uromitexan). The dose should be adjusted according to changes in the total leukocyte count, and CYC should be applied only under the supervision of an experienced physician.
Interferon-beta/glatiramer acetate
Interferon (INF)-beta should not be used in patients with NMO, as several retrospective studies have shown that INF-beta treatment frequently results in NMO disease exacerbation [100, 204–208]. Glatiramer acetate has not been shown to have detrimental effects in NMO patients to date; with only three cases reported, however, insufficient data exist on glatiramer acetate as NMO treatment [100, 209, 210].
Methotrexate
In a retrospective study of 14 AQP4-Ab-positive patients, treatment with methotrexate, mainly prescribed as a second-line drug, was associated with a significant decrease in the median annualized relapse rate (ARR) and was relatively well-tolerated. After exclusion of relapses within the first 3 months of treatment or on subtherapeutic doses, the proportion of relapse-free patients was 64 %. Disability stabilized or improved in 79 % [211]. In 13 of 14 cases, however, concomitant immunosuppression with oral prednisolone (n = 11), rituximab (n = 1), or tacrolimus (n = 1) was applied, and the impact of this remains unclear. Treatment with methotrexate and prednisone also resulted in disease stabilization in a smaller and less well-documented retrospective case series (n = 7) [203], and in a pediatric patient on methotrexate monotherapy [37].
Natalizumab
The treatment of NMO with natalizumab should be avoided; a recent retrospective study reported clinical deterioration after natalizumab treatment in five NMO patients initially misdiagnosed with MS [212]. In line with this, Barnett et al. [213] and Jacob et al. [214] have also described natalizumab treatment as causing disease exacerbation in NMO patients.
Fingolimod
Min et al. [215] reported a patient who had been diagnosed with MS due to an MS-typical brain MRI (which met the criteria of Barkhof et al.), but without OCB and with a normal IgG index. The patient had been enrolled in a clinical trial with fingolimod after experiencing relapses during 2 years of INF-beta treatment. Clinical deterioration and increased MRI activity was found 2 weeks after initiation of fingolimod. Diagnosis re-evaluation showed anti-AQP4 antibodies, indicating NMOSD, and determined that the patient met the American–European Consensus Group Criteria (US-EU criteria) for Sjögren’s syndrome, based on anti-SSA antibody detection, a positive Schirmer’s test, and a lip biopsy with focal lymphocytic sialoadenitis.
Combination therapies
Combination therapy is a potential option for NMO patients who have a refractory course. Oral steroids combined with AZA led to a decrease in ARR in two more recent studies [177, 178]. Similarly, methotrexate in combination with oral steroids resulted in disease stabilization in two studies [203, 211]. Another recent study showed that cyclosporin A in combination with low-dose oral steroids is effective in NMO patients [216]. Methotrexate may be also combined with RX therapy as in rheumatoid arthritis. Individual case reports have also shown that intermittent plasmapheresis combined with immunosuppressive treatment reduces attacks of NMO [217].
Anti-IL-6 therapy and other new therapies
Recent reports have suggested that IL-6 plays a role in NMO, contributing to the persistence of NMO-IgG-producing plasmablasts in patients with NMO [218]. The hypothesis has been lent weight by studies showing a favorable effect of the IL-6 receptor-blocking antibody tocilizumab, already licensed for therapy of rheumatoid arthritis, in NMO patients who have failed to respond to other therapies [219–221]. Thus, tocilizumab may be another therapeutic option for such patients.
The monoclonal antibody eculizumab, which is directed against the complement component 5, showed considerable efficacy in a small, open-label study of 14 NMO/NMOSD patients with disease activity [222, 223]. Of the 14 treated patients, 12 remained relapse-free and two showed disease activity. Apart from meningococcal sepsis and sterile meningitis in one patient approximately 2 months after the first eculizumab infusion, no other drug-related serious adverse events were reported. However, confirmation from larger, phase III studies is needed; moreover, broad administration of eculizumab would be hampered by its presently exorbitant cost.
Recent experimental strategies, which showed some beneficial effect in animal models in vitro and in vivo, include the use of competitive, non-pathogenic AQP4-specific antibodies (e.g., aquaporumab) [224, 225], neutrophil elastase inhibitors [226], antihistamines with eosinophil-stabilizing actions [227], and enzymatic AQP4-IgG deglycosylation or cleavage [228, 229].
An isolated case report showing that autologous hematopoietic stem cell transplantation (AHSCT) failed to prevent further relapses in a NMO patient raised concerns about the treatment’s efficacy in NMO. However, an ongoing AHSCT trial involving 10 NMO patients is expected to shed light on whether some patients do benefit from the therapy [230]. Alemtuzumab, a B- and T-cell-depleting antibody previously used in MS trials with favorable outcome, did not show beneficial effects when used in individual NMO patients [220, 231].
Summary for treatment recommendations
Based on the currently available evidence as summarized above, the NEMOS group gives the following treatment recommendations:
The frequently severe disease course of NMO calls for prompt initiation of immunosuppressive treatment once the diagnosis of NMO or AQP4-Ab-positive NMOSD has been confirmed, with azathioprine or rituximab as first-line treatment (see Fig. 1). In children or in patients with contraindications to immunosuppressive therapies, IVIg may be used as first-line therapy. In patients with NMOSD who are AQP4-Ab negative, therapy initiation depends on the severity and remission of the first relapse and the clinical course.

Long-term therapy of NMO
In the case of side effects or poor response, treatment can be switched from azathioprine to rituximab or vice versa, or to mycophenolate mofetil, methotrexate, or mitoxantrone. If disease progression occurs and if the above treatments fail, combination therapy or newer agents such as tocilizumab may be applied. Treatment with interferon-beta, natalizumab, and fingolimod should be avoided. How and whether treatment with the recently approved therapies for MS, teriflunomide and fumaric acid, influences the disease course in NMO patients remains to be elucidated.
In general, physicians must inform patients about the risks of side effects, such as malignancy, infertility, cytotoxicity and myelotoxicity, infections including PML, vaccination issues, and the need for contraception before initiating immunosuppressive therapies. Tests for pregnancy and chronic infections (HIV, hepatitis B and C) before treatment commencement are recommended.
Future directions
The pathogenesis, diagnosis, and treatment of NMO are rapidly expanding research areas, as reflected by the steep increase in the number of publications on NMO since AQP4 antibodies were first described. Consequently, we expect major advances in all three areas over the next few years. Research on pathogenesis has progressed to studying the role of T-cells, neutrophils, eosinophils, and other cellular components of the immune system [27, 227, 232–234]. Several new potential therapeutic approaches have resulted from recent insights in NMO pathogenesis, including complement and neutrophil elastase inhibition [226] (eculizumab, sivelestat [235]), and the blocking of antibodies to AQP4 with monoclonal antibodies (aquaporumab), among others. The challenges in finding new and better medicines for NMO are the rareness of the disease and the unfavorable prognosis in many cases, which make clinical studies with placebo groups difficult. Although designing meaningful and clinically relevant NMO therapy studies is laborious, these trials will eventually increase our options for treating NMO.
Notes
Four or more white matter lesions, or more than three white matter lesions if one of these is located in the periventricular region.
If no cranial MRI was performed at disease onset, or the findings are unknown, the earliest available MRI should be used [28].
Detection of AQP4-Ab using recombinant methods can replace immunohistochemical detection of NMO-IgG [18], provided that the respective recombinant test has been demonstrated to yield equal or better sensitivity and specificity in clinically well-defined and sufficiently large patient and control collectives, and has been successfully validated using an NMO-IgG-positive patient collective. Of the testing systems described in the literature and currently available for diagnosis, mainly cell-based assays meet these requirements. Alternatively, seropositivity for AQP4-Ab in two methodologically independent immunoassays is considered by some to be a valid substitute for NMO-IgG seropositivity (expert opinion).
References
Jarius S, Ruprecht K, Wildemann B et al (2012) Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 9:14
Trebst C, Berthele A, Jarius S, Kümpfel T, Schippling S, Wildemann B, Wilke C (2011) Diagnosis and treatment of neuromyelitis optica. Consensus recommendations of the Neuromyelitis Optica Study Group. Nervenarzt 82:768–777
Kimbrough DJ, Fujihara K, Jacob A et al (2012) Treatment of neuromyelitis optica: review and recommendations. Mult Scler Relat Disord 1:180–187
Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, Du Pasquier RA, Polman CH, Sorensen PS, Hemmer B (2010) EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol 17:1019–1032
Scott TF, Frohman EM, De Seze J, Gronseth GS, Weinshenker BG (2011) Evidence-based guideline: clinical evaluation and treatment of transverse myelitis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 77:2128–2134
Jarius S, Wildemann B (2007) Neuromyelitis optica. Nervenarzt 78:1365–1377
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG (1999) The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 53:1107–1114
Jarius S, Wildemann B (2013) The history of neuromyelitis optica. J Neuroinflammation 10:8
Jarius S, Wildemann B (2012) The case of the Marquis de Causan (1804): an early account of visual loss associated with spinal cord inflammation. J Neurol 259:1354–1357
Jarius S, Wildemann B (2012) “Noteomielite” accompanied by acute amaurosis (1844). An early case of neuromyelitis optica. J Neurol Sci 313:182–184
Jarius S, Wildemann B (2012) An early British case of neuromyelitis optica (1850). BMJ 345:e6430
Jarius S, Wildemann B (2011) An early case of neuromyelitis optica: on a forgotten report by Jacob Lockhart Clarke, FRS. Mult Scler 17:1384–1386
Jarius S, Wildemann B (2013) On the contribution of Thomas Clifford Allbutt, F.R.S., to the early history of neuromyelitis optica. J Neurol 260:100–104
Mandler RN, Davis LE, Jeffery DR, Kornfeld M (1993) Devic’s neuromyelitis optica: a clinicopathological study of 8 patients. Ann Neurol 34:162–168
Lucchinetti CF, Mandler RN, McGavern D et al (2002) A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 125:1450–1461
Lee D-H, Metz I, Berthele A, Stadelmann C, Brück W, Linker RA, Gold R, Schroeder A (2010) Supraspinal demyelinating lesions in neuromyelitis optica display a typical astrocyte pathology. Neuropathol Appl Neurobiol 36:685–687
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 202:473–477
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112
Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, Vincent A, Wildemann B (2008) Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol 4:202–214
Jarius S, Wildemann B (2010) AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol 6:383–392
Graber DJ, Levy M, Kerr D, Wade WF (2008) Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflammation 5:22
Bennett JL, Lam C, Kalluri SR et al (2009) Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629
Bradl M, Misu T, Takahashi T et al (2009) Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 66:630–643
Ratelade J, Bennett JL, Verkman AS (2011) Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One 6:e27412
Mader S, Lutterotti A, Di Pauli F et al (2010) Patterns of antibody binding to aquaporin-4 isoforms in neuromyelitis optica. PLoS One 5:e10455
Nelson PA, Khodadoust M, Prodhomme T, Spencer C, Patarroyo JC, Varrin-Doyer M, Ho JD, Stroud RM, Zamvil SS (2010) Immunodominant T cell determinants of aquaporin-4, the autoantigen associated with neuromyelitis optica. PLoS One 5:e15050
Kalluri SR, Rothhammer V, Staszewski O, Srivastava R, Petermann F, Prinz M, Hemmer B, Korn T (2011) Functional characterization of aquaporin-4 specific T cells: towards a model for neuromyelitis optica. PLoS One 6:e16083
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66:1485–1489
Miller DH, Weinshenker BG, Filippi M et al (2008) Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler 14:1157–1174
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG (2007) The spectrum of neuromyelitis optica. Lancet Neurol 6:805–815
Bizzoco E, Lolli F, Repice AM et al (2009) Prevalence of neuromyelitis optica spectrum disorder and phenotype distribution. J Neurol 256:1891–1898
Jacob A, Panicker J, Lythgoe D, Elsone L, Mutch K, Wilson M, Das K, Boggild M (2013) The epidemiology of neuromyelitis optica amongst adults in the Merseyside county of United Kingdom. J Neurol 260:2134–2137
Asgari N, Lillevang ST, Skejoe HPB, Falah M, Stenager E, Kyvik KO (2011) A population-based study of neuromyelitis optica in Caucasians. Neurology 76:1589–1595
Wingerchuk DM (2009) Neuromyelitis optica: effect of gender. J Neurol Sci 286:18–23
Barbieri F, Buscaino GA (1989) Neuromyelitis optica in the elderly. Acta Neurol (Napoli) 11:247–251
Banwell B, Tenembaum S, Lennon VA, Ursell E, Kennedy J, Bar-Or A, Weinshenker BG, Lucchinetti CF, Pittock SJ (2008) Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology 70:344–352
McKeon A, Lennon VA, Lotze T et al (2008) CNS aquaporin-4 autoimmunity in children. Neurology 71:93–100
Lotze TE, Northrop JL, Hutton GJ, Ross B, Schiffman JS, Hunter JV (2008) Spectrum of pediatric neuromyelitis optica. Pediatrics 122:e1039–e1047
Bergamaschi R, Jarius S, Robotti M, Pichiecchio A, Wildemann B, Meola G (2009) Two cases of benign neuromyelitis optica in patients with celiac disease. J Neurol 256:2097–2099
Jarius S, Jacob S, Waters P, Jacob A, Littleton E, Vincent A (2008) Neuromyelitis optica in patients with gluten sensitivity associated with antibodies to aquaporin-4. J Neurol Neurosurg Psychiatr 79:1084
Kay CSK, Scola RH, Lorenzoni PJ, Jarius S, Arruda WO, Werneck LC (2008) NMO-IgG positive neuromyelitis optica in a patient with myasthenia gravis but no thymectomy. J Neurol Sci 275:148–150
McKeon A, Lennon VA, Jacob A et al (2009) Coexistence of myasthenia gravis and serological markers of neurological autoimmunity in neuromyelitis optica. Muscle Nerve 39:87–90
Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, Lucchinetti CF, Zéphir H, Moder K, Weinshenker BG (2008) Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 65:78–83
Wandinger K-P, Stangel M, Witte T, Venables P, Charles P, Jarius S, Wildemann B, Probst C, Iking-Konert C, Schneider M (2010) Autoantibodies against aquaporin-4 in patients with neuropsychiatric systemic lupus erythematosus and primary Sjögren’s syndrome. Arthritis Rheum 62:1198–1200
Jarius S, Jacobi C, de Seze J et al (2011) Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler 17:1067–1073
Závada J, Nytrová P, Wandinger KP, Jarius S, Svobodová R, Probst C, Peterová V, Tegzová D, Pavelka K, Vencovský J (2013) Seroprevalence and specificity of NMO-IgG (anti-aquaporin 4 antibodies) in patients with neuropsychiatric systemic lupus erythematosus. Rheumatol Int 33:259–263
Jarius S, Paul F, Franciotta D et al (2012) Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler 18:1135–1143
Nagaishi A, Takagi M, Umemura A, Tanaka M, Kitagawa Y, Matsui M, Nishizawa M, Sakimura K, Tanaka K (2011) Clinical features of neuromyelitis optica in a large Japanese cohort: comparison between phenotypes. J Neurol Neurosurg Psychiatr 82:1360–1364
Kister I, Gulati S, Boz C, Bergamaschi R, Piccolo G, Piccolo G, Oger J, Swerdlow ML (2006) Neuromyelitis optica in patients with myasthenia gravis who underwent thymectomy. Arch Neurol 63:851–856
Gotkine M, Fellig Y, Abramsky O (2006) Occurrence of CNS demyelinating disease in patients with myasthenia gravis. Neurology 67:881–883
Vaknin-Dembinsky A, Abramsky O, Petrou P, Ben-Hur T, Gotkine M, Brill L, Brenner T, Argov Z, Karussis D (2011) Myasthenia gravis-associated neuromyelitis optica-like disease: an immunological link between the central nervous system and muscle? Arch Neurol 68:1557–1561
Wingerchuk DM, Weinshenker BG (2012) The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler 18:5–10
Kitley J, Leite MI, Nakashima I et al (2012) Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 135:1834–1849
Fragoso YD, Adoni T, Bichuetti DB, Brooks JBB, Ferreira MLB, Oliveira EML, Oliveira CLS, Ribeiro SBF, Silva AE, Siquineli F (2013) Neuromyelitis optica and pregnancy. J Neurol 260:2614–2619
Bourre B, Marignier R, Zéphir H et al (2012) Neuromyelitis optica and pregnancy. Neurology 78:875–879
Paty DW, Oger JJ, Kastrukoff LF, Hashimoto SA, Hooge JP, Eisen AA, Eisen KA, Purves SJ, Low MD, Brandejs V (1988) MRI in the diagnosis of MS: a prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal banding, and CT. Neurology 38:180–185
Lu Z, Zhang B, Qiu W, Kang Z, Shen L, Long Y, Huang J, Hu X (2011) Comparative brain stem lesions on MRI of acute disseminated encephalomyelitis, neuromyelitis optica, and multiple sclerosis. PLoS One 6:e22766
Apiwattanakul M, Popescu BF, Matiello M, Weinshenker BG, Lucchinetti CF, Lennon VA, McKeon A, Carpenter AF, Miller GM, Pittock SJ (2010) Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol 68:757–761
Popescu BFG, Lennon VA, Parisi JE et al (2011) Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 76:1229–1237
Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y (2005) Intractable hiccup and nausea with periaqueductal lesions in neuromyelitis optica. Neurology 65:1479–1482
Takahashi T, Miyazawa I, Misu T, Takano R, Nakashima I, Fujihara K, Tobita M, Itoyama Y (2008) Intractable hiccup and nausea in neuromyelitis optica with anti-aquaporin-4 antibody: a herald of acute exacerbations. J Neurol Neurosurg Psychiatr 79:1075–1078
Kanbayashi T, Shimohata T, Nakashima I, Yaguchi H, Yabe I, Nishizawa M, Shimizu T, Nishino S (2009) Symptomatic narcolepsy in patients with neuromyelitis optica and multiple sclerosis: new neurochemical and immunological implications. Arch Neurol 66:1563–1566
Suzuki K, Nakamura T, Hashimoto K, Miyamoto M, Komagamine T, Nagashima T, Izawa N, Kanbayashi T, Takahashi T, Hirata K (2012) Hypothermia, hypotension, hypersomnia, and obesity associated with hypothalamic lesions in a patient positive for the anti-aquaporin 4 antibody: a case report and literature review. Arch Neurol 69:1355–1359
Magaña SM, Matiello M, Pittock SJ, McKeon A, Lennon VA, Rabinstein AA, Shuster E, Kantarci OH, Lucchinetti CF, Weinshenker BG (2009) Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 72:712–717
Schmidt F, Önder G, Jarius S, Wildemann B, Ruprecht K, Paul F, Harms L (2013) Olfactory dysfunction in patients with neuromyelitis optica. Multiple Sclerosis International: ID 654501
Saadoun S, Waters P, Leite MI, Bennett JL, Vincent A, Papadopoulos MC (2013) Neuromyelitis optica IgG causes placental inflammation and fetal death. J Immunol 191:2999–3005
Reuss R, Rommer PS, Brück W et al (2009) A woman with acute myelopathy in pregnancy: case outcome. BMJ 339:b4026
Reuss R, Rommer P, Brück W, Jarius S, Bolz M, Zettl UK (2010) Anti-AQP4 ab might be relevant in pregnancy. BMJ reply
Cosgrove J, Alli S, Ramadan H, Ford HL (2013) Myocarditis and diffuse skeletal muscle oedema: new features of neuromyelitis optica spectrum disorder? A case report. Mult Scler. doi:10.1177/1352458513495939
Deguchi S, Deguchi K, Sato K et al (2012) HyperCKemia related to the initial and recurrent attacks of neuromyelitis optica. Intern Med 51:2617–2620
Di Filippo M, Franciotta D, Massa R et al (2012) Recurrent hyperCKemia with normal muscle biopsy in a pediatric patient with neuromyelitis optica. Neurology 79:1182–1184
Suzuki N, Takahashi T, Aoki M et al (2010) Neuromyelitis optica preceded by hyperCKemia episode. Neurology 74:1543–1545
Jarius S, Lauda F, Wildemann B, Tumani H (2013) Steroid-responsive hearing impairment in NMO-IgG/aquaporin-4-antibody-positive neuromyelitis optica. J Neurol 260:663–664
Jarius S, Frederikson J, Waters P et al (2010) Frequency and prognostic impact of antibodies to aquaporin-4 in patients with optic neuritis. J Neurol Sci 298:158–162
Matiello M, Lennon VA, Jacob A, Pittock SJ, Lucchinetti CF, Wingerchuk DM, Weinshenker BG (2008) NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology 70:2197–2200
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT (2010) Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. J Neurol Neurosurg Psychiatr 81:109–111
Weinshenker BG, Wingerchuk DM, Vukusic S, Linbo L, Pittock SJ, Lucchinetti CF, Lennon VA (2006) Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 59:566–569
Kim S-M, Go MJ, Sung J-J, Park KS, Lee K-W (2012) Painful tonic spasm in neuromyelitis optica: incidence, diagnostic utility, and clinical characteristics. Arch Neurol 69:1026–1031
Kanamori Y, Nakashima I, Takai Y, Nishiyama S, Kuroda H, Takahashi T, Kanaoka-Suzuki C, Misu T, Fujihara K, Itoyama Y (2011) Pain in neuromyelitis optica and its effect on quality of life: a cross-sectional study. Neurology 77:652–658
Qian P, Lancia S, Alvarez E, Klawiter EC, Cross AH, Naismith RT (2012) Association of neuromyelitis optica with severe and intractable pain. Arch Neurol 69:1482–1487
Jarius S, Paul F, Ruprecht K, Wildemann B (2012) Low vitamin B12 levels and gastric parietal cell antibodies in patients with aquaporin-4 antibody-positive neuromyelitis optica spectrum disorders. J Neurol 259:2743–2745
Jarius S, Wandinger KP, Borowski K, Stoecker W, Wildemann B (2012) Antibodies to CV2/CRMP5 in neuromyelitis optica-like disease: case report and review of the literature. Clin Neurol Neurosurg 114:331–335
Jaiser SR, Winston GP (2010) Copper deficiency myelopathy. J Neurol 257:869–881
Mader S, Gredler V, Schanda K et al (2011) Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 8:184
Rostasy K, Mader S, Schanda K et al (2012) Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol 69:752–756
Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, Palace J, Vincent A (2012) Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 79:1273–1277
Reindl M, Di Pauli F, Rostásy K, Berger T (2013) The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 9:455–461
Waters P, Jarius S, Littleton E et al (2008) Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol 65:913–919
Hayakawa S, Mori M, Okuta A et al (2008) Neuromyelitis optica and anti-aquaporin-4 antibodies measured by an enzyme-linked immunosorbent assay. J Neuroimmunol 196:181–187
Jarius S, Franciotta D, Bergamaschi R, Wright H, Littleton E, Palace J, Hohlfeld R, Vincent A (2007) NMO-IgG in the diagnosis of neuromyelitis optica. Neurology 68:1076–1077
Jarius S, Probst C, Borowski K, Franciotta D, Wildemann B, Stoecker W, Wandinger KP (2010) Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J Neurol Sci 291:52–56
Kalluri SR, Illes Z, Srivastava R, Cree B, Menge T, Bennett JL, Berthele A, Hemmer B (2010) Quantification and functional characterization of antibodies to native aquaporin 4 in neuromyelitis optica. Arch Neurol 67:1201–1208
Paul F, Jarius S, Aktas O et al (2007) Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS Med 4:e133
Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, Watanabe S, Ishii N, Itoyama Y (2006) Establishment of a new sensitive assay for anti-human aquaporin-4 antibody in neuromyelitis optica. Tohoku J Exp Med 210:307–313
Jarius S, Franciotta D, Paul F, Bergamaschi R, Rommer PS, Ruprecht K, Ringelstein M, Aktas O, Kristoferitsch W, Wildemann B (2012) Testing for antibodies to human aquaporin-4 by ELISA: sensitivity, specificity, and direct comparison with immunohistochemistry. J Neurol Sci 320:32–37
Kim W, Lee J-E, Li XF, Kim S-H, Han B-G, Lee BI, Kim JK, Choi K, Kim HJ (2012) Quantitative measurement of anti-aquaporin-4 antibodies by enzyme-linked immunosorbent assay using purified recombinant human aquaporin-4. Mult Scler 18:578–586
Jarius S, Wildemann B (2013) Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 23:661–683
Takahashi T, Fujihara K, Nakashima I et al (2007) Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 130:1235–1243
Ketelslegers IA, Modderman PW, Vennegoor A, Killestein J, Hamann D, Hintzen RQ (2011) Antibodies against aquaporin-4 in neuromyelitis optica: distinction between recurrent and monophasic patients. Mult Scler 17:1527–1530
Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, Lang W, Reindl M, Vincent A, Kristoferitsch W (2008) Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 131:3072–3080
Heerlein K, Jarius S, Jacobi C, Rohde S, Storch-Hagenlocher B, Wildemann B (2009) Aquaporin-4 antibody positive longitudinally extensive transverse myelitis following varicella zoster infection. J Neurol Sci 276:184–186
Pellkofer HL, Krumbholz M, Berthele A et al (2011) Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 76:1310–1315
Jarius S, Franciotta D, Bergamaschi R, Wildemann B, Wandinger K-P (2010) Immunoglobulin M antibodies to aquaporin-4 in neuromyelitis optica and related disorders. Clin Chem Lab Med 48:659–663
Jarius S, Franciotta D, Paul F et al (2010) Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation 7:52
Klawiter EC, Alvarez E 3rd, Xu J, Paciorkowski AR, Zhu L, Parks BJ, Cross AH, Naismith RT (2009) NMO-IgG detected in CSF in seronegative neuromyelitis optica. Neurology 72:1101–1103
Jarius S, Wildemann B (2011) Effect of storage conditions and freeze/thaw cycles on aquaporin-4 antibody (NMO-IgG) serum levels. Clin Chem Lab Med 49:2121–2122
Waters PJ, McKeon A, Leite MI et al (2012) Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 78:665–671 discussion 669
De Seze J, Stojkovic T, Ferriby D, Gauvrit J-Y, Montagne C, Mounier-Vehier F, Verier A, Pruvo J-P, Hache J-C, Vermersch P (2002) Devic’s neuromyelitis optica: clinical, laboratory, MRI and outcome profile. J Neurol Sci 197:57–61
Ghezzi A, Bergamaschi R, Martinelli V et al (2004) Clinical characteristics, course and prognosis of relapsing Devic’s Neuromyelitis Optica. J Neurol 251:47–52
O’Riordan JI, Gallagher HL, Thompson AJ, Howard RS, Kingsley DP, Thompson EJ, McDonald WI, Miller DH (1996) Clinical, CSF, and MRI findings in Devic’s neuromyelitis optica. J Neurol Neurosurg Psychiatr 60:382–387
Jarius S, Paul F, Franciotta D et al (2011) Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 306:82–90
Lepur D, Peterković V, Kalabrić-Lepur N (2009) Neuromyelitis optica with CSF examination mimicking bacterial meningomyelitis. Neurol Sci 30:51–54
Jarius S, Wildemann B (2013) Aquaporin-4 antibodies, CNS acidosis and neuromyelitis optica: A potential link. Med Hypotheses. doi:10.1016/j.mehy.2013.10.011
Bergamaschi R, Tonietti S, Franciotta D, Candeloro E, Tavazzi E, Piccolo G, Romani A, Cosi V (2004) Oligoclonal bands in Devic’s neuromyelitis optica and multiple sclerosis: differences in repeated cerebrospinal fluid examinations. Mult Scler 10:2–4
Jarius S, Franciotta D, Bergamaschi R et al (2008) Polyspecific, antiviral immune response distinguishes multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatr 79:1134–1136
Uzawa A, Mori M, Arai K, Sato Y, Hayakawa S, Masuda S, Taniguchi J, Kuwabara S (2010) Cytokine and chemokine profiles in neuromyelitis optica: signifiance of interleukin-6. Mult Scler 16:1443–1452
Içöz S, Tüzün E, Kürtüncü M, Durmuş H, Mutlu M, Eraksoy M, Akman-Demir G (2010) Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int J Neurosci 120:71–75
Wang H, Wang K, Zhong X, Dai Y, Qiu W, Wu A, Hu X (2012) Notable increased cerebrospinal fluid levels of soluble interleukin-6 receptors in neuromyelitis optica. Neuroimmunomodulation 19:304–308
Storoni M, Petzold A, Plant GT (2011) The use of serum glial fibrillary acidic protein measurements in the diagnosis of neuromyelitis optica spectrum optic neuritis. PLoS One 6:e23489
Petzold A, Eikelenboom MJ, Gveric D et al (2002) Markers for different glial cell responses in multiple sclerosis: clinical and pathological correlations. Brain 125:1462–1473
Misu T, Takano R, Fujihara K, Takahashi T, Sato S, Itoyama Y (2009) Marked increase in cerebrospinal fluid glial fibrillar acidic protein in neuromyelitis optica: an astrocytic damage marker. J Neurol Neurosurg Psychiatr 80:575–577
Takano R, Misu T, Takahashi T, Izumiyama M, Fujihara K, Itoyama Y (2008) A prominent elevation of glial fibrillary acidic protein in the cerebrospinal fluid during relapse in neuromyelitis optica. Tohoku J Exp Med 215:55–59
Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y (2010) Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 75:208–216
Uzawa A, Mori M, Sawai S, Masuda S, Muto M, Uchida T, Ito S, Nomura F, Kuwabara S (2013) Cerebrospinal fluid interleukin-6 and glial fibrillary acidic protein levels are increased during initial neuromyelitis optica attacks. Clin Chim Acta 421:181–183
Ringelstein M, Kleiter I, Ayzenberg I et al (2013) Visual evoked potentials in neuromyelitis optica and its spectrum disorders. Mult Scler. doi:10.1177/1352458513503053
Neto SP, Alvarenga RMP, Vasconcelos CCF, Alvarenga MP, Pinto LC, Pinto VLR (2013) Evaluation of pattern-reversal visual evoked potential in patients with neuromyelitis optica. Mult Scler 19:173–178
Filippi M, Rocca MA (2004) MR imaging of Devic’s neuromyelitis optica. Neurol Sci 25(Suppl 4):S371–S373
Lim BC, Hwang H, Kim KJ, Hwang YS, Cheon J-E, Kim I-O, Kim HJ, Chae J-H (2011) Relapsing demyelinating CNS disease in a Korean pediatric population: multiple sclerosis versus neuromyelitis optica. Mult Scler 17:67–73
Krampla W, Aboul-Enein F, Jecel J, Lang W, Fertl E, Hruby W, Kristoferitsch W (2009) Spinal cord lesions in patients with neuromyelitis optica: a retrospective long-term MRI follow-up study. Eur Radiol 19:2535–2543
Ringelstein M, Metz I, Ruprecht K, Koch A, Rappold J, Ingwersen J, Mathys C, Jarius S, Bruck W, Hartung HP, Paul F, Aktas O (2013) Contribution of spinal cord biopsy to diagnosis of aquaporin-4 antibody positive neuromyelitis optica spectrum disorder. Mult Scler. doi:10.1177/1352458513510981
Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG (2006) Brain abnormalities in neuromyelitis optica. Arch Neurol 63:390–396
Matthews L, Marasco R, Jenkinson M et al (2013) Distinction of seropositive NMO spectrum disorder and MS brain lesion distribution. Neurology 80:1330–1337
Sinnecker T, Dörr J, Pfueller CF, Harms L, Ruprecht K, Jarius S, Brück W, Niendorf T, Wuerfel J, Paul F (2012) Distinct lesion morphology at 7-T MRI differentiates neuromyelitis optica from multiple sclerosis. Neurology 79:708–714
Kister I, Herbert J, Zhou Y, Ge Y (2013) Ultrahigh-Field MR (7 T) Imaging of brain lesions in Neuromyelitis Optica. Mult Scler Int 2013:398259
Nakamura M, Misu T, Fujihara K, Miyazawa I, Nakashima I, Takahashi T, Watanabe S, Itoyama Y (2009) Occurrence of acute large and edematous callosal lesions in neuromyelitis optica. Mult Scler 15:695–700
Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA (2006) Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 63:964–968
Nakajima H, Fujiki Y, Ito T, Kitaoka H, Takahashi T (2011) Anti-aquaporin-4 antibody-positive neuromyelitis optica presenting with syndrome of inappropriate antidiuretic hormone secretion as an initial manifestation. Case Rep Neurol 3:263–267
Nakashima I, Fujihara K, Miyazawa I et al (2006) Clinical and MRI features of Japanese patients with multiple sclerosis positive for NMO-IgG. J Neurol Neurosurg Psychiatr 77:1073–1075
Poppe AY, Lapierre Y, Melançon D, Lowden D, Wardell L, Fullerton LM, Bar-Or A (2005) Neuromyelitis optica with hypothalamic involvement. Mult Scler 11:617–621
Banker P, Sonni S, Kister I, Loh JP, Lui YW (2012) Pencil-thin ependymal enhancement in neuromyelitis optica spectrum disorders. Mult Scler 18:1050–1053
Kim W, Park MS, Lee SH, Kim S-H, Jung IJ, Takahashi T, Misu T, Fujihara K, Kim HJ (2010) Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Mult Scler 16:1229–1236
Kim W, Kim S-H, Huh S-Y, Kim HJ (2012) Brain abnormalities in neuromyelitis optica spectrum disorder. Mult Scler Int 2012:735486
Bock M, Brandt AU, Dörr J, Pfueller CF, Ohlraun S, Zipp F, Paul F (2010) Time domain and spectral domain optical coherence tomography in multiple sclerosis: a comparative cross-sectional study. Mult Scler 16:893–896
Oberwahrenbrock T, Schippling S, Ringelstein M et al (2012) Retinal damage in multiple sclerosis disease subtypes measured by high-resolution optical coherence tomography. Mult Scler Int 2012:530305
Brandt AU, Oberwahrenbrock T, Ringelstein M, Young KL, Tiede M, Hartung HP, Martin R, Aktas O, Paul F, Schippling S (2011) Primary retinal pathology in multiple sclerosis as detected by optical coherence tomography. Brain 134:e193 author reply e194
Stricker S, Oberwahrenbrock T, Zimmermann H, Schroeter J, Endres M, Brandt AU, Paul F (2011) Temporal retinal nerve fiber loss in patients with spinocerebellar ataxia type 1. PLoS One 6:e23024
Zimmermann H, Freing A, Kaufhold F et al (2013) Optic neuritis interferes with optical coherence tomography and magnetic resonance imaging correlations. Mult Scler 19:443–450
Dörr J, Wernecke KD, Bock M, Gaede G, Wuerfel JT, Pfueller CF, Bellmann-Strobl J, Freing A, Brandt AU, Friedemann P (2011) Association of retinal and macular damage with brain atrophy in multiple sclerosis. PLoS One 6:e18132
Albrecht P, Müller A-K, Südmeyer M et al (2012) Optical coherence tomography in parkinsonian syndromes. PLoS One 7:e34891
Oberwahrenbrock T, Ringelstein M, Jentschke S et al (2013) Retinal ganglion cell and inner plexiform layer thinning in clinically isolated syndrome. Mult Scler. doi:10.1177/1352458513489757
Pfueller CF, Paul F (2011) Imaging the visual pathway in neuromyelitis optica. Mult Scler Int 2011:869814
Bichuetti DB, de Camargo AS, Falcão AB, Gonçalves FF, Tavares IM, de Oliveira EML (2013) The retinal nerve fiber layer of patients with neuromyelitis optica and chronic relapsing optic neuritis is more severely damaged than patients with multiple sclerosis. J Neuroophthalmol 33:220–224
De Seze J, Blanc F, Jeanjean L et al (2008) Optical coherence tomography in neuromyelitis optica. Arch Neurol 65:920–923
Naismith RT, Tutlam NT, Xu J, Klawiter EC, Shepherd J, Trinkaus K, Song S-K, Cross AH (2009) Optical coherence tomography differs in neuromyelitis optica compared with multiple sclerosis. Neurology 72:1077–1082
Ratchford JN, Quigg ME, Conger A, Frohman T, Frohman E, Balcer LJ, Calabresi PA, Kerr DA (2009) Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology 73:302–308
Green AJ, Cree BAC (2009) Distinctive retinal nerve fibre layer and vascular changes in neuromyelitis optica following optic neuritis. J Neurol Neurosurg Psychiatr 80:1002–1005
Gelfand JM, Cree BA, Nolan R, Arnow S, Green AJ (2013) Microcystic inner nuclear layer abnormalities and neuromyelitis optica. JAMA Neurol pp 1–5
Sotirchos ES, Saidha S, Byraiah G et al (2013) In vivo identification of morphologic retinal abnormalities in neuromyelitis optica. Neurology 80:1406–1414
Bouyon M, Collongues N, Zéphir H et al (2013) Longitudinal follow-up of vision in a neuromyelitis optica cohort. Mult Scler 19:1320–1322
Schneider E, Zimmermann H, Oberwahrenbrock T et al (2013) Optical coherence tomography reveals distinct patterns of retinal damage in neuromyelitis optica and multiple sclerosis. PLoS One p e66151
De Sa JCC, Airas L, Bartholome E et al (2011) Symptomatic therapy in multiple sclerosis: a review for a multimodal approach in clinical practice. Ther Adv Neurol Disord 4:139–168
Samkoff LM, Goodman AD (2011) Symptomatic management in multiple sclerosis. Neurol Clin 29:449–463
Chang K-H, Lyu R-K, Chen C-M, Wu Y-R, Chang H-S, Huang C-C, Kuo H-C, Chu C-C, Hsu W-C, Ro L-S (2013) Distinct features between longitudinally extensive transverse myelitis presenting with and without anti-aquaporin 4 antibodies. Mult Scler 19:299–307
Wingerchuk DM, Weinshenker BG (2008) Neuromyelitis optica. Curr Treat Options Neurol 10:55–66
Keegan M, Pineda AA, McClelland RL, Darby CH, Rodriguez M, Weinshenker BG (2002) Plasma exchange for severe attacks of CNS demyelination: predictors of response. Neurology 58:143–146
Bonnan M, Valentino R, Olindo S, Mehdaoui H, Smadja D, Cabre P (2009) Plasma exchange in severe spinal attacks associated with neuromyelitis optica spectrum disorder. Mult Scler 15:487–492
Roesner S, Appel R, Gbadamosi J, Martin R, Heesen C (2012) Treatment of steroid-unresponsive optic neuritis with plasma exchange. Acta Neurol Scand 126:103–108
Bonnan M, Cabre P (2012) Plasma exchange in severe attacks of neuromyelitis optica. Mult Scler Int 2012:787630
Merle H, Olindo S, Jeannin S et al (2012) Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol 130:858–862
Magaña SM, Keegan BM, Weinshenker BG et al (2011) Beneficial plasma exchange response in central nervous system inflammatory demyelination. Arch Neurol 68:870–878
Weinshenker BG, O’Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW, Pineda AA, Stevens LN, Rodriguez M (1999) A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol 46:878–886
Nakashima I, Takahashi T, Nishiyama S, Kasahara S, Suzuki C, Misu T, Itoyama Y, Fuijihara K (2009) Plasma Exchange for Neuromyelitis Optica with Aquaporin-4 Antibody. Neurology 72:A187
Llufriu S, Castillo J, Blanco Y et al (2009) Plasma exchange for acute attacks of CNS demyelination: predictors of improvement at 6 months. Neurology 73:949–953
Watanabe S, Nakashima I, Misu T, Miyazawa I, Shiga Y, Fujihara K, Itoyama Y (2007) Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler 13:128–132
Elsone L, Panicker J, Mutch K, Boggild M, Appleton R, Jacob A (2013) Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler. doi:10.1177/1352458513495938
Greenberg BM, Thomas KP, Krishnan C, Kaplin AI, Calabresi PA, Kerr DA (2007) Idiopathic transverse myelitis: corticosteroids, plasma exchange, or cyclophosphamide. Neurology 68:1614–1617
Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, Thapa P, McKeon A (2011) Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 77:659–666
Bichuetti DB, Lobato de Oliveira EM, Oliveira DM, Amorin de Souza N, Gabbai AA (2010) Neuromyelitis optica treatment: analysis of 36 patients. Arch Neurol 67:1131–1136
Watanabe S, Misu T, Miyazawa I, Nakashima I, Shiga Y, Fujihara K, Itoyama Y (2007) Low-dose corticosteroids reduce relapses in neuromyelitis optica: a retrospective analysis. Mult Scler 13:968–974
Higgs JE, Payne K, Roberts C, Newman WG (2010) Are patients with intermediate TPMT activity at increased risk of myelosuppression when taking thiopurine medications? Pharmacogenomics 11:177–188
Cree BAC, Lamb S, Morgan K, Chen A, Waubant E, Genain C (2005) An open label study of the effects of rituximab in neuromyelitis optica. Neurology 64:1270–1272
Jacob A, Weinshenker BG, Violich I et al (2008) Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 65:1443–1448
Kim S-H, Kim W, Li XF, Jung I-J, Kim HJ (2011) Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol 68:1412–1420
Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA (2011) Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler 17:1225–1230
Kim S-H, Huh S-Y, Lee SJ, Joung A, Kim HJ (2013) A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. doi:10.1001/jamaneurol.2013.3071
Greenberg BM, Graves D, Remington G, Hardeman P, Mann M, Karandikar N, Stuve O, Monson N, Frohman E (2012) Rituximab dosing and monitoring strategies in neuromyelitis optica patients: creating strategies for therapeutic success. Mult Scler 18:1022–1026
Yang C-S, Yang L, Li T et al (2013) Responsiveness to reduced dosage of rituximab in Chinese patients with neuromyelitis optica. Neurology 81:710–713
Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, Carter J, Keegan BM, Kantarci OH, Pittock SJ (2009) Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol 66:1128–1133
Neff RT, Hurst FP, Falta EM, Bohen EM, Lentine KL, Dharnidharka VR, Agodoa LY, Jindal RM, Yuan CM, Abbott KC (2008) Progressive multifocal leukoencephalopathy and use of mycophenolate mofetil after kidney transplantation. Transplantation 86:1474–1478
Bakker J, Metz L (2004) Devic’s neuromyelitis optica treated with intravenous gamma globulin (IVIG). Can J Neurol Sci 31:265–267
Okada K, Tsuji S, Tanaka K (2007) Intermittent intravenous immunoglobulin successfully prevents relapses of neuromyelitis optica. Intern Med 46:1671–1672
Magraner MJ, Coret F, Casanova B (2012) The effect of intravenous immunoglobulin on neuromyelitis optica. Neurologia 28:65–72
Kim S-H, Kim W, Park MS, Sohn EH, Li XF, Kim HJ (2011) Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol 68:473–479
Cabre P, Olindo S, Marignier R, Jeannin S, Merle H, Smadja D, Aegis of French National Observatory of Multiple Sclerosis (2013) Efficacy of mitoxantrone in neuromyelitis optica spectrum: clinical and neuroradiological study. J Neurol Neurosurg Psychiatr 84:511–516
Dörr J, Bitsch A, Schmailzl KJG et al (2009) Severe cardiac failure in a patient with multiple sclerosis following low-dose mitoxantrone treatment. Neurology 73:991–993
Stroet A, Hemmelmann C, Starck M et al (2012) Incidence of therapy-related acute leukaemia in mitoxantrone-treated multiple sclerosis patients in Germany. Ther Adv Neurol Disord 5:75–79
Martinelli V, Cocco E, Capra R et al (2011) Acute myeloid leukemia in Italian patients with multiple sclerosis treated with mitoxantrone. Neurology 77:1887–1895
Bonnet F, Mercié P, Morlat P et al (1999) Devic’s neuromyelitis optica during pregnancy in a patient with systemic lupus erythematosus. Lupus 8:244–247
Jacobi C, Stingele K, Kretz R, Hartmann M, Storch-Hagenlocher B, Breitbart A, Wildemann B (2006) Neuromyelitis optica (Devic’s syndrome) as first manifestation of systemic lupus erythematosus. Lupus 15:107–109
Mok CC, To CH, Mak A, Poon WL (2008) Immunoablative cyclophosphamide for refractory lupus-related neuromyelitis optica. J Rheumatol 35:172–174
Yaguchi H, Sakushima K, Takahashi I et al (2013) Efficacy of intravenous cyclophosphamide therapy for neuromyelitis optica spectrum disorder. Intern Med 52:969–972
Bichuetti DB, Oliveira EML, Boulos F de C, Gabbai AA (2012) Lack of response to pulse cyclophosphamide in neuromyelitis optica: evaluation of 7 patients. Arch Neurol 69:938–939
Minagar A, Sheremara W (2000) Treatment of Devic’s disease with methotrexate and prednisone. Int J MS Care 2:39–43
Papeix C, Vidal J-S, de Seze J et al (2007) Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler 13:256–259
Shimizu J, Hatanaka Y, Hasegawa M et al (2010) IFNβ-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology 75:1423–1427
Palace J, Leite MI, Nairne A, Vincent A (2010) Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 67:1016–1017
Kim S-H, Kim W, Li XF, Jung I-J, Kim HJ (2012) Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler 18:1480–1483
Uzawa A, Mori M, Hayakawa S, Masuda S, Kuwabara S (2010) Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol 17:672–676
Gartzen K, Limmroth V, Putzki N (2007) Relapsing neuromyelitis optica responsive to glatiramer acetate treatment. Eur J Neurol 14:e12–e13
Bergamaschi R, Uggetti C, Tonietti S, Egitto MG, Cosi V (2003) A case of relapsing neuromyelitis optica treated with glatiramer acetate. J Neurol 250:359–361
Kitley J, Elsone L, George J, Waters P, Woodhall M, Vincent A, Jacob A, Leite MI, Palace J (2013) Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J Neurol Neurosurg Psychiatry 84:918–921
Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung H-P, Paul F, Aktas O (2012) Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 69:239–245
Barnett MH, Prineas JW, Buckland ME, Parratt JDE, Pollard JD (2012) Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler 18:108–112
Jacob A, Hutchinson M, Elsone L, Kelly S, Ali R, Saukans I, Tubridy N, Boggild M (2012) Does natalizumab therapy worsen neuromyelitis optica? Neurology 79:1065–1066
Min J-H, Kim BJ, Lee KH (2012) Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler 18:113–115
Kageyama T, Komori M, Miyamoto K, Ozaki A, Suenaga T, Takahashi R, Kusunoki S, Matsumoto S, Kondo T (2013) Combination of cyclosporine A with corticosteroids is effective for the treatment of neuromyelitis optica. J Neurol 260:627–634
Miyamoto K, Kusunoki S (2009) Intermittent plasmapheresis prevents recurrence in neuromyelitis optica. Ther Apher Dial 13:505–508
Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, Ogawa M, Toda T, Yamamura T (2011) Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA 108:3701–3706
Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T (2013) Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol 23:827–831
Kieseier BC, Stüve O, Dehmel T et al (2012) Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol 70:390–393
Ayzenberg I, Kleiter I, Schröder A, Hellwig K, Chan A, Yamamura T, Gold R (2013) Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol 70:394–397
Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, O’Toole O, Wingerchuk DM (2013) Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol 12:554–562
Paul F (2013) Hope for a rare disease: eculizumab in neuromyelitis optica. Lancet Neurol 12:529–531
Tradtrantip L, Zhang H, Saadoun S, Phuan P-W, Lam C, Papadopoulos MC, Bennett JL, Verkman AS (2012) Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol 71:314–322
Miyazaki K, Abe Y, Iwanari H et al (2013) Establishment of monoclonal antibodies against the extracellular domain that block binding of NMO-IgG to AQP4. J Neuroimmunol 260:107–116
Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2012) Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol 71:323–333
Zhang H, Verkman AS (2013) Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest 123:2306–2316
Tradtrantip L, Asavapanumas N, Verkman AS (2013) Therapeutic cleavage of anti-aquaporin-4 autoantibody in neuromyelitis optica by an IgG-selective proteinase. Mol Pharmacol 83:1268–1275
Tradtrantip L, Ratelade J, Zhang H, Verkman AS (2013) Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin G into therapeutic antibody. Ann Neurol 73:77–85
Matiello M, Pittock SJ, Porrata L, Weinshenker BG (2011) Failure of autologous hematopoietic stem cell transplantation to prevent relapse of neuromyelitis optica. Arch Neurol 68:953–955
Qian P, Cross AH, Naismith RT (2011) Lack of response to monoclonal antibody therapy in neuromyelitis optica. Arch Neurol 68:1207–1209
Vaknin-Dembinsky A, Brill L, Kassis I, Petrou P, Ovadia H, Ben-Hur T, Abramsky O, Karussis D (2012) T-cell reactivity against AQP4 in neuromyelitis optica. Neurology 79:945–946
Weiner HL (2012) Role of T cells in neuromyelitis optica. Ann Neurol 72:6–8
Herges K, de Jong BA, Kolkowitz I et al (2012) Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult Scler 18:398–408
Papadopoulos MC, Verkman AS (2012) Aquaporin 4 and neuromyelitis optica. Lancet Neurol 11:535–544
Conflicts of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Members of Neuromyelitis Optica Study Group (NEMOS) are listed in the appendix.
Members of the Neuromyelitis Optica Study Group (in alphabetical order)
Members of the Neuromyelitis Optica Study Group (in alphabetical order)
P. Albrecht, Universität Düsseldorf; O. Aktas, Universität Düsseldorf; K. Angstwurm, Universität Regensburg; A. Berthele, Technische Universität München; F. Bischof, Zentrum für Neurologie Tübingen; N. Borisow, Charite Berlin; T. Böttcher, Bonhoeffer Klinikum Neubrandenburg; J. Brettschneider, Universität Ulm; M. Buttmann, Universitätsklinik Würzburg; B. Ettrich, Universität Leipzig; J. Faiss, Asklepios Klinik Teupitz; A. Gass, Universitätsklinikum Mannheim; C. Geis, Universitätsklinikum Jena; K. Guthke, Klinikum Görlitz; J. Havla, Institut für Klinische Neuroimmunologie LMU-München; H-P. Hartung, Universität Düsseldorf, K. Hellwig, Ruhr-Universität Bochum; B. Hemmer, Technische Universität München; F. Hoffmann, Krankenhaus Martha-Maria Halle; U. Hofstadt-van Oy, Klinikum Bayreuth; S. Jarius, Universität Heidelberg; P. Kern, Asklepios Klinik Teupitz; C. Kleinschnitz, Universitätsklinik Würzburg; I. Kleiter, Ruhr-Universität Bochum; W. Köhler, Fachkrankenhaus Hubertusburg; E. Kolesilova, Asklepios Klinik Teupitz; M. Krumbholz, Institut für Klinische Neuroimmunologie LMU-München; T. Kümpfel, Institut für Klinische Neuroimmunologie LMU-München; S. Langel, Landeskrankenhaus Rheinhessen; F. Lauda, Universität Ulm; M. Liebetrau, Agaplesion Ev. Bathildiskrankenhaus Bad Pyrmont; R. Linker, Universität Erlangen; W. Marouf, Heliosklinik Stralsund; M. Marziniak, Isar-Amper Klinik Ost München; I. Metz, Universität Göttingen; C. Mayer, Universität Frankfurt; A. Melms, Universität Erlangen; C. Münch, Charité Berlin; O. Neuhaus, Kreiskrankenhaus Sigmaringen; S. Niehaus, Klinikum Dortmund; F. Pache, Charité Berlin; F. Paul, Charité Berlin, H. Pellkofer, Universität Göttingen; R. Reuss, Bezirkskrankenhaus Bayreuth; A. Riedlinger, Asklepios Klinik Teupitz; M. Ringelstein, Universität Düsseldorf; S.P. Rommer, Universität Rostock; K. Ruprecht, Charité Berlin; S. Schippling, Universität Zürich (Schweiz); M. Schwab, Universität Jena; M. Stangel, Medizinische Hochschule Hannover, J. Stellmann, Universität Hamburg; F. Then-Bergh, Universität Leipzig; C. Trebst, Medizinische Hochschule Hannover; H. Tumani, Universität Ulm; C. Veauthier, Heliosklinik Stralsund; KP. Wandinger, Universitätsklinikum Schleswig–Holstein; R. Weissert, Universität Regensburg; B. Wildemann, Universität Heidelberg; C. Wilke, Nervenzentrum Potsdam; A. Winkelmann, Universität Rostock; L. Zeltner, Universität Tübingen; C. Zentner, Krankenhaus Martha Maria, Halle; U. Zettl, Universität Rostock; U. Ziemann, Universität Tübingen.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Trebst, C., Jarius, S., Berthele, A. et al. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 261, 1–16 (2014). https://doi.org/10.1007/s00415-013-7169-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-013-7169-7