
Abstract
To determine the factors that control the oxygen content of sulphide melts in the upper mantle, mono-sulphides were equilibrated with mantle peridotite assemblages, with varying FeO and NiO contents, between 3 and 13 GPa and 1300 to 1819 °C. Carbonates were added as a flux and the use of graphite capsules allowed oxygen fugacities (fo2) to be estimated for most of the experiments. Sulphide melt O concentrations were in the range 0.2–3.7 wt. %, coexisting with silicate assemblages with olivine Fe/(Fe + Mg) ratios between 0.04 and 0.25. Except in Ni-bearing experiments, variations in the sulphur/metal cation ratio did not affect the sulphide melt O contents, which also appeared to be independent of fo2. The silicate FeO contents, temperature and pressure were found to be the main controls on O contents. Nickel lowers the O content, although all Ni-bearing experiments produced sulphides with lower sulphur/metal ratios, making it difficult to categorically separate the effect of Ni alone. A preliminary geothermometer expression was developed based on the O content of the sulphide melts and the iron oxide concentrations of coexisting olivine and orthopyroxene. With this expression, the experimental temperatures are reproduced to within 74 K for Ni-free experiments and within 135 K for Ni-bearing samples. Using measurements of the O contents of sulphide inclusions of peridotite affinity in diamonds from the Lac de Gras kimberlite field, a plausible average entrapment temperature of 1318 ± 48 °C is calculated. Mantle peridotite assemblages would be expected to contain sulphide melts with approximately 0.4–0.6 wt. % O along a typical mantle adiabat down to 200 km depth.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Base metal, iron–nickel sulphides are found as accessory phases in most upper-mantle rocks and also form the most common inclusions in diamonds (Sharp 1966; Pearson et al. 1998; Richardson et al. 2004). Sulphides exert an important control over the distribution of highly siderophile elements in the mantle (Pearson et al. 1998; Kiseeva et al. 2017; Zhang et al. 2018), are commonly employed in geochronology due to their high Re and Pb contents (Rudnick et al. 1993; Pearson et al. 1999; Wiggers de Vries et al. 2013; Harvey et al. 2016; Smit et al. 2016), and are also, used to estimate mantle sulphur fugacities fs2 (Eggler and Lorand 1993). Along a typical adiabat, mantle sulphides should remain molten, or at least partially molten, up to pressures of 8 GPa, i.e. approximately up to 250 km depth (Bockrath et al. 2004; Zhang and Hirschmann 2016). Some mantle sulphides found as inclusions in xenoliths have textures consistent with them being molten at some point (e.g. Aulbach et al. 2004; Harvey et al. 2016). Upon cooling, however, they undergo a relatively complex crystallisation process. This starts with the crystallisation of (Fe,Ni)(1-x)S monosulphide solid solution (MSS) and ends, after a series of crystallisation and exsolution reactions, with the formation of pentlandite ((Fe,Ni)9S8), chalcopyrite (CuFeS2) and pyrrhotite (Fe1-xS) (Bulanova et al. 1996; Holwell and Mcdonald 2010; Harvey et al. 2016). It is often assumed that sulphide inclusions in diamond were trapped as MSS, from which a similar sulphide assemblage forms through solid state exsolution during cooling (Taylor and Liu 2009). Geothermometry estimates for cratonic geotherms and diamond formation yield temperatures mainly below the solidus of MSS (Shirey et al. 2013; Zhang and Hirschmann 2016). The common occurrence of sulphide inclusions in diamond, however, has led many researchers to propose a link between sulphide melts and diamond formation, either through the provision of a carbon-saturated source or by acting as a reducing agent for carbonate melts (Bulanova 1995; Gunn and Luth 2006; Palyanov et al. 2007; Shushkanova and Litvin 2008). Furthermore, some cratonic lithosphere regions appear to encompass conditions where sulphide melts would be stable (Ashchepkov et al. 2010; Shirey et al. 2013; Zhang and Hirschmann 2016), and more Fe-metal-rich sulphide-bearing melts certainly appear to have played a role in the formation of some sublithospheric diamonds (Smith et al. 2016). The extent to which other components, such as Cu, O or H, may lower MSS melting temperatures is also unclear.
One potential indicator for the initial entrapment of sulphide melt is the presence of magnetite in recovered sulphide assemblages (Bulanova et al. 1996), which is an indicator of dissolved O in sulphide melt. Whilst O appears to be slightly soluble in MSS at high temperatures, concentrations are relatively low (< 0.4 wt. % O) in comparison to sulphide melts (Graham and McKenzie 1987; Graham et al. 1987; Zhang and Hirschmann 2016). The 1 bar Fe–FeO–FeS ternary eutectic melt, for example, contains approximately 7.5 wt. % O and is essentially a binary composition of 62 mol % FeS and 38 mol % FeO, with a melting temperature of 915 °C, i.e. approximately 270 °C lower than the FeS liquidus (Naldrett 1969). In equilibrium with silicate melts, sulphide liquid O contents have been reported from approximately 1 to over 10 wt. % (Wendlandt 1982; Doyle and Naldrett 1987; Terasaki et al. 2005; Brenan 2015; Boujibar et al. 2019). A number of factors are likely important in controlling sulphide liquid O contents such as pressure, temperature, fo2, fs2 and Ni contents (Wendlandt 1982; Doyle and Naldrett 1987; Fonseca et al. 2008). As yet, however, no model exists for determining the O contents of sulphide melts in equilibrium with mantle assemblages. The O content may be an important factor controlling the mobility and geochemistry of sulphides. Oxygen exerts the main influence on the dihedral angle between sulphide melts and olivine, such that high O-bearing melts may potentially form interconnected networks (Gaetani and Grove 1999; Terasaki et al. 2005; Zhang et al. 2018). Kiseeva and Wood (2015) showed that sulphide melt O contents also control the partitioning of many chalcophile elements with coexisting silicate melts.
Although rarely reported, O contents of sulphide melts hosted by olivine xenocrysts range up to approximately 16 wt. %, with samples showing O concentrations higher than around 4 wt. % generally also showing signs of alteration (Aulbach et al. 2004; Alard et al. 2011; Delpech et al. 2012). Olivine is unlikely to be an inert container with respect to sulphide O contents, however, which may well re-equilibrate during ascent to the surface and cooling. Diamonds, on the other hand, should provide a more unreactive host, as long as they remain crack free. Oxygen concentrations of sulphide inclusions in diamonds are rarely measured, however, either because studies focus more on the sulphide trace element concentrations, or radiogenic isotopes, or because analysing the bulk composition is challenging, due to the initially homogeneous sulphides recrystallizing into the previously described phases on cooling (McDonald et al. 2017). The presence of magnetite in diamond sulphide inclusion assemblages has been reported, however, (Bulanova et al. 1996; Jacob et al. 2016) and measurements of the bulk O contents do exist. Davies et al., (1999, 2004) measured O concentrations in the range of 0.2–12 wt. % for sulphide diamond inclusions of both peridotitic and eclogitic associations. Sulphide inclusion Ni contents are used to characterise this association as either peridotite (> 12 wt. % Ni), pyroxenitic (8–12 wt. % Ni) or eclogitic (< 8 wt. % Ni) (Yefimova et al. 1983; Deines and Harris 1995; Aulbach et al. 2009). On the other hand, Bulanova et al. (1996) found magnetite to be rare in sulphide inclusions within a suite of Yakutian diamonds and, in what can be viewed as a cautionary note, observed that assemblages that did contain magnetite appeared to be associated with cracks, implying an epigenetic origin.
In an attempt to clarify what factors may control the O contents of sulphide assemblages in the mantle, and particularly under conditions where lithospheric diamonds may form, we have performed high-pressure and high-temperature experiments on sulphide melts in equilibrium with a peridotite assemblage. We examine how factors such as pressure, temperature, fo2, silicate iron oxide content, sulphide Ni content and metal/sulphur ratio influence the O contents and, with the aid of further studies from the literature, we propose a preliminary model for describing sulphide O contents in mantle peridotite assemblages. We also demonstrate that Mössbauer spectroscopy can be used as a non-destructive method to determine the O content of sulphide inclusions in diamonds (McCammon et al. 1997, 1998).
Methods
Starting materials were prepared using reagent-grade oxide and carbonate powders. The silicate bulk composition employed is close to KLB-1 peridotite (Davis et al. 2009), but the FeO content was varied between 8.1 and 17.6 wt. % (Table S1). Each silicate mixture was ground under ethanol in an agate mortar for 30–40 min and subsequently reduced and decarbonated for 24 h in a 1 atm CO:CO2 gas mixing furnace using a Pt cage at 1100 °C and an fo2 2 log units below the fayalite–magnetite–quartz buffer (FMQ; Ballhaus et al. 1991).
Sulphides were added to the silicate material as reagent-grade FeS powder in a 30:70 weight proportion of sulphide to silicate. To recreate natural sulphide inclusion compositions, Ni and Cu were added as metals (mixed with the sulphide) to some experiments. Additionally, we added Ir metal powder as a redox sensor to calculate the fo2 of the experiments (Stagno and Frost 2010). To saturate the sulphide phase and have a coexisting Fe–Ir alloy, the Ir content of some experiments had to be raised up to 5 wt. %. To promote equilibrium between sulphide melt and silicate mineral phases, between 1 and 10 wt. % of either Mg(OH)2 or MgCO3 were added to most of the starting materials as a flux to stabilise the formation of a silicate-bearing melt phase. Details of the starting mixtures employed in each experiment are reported in Table 1.
Experiments were performed using a 1000 tonne and a 5000 tonne multi-anvil press. A Cr2O3-doped MgO octahedral pressure medium was used with an edge length of 18 mm. Stepped graphite (< 6 GPa) or LaCrO3 (> 6 GPa) resistive heaters were used inside a thermally insulating sleeve of ZrO2. Samples were placed in a 2 mm outer and 1 mm inner diameter graphite or single crystal MgO (above 8 GPa) capsule, with a 1.5 mm sample length, which was inserted into the furnace inside an MgO sleeve. A type D (W97Re3–W75Re25) thermocouple inserted axially was in contact with a thin (~ 0.3 mm) MgO disk placed on the top of the sample capsule. The MgO disk prevents damage to the soft graphite capsule by the thermocouple. The estimated uncertainty of the temperature measurements due to the length of the sample is ± 50 K (Rubie 1999; Hernlund et al. 2006). A sketch of the high-pressure assembly is given in Supplementary Figure S1.
Following the compression to targeted pressures between 3 and 13 GPa, samples were heated up to target temperatures (1300–1819 °C) by increasing the electric power to the heater. When using graphite capsules, experiments were first heated to 800–900 °C for 4 h to sinter the graphite capsules, before being raised to the target temperature. This step helps to prevent the escape of sulphide melt during the experimental run. Run times of experiments at target temperature ranged from 2 to 24 h depending on the temperature. Details of the experimental conditions are given in Table 1. The experiments were quenched by switching off the electrical power to the heater and then decompressed overnight to 1 bar pressure (0.1 MPa). Recovered samples were mounted in epoxy resin and ground and polished for chemical analysis.
Chemical analyses were performed with a JEOL JXA 8200 electron probe micro analyser (EPMA) in wavelength dispersive X-ray spectroscopy (WDS) mode. For silicate phases, a 15 kV accelerating voltage and 15 nA probe current were employed. Sulphides and Ir–Fe alloy were analysed with a 20 kV acceleration voltage and 20 nA probe current. Oxygen in the sulphides was measured using a LDE 1 (Kα) crystal in the WD spectrometer. Counting times were 20 s for major elements and 60 s for minor elements, with half of each counting time used for the respective backgrounds. We used a focussed electron beam to analyse the mineral phases, and a defocused beam with a diameter of 10–30 µm for the sulphide and silicate melt, which commonly show a spatially inhomogeneous quench texture. The beam diameter was varied for different samples depending on the size of the quench textures. Standards employed were natural silicate minerals and oxides for silicate analyses and pure metals and sulphides for sulphide and alloy phases (Table S2). To check the O-free background level, we performed analyses of freshly polished O-free standards of FeS2, pyrrhotite, Fe metal and FeSi alloy, following the methodology of Fonseca et al. (2008). In contrast to Fonseca et al. (2008), however, we found background O levels of 0.06 ± 0.04 wt. % and so applied no correction to the O analyses to account for an apparent O contribution from the background. We also analysed a series of secondary standards during each session to ensure internal consistency. Matrix corrections were performed using the “φρ-z” method for silicates, whereas the “ZAF” method was used for sulphide melt and alloy phases.
A Mössbauer spectroscopy measurement was performed on a mass of quenched sulphide melt with a diameter of approximately 200 µm separated from sample Z1778 (7 GPa, 1600 °C). The biggest sulphide mass in the sample (approximately 200 µm diameter) was liberated from the rest of the silicate assemblage and mounted in epoxy resin. The separated sulphide was then parallel polished to create a section that was approximately 100 µm thick. A 25 µm thick Ta foil was placed over the section with the sample exposed through a 200 µm diameter hole. The Ta foil absorbs more than 99% of the 14.4 keV gamma rays, which acts as a collimator. A point source Mössbauer spectrometer (McCammon et al. 1997; 1998) (specific activity ≥ 2000 mCi/cm2) was employed with a collection time of just over 6 days.
Transmission electron microscopy (TEM) measurements were also performed on a quenched sulphide melt sample synthesised at 3 GPa and 1400 °C using similar methods as described above (Sample number V1045; Armstrong 2018). A thick section (~ 30 µm) of the sample was glued onto a 3 mm sized Mo grid and subsequently thinned to electron transparency using Ar ion milling. The sample was observed in a 200 kV analytical transmission electron microscope (ATEM, FEI Titan G2 80–200 S/TEM) equipped with an energy-dispersive X-ray spectrometer (EDXS, Bruker QUANTAX silicon drift detector). For phase identification, selected area electron diffraction (SAED), bright field TEM images, high-angle annular dark field (HAADF) scanning TEM images and EDX element maps were obtained.
Results
The experimental run products consisted of silicate mineral phases, mainly olivine and orthopyroxene, quenched silicate melt and quenched sulphide melt. In some experiments an Fe–Ir alloy was also present. A complete list of phases in each experiment is given in Table 1, and the full chemical analyses in the Supplementary Table S3 and S4. Olivine Fe/(Fe + Mg) ratios (\({X}_\text{Fe}^\text{Ol}\)) vary between the experiments from approximately 0.04 to 0.25. The iron contents of coexisting orthopyroxene are in excellent agreement with the predictions of previous models (Von Seckendorff and O’Neill 1993), with the olivine–orthopyroxene Fe–Mg exchange coefficient (\({K}_{D}^\text{Fe-Mg}= {[X}_\text{Fe}^\text{Ol}{ X}_\text{Mg}^\text{Opx}]/{[X}_\text{Fe}^\text{Opx}{ X}_\text{Mg}^\text{Ol}]\)) showing very little variation with either pressure or temperature over the conditions of this study. Proportions of silicate melt fractions varied with temperature and with the amount of flux added to the experiments. In most samples, silicate melt accumulated in the upper section of the capsule but as shown in Fig. 1a, the capsule length was < 1.5 mm, so thermal gradients were likely to be < 50 °C (Rubie 1999; Hernlund et al. 2006).

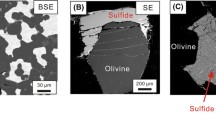
a Sample, Z1780 (5 GPa, 1450 °C), in a graphite capsule containing sulphide melt globules within both quenched silicate melt and crystals of a subsolidus peridotite assemblage. Ir–Fe alloy redox sensor is also present in some sulphide melts. This is a composite of two back scattered electron images with different brightness and contrast settings to show the textural characteristics of both silicate and sulphide phases. b A quenched sulphide globule showing a typical quench texture with crystallised sulphide (light grey), an interstitial O-rich phase (dark grey), and quenched Fe–Ir alloy (white). c Silicate grains of olivine and orthopyroxene coexisting with quenched sulphide melt
Quenched sulphides generally formed rounded melt globules, which become more irregular in shape when surrounded by silicate minerals (Fig. 1c). Quenched sulphide melts viewed in backscattered electron images (Fig. 1b) are heterogeneous and contain regularly spaced regions of a lighter grey, often elongated, sulphide phase separated by dark grey interstitial O-rich material. If sufficient Ir was added to the experiments, an additional Fe–Ir alloy phase formed, which can be distinguished from quenched alloy originally dissolved in the sulphide melt because it forms euhedral crystals with sizes up to 30 μm (Fig. 1a). This alloy phase was, nonetheless, generally always found in contact with blobs of sulphide melt (Fig. 1a). To capture the average composition of the heterogeneously quenched sulphide melt globules, they were analysed with a defocused beam, generally of 10–20 µm diameter. Using a defocused beam, average compositions of the quenched melts were obtained, with the choice of beam diameter (10–20 µm) depending on the grain size of the quenched crystals within the sulphides. The same method was applied to the silicate melt analyses, where in some cases the beam diameter was increased up to 30 µm. EPMA chemical analyses of all phases are reported in the Supplementary Tables (S3 and S4). However, the experiments were not optimised to obtain silicate melt analyses, which due to the often small melt fractions and heterogeneous quench textures, show large uncertainties and are reported only for completeness. Sulphide melt globules smaller than 10 µm were avoided because they could potentially have re-equilibrated with surrounding silicates during quenching (O’Neill et al. 1998). The standard deviation (1σ) reported for each sulphide melt composition is determined from the average of at least 14 but up to 69 individual melt analyses (Table S4). Carbon contents in sulphide melt have been measured by Zhang and Hirschmann (2012) from experiments performed under similar conditions. They reported negligible C concentrations for sulphide melts with S contents > 20 wt. %. Sulphur contents in the present study were above 30 wt. %, so that the sulphide melts should not contain C, as reflected by overall good totals (99.01–101.23 wt. %) of EPMA analyses.
Sulphide melting relations are in good agreement with those of Zhang and Hirschmann (2016). Experiments Z1912 and Z1915 at 8 and 11 GPa and 1300 °C are likely subsolidus but their O contents actually agree with the resulting temperature trend. As shown in Fig. 2, the variation in excess O and S in the sulphide melts from this study, determined as the mole fractions of S + O-M, where M is the total mole fraction of metal atoms, varied from 0.03 to 0.12, which is a similar range to that found in many sulphide inclusions in diamonds (Davies et al. 1999; 2004). The highest sulphide O concentration corresponds to 3.7 wt. % in sample Z1864 from 5 GPa and 1618 °C. When the O/(O + S) ratio is examined, along with numerous other high-pressure studies from the literature, which also contained either coexisting olivine or silicate melts, there appears to be no clear dependence of O content on the excess anion content. Although some relationship probably exists, as seen in 1 bar pressure (0.1 MPa) studies (Kress 1997; Fonseca et al. 2008), it appears not to be a dominant factor at least for S + O-M > 0. Although possibly arising from incomplete data coverage, it does appear that metal-rich samples have generally lower O contents, and certainly the Ni-bearing samples appear to have consistently lower O contents, aside from a few outliers. Some of the Ni-bearing samples extend to metal-rich values as a result of adding Ni to the experiments as metal rather than NiS. This raises some uncertainty as to whether the low O contents may be related to high metal or high Ni contents.

The variation of excess anions (mole fractions of sulphur + oxygen – metal) as a function of the normalised O content (expressed as oxygen/total anions) in sulphide. This study filled symbols; literature data—open symbols. Green symbols (both open and filled)—Ni-bearing samples. Individual error bars are shown only for this study, with a typical uncertainty for literature data indicated
The use of graphite capsules and the presence of carbonate-bearing melts in many of the experiments constrain the experimental fo2 levels to be below the enstatite–magnesite–olivine-diamond (EMOD) buffer, defined by the equilibrium MgSiO3 + MgCO3 = Mg2SiO4 + C + O2. Dilution of the carbonate component of the melt by silicate components lowers the fo2 below EMOD. Using the expression given by Stagno and Frost (2010), the fo2 can be estimated from the melt CO2 content (Table S3), which can, in turn, be approximated from the deficit in EPMA analysis totals (e.g. Stagno and Frost 2010). In four experiments sufficient, Ir was added to saturate the sulphide phase and force the existence of an Ir-dominated alloy. The Fe content of this alloy is then sensitive to the fo2 (Woodland and O’Neill 1997), which can be accurately calculated using the equilibrium 2Fe2SiO4 = 2FeSiO3 + 2Fe + O2 and employing thermodynamic data given in Stagno and Frost (2010). All values are calculated relative to the FMQ buffer (Ballhaus et al. 1991) to remove P and T effects.
It is interesting to note that in comparison to 1 bar pressure (0.1 MPa) experiments (Fonseca et al. 2011), much greater sulphide Ir contents are required to reach Ir alloy saturation at 5 GPa. The solubility of Ir in 1 bar pressure (0.1 MPa) sulphide melt has been shown to depend on temperature, fs2 and sulphide O content but values are reported to be less than ~ 1 wt. % at temperatures up to 1400 °C (Fonseca et al. 2011). At 5 GPa, however, our experiments show that sulphide Ir concentrations varied between 3.8 and 19.9 wt. % at Ir alloy saturation, which implies an important role of pressure on the exsolution of noble metal alloys from sulphides. The main cause of the Ir variation in the sulphide melt at these high-pressure conditions appears to be an inverse relationship with the sulphide O content, which varied between 3.7 and 1.1 wt. %. This is a similar but stronger O relationship to that found at 1 bar pressure (0.1 MPa) (Fonseca et al. 2011). The mole fractions of Ir in the sulphide melt are still only ~ 0.05 at maximum, however, and there is no indication that this Ir concentration significantly influences the sulphide O contents.
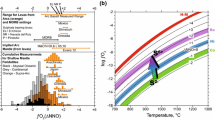
Oxygen fugacities calculated with both methods are reported in Table 2 and shown in Fig. 3a. Where both methods can be employed, the carbonate–graphite equilibrium gives values which are consistently around 0.6 log units lower, which probably reflects inaccuracies in the CO2 concentration estimated using the EMPA totals. The experimental fo2 range from approximately 1.8 to 4.0 log units below the FMQ buffer, is similar to the range obtained for the majority of peridotite xenoliths from the subcratonic lithospheric mantle (Stagno et al. 2013) and most likely covers the range relevant to diamond formation in the upper mantle. Within this range, which spans over 3 log units together with the results of Brenan (2015), there is no obvious dependence with the sulphide O content.

a Experimental O contents (mole fraction) in sulphide melts as a function of fo2 normalised to the FMQ buffer. Experimental conditions—this study P = 3–8 GPa, T = 1300–1819 °C; Brenan (2015) P = 0.9–0.5 GPa, T = 1200–1300 °C. The curve is calculated from the 1 bar O equation of Fonseca et al. (2008) at a constant log fs2 of -2 and at 1500 °C. b Experimental sulphide O contents as a function of olivine molar Fe/(Fe + Mg) ratio. Uncertainties indicated are 1σ
A correlation between the O content of sulphide melts and the FeO concentration in coexisting silicate melts has been previously reported (Kiseeva and Wood 2013, 2015; Boujibar et al. 2019). A broadly similar relationship can be observed in the results of this study when sulphide O contents are plotted as a function of the iron content of coexisting olivine, as shown in Fig. 3b. The increase in Fe/(Fe + Mg) ratio from approximately 0.03 to 0.25 results in a clear increase in the mole fraction of O in the sulphide melt from near zero to approximately 0.1. The experiments of Terasaki et al. (2005), which were performed up to an Fe/(Fe + Mg) ratio of over 0.5 are generally consistent with this trend. Nonetheless, for a given olivine Fe content, there is still a considerable scatter in sulphide O contents, at least between 3 and 11 GPa. As will be seen, most of this variation can be explained by changing temperature. Previous experimental studies have proposed that the presence of Ni reduces the O content of sulphide melts at otherwise comparable conditions (Fonseca et al. 2008; Kiseeva and Wood 2015; Boujibar et al. 2019), this is also apparent in the current results, as discussed later. As peridotitic sulphide inclusions in natural diamonds have Ni contents in the range 12–60 wt. %, this effect is important.
TEM and Mössbauer spectroscopy analyses were performed to examine how O might be accommodated in sulphide melt, i.e. its oxidation state, by determining which phases are present in the quench assemblage. A Mössbauer spectroscopy measurement was performed on a globule of quenched sulphide with a diameter of approximately 200 µm separated from the silicate melt of sample Z1778 (7 GPa, 1600 °C). The resulting spectrum of the Ni-free sulphide assemblage is dominated by a magnetic sextet signal from FeS, but a signal from magnetite is also clearly distinguishable (Fig. 4a), from which a sample magnetite content of 8 ± 2 wt. % can be determined from the intensity ratios. This corresponds to an O content of 2.28 ± 0.55 wt. %, which is within the combined error of the EPMA measurement for this sample of 1.70 ± 0.49 wt. % (i.e. 6.16 ± 1.78 wt. % magnetite). The EPMA O value is an average of multiple analyses (30 points) distributed in several sulphide globules, whereas the Mössbauer measurement is from only a single globule. Thus, a level of inhomogeneity in the O concentration likely contributes to this mismatch. To confirm that O is hosted only by magnetite, a TEM analysis was performed on another quenched sulphide assemblage, this time Ni-bearing, produced in a similar experiment (V1045) conducted at 3 GPa, 1400 °C (Armstrong 2018). Selected area electron diffraction (SAED) and bright field imaging (Fig. 4b), as well as high-angle annular dark field imaging, and element maps (Fig. S2) show that O is indeed hosted by quenched magnetite, which is closely associated with Fe–Ni alloy.

a Mössbauer spectrum collected for sample Z1778 (Ni free). The sextet in green is FeS and in blue is magnetite. b Bright field TEM image of a quenched sulphide melt assemblage (V1045) containing magnetite and Fe–Ni alloy. Inset in b is a selected area electron diffraction (SAED) pattern of magnetite. See also Supplementary Figure S3
Discussion
Thermodynamic model
The sulphide melt O contents determined in this study show no apparent dependence with fo2, in contrast to the results of Kress (1997) and Fonseca et al. (2008), who found sulphide O contents to increase quite strongly with log fo2 at 1 bar. In fact, the expression derived by Fonseca et al. (2008) at 1 bar predicts an order of magnitude increase in O over the same range of fo2 shown by the experiments in Fig. 3a, for a log fs2 of -2 and a temperature of 1500 °C. A possible explanation is that the effect of fo2 is subtler at high-pressure conditions and not apparent in Fig. 3a due to the fact that the data are not collected at either constant fs2 or temperature. Furthermore, as the fo2 in this study is in most instances governed by the coexistence of graphite and carbonate-bearing melt, higher temperatures result in generally lower relative fo2, as the melt carbonate component becomes diluted, so it is possible that T and fo2 effects cancel out to some extent. However, an fo2-dependant O content implies that O is associated with an Fe3+-bearing sulphide melt component (Fonseca et al. 2008). This appears, at first sight, to be in agreement with the observation that magnetite is the only oxide phase that we identify in sulphide melts using both Mössbauer and TEM measurements. However, the TEM imaging (Fig. 4b) indicates that magnetite coexists in the quenched sample with Fe–Ni alloy and it seems that both phases were formed during quenching from an initial (Fe,Ni)O component that is not stable as low temperatures. A sulphide melt FeO component would explain the lack of fo2 dependence at the higher-pressure conditions. Given the lack of fo2 dependence, at least at graphite saturation, it seems appropriate, therefore, to describe the variation in sulphide O content with the equilibrium:
The FeO distribution coefficient, KD, from Eq. (1) is,
where \({X}_\text{FeO}^\text{Sul}\) is the mole fraction of FeO in the sulphide melt and \({X}_\text{Fe}^\text{Opx}\) and \({X}_\text{Fe}^\text{Ol}\) are the molar \(\mathrm{Fe}/(\mathrm{Fe}+\mathrm{Mg})\) ratios of orthopyroxene and olivine, respectively. The sulphide can be described as a mixture of the components FeS, FeS2 and FeO, in addition to Fe and NiS and other minor metal sulphides when necessary. Figure 5 shows the dependence of KD with temperature for the experiments in this study, with potentially a minor dependence also on pressure. Ni-bearing data show a greater variability due to the differences in Ni concentration. There are no previous experimental studies that we are aware of, that report sulphide O contents and coexisting olivine and orthopyroxene compositions. However, both Terasaki et al. (2005) and Zhang et al. (2018) report sulphide O contents coexisting with olivine and using the relationship between olivine and orthopyroxene Fe–Mg partitioning, we can estimate fictive orthopyroxene compositions. We do this also for a few experiments performed in this study where melting resulted in the loss of orthopyroxene from the solid assemblage. The study of Terasaki et al. (2005) was also performed using graphite capsules and for the study of Zhang et al. (2018), we only plot data where graphite capsules were employed, as it is possible that different O dependencies occur at significantly higher fo2. This compositional range, however, is at least consistent with most sulphide inclusions reported for lithospheric diamonds (Davies et al. 1999; 2004). Although most of the data from Terasaki et al. (2005) are in good agreement with the trend from this study, even though they cover very large ranges of olivine Fe content, one data point is an outlier and the data of Zhang et al. (2018) from measurements at 2 GPa are also in poor agreement. It is difficult to understand how this variation arises. One possibility might be that it is difficult to achieve an equilibrium O concentration in the sulphide in the absence of a silicate-bearing melt phase.

Distribution coefficient (KD) dependence on experimental temperature. The KD of FeO between the sulphide and silicate assemblage, calculated using Eq. 2, correlates positively with temperature. Literature data from experiments performed in graphite capsules
We can describe the experimental KD with a simple thermodynamic model where,
where \({\gamma }_\text{FeO}^\text{Sul}\) is the activity coefficient for FeO in the sulphide melt and \({\Delta G}^\text{o}\), \({\Delta H}^\text{o}\), \({\Delta S}^\text{o}\) and \(\Delta V\) are the standard state Gibbs free energy, enthalpy, entropy and volume changes, respectively, of the pure end members in equilibrium 1. We assume a symmetric mixing model for both olivine and enstatite solid solutions, e.g.
where \({W}_\text{Fe-Mg}^\text{Opx}\) is a Margules interaction parameter. We find good agreement with the Fe–Mg partitioning data between olivine and orthopyroxene with interaction parameters for the two phases of 2000 and 1000 J/mol, respectively, on a single site basis (Fig. S3). The Fe–Mg partitioning data are in very good agreement with previous work (von Seckendorff and O’Neill 1993).
We examined different models for \({\gamma }_\text{FeO}^\text{Sul}.\) Initially the effect of Ni was ignored and a simple binary mixing model was used that assumed the sulphide melt was simply a mixture of FeO and FeS, i.e.
The value of \({W}_\text{FeO-FeS}^\text{Sul}\) was obtained by performing a least-squares fit to the Ni-free experimental data of this study, whilst simultaneously fitting \(\Delta {H}^\text{o}\), \(\Delta {S}^\text{o}\) and \(\Delta V\) in Eq. 3. No improvement in the quality of the fit was obtained by considering the presence of an FeS2 component in the liquid and extending the mixing model to that of a symmetric ternary. To include the effects of Ni, however, a symmetric ternary model was employed, i.e.
where the first term is taken from the fit of Eq. 5. Ni-bearing data where S + O-M was significantly below zero were not included in the fitting to ensure that we only consider the effect of Ni, rather than the S/M ratio, which limited the Ni-bearing data to only four data points (H5560-1, H5560-2, H5561 and Z1999). The Ni data were then fitted to obtain \({W}_\text{FeO-FeS}^\text{Sul}\). Attempts to fit \({W}_\text{FeS-NiS}^\text{Sul}\) always returned a value of zero. Figure 6 shows the value of \(\Delta {G}^\text{o}\) calculated for each experimental data point using Eq. (3) and the terms given in Table 3. The resulting fitting of these data using the values of \(\Delta {H}^\text{o}\), \(\Delta {S}^\text{o}\) and \(\Delta V\) given in Table 3 is shown calculated at 3 and 11 GPa in Fig. 6. Although the majority of the Ni-bearing data were not used in the fitting due to the data extending to values of S + O-M that were significantly less than zero, in Fig. 7a, it can be seen that when the resulting model is compared with these data and with those from the study of Zhang et al. (2018), there is a generally good agreement. However, it should be noted that here again there are multiple outliers, which are in poor agreement with the model and for which no explanation can be currently found.

Calculated \(\Delta {G}_\text{(eq.1)}^\text{o}\) of all experiments plotted as a function of the temperature. The lines show the P- and T-dependent fit of Eq. 3. Uncertainties calculated from the 1σ errors on the \({X}_\text{FeO}^\text{Sul}\) determinations

a Distribution coefficients (KD—Eq. 2) as a function of Ni mole fraction in sulphide melt. Diamonds with red outline indicate data from this study between 5 and 13 GPa, 1400–1600 °C, that were used in the fitting procedure due to having S + O-M greater than or close to zero. Other Ni-bearing data with S + O-M < 0 are shown in green. Small black squares are from Zhang et al. (2018) experiments at 2 GPa and 1400 °C. The black curve is calculated from the model at 5 GPa and 1500 °C. Uncertainties reported are 2σ. b Comparison between the experimental temperatures and those calculated using Eq. 7. Only Ni-bearing data where S + O-M is close to 0 are plotted. Uncertainties are propagated from the errors on the sulphide melt FeO contents
By substituting Eqs. 2 and 6 into Eq. 3 and rearranging, we can obtain an expression for the temperature in terms of pressure, \({X}_\text{Fe}^\text{Ol}\), \({X}_\text{Fe}^\text{Opx}\) \({X}_\text{FeO}^\text{Sul}\) and \({X}_\text{NiS}^\text{Sul}\) i.e.,
where T is in K and P in GPa. For convenience \({X}_\text{Fe}^\text{Opx}\) can be replaced by (0.0039 + 0.8833 \({X}_\text{Fe}^\text{Ol}\)). In Fig. 7b, the temperature calculated using this equation is compared to the experimental temperatures. For the Ni-free experiments, the largest deviation between the model and the experiments is 74 K, whilst for the Ni-bearing samples, the highest deviation amongst the low S + O-M samples is 135 K, although this sample is from the highest pressure investigated of 13 GPa. The largest uncertainty arises from the sulphide O contents, with typical uncertainties of approximately 80 K. However, when sulphide O contents drop below 1 wt. %, i.e. \({X}_\text{FeO}^\text{Sul}\)< 0.05, which corresponds to samples with low olivine Fe content (Fe/[Fe + Mg] < 0.05), uncertainties start to rise and approach 300 K as O contents reach approximately 0.2 wt %, i.e. \({X}_\text{FeO}^\text{Sul}\)~ 0.01. Much of the variation in the O standard deviation arises from the inhomogeneity of the O distribution in the samples, which is an inevitable consequence of analysing samples that crystallise on quenching. Uncertainties arising from the olivine or enstatite chemical compositions are small and at most 20 K and similar to assuming a pressure uncertainty of 1 GPa.
The effect of Ni introduces a large uncertainty because only data points with quite low Ni contents have S + O-M values close to or above zero. This appears to be also the case for the majority of literature data, as shown in Fig. 2, i.e. whenever Ni is added to such experiments, the excess metal cation content increases. This seems to have little to do with the way Ni is added to the experiments as we also added Ni as NiS. Although the model reproduces the O concentrations of samples with higher Ni contents, there is considerable scatter and some uncertainty remains as to whether the Ni or the excess metal cation content controls the O concentration. Sulphides in diamond inclusions do not show the same relationship between Ni and excess metal content as seen in the experiments (Davies et al. 1999; 2004). If we make rather extreme alternative assumption that Ni does not affect the O content at all for samples with S + O-M > 0, then, for a sulphide with Ni/(Ni + Fe) ratio of 0.5, the temperature calculated with Eq. 7 would change by approximately 250 K.
The variation in oxygen content of sulphide melts in the mantle
We can use our model to calculate the O concentration that we expect for a sulphide melt in equilibrium with peridotite along a typical mantle adiabat within the top 200 km of the mantle. The main factor affecting the change in O content with depth is the sulphide Ni concentration, which can be determined from the sulphide–olivine Fe–Ni exchange experiments of Zhang et al. (2018). Zhang et al. (2018) propose a series of equations to describe this exchange, which results in the mantle sulphide Ni/(Ni + Fe) ratio reaching a maximum of approximately 0.65 at a depth of around 100 km and then subsequently decreasing to a value of approximately 0.3 at around 180 km. Zhang et al. (2018) proposed that the Ni/(Ni + Fe) ratio decreases as a result of a decrease in the fo2 of the mantle and approaches 0.3 as the mantle fo2 approaches ΔFMQ-3.
Figure 8 shows the sulphide O concentrations in wt. % calculated for a bulk silicate Earth composition (McDonough and Sun 1995) along a 1320 °C mantle adiabat, using the mantle fo2 profile of Stagno et al. (2013), which results in a mantle fo2 of ΔFMQ -3 at approximately 230 km. The curvature results purely from the change in sulphide Ni concentration and although it might appear that the O content would rise further, beyond this depth, the metal cation content of the sulphide is likely to increase as the mantle fo2 is projected to drop further (Zhang et al. 2018) and the O model developed here may no longer be valid. For comparison, the dotted line in Fig. 8 shows the O content if the Ni effect is ignored, which provides a prudent estimate of the uncertainty. Over this depth interval, typical peridotite rocks would not be expected to contain sulphides with O contents greater than approximately 0.5 ± 0.3 wt. %. At depths shallower than 100 km, graphite is likely exhausted from a typical mantle assemblage (Stagno et al. 2013) and our model may also not hold at the resulting higher fo2 levels, potentially explaining higher O contents in sulphide melts found inside olivines from some mantle xenoliths (Aulbach et al. 2004). Although the variation in sulphide O contents throughout this depth range is small, the results of Kiseeva and Wood (2015) imply that even this change would have a significant effect on the partitioning between sulphide and silicate melts of elements with high charges in the silicates such as Ga and Ge.

Variation in sulphide melt O contents in equilibrium with mantle peridotite. Depth variation is mainly due to changing Ni concentrations in sulphide (dashed curve) due to partitioning with olivine. The dotted curve shows the same calculation if the Ni in sulphide is ignored. The calculation is made along a 1320 °C mantle adiabat, with the mantle fo2 decreasing with depth to reach FMQ -3 at approximately 230 km depth (Stagno et al. 2013). In the shaded area (< 100 km depth), graphite may no longer be stable (Stagno et al. 2013) and the higher O fugacities could lead to higher O concentrations in sulphide melts
The interpretation of oxygen contents of natural sulphide assemblages
Although the experiments in this study were performed principally to determine the factors important in controlling mantle sulphide O contents, we can use the model described above to interpret variations in sulphide O contents reported for natural samples. The best measurements of mantle sulphide O contents should come from inclusions in diamond, which are not only chemically isolated but also have an upper-limiting constraint on the fo2 imposed by carbon saturation, as in our experiments. Unfortunately, analyses of such inclusions rarely include O contents, and in some instances do not even report S contents, which are instead often determined by stoichiometry (Westerlund et al. 2006; Aulbach et al. 2009). Many studies simply dissolve sulphides broken out of diamonds to perform Re–Os age determinations. Nevertheless, a few sulphide inclusion O contents have been reported from diamonds recovered from the Lac de Gras kimberlite field (central Canadian Slave Craton; Davies et al. 1999; 2004). Oxygen contents range from 0.27 to 12 wt. % in these sulphides, which have been divided into both peridotitic and eclogitic parageneses, based on their Ni contents.
There are significant uncertainties in using published sulphide inclusion O concentrations. It cannot be determined from the publications, for example, whether cracks have exposed the inclusions to post-entrapment alteration. Furthermore, the assemblage may have been coarse grained, making a determination of the average composition difficult, as surfaces may not represent bulk compositions. In the only such publication available (Davies et al. 1999; 2004), there is little description as to how the sulphide analyses were made, so it is difficult to assess how accurate and representative the O analyses are likely to be. We note, however, that Mössbauer measurements would provide a potentially effective and non-destructive method for determining the bulk O concentration of sulphide assemblages. Although the Mössbauer O estimate made for the sample in Fig. 4a has a large uncertainty, this could be reduced by utilizing Synchrotron Mössbauer Source (SMS) spectroscopy, which has a much higher source flux (Rüffer and Chumakov 1996; Potapkin et al. 2012). A high brilliance, small divergence beam would allow sulphide inclusions of the order of 10 µm to be analysed for their magnetite content whilst potentially still trapped in the diamonds.
Taken at face value, however, the available sulphide inclusion O concentrations (Davies et al. 1999; 2004) are those expected for sulphide liquids rather than MSS. The range is also consistent, in a qualitative fashion, with our experimentally produced sulphide melts, apart from two samples with O contents above 4 wt. %, which may well have been oxidised post-entrapment (Aulbach et al. 2004).
To apply our model to the peridotitic inclusions of Davies et al. (1999; 2004) we assume the sulphides were in equilibrium with mantle rocks of a particular olivine Fe content, adding further uncertainty. Olivine inclusions are also found in the diamonds studied by Davies et al. (1999, 2004), with Fe contents of \({X}_\text{Fe}^\text{Ol}\) = 0.075 ± 0.01. This quite narrow range is in good agreement with measurements made on mantle xenoliths from the central Slave Craton (Kopylova and Caro 2004) and corresponds to a temperature uncertainty from Eq. 7 of 100 K.
As shown in Fig. 9, many high Ni peridotite-associated inclusions (NiS > 0.6) give calculated temperatures at 6 GPa, which are unrealistically high. However, based on the expected Ni–Fe exchange coefficient between olivine and sulphide melt (Zhang et al. 2018), these sulphides have Ni concentrations too high to be in equilibrium with typical peridotitic olivines, such as those found as inclusions in the same diamonds or in xenoliths from the Slave Craton lithosphere. The high Co contents (up to 14.7 wt. %) of several of these inclusions also imply dis-equilibrium with typical mantle assemblages. If the O concentrations are reliable, then the anomalously high temperatures calculated are a result of equilibrium with mantle rocks enriched in both Ni and Fe. No experiments performed have accessed the compositions and conditions required to produce the simultaneously high Ni and O contents of these samples and contrary to Ni-rich experiments, the inclusions all have positive values of S + O-M. The inclusions may, therefore, have fractionally crystallised from sulphides that were far from equilibrium with mantle silicates.

Five peridotitic sulphide inclusions reported by Davies et al. (1999; 2004) do have Ni contents close to what would be expected for typical mantle, (Zhang et al. 2018) and although one of these gives an anomalously high temperature (1825 °C), the other four give a narrow range of temperatures with an average of 1318 ± 48 °C. This is plausible for lithospheric diamond formation (Nimis 2002; Stachel and Harris 2009), is above the nominal FeS solidus (Zhang and Hirschmann 2016), and also agrees with the upper temperature limit obtained by Davies et al. (2004), using garnet–clinopyroxene inclusions in the same diamonds, which yield temperatures between 1040 and 1300 °C (Ellis and Green 1979).
Half of the sulphide inclusions reported by Davies et al. (1999; 2004) are of eclogitic paragenesis and experiments in eclogitic compositions would be required to interpret the O contents of these samples. However, although our model cannot be applied directly to such assemblages, we can very tentatively calculate fictive olivine and opx iron contents using a model for olivine–garnet Fe–Mg exchange (O’Neill and Wood 1979) and then apply our model. Eclogitic garnet inclusions from the same diamonds have an average Fe/(Fe + Mg + Ca) ratio of 0.30 ± 0.04, from which an equivalent olivine Fe/(Fe + Mg) ratio of approximately 0.20 ± 0.08 can be calculated, depending on temperature. When this fictive assemblage of olivine and opx is combined with the O contents of the eclogitic sulphide inclusions, a narrow range of entrapment temperatures is calculated, 978 ± 50 °C, which is close to the minimum temperature of entrapment of 1040 °C determined by Davies et al. (2004) using garnet–clinopyroxene inclusion pairs. This temperature is, however, below the FeS solidus and would require an additional sulphide component, such as H2O or H2 (Wykes and Mavrogenes 2005; Shibazaki et al. 2011), to further depress the melting temperature. To get temperatures compatible with the FeS solidus would require the silicate assemblage to have an FeO content similar to peridotitic values.
Summary and conclusion
To understand the factors that control the O concentration of sulphide melts in the upper mantle, sulphide liquids were experimentally equilibrated with mantle peridotite assemblages with varying FeO and NiO contents at P, T conditions between 3 and 13 GPa and from 1300 to 1819 °C, mainly in graphite capsules. Carbonates were added as a flux, which allowed fo2 to be estimated from the concentration of CO2 in the resulting carbonate–silicate melts in equilibrium with graphite (Stagno et al. 2010). In some experiments, an additional saturating Ir–Fe alloy was added to provide fo2 determinations from the proportion of iron in the alloy. Sulphide O contents varied from 0.2 to 3.7 wt. %, which is in qualitative agreement with the range reported for some sulphide inclusion assemblages in diamonds (Davies et al. 1999; 2004). The resulting silicate assemblage comprised mainly olivine and orthopyroxene with olivine Fe/(Fe + Mg) in the range 0.04–0.25. Neither variation in the sulphur to metal ratio nor fo2, over a range from 1.8 to 4.0 log units below the FMQ buffer, appear to influence the sulphide melt O content. Rather, the FeO content of coexisting silicates and the temperature and pressure appear to be the main controlling factors. Further experiments show that Ni appears to lower the O content of sulphide melts, although all Ni-bearing experiments, and those in the literature, also show a decrease in the sulphur/metal ratio as Ni is added that is hard to separate from the effect of Ni alone, and thus causes some uncertainty.
A thermodynamic model was developed based on the exchange of FeO between olivine, orthopyroxene and sulphide melt (equilibrium 1). The resulting relationship reproduces the experimental temperatures ± 74 K for Ni-free experiments and ± 135 K for Ni-bearing samples. The increased uncertainty for the Ni-bearing data may be an indication that other factors such as the sulphur/metal ratio may also influence the O content. Applying this model, the expected O contents of sulphide melts in equilibrium with typical peridotite along a mantle adiabat between 100 and 200 km depth is determined to be in the range 0.4–0.6 wt. %.
Temperatures can be estimated for a series of peridotitic sulphide inclusions in diamonds from the Lac de Gras kimberlite field (Davies et al. 1999, 2004), from their O contents using our thermodynamic model. It is assumed that the inclusions were in equilibrium with a silicate assemblage with an olivine Fe/(Fe + Mg) ratio of 0.075 ± 0.01, which is the average value of olivine inclusions found in the same diamonds and is typical for these xenoliths (Davies et al. 1999, 2004; Kopylova and Caro 2004). A group of peridotitic inclusions with NiS mole fractions < 0.6, give plausible temperatures (1318 ± 48 °C) for lithospheric diamond formation. Unrealistically high temperatures are found for other inclusions, however, which have NiS mole fractions > 0.6, which is higher than expected for sulphides in equilibrium with peridotites with typical mantle Ni concentrations (Zhang et al. 2018). These sulphides have much higher than expected O contents and may have equilibrated with silicates with very high Ni and Fe contents or fractionally crystallised from sulphide melts that were no longer in equilibrium with mantle silicates.
Temperatures for eclogitic sulphide inclusions in diamonds from the same locality were calculated using Fe–Mg partitioning data to calculate fictive olivine and orthopyroxene iron contents assuming a typical eclogitic garnet iron content. These calculations result in entrapment temperatures, 978 ± 50 °C, far below the sulphide solidus temperature, although other components such as H2 or H2O may have lowered the sulphide solidus or these inclusions were in equilibrium with assemblages with lower iron contents, i.e. more similar to peridotites, that would yield temperatures above the sulphide solidus.
The model produced here is preliminary and further work is required to ensure that the effect of Ni on the O contents has been suitably described, for which it would be necessary to produce Ni-bearing sulphide assemblages that do not also have low sulphur/metal ratios. Although the temperatures calculated for some sulphide assemblages from diamonds appear reasonable, this could simply be a coincidence and the inclusions themselves may contain O as a result of post-entrapment (epigenetic) alteration or the O analyses might not be representative of the entire inclusion as a result of a heterogeneous distribution of phases. Therefore, perhaps the most important further step would be to study sulphide inclusions to determine whether the O contents are epigenetic, to examine if they are captured melts or fractionally crystallised products from melts and to obtain representative O analyses. Finally, we note that Mössbauer spectroscopy could be a useful non-destructive method to determine the O content in sulphide inclusions, potentially whilst they are still trapped inside the host.
Data availability
All the data are included within the article and as online supplementary materials.
References
Alard O, Lorand JP, Reisberg L et al (2011) Volatile-rich metasomatism in montferrier xenoliths (Southern France): Implications for the abundances of chalcophile and highly siderophile elements in the subcontinental mantle. J Petrol 52:2009–2045. https://doi.org/10.1093/petrology/egr038
Armstrong K (2018) Redox Evolution of the Early Earth’s Mantle. Doctoral dissertation, University of Bayreuth
Ashchepkov IV, Pokhilenko NP, Vladykin NV et al (2010) Structure and evolution of the lithospheric mantle beneath Siberian craton, thermobarometric study. Tectonophysics 485:17–41. https://doi.org/10.1016/j.tecto.2009.11.013
Aulbach S, Griffin WL, Pearson NJ et al (2004) Mantle formation and evolution, slave craton: constraints from HSE abundances and Re-Os isotope systematics of sulfide inclusions in mantle xenocrysts. Chem Geol 208:61–88. https://doi.org/10.1016/j.chemgeo.2004.04.006
Aulbach S, Stachel T, Creaser RA et al (2009) Sulphide survival and diamond genesis during formation and evolution of archaean subcontinental lithosphere : a comparison between the slave and Kaapvaal cratons. Lithos 112:747–757. https://doi.org/10.1016/j.lithos.2009.03.048
Ballhaus C, Berry RF, Green DH (1991) High pressure experimental calibration of the olivine-orthopyroxene-spinel oxygen geobarometer: implications for the oxidation state of the upper mantle. Contrib to Mineral Petrol 107:27–40. https://doi.org/10.1007/BF00311183
Bockrath C, Ballhaus C, Holzheid A (2004) Fractionation of the platinum-group elements during mantle melting. Science 305:1951–1953. https://doi.org/10.1126/science.1100160
Boujibar A, Righter K, Bullock ES et al (2019) Segregation of Na, K, Rb and Cs into the cores of Earth, Mars and Vesta constrained with partitioning experiments. Geochim Cosmochim Acta 269:622–638. https://doi.org/10.1016/j.gca.2019.11.014
Brenan JM (2015) Se–Te fractionation by sulfide–silicate melt partitioning : Implications for the composition of mantlederived magmas and their melting residues. Earth Planet Sci Lett 422:45–57
Bulanova GP (1995) The formation of diamond. J Geochemical Explor 53:1–23. https://doi.org/10.1016/0375-6742(94)00016-5
Bulanova GP, Griffin WL, Ryan CG et al (1996) Trace elements in sulfide inclusions from yakutian diamonds. Contrib to Mineral Petrol 124:111–125. https://doi.org/10.1007/s004100050179
Davies RM, Griffin WL, O’Reilly SY, Doyle BJ (2004) Mineral inclusions and geochemical characteristics of microdiamonds from the DO27, A154, A21, A418, DO18, DD17 and Ranch Lake kimberlites at Lac de Gras, Slave Craton, Canada. Lithos 77:39–55. https://doi.org/10.1016/j.lithos.2004.04.016
Davies RM, Griffin WL, Pearson NJ, et al (1999) Diamonds from the deep: pipe DO27, Slave craton, Canada. In: Gurney JJ, Gurney JL, Pascoe MD, Richardson SH (eds) 7th international Kimberlite conference. Red Roof Designs 148–155
Davis FA, Tangeman JA, Tenner TJ, Hirschmann MM (2009) The composition of KLB-1 peridotite. Am Mineral 94:176–180. https://doi.org/10.2138/am.2009.2984
Deines P, Harris JW (1995) Sulfide inclusion chemistry and carbon isotopes of African diamonds. Geochim Cosmochim Acta 59:3173–3188. https://doi.org/10.1016/0016-7037(95)00205-E
Delpech G, Lorand JP, Grégoire M et al (2012) In-situ geochemistry of sulfides in highly metasomatized mantle xenoliths from Kerguelen, southern Indian Ocean. Lithos 154:296–314. https://doi.org/10.1016/j.lithos.2012.07.018
Doyle CD, Naldrett AJ (1987) The oxygen content of sulfide magma and it’s effect on the partitioning of nickel between coexisting olivine and molten ores. Econ Geol 82:208–211
Eggler DH, Lorand JP (1993) Mantle sulfide geobarometry. Geochim Cosmochim Acta 57:2213–2222. https://doi.org/10.1016/0016-7037(93)90563-C
Ellis DJ, Green DH (1979) An experimental study of the effect of Ca upon garnet-clinopyroxene Fe-Mg exchange equilibria. Contrib to Mineral Petrol 71:13–22. https://doi.org/10.1007/BF00371878
Fonseca ROC, Campbell IH, O’Neill HSC, Fitzgerald JD (2008) Oxygen solubility and speciation in sulphide-rich mattes. Geochim Cosmochim Acta 72:2619–2635. https://doi.org/10.1016/j.gca.2008.03.009
Fonseca ROC, Mallmann G, O’Neill HSC et al (2011) Solubility of Os and Ir in sulfide melt: Implications for Re/Os fractionation during mantle melting. Earth Planet Sci Lett 311:339–350. https://doi.org/10.1016/j.epsl.2011.09.035
Frost DJ (2003) Fe2+ -Mg partitioning between garnet, magnesiowüstite, and (Mg, Fe)2SiO4 phases of the transition zone. Am Mineral 88:387–397
Gaetani GA, Grove TL (1999) Wetting of mantle olivine by sulfide melt: Implications for Re/Os ratios in mantle peridotite and late-stage core formation. Earth Planet Sci Lett 169:147–163. https://doi.org/10.1016/S0012-821X(99)00062-X
Graham J, McKenzie CD (1987) Oxygen in pyrrhotite: 2. Determination of oxygen in natural pyrrhotites. Am Mineral 72:605–609
Graham J, Bennett CEG, Van Riessen A (1987) Oxygen in pyrrhotite: 1. Thermomagnetic behavior and annealing of pyrrhotites containing small quantities of oxygen. Am Mineral 72:599–604
Gunn SC, Luth RW (2006) Carbonate reduction by Fe-S-O melts at high pressure and high temperature. Am Mineral 91:1110–1116. https://doi.org/10.2138/am.2006.2009
Harvey J, Warren JM, Shirey SB (2016) Mantle Sulfides and their Role in Re-Os and Pb Isotope geochronology. Rev Mineral Geochemistry 81:579–649
Hernlund J, Leinenweber K, Locke D, Tyburczy JA (2006) A numerical model for steady-state temperature distributions in solid-medium high-pressure cell assemblies. Am Mineral 91:295–305. https://doi.org/10.2138/am.2006.1938
Holwell DA, Mcdonald I (2010) A review of the behaviour of platinum group elements within natural magmatic sulfide ore systems. Platin Met Rev 54:26–36. https://doi.org/10.1595/147106709X480913
Jacob DE, Piazolo S, Schreiber A, Trimby P (2016) Redox-freezing and nucleation of diamond via magnetite formation in the Earth’s mantle. Nat Commun 7:11897. https://doi.org/10.1038/ncomms11891
Kiseeva ES, Wood BJ (2013) A simple model for chalcophile element partitioning between sulphide and silicate liquids with geochemical applications. Earth Planet Sci Lett 383:68–81. https://doi.org/10.1016/j.epsl.2013.09.034
Kiseeva ES, Wood BJ (2015) The effects of composition and temperature on chalcophile and lithophile element partitioning into magmatic sulphides. Earth Planet Sci Lett 424:280–294. https://doi.org/10.1016/j.epsl.2015.05.012
Kiseeva ES, Fonseca ROC, Smythe DJ (2017) Chalcophile elements and sulfides in the upper mantle. Elements 13:111–116. https://doi.org/10.2113/gselements.13.2.111
Kopylova MG, Caro G (2004) Mantle xenoliths from the Southeastern slave craton: evidence for chemical zonation in a thick, cold lithosphere. J Petrol 45:1045–1067. https://doi.org/10.1093/petrology/egh003
Kress V (1997) Thermochemistry of sulfide liquids. I. the system O-S-Fe at 1 bar. Contrib Mineral Petrol 127:176–186
Lehlooh A, Mahmood S (2002) Mössbauer spectroscopy study of iron nickel alloys. Hyperfine Interact 139(140):387–392
Lyubutin IS, Lin CR, Korzhetskit YV et al (2009) Mössbauer spectroscopy and magnetic properties of hematite / magnetite nanocomposites. J Appl Phys. https://doi.org/10.1063/1.3194316
McCammon CA, Mccammon C, Hutchison M, Harris J (1997) Ferric iron content of mineral inclusions in diamonds from São Luiz : a view into the lower ferric iron content of mineral inclusions in diamonds from são luiz : a view into the lower mantle. Science. https://doi.org/10.1126/science.278.5337.434
McCammon CA, Chin IL, Gurney JJ, Mccallum ME (1998) Ferric iron content of mineral inclusions in diamonds from George creek, colorado determined using Mössbauer spectroscopy. Contrib to Mineral Petrol 133:30–37
McDonald I, Hughes HSR, Butler IB et al (2017) Homogenisation of sulphide inclusions within diamonds: a new approach to diamond inclusion geochemistry. Geochim Cosmochim Acta 216:335–357. https://doi.org/10.1016/j.gca.2017.04.039
McDonough WF, Sun S (1995) The composition of the Earth. Chem Geol 120:223–253. https://doi.org/10.1016/0009-2541(94)00140-4
Naldrett AJ (1969) A portion of the system Fe-S-O between 900 and 1080 °C and its application to sulfide ore magmas. J Petrol 10:171–201. https://doi.org/10.1093/petrology/10.2.171
Nimis P (2002) The pressures and temperatures of formation of diamond based on thermobarometry of chromian diopside inclusions. Can Mineral 40:871–884. https://doi.org/10.2113/gscanmin.40.3.871
O’Neill HSC, Wood BJ (1979) An experimental study of Fe-Mg partitioning between garnet and olivine and its calibration as a geothermometer. Contrib to Mineral Petrol 70:59–70. https://doi.org/10.1007/BF00371872
O’Neill HSC, Canil D, Rubie DC (1998) Oxide- metal equilibria to 2500 °C and 25 GPa: Implications for core formation and the light component in the Earth’s core. J Geophys Res 103:12239–12260
Palyanov YN, Borzdov YM, Bataleva YV et al (2007) Reducing role of sulfides and diamond formation in the Earth’s mantle. Earth Planet Sci Lett 260:242–256. https://doi.org/10.1016/j.epsl.2007.05.033
Pearson DG, Shirey SB, Harris JW, Carlson RW (1998) Sulphide inclusions in diamonds from the Koffiefontein kimberlite, S Africa: Constraints on diamond ages and mantle Re-Os systematics. Earth Planet Sci Lett 160:311–326. https://doi.org/10.1016/S0012-821X(98)00092-2
Pearson DG, Shirey SB, Bulanova GP et al (1999) Re-Os isotope measurements of single sulfide inclusions in a Siberian diamond and its nitrogen aggregation systematics. Geochim Cosmochim Acta 63:703–711. https://doi.org/10.1016/S0016-7037(99)00042-3
Potapkin V, Chumakov AI, Smirnov GV et al (2012) The 57 Fe synchrotron mössbauer source at the ESRF. J Synchrotron Radiat 19:559–569. https://doi.org/10.1107/S0909049512015579
Richardson SH, Shirey SB, Harris JW (2004) Episodic diamond genesis at Jwaneng, Botswana, and implications for Kaapvaal craton evolution. Lithos 77:143–154. https://doi.org/10.1016/j.lithos.2004.04.027
Rubie DC (1999) Characterising the sample environment in multianvil high-pressure experiments. Phase Transitions 68:431–451. https://doi.org/10.1080/01411599908224526
Rudnick RL, Eldridge CS, Bulanova GP (1993) Diamond growth history from in situ measurement of Pb and S isotopic compositions of sulfide inclusions. Geology 21:13. https://doi.org/10.1130/0091-7613(1993)021%3c0013:DGHFIS%3e2.3.CO;2
Rüffer R, Chumakov AI (1996) Nuclear-resonance beamline at ESRF. Hyperfine Interact 97:589–604. https://doi.org/10.1007/BF02458921
Sharp WE (1966) Pyrrhotite: a common inclusion in South African diamonds. Nature 211:402–403
Shibazaki Y, Ohtani E, Terasaki H et al (2011) Effect of hydrogen on the melting temperature of FeS at high pressure: implications for the core of Ganymede. Earth Planet Sci Lett 301:153–158. https://doi.org/10.1016/j.epsl.2010.10.033
Shirey SB, Cartigny P, Frost DJ et al (2013) Diamonds and the geology of mantle carbon. Rev Mineral Geochem 75:355–421. https://doi.org/10.2138/rmg.2013.75.12
Shushkanova AV, Litvin YA (2008) Experimental evidence for liquid immiscibility in the model system CaCO3-pyrope-pyrrhotite at 7.0 GPa: The role of carbonatite and sulfide melts in diamond genesis. Can Mineral 46:991–1005. https://doi.org/10.3749/canmin.46.4.991
Smit KV, Shirey SB, Wang W (2016) Type Ib diamond formation and preservation in the West African lithospheric mantle: Re–Os age constraints from sulphide inclusions in Zimmi diamonds. Precambrian Res 286:152–166. https://doi.org/10.1016/j.precamres.2016.09.022
Smith EE, Shirey SB, Nestola F et al (2016) Large gem diamonds from metallic liquid in Earth’s deep mantle. Science 354:1403–1406
Stachel T, Harris JW (2009) Formation of diamond in the Earth’s mantle. J Phys Condens Matter. https://doi.org/10.1088/0953-8984/21/36/364206
Stagno V, Frost DJ (2010) Carbon speciation in the asthenosphere: experimental measurements of the redox conditions at which carbonate-bearing melts coexist with graphite or diamond in peridotite assemblages. Earth Planet Sci Lett 300:72–84. https://doi.org/10.1016/j.epsl.2010.09.038
Stagno V, Ojwang D, McCammon CA, Frost DJ (2013) The oxidation state of the mantle and the extraction of carbon from Earth’s interior. Nature 493:84–88. https://doi.org/10.1038/nature11679
Taylor LA, Liu Y (2009) Sulfide inclusions in diamonds: not monosulfide solid solution. Russ Geol Geophys 50:1201–1211. https://doi.org/10.1016/j.rgg.2009.11.018
Terasaki H, Frost DJ, Rubie DC, Langenhorst F (2005) The effect of oxygen and sulphur on the dihedral angle between Fe-O-S melt and silicate minerals at high pressure: Implications for Martian core formation. Earth Planet Sci Lett 232:379–392. https://doi.org/10.1016/j.epsl.2005.01.030
von Seckendorff V, O’Neill HSC (1993) An experimental study of Fe-Mg partitioning between olivine and orthopyroxene at 1173, 1273 and 1423 K and 1.6 GPa. Contrib to Mineral Petrol 113:196–207. https://doi.org/10.1007/BF00283228
Wendlandt RF (1982) Sulfide saturation of basalt and andesite melts at high pressures and temperatures. Am Mineral 67:877–885
Westerlund KJ, Shirey SB, Richardson SH et al (2006) A subduction wedge origin for Paleoarchean peridotitic diamonds and harzburgites from the panda kimberlite, Slave craton: evidence from Re-Os isotope systematics. Contrib to Mineral Petrol 152:275–294. https://doi.org/10.1007/s00410-006-0101-8
Wiggers de Vries DF, Pearson DG, Bulanova GP et al (2013) Re-Os dating of sulphide inclusions zonally distributed in single Yakutian diamonds: evidence for multiple episodes of proterozoic formation and protracted timescales of diamond growth. Geochim Cosmochim Acta 120:363–394. https://doi.org/10.1016/j.gca.2013.06.035
Woodland AB, O’Neill HSC (1997) Thermodynamic data for Fe-bearing phases obtained using noble metal alloys as redox sensors. Geochim Cosmochim Acta 61:4359–4366. https://doi.org/10.1016/S0016-7037(97)00247-0
Wykes JL, Mavrogenes JA (2005) Hydrous sulfide melting: experimental evidence for the solubility of H2O in sulfide melts. Econ Geol 100:157–164. https://doi.org/10.2113/100.1.0157
Yefimova ES, Sobolev NV, Pospelova LN (1983) Sulfide inclusions in diamonds and specific features of their paragenesis. Zap Vsesoyuznogo Mineral Obs 112:300–310
Zhang Z, Hirschmann MM (2016) Experimental constraints on mantle sulfide melting up to 8 GPa. Am Mineral 101:181–192. https://doi.org/10.2138/am-2016-5308
Zhang Z, von der Handt A, Hirschmann MM (2018) An experimental study of Fe–Ni exchange between sulfide melt and olivine at upper mantle conditions: implications for mantle sulfide compositions and phase equilibria. Contrib to Mineral Petrol. https://doi.org/10.1007/s00410-018-1444-7
Zhang Z, Hirschmann MM (2012) Carbon Solubility of Molten Sulfides at 2–3 GPa. In: American Geophysical Union, Fall Meeting 2012. p abstract id. DI13D-2454
Acknowledgements
We would like to thank Detlef Krausse and Anke Potzel for help with EPMA analyses, Hubert Schulze, Raphael Njul and Alexander Rother for sample preparation and Heinz Fischer for machining high-pressure cell assembly parts. We appreciate the very thorough reviews of Kate Kiseeva, John A. Mavrogenes, and editorial handling and comments by Dante Canil. This project was conducted under the “Deep volatile cycles – International Research Training Group” funded by German Research Foundation (Deutsche Forschungsgemeinschaft – DFG, GRK 2156/1) and DFG grant FR1555/11.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Dante Canil.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abeykoon, S., Laurenz, V., Frost, D.J. et al. An experimental investigation of factors controlling the oxygen content of sulphide melts in the Earth’s upper mantle. Contrib Mineral Petrol 178, 13 (2023). https://doi.org/10.1007/s00410-023-01992-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-023-01992-3